

Abstract
Protein modification by the ubiquitin-like SUMO protein contributes to many cellular regulatory mechanisms. In Saccharomyces cerevisiae, both sumoylating and desumoylating activities are essential for viability. Of its two known desumoylating enzymes, Ubl-specific protease (Ulp)1 and Ulp2/Smt4, Ulp1 is specifically required for cell cycle progression. A ∼200-residue segment, the Ulp domain (UD), is conserved among Ulps and includes a core cysteine protease domain that is even more widespread. Here we demonstrate that the Ulp1 UD by itself can support wild-type growth rates and in vitro can cleave SUMO from substrates. However, in cells expressing only the UD of Ulp1, many SUMO conjugates accumulate to high levels, indicating that the nonessential Ulp1 NH2-terminal domain is important for activity against a substantial fraction of sumoylated targets. The NH2-terminal domain also includes sequences necessary and sufficient to concentrate Ulp1 at nuclear envelope sites. Remarkably, NH2-terminally deleted Ulp1 variants are able, unlike full-length Ulp1, to suppress defects of cells lacking the divergent Ulp2 isopeptidase. Thus, the NH2-terminal regulatory domain of Ulp1 restricts Ulp1 activity toward certain sumoylated proteins while enabling the cleavage of others. These data define key functional elements of Ulp1 and strongly suggest that subcellular localization is a physiologically significant constraint on SUMO isopeptidase specificity.
Keywords: ubiquitin; SUMO; Ulp1; cell cycle; nuclear pore complex
Introduction
Many cellular processes in eukaryotes depend on covalent modification of specific proteins by ubiquitin (for review see Hochstrasser, 1996; Pickart, 2001; Weissman, 2001). Attachment of ubiquitin to proteins changes their functional properties in ways that are only partly understood, but in general it appears to alter the ability of the modified protein to interact with other macromolecules. For example, when a protein is modified by a polymeric chain of ubiquitins, its affinity for the proteasome is greatly increased. This large protease cleaves the protein into short peptides and releases the ubiquitin tag. Eukaryotes also express a set of functionally diverse ubiquitin-like proteins (Ubls).* Ubls are significantly diverged from ubiquitin yet are also ligated to other proteins (for review see Hochstrasser, 2000; Jentsch and Pyrowolakis, 2000). An intensely studied Ubl is the vertebrate SUMO-1 protein (Melchior, 2000; Hochstrasser, 2001; Kim et al., 2002). Human SUMO-1 shares <20% identity with ubiquitin but is 48% identical to a yeast protein called Smt3. SUMO-1 and Smt3 both have the ubiquitin fold (Bayer et al., 1998; Mossessova and Lima, 2000). Analogous to ubiquitin ligation, a cascade of three enzymes (E1, E2, and E3) catalyzes amide (isopeptide) bond formation between the COOH-terminal carboxyl group of SUMO and lysine side chains of acceptor proteins. Conjugation of substrates to Smt3/SUMO has been shown to depend on an E1-related enzyme, the Uba2-Aos1 heterodimer, an E2-like enzyme, Ubc9, and several distinct E3-like factors (Kim et al., 2002). Vertebrates have two additional highly similar proteins, SUMO-2 and SUMO-3, which are ∼45% identical to SUMO-1 and are thought to utilize the same E1 and E2 enzymes that act on SUMO-1 (Saitoh and Hinchey, 2000).
The SUMO system has been implicated in multiple physiological pathways in both yeast and other eukaryotes (Melchior, 2000; Hochstrasser, 2001; Kim et al., 2002). In the yeast Saccharomyces cerevisiae, many SUMO system components are essential for viability, principally because of their requirement for cell cycle progression (Seufert et al., 1995; Li and Hochstrasser, 1999). Although several dozen substrates for SUMO attachment have been identified in metazoan cells, the physiological importance of SUMO attachment to most of these specific proteins is unknown (Melchior, 2000). Only a handful of yeast Smt3 substrates are known. The first was a subset of the septins, which are proteins essential for cytokinesis; however, eliminating septin sumoylation had no detectable phenotypic effect (Johnson and Blobel, 1999; Takahashi et al., 1999; Johnson and Gupta, 2001). Yeast topoisomerase II can be sumoylated, and this modification, although not required for the essential function of the enzyme, appears to contribute in some way to the elastic or cohesive properties of centromeres (Bachant et al., 2002). Smt3 can also be ligated to the DNA replication/repair protein PCNA, and this may negatively regulate the DNA repair activity of PCNA (Hoege et al., 2002).
Modification of proteins by ubiquitin and Ubls is usually reversible. Moreover, ubiquitin and most Ubls are synthesized in precursor forms, with one or more amino acids following what will become the mature COOH terminus. Ubiquitin substrate deconjugation and precursor processing are performed by members of a diverse group of specialized proteases called deubiquitinating enzymes or Dubs (Wilkinson and Hochstrasser, 1998; Chung and Baek, 1999). Analyses of these enzymes indicate that they have diverse regulatory roles in the ubiquitin system. Much less is known about the analogous enzymes that act on the SUMO proteins. We initially identified a S. cerevisiae enzyme, Ubl-specific protease (Ulp)1, which specifically removes SUMO from proteins and is required for cell cycle progression (Li and Hochstrasser, 1999). Ulp1 lacks obvious sequence similarity to any known Dub and is unable to process ubiquitin-linked substrates.
Numerous proven or putative desumoylating enzymes have been identified in the sequence databases based on their similarity to an ∼200-residue COOH-terminal domain in Ulp1 (the Ulp domain [UD]). These enzymes include multiple proteins from humans and one additional protein in S. cerevisiae (Li and Hochstrasser, 1999; Gong et al., 2000; Li and Hochstrasser, 2000). The Ulp1-related yeast protein, Ulp2/Smt4, was subsequently shown to have desumoylating activity (Li and Hochstrasser, 2000). Within the UD is a more widely conserved segment referred to as the core domain, which shares limited sequence similarity to viral cysteine proteases (Ding et al., 1996; Stephens et al., 1998; Li and Hochstrasser, 1999). The active site residues within this core domain of Ulp1 and Ulp2 are indeed necessary for catalytic activity and in vivo function (Li and Hochstrasser, 1999; Strunnikov et al., 2001). Ulp1 is required for cell cycle progression, whereas Ulp2, although not essential, is required for normal chromosome stability and for recovery from cell cycle checkpoint arrest (Li and Hochstrasser, 2000; Strunnikov et al., 2001; Bachant et al., 2002).
An important unanswered question about the desumoylating enzymes concerns the basis of their in vivo substrate specificities. The patterns of SUMO–protein conjugates in ulp1-ts and ulp2Δ mutants are dissimilar, as are their cellular phenotypes, indicating that the two yeast SUMO isopeptidases have distinct specificities (Li and Hochstrasser, 2000). Ulp2 has very weak activity in vitro and is unable to compensate for loss of Ulp1 in vivo even when overproduced. Ulp1, on the other hand, can act in vitro on the Smt3 conjugates that accumulate to high levels in ulp2Δ cells, and at moderately high dosage ULP1 can suppress some phenotypic abnormalities of the ulp2Δ mutant (Li and Hochstrasser, 2000). This suggests that Ulp1 activity is somehow constrained in the cell to prevent it from acting on Ulp2 substrates.
Here we show that the conserved UD of Ulp1 is sufficient for its essential in vivo function. However, the NH2-terminal domain is required both for localization of Ulp1 to the nuclear pore complex (NPC) and for desumoylation of many Ulp1 substrates. Remarkably, when Ulp1 is delocalized in the cell by NH2-terminal deletion, it acquires the ability to desumoylate proteins that are normally targets of the Ulp2 enzyme and to suppress phenotypic abnormalities associated with loss of Ulp2. Thus, the net effect in vivo of removing the NH2-terminal domain of Ulp1 is a broad switch in substrate specificity, with loss of targeting to a significant subset of Ulp1 substrates and gain of activity toward what are normally Ulp2-restricted substrates.
Results
The UD of Ulp1 is sufficient for cell viability and catalytic activity
Loss of Ulp1 is lethal in S. cerevisiae because the mutant cells are unable to traverse the G2/M phase of the cell cycle efficiently (Li and Hochstrasser, 1999). The Smt3-cleaving activity of the 621-residue Ulp1 enzyme is necessary for this essential in vivo function (Fig. 1). We asked whether any segments NH2-terminal to the catalytic domain (UD) could be deleted without loss of viability. A previous study had suggested that the UD by itself was insufficient for viability and, moreover, behaved as a dominant lethal (Mossessova and Lima, 2000). We were therefore surprised when we found that deletions could be extended as far as residue 417 (ulp1-C204) without a significant effect on growth rate at 30°C (Fig. 1). Mutant ulp1-C204 protein consists of the UD and little else. These experiments were performed with ulp1 alleles on low copy plasmids. When ulp1-C204 was integrated into the chromosome (at the LEU2 locus), viability was still seen although growth was slower than for wild-type cells, presumably because of lower ulp1-C204 expression levels (unpublished data). The differences in viability observed between studies appear to be due to properties of the specific constructs used (see Discussion).
Figure 1.

Deletion analysis of the Ulp1 protein. (A) The set of terminal Ulp1 deletions used in the present work. Gray boxes represent the UD. Positions of the catalytic His514 and Cys580 residues and the C580S mutation are indicated. CC, potential coiled-coil region (aa 346–404). Catalytic activity of recombinant GST-Ulp1 deletion proteins purified from bacterial cells was determined by in vitro processing of an 35S-labeled His6-ubiquitin-Smt3-HA chimeric substrate (see Fig. 2 A). (B) Complementation analysis of each deletion allele was done in a ulp1Δ strain by plasmid shuffling. Strain MHY1321 (ulp1Δ), which carries the wild-type ULP1 gene in a YCp50 (CEN, URA3) plasmid, was transformed with YCplac22 (TRP1)-based plasmids bearing the various ulp1 deletion alleles. Trp+ transformants were then streaked on 5-FOA plates and incubated at 30°C for 3 d to determine the ability of the different ULP1 deletions to support growth.
The growth-complementing activity of ulp1-C204 required a catalytically active UD. When the active site cysteine in the ulp1-C204 construct was mutated to create ulp1-C204(C580S), viability of ulp1Δ cells was no longer rescued. If the region of the UD upstream of the active site catalytic residues was deleted (ulp1-C173), the mutant also behaved as a null (Fig. 1). This part of the UD makes extensive contacts with Smt3 in the Ulp1-Smt3 cocrystal structure as had been predicted from sequence conservation among the diverse Ulps (Li and Hochstrasser, 1999; Mossessova and Lima, 2000). Therefore, the ulp1-C173 protein would be predicted to lack desumoylating activity as was found (Fig. 2 A). A deletion allele expressing only the NH2-terminal domain of Ulp1, ulp1-N417, also failed to complement the ulp1Δ strain (Fig. 1). Protein expression of the various deletion constructs varied over a very small range (Fig. 2 D and unpublished data).
Figure 2.

Comparison of Ulp1 catalytic activity in vitro and function in vivo. (A) Enzymatic activity of selected GST-Ulp1 derivatives. Cleavage of purified 35S-labeled His6-ubiquitin-Smt3-HA by purified GST fusions was assayed at 30°C for 30 min and stopped by addition of SDS-loading buffer. (B) In vivo processing of HA-Smt3-aty. MHY1321 cells carrying pRS425-HA-Smt3-aty and either wild-type ULP1 or ulp1-C204 in YCplac22 were pulse labeled with 35S-Translabel for 5 min and chased in buffer containing cycloheximide and an excess of unlabeled methionine. At the indicated time points, aliquots of cells were withdrawn and processed for immunoprecipitation with anti-HA antibodies. The positions of precursor HA-Smt3-aty and the processed product are indicated. Because of the efficiency of processing, it is often difficult to detect precursor in ULP1 cells at the initial time point. (C) Smt3 protein profiles of ulp1Δ (MHY1321) cells carrying the indicated plasmids with different Ulp1 deletions. Cell extracts were fractionated through an 11–14% gradient SDS gel and assayed by immunoblotting with a polyclonal anti-Smt3 antiserum. The membrane was reprobed with anti-PGK (phosphoglycerate kinase) to compare the protein loading (bottom). Positions of marker proteins (kD) are indicated on the left. (D) Anti-MYC immunoblot showing expression levels of MYC9-tagged Ulp1 derivatives. ULP1 alleles used the ULP1 promoter and were based in YCplac22. COOH-terminally MYC9-tagged Ulp1, C478, C459, C449, C275, and C204 proteins, which were indistinguishable in growth from the untagged alleles use in C, were expressed in strain MHY1321 (ulp1Δ). The recessive lethal alleles encoding the catalytically inactive Ulp1-N417-MYC9 and Ulp1-C173-MYC9 proteins were expressed in congenic wild-type MHY500 cells. (Bottom) The same membrane reprobed with anti-PGK antibody.
We assayed Smt3-cleaving activity of all the Ulp1 deletion proteins. The alleles were fused with the coding sequence for GST, and the fusion proteins were expressed in Escherichia coli and purified by glutathione-Sepharose affinity chromatography. Processing of an 35S-labeled His6-ubiquitin-Smt3-HA chimera was assayed by SDS-PAGE. Smt3 cleavage in vitro and growth complementation of ulp1Δ yeast correlated exactly (Fig. 1 and Fig. 2 A). Ulp1-C204 activity was lower than that of the full-length protein (Fig. 2 A). Kinetic analysis of His6-ubiquitin-Smt3-HA processing between the Smt3 and HA segments yielded values for vobs of 66 min−1 for GST-Ulp1 and 7.3 min−1 for GST-ulp1-C204. Processing in yeast cells of the natural COOH-terminal tripeptide Ala-Thr-Tyr from the Smt3 precursor was also tested (Fig. 2 B). This cleavage, which is due primarily to Ulp1 and not Ulp2 (Li and Hochstrasser, 2000), was slower in cells expressing ulp1-C204 than in those with full-length Ulp1 (Fig. 2 B and Fig. 3 B). On the other hand, Smt3 cleavage in vitro by all the other Ulp1 COOH-terminal fragments shown in Fig. 1 A was indistinguishable from cleavage by full-length Ulp1 with the possible exception of ulp1-C275, which was slightly slower (unpublished data). The activity of these fragments when overproduced in yeast also appeared to be comparable to that of full-length Ulp1 (see Fig. 4 A). In summary, the COOH-terminal catalytic domain of Ulp1 is both necessary and sufficient for the essential function of the protein in cell cycle progression and for Smt3 precursor cleavage.
Figure 3.

Contribution of the Ulp1 NH2 terminus to substrate recognition. (A) Overproduction of GST-Ulp1N417 and GST-Ulp1N255i leads to accumulation of sumoylated proteins. Strains and growth conditions as in B, but induction was for 6 h. Molecular mass markers are indicated. The vertical line denotes Smt3 protein conjugates. In the bottom panel, the filter was reprobed with anti-PGK antibody. (B) Overproduction of GST-Ulp1N417 inhibits Smt3 precursor processing. Mid-log phase cultures of wild-type MHY500 cells carrying pEMBL-GST-Ulp1N417, pGal4-ER-VP16 and pRS425-HA-Smt3-aty in selective medium were either induced with 100 nM β-estradiol (+N417) or not induced (–N417) for GST-Ulp1-N417 overproduction. After 4 h, the cells were collected by centrifugation and subjected to pulse–chase analysis. (C) Aliquots of 10-fold serially diluted cells carrying either empty vector (pEMBL-GST) or GAL1-driven alleles encoding Ulp1 NH2-terminal deletions were spotted to minimal plates containing glucose or galactose and incubated at 30°C for 4 d.
Figure 4.
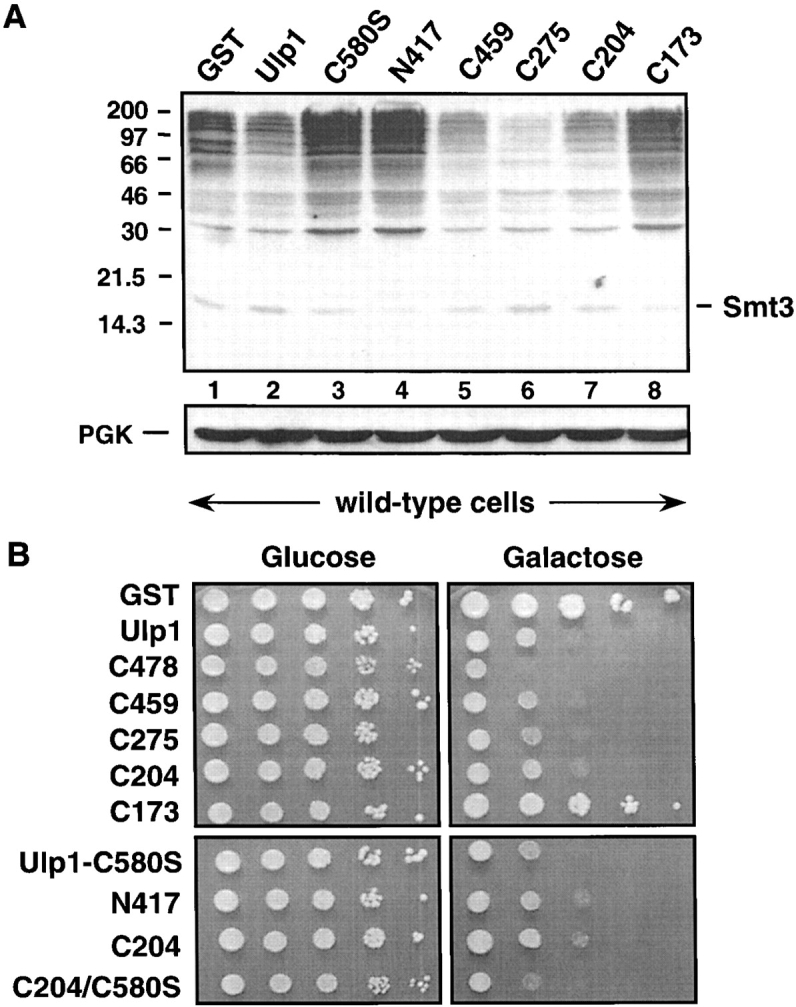
High level expression of Ulp1 or Ulp1 mutants is detrimental to cell growth. (A) Anti-Smt3 immunoblot of a subset of strains shown in B except that cells also carried a plasmid encoding a Gal4-ER-VP16 fusion protein, which allowed high level expression of the GST fusions in glucose medium through gratuitous induction of GAL1-driven genes by addition of β-estradiol. The cells were induced for 4 h at 30°C, and similar expression levels of the GST fusions were confirmed by anti-GST immunoblotting (not depicted). Positions of the molecular mass standards are shown on the left. (Bottom) Anti-PGK immunoblot of the same filter. (B) 10-fold serial dilutions of mid-log phase wild-type MHY500 cells transformed with plasmids expressing full-length GST-Ulp1 or different GST-Ulp1 deletions from plasmid-born alleles under the control of a GAL1 promoter. Cells were spotted onto URA drop-out plates containing either glucose or galactose and incubated for 4 d at 30°C.
The noncatalytic domain of Ulp1 contributes to substrate specificity
Although the NH2-terminal two-thirds of Ulp1 was dispensable for viability, loss of this region led to a striking accumulation of Smt3–protein conjugates (Fig. 2 C). Importantly, progressive deletion of segments from the NH2 terminus of Ulp1 caused a generally progressive increase in levels of high molecular mass Smt3–protein species. Even the smallest deletion, ulp1-C478, resulted in a mild accumulation of conjugates above the wild-type level despite the fact that, as noted above, this Ulp1 derivative has wild-type levels of Smt3 processing activity, at least in vitro. A sharp increase in the levels of Smt3 conjugates with many distinct new species was seen between the ulp1-C459 and -C449 transformants. The data cannot be explained by differences in protein expression, since these were all fairly small (Fig. 2 D). Therefore, the results imply that the NH2-terminal domain of Ulp1 bears multiple determinants for substrate discrimination, which might act either directly or indirectly in substrate binding. A few exceptions to the monotonic increase in Smt3 conjugates can be seen in Fig. 2 C. For instance, a 30-kD species accumulated in cells with ulp1-C449 but not in -C275 or -C204, even though the latter two proteins were missing larger NH2-terminal segments. This gave a first hint that the regulatory NH2-terminal domain of Ulp1 might contain both elements that act positively and ones that act negatively on Smt3–protein cleavage, depending on the conjugate (see below).
If the NH2-terminal domain of Ulp1 (ulp1-N417) contains sites for substrate interaction, then overproduction of this protein fragment might be able to compete with endogenous full-length Ulp1 for Smt3–conjugate binding. A sequence encoding a fusion between GST and N417 was placed under the control of the galactose-inducible GAL1 promoter and expressed in wild-type yeast cells. As can be seen in Fig. 3 A (lane 2), this resulted in a pronounced accumulation of Smt3–protein conjugates. This was not seen in cells overproducing only GST (lane 1). Overproduction of ulp1-N417 also impaired Smt3 precursor processing (Fig. 3 B). Smaller Ulp1 subfragments (Fig. 3 C) tested in the same way localized the region responsible for this dominant-negative effect to residues 163–417 (N255i; Fig. 3 A, lane 3). The putative coiled coil region was necessary for strong growth inhibition. Competition between ulp1-N417 or ulp1-N255i and endogenous Ulp1 for Smt3 or Smt3–protein substrates would be expected to be detrimental to cell growth. In fact, strong inhibition of cell growth correlated closely with the ability of the NH2-terminal Ulp1 fragments to impair Smt3–protein deconjugation in vivo (Fig. 3 C).
The ability of the overproduced noncatalytic domain of Ulp1 to slow Smt3 precursor processing suggested either that this domain could bind to and inhibit the function of Ulp1 or that it could bind Smt3 itself (or both). (The possibility that this overproduced domain could compete with Ulp1 for NPC binding [see below] was not supported by immunofluorescence analysis of Ulp1-MYC in cells overproducing ulp1-N417 [unpublished data], although interference with another aspect of NPC function is possible.) Neither two-hybrid analysis nor GST pull-down assays suggested a stable interaction between the NH2-terminal domain and either the COOH-terminal domain or full-length Ulp1 (unpublished data). Based on GST pull-down assays, ulp1-N417 was able to bind what appeared to be free Smt3 and a small number of Smt3 conjugates in yeast cells, but the amount of copurified Smt3 proteins varied between experiments (unpublished data). Therefore, clarification of the exact mechanism behind the dominant-negative effect seen with the Ulp1 NH2-terminal domain will require additional studies.
We conclude that the noncatalytic domain of Ulp1 makes a significant contribution to substrate recognition in vivo. This may depend in part on the ability of this domain of the enzyme to bind Smt3, but other more indirect roles in modulating substrate specificity would be consistent with the data as well (see below).
Overproduction of active Ulp1 is toxic to yeast
Interestingly, high levels of full-length Ulp1 also impaired yeast growth (Fig. 3 C and Fig. 4 B). However, in this case a depression of Smt3–protein conjugates was observed (Fig. 4 A, lanes 1 and 2). This reduction in conjugates upon acute induction of Ulp1 expression required the desumoylating activity of Ulp1, since hyperaccumulation of these species, rather than a reduction, was observed with the catalytically inactive ulp1-C580S mutant (Fig. 4 A, lane 3). Chronic overproduction of catalytically active COOH-terminal fragments of Ulp1 also strongly inhibited cell growth (Fig. 4 B). These overproduced fragments strongly reduced levels of bulk sumoylated proteins (Fig. 4 A, lanes 5–7). This contrasted with the observation that these same Ulp1 derivatives, when present at near endogenous levels of expression, were greatly impaired in their ability to act on the bulk of detectable Ulp1 targets (Fig. 2 C).
The implication is that at a high enough concentration, the catalytic domain of Ulp1 by itself and the full-length protein can act relatively promiscuously without the usual constraints on its activity that are imposed at normal Ulp1 expression levels. These constraints might include endogenous inhibitors of Ulp1 and/or a restricted subcellular distribution. Loss of this restriction on Ulp1 activity is also evident in lysates from ulp2Δ cells where the endogenous Ulp1 can cleave the ulp2Δ-specific Smt3 conjugates that had accumulated in vivo (Li and Hochstrasser, 2000).
The NH2-terminal domain of Ulp1 localizes it to the nuclear envelope
As noted above, the isolated catalytic domain of Ulp1 is unable to cleave many cellular Smt3–protein conjugates efficiently (Fig. 2 C); yet at high concentration, it can still strongly reduce levels of most of these conjugates (Fig. 4 A). A possible explanation of these unexpected results is that the noncatalytic domain normally localizes the enzyme to specific sites in the cell where these Smt3 conjugates are also concentrated. However, at sufficiently high levels the delocalized enzyme lacking the noncatalytic domain could still encounter its normal targets. We examined this possibility by tagging Ulp1 and its derivatives with nine copies of the MYC epitope (Li and Hochstrasser, 2000), which allowed localization by immunofluorescence microscopy. All MYC-tagged Ulp1 constructs that had an intact UD fully complemented the lethality of a ulp1Δ strain.
Full-length Ulp1 and ulp1-C478 proteins contained a consensus bipartite nuclear localization signal (NLS) and localized predominantly to the nuclear envelope, which was visible as a ring-like perinuclear fluorescence (Fig. 5). As seen previously, this rim staining was punctate (Li and Hochstrasser, 2000), which reflects Ulp1 binding to NPCs (Schwienhorst et al., 2000; Takahashi et al., 2000). The ulp1-C459 protein showed a detectable but less pronounced nuclear envelope localization, whereas ulp1-C449, which lacks the entire NLS, no longer concentrated clearly in a rim around the nucleus, although bright spots of stain on the perimeter of the nucleus were often seen. This transition from strong localization at NPCs to a more disperse distribution coincided with the strong increase in Smt3 conjugate accumulation seen with the same deletions (Fig. 2 C). Moreover, the ulp1-C275 and ulp1-C204 proteins no longer localized detectably at the nuclear rim (Fig. 5), and high levels of Smt3 conjugates accumulated in ulp1Δ cells expressing these derivatives (Fig. 2 C).
Figure 5.
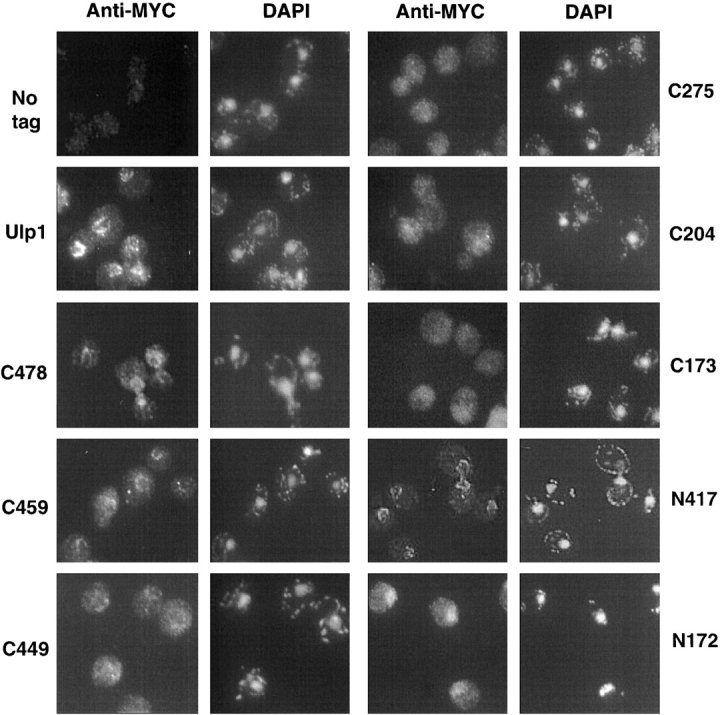
Subcellular localization of Ulp1 deletion derivatives. Localization of MYC9-tagged Ulp1 proteins by indirect immunofluorescence. Strains and plasmids were the same as in the legend to Fig. 2 D. Nuclei were stained with DAPI.
We conclude that the NH2-terminal domain of Ulp1, particularly residues between positions 144–346, are necessary for nuclear envelope localization. The ability to localize to the envelope correlated closely with the ability of the Ulp1 derivative to cleave many Smt3 conjugates efficiently, suggesting a close link between these two properties. However, localization of Ulp1 to NPCs cannot explain all of its substrate-targeting features. First, the COOH-terminal UD retains the capacity to act on at least the essential targets of Ulp1 in vivo despite failing to localize appreciably to NPCs. Second, as noted earlier the accumulation of a small subset of Smt3 conjugates did not correlate with Ulp1 delocalization.
The above results indicate that the NH2-terminal domain is necessary for Ulp1 localization to the NPC. Conversely, ulp1-N417 (Fig. 5) and ulp1-N346 (unpublished data), which were missing the entire UD, were still able to concentrate at the nuclear envelope like the full-length protein, indicating that the first 346 residues of Ulp1 are sufficient for NPC localization. In contrast, a protein with just the NH2-terminal 172 residues of Ulp1, ulp1-N172, which included the intact NLS, concentrated primarily in the nucleus without obvious nuclear rim staining. This suggests that in addition to the NLS, information within residues 173–346 also contributes to proper nuclear envelope localization.
Together, these localization data indicate that information within the NH2-terminal 346 residues of Ulp1 is both necessary and sufficient for nuclear envelope localization of this SUMO isopeptidase. This localization may be important for modulating Ulp1 activity toward specific substrates (see Discussion). Interestingly, the NH2-terminal noncatalytic domains of some Dubs can also affect their localization and functional activity (Lin et al., 2000; Park et al., 2002).
Enhanced activity of Ulp1 COOH-terminal fragments toward Ulp2 substrates
When expressed at roughly endogenous levels, Ulp1 derivatives lacking all or some of the NH2-terminal noncatalytic domain failed to cleave many Smt3–protein conjugates efficiently (Fig. 2 C). This suggested a positive regulatory role(s) for the noncatalytic domain in substrate targeting. Remarkably, however, when we expressed these same NH2-terminally truncated versions of Ulp1 at low copy in strains lacking the other known desumoylating enzyme of yeast, Ulp2/Smt4, a substantial reduction in Smt3–protein conjugates was seen (Fig. 6 A). This was not true for full-length Ulp1 (or ulp1-C478) that was also expressed from a low copy plasmid. Most noticeably, the ulp1-C275 and ulp1–204 proteins eliminated the majority of the Smt3–protein species that characterize the ulp2Δ strain. Reduction of these sumoylated species correlated with substantial suppression of the ulp2Δ cellular phenotype, including temperature-sensitive growth and hypersensitivity to hydroxyurea (HU) or benomyl (Fig. 6 B). Growth on 0.1 M HU of ulp2Δ cells transformed with ulp1-C275 or ulp1-C204 was indistinguishable from the ULP2 transformants; ulp1-C204 was a slightly less effective suppressor of the high temperature and benomyl sensitivities. Catalytic activity was required inasmuch as the inactive ulp1-C173 protein failed to reduce the ulp2Δ-specific Smt3 conjugates or to restore growth under the tested conditions.
Figure 6.

The NH2-terminal domain of Ulp1 constrains its in vivo substrate specificity. (A) Smt3 protein conjugates in MHY1380 ulp2Δ cells carrying YCplac22-based plasmids with different ULP1 deletions or full-length ULP1 or ULP2. Cell lysates were fractionated by gradient SDS-PAGE. Positions of molecular mass markers are shown at left. The asterisks on the right indicate prominent sumoylated protein species observed in the ulp2Δ/vector lane but not in the lanes from cells transformed with certain ULP1 deletions (C459, C275, and/or C204). The double asterisk highlights a sumoylated protein found at higher levels upon expression of the above three ULP1 deletions. This might reflect incomplete removal of Smt3 from a substrate modified with multiple Smt3 molecules. (Bottom) The same filter reprobed with anti-PGK antibody. (B) Ulp1 NH2-terminal deletions can suppress ulp2Δ phenotypic defects. Strains used are the same as in A. For each strain, aliquots from 10-fold serial dilutions were spotted onto YPD plates (rich media) incubated at 30°C or 37°C and onto plates containing 0.1 M HU, or 20 μg/ml benomyl sulfate and incubated at 30°C for 5 d.
We conclude that the noncatalytic NH2-terminal domain of Ulp1 limits the activity of this enzyme toward the subset of sumoylated proteins that are normally targeted by Ulp2. This inhibitory function of the Ulp1 noncatalytic domain, together with its positive role in targeting Ulp1 to other Smt3–protein conjugates, allows for a sharp separation of substrate specificities between these two SUMO isopeptidases in vivo.
Discussion
The two yeast SUMO isopeptidases, Ulp1 and Ulp2, have distinct in vivo substrate specificities, and the data presented here provide several important clues about the molecular underpinnings of these differences. Based on both the present results (summarized in Fig. 7) and our earlier work, it is clear that Ulp1 has the ability to cleave a very broad range of sumoylated substrates in vitro. However, at its normal levels of expression in vivo the spectrum of proteins acted upon by Ulp1 is more limited. A critical element in controlling Ulp1 activity appears to be its restricted localization: high levels are normally only found at the nuclear envelope (NPC), and loss of this constraint correlates with the ability to act on additional sumoylated proteins. On the other hand, a failure to localize to NPCs also may impair Ulp1 action toward many of its normal targets, suggesting a specific requirement for Ulp1 at the NPC. The noncatalytic domain of Ulp1 may also control targeting to at least a subset of substrates by means other than NPC binding. Surprisingly, the essential function of Ulp1 in cell cycle progression does not require the noncatalytic domain, indicating that certain substrates can be recognized by the catalytic domain alone. We discuss the implications of these data for both the evolution of multiple SUMO isopeptidases with diverse specificities and the potential relationship between the yeast Ulps and those from other eukaryotes.
Figure 7.

Summary of Ulp1 functional elements. Boundaries represent maximal limits of segments necessary for the indicated property based on the deletions analyzed in the present work. See Results for details.
Essential function of Ulp1
A significant finding of the current work is that the requirement of Ulp1 for cell viability can be separated from the ability of the enzyme to localize to NPCs. This strongly suggests that the essential cell cycle function of Ulp1 does not involve regulation of nucleocytoplasmic transport at the NPC, although we cannot completely exclude the possibility that a small amount of the NH2-terminally truncated Ulp1 enzyme can still access the NPC, permitting an essential target there to be desumoylated. It seems more likely that the protein or proteins that must by desumoylated by Ulp1 for progression through G2/M phase need not be targeted at the NPC. The catalytic domain (UD) is sufficient to recognize and cleave these SUMO conjugates, which remain to be identified, although the N domain may contribute to their recognition.
When expressed as a separate domain, the catalytically inactive UD of Ulp1 (ulp1-C204-C580S) behaves as a dominant-negative lethal in yeast (Fig. 4 B). The degree of growth inhibition is comparable to that seen with overproduced full-length ulp1-C580S. These data imply either that high levels of the inactive UD can titrate an essential modulator of Ulp1, which might only control activity against a specific subset of targets, or that the inactive UD binds to and blocks access to SUMO conjugates that must be cleaved by Ulp1 for the cell cycle to progress. The UD of Ulp1 can bind SUMO directly based on in vitro binding with bacterially expressed recombinant proteins (unpublished data).
Regulation of SUMO isopeptidase activity by the noncatalytic domain
When expressed at high levels, the isolated NH2-terminal noncatalytic domain of Ulp1 also has a profound impact on growth, even though this domain is not required for the essential function of Ulp1 (Fig. 4). Growth impairment correlates with excessive accumulation of sumoylated proteins in these cells. Several distinct protein interaction techniques failed to detect binding of the NH2-terminal domain of Ulp1 with full-length Ulp1 or the COOH-terminal domain. We have preliminary data for interaction of the NH2-terminal domain of Ulp1 with SUMO, but this interaction appears to be weak (unpublished data). Weak binding to specific substrates is also possible. A plausible explanation of these results is that the noncatalytic domain can compete with endogenous Ulp1 for SUMO-modified proteins (or possibly for free SUMO or a regulator of Ulp1) and thereby impede Ulp1-mediated cleavage.
Among the most surprising findings of the current work was the robust suppression of ulp2Δ mutant defects by NH2-terminally truncated forms of Ulp1 (Fig. 6). The ulp2Δ complementation by ulp1-C275 and ulp1-C204 could be accounted for by their delocalized distribution within the cell (Fig. 5), which would allow them to act on Smt3 conjugates that are normally accessible only to Ulp2, an enzyme that concentrates in the nucleus (Li and Hochstrasser, 2000). On the other hand, results with the ulp1-C459 and ulp1-C449 proteins are not as easily explained by this model. The ulp1-C459 protein, which still largely concentrates at the nuclear envelope, reduced certain Smt3–protein species in ulp2Δ cells and partially suppresses the HU sensitivity of the mutant. In contrast, ulp1-C449, which shows much less nuclear envelope localization, cannot suppress any ulp2Δ phenotypic defects. One potential explanation is that ulp1-C459, by virtue of being transported into the nucleus but only loosely associated with the NPC, can access many Ulp2 substrates in the nucleus. It is interesting that ulp1-C459 can suppress the HU sensitivity of ulp2Δ cells but not its benomyl sensitivity. This might reflect differences in the sumoylated proteins that must be deconjugated, or in the extent of desumoylation of the same protein that must occur, for cell recovery from these two different kinds of cell cycle checkpoint arrest (Li and Hochstrasser, 2000).
Earlier studies demonstrated that certain defects associated with the ulp2Δ single mutant are suppressed in ulp1-ts ulp2Δ double mutants (Li and Hochstrasser, 2000). Therefore, another interpretation of ulp2Δ suppression by alleles encoding NH2-terminally truncated Ulp1 is that these alleles exert a dominant-negative effect on Ulp1 action in the ulp2Δ cells. However, several results argue against this view. First, we see no growth defect or increase in SUMO conjugates in our wild-type strains that express ulp1-C204 or ulp1-C275. Moreover, where these truncated Ulp1 proteins do cause an increase in SUMO conjugates is in ulp1Δ cells (Fig. 2 C), an effect that cannot be due to interference with endogenous Ulp1. Finally, when cells bearing GAL-driven alleles of GST–ulp1-C204 were grown on glucose (i.e., conditions that allow very low levels of transcription), this rescues rather than impedes the growth of ulp1-ts cells at 37°C, and GST–ulp1-C204 is not deleterious to growth under these conditions in wild-type cells (Fig. 4 B), again arguing against a dominant interfering effect.
The Ulp1 subfamily of desumoylating enzymes
Database searches with full-length yeast Ulp1 protein reveal substantial similarity with the mammalian SENP1 and SENP2/AXAM groups of Ulp isozymes, with up to 23% identity/ 39% similarity over 580 residues. SENP1 and SENP2 are ∼32% identical to one another over their entire lengths. The similarities among these proteins extend beyond the UD itself. Intriguingly, full-length SENP2 is localized to the NPC in mammalian cells (Hang and Dasso, 2002; Zhang et al., 2002). SENP2 localizes specifically to the nuclear basket on the intranuclear side of the NPC. Together, the sequence similarity of Ulp1 to SENP1 and SENP2/AXAM, the related cellular localization patterns of Ulp1 and SENP2, and the strong activity of this subgroup of Ulp enzymes against a broad range of substrates (at least when overexpressed) suggest that this group of enzymes represents a distinct functional subfamily of SUMO isopeptidases. In addition, alternative splicing generates at least three distinct SENP2-related proteins in mammalian cells (Gong et al., 2000; Nishida et al., 2001; Best et al., 2002). These isoforms localize in strikingly different patterns within the cell, underscoring the idea that subcellular distribution could provide an important constraint on the activity of the Ulp1 subfamily of enzymes.
Recently, it was reported that expression of one of the human SENP2-related splice variants, called SuPr-1, in yeast ulp2Δ cells suppressed at least some of the phenotypic abnormalities of the mutant (Best et al., 2002). Although this might be interpreted to mean that SuPr-1 is the functional counterpart of Ulp2 (even though the SuPr-1 and Ulp2 sequences are highly diverged), our results suggest a different possibility. The SuPr-1 protein is identical to SENP2 except that it lacks the first 82 residues of the latter; the missing NH2-terminal segment includes sequences necessary for NPC localization (Hang and Dasso, 2002). Therefore, SuPr-1 localizes more broadly, analogous to the NH2-terminally truncated versions of Ulp1 that we generated. Some of these truncated Ulp1 enzymes are able to suppress the ulp2Δ growth abnormalities very efficiently (Fig. 6), as can moderately overproduced full-length Ulp1 (Li and Hochstrasser, 2000). From these observations, we would suggest an alternative model for SuPr-1 suppression of ulp2Δ in which SuPr-1 is mimicking NH2-terminally truncated Ulp1 in these trans-species complementation assays rather than being the human version of Ulp2.
It is also instructive to compare some of the recent structure-function data on vertebrate SENP2 with our data on yeast Ulp1. Hang and Dasso (2002) found that transient transfection of an NH2-terminally truncated SENP2 that no longer concentrated at the nuclear envelope caused a much stronger drop in total cellular SUMO conjugates than did the full-length protein. These conditions are roughly akin to the overexpression of NH2-terminally truncated Ulp1 derivatives in wild-type yeast cells (Fig. 4 A), which also greatly reduced the concentration of sumoylated proteins. Although Hang and Dasso (2002) inferred that NPC localization negatively regulates SENP2 activity against bulk SUMO conjugates, our data suggest a more complex picture at least for Ulp1. At roughly physiological levels of expression (Fig. 2), loss of NPC localization of Ulp1 does correlate with enhanced activity against one set of sumoylated substrates, those normally cleaved by Ulp2 (Fig. 6 A), but it also correlates closely with decreased activity against many of the normal targets of Ulp1 (Fig. 2 C). Hence, Ulp1 localization to the nuclear envelope could positively regulate Ulp1 activity, particularly toward many of its normal physiological substrates.
The simplest model to explain this positive role of Ulp1 localization to NPCs is that many of its substrates are only accessible at these sites. The amount of Ulp1 in yeast cells is relatively low (Li and Hochstrasser, 2000), so concentration of the enzyme at places where its targets also congregate could well increase its ability to process these SUMO conjugates efficiently. NPC localization raises the obvious possibility that Ulp1 might play a direct role in NPC function and nucleocytoplasmic trafficking. Indeed, a recent study suggests a link between protein sumoylation in yeast and transport through the NPC: both a ulp1 mutant and uba2 mutant were found to be impaired in nuclear protein import (Stade et al., 2002). The authors concluded that Ulp1 only contributes indirectly to nuclear transport by processing the SUMO precursor because provision of processed SUMO to the ulp1 mutant rescued nuclear import of the test substrate. Because ulp1 suppression by supplemented SUMO was only tested with an artificial transport substrate, it remains possible that natural transport intermediates, which might need to go through additional steps, such as release from NPC binding sites, might still require SUMO deconjugation events at the NPC. Overproduced SUMO might also titrate a negatively acting factor(s); for example, this factor could bind sumoylated transport substrates tightly and prevent their transport, an effect normally countered by Ulp1-mediated desumoylation. This interpretation would predict that provision of SUMO precursor (instead of the mature form) would still suppress the transport defect of the ulp1 mutant, but this was not tested.
While the present study was under review, another group also reported that Ulp1 localizes to nuclear pores via its NH2-terminal domain and showed that this is mediated by specific karyopherins (Panse et al., 2003). Their localization data are in general agreement with ours. However, Panse et al. (2003) like Mossessova and Lima (2000) found that expression of their Ulp1 COOH-terminal domain is dominant lethal. It is likely that this difference from our data reflects differences in the activity of the different C domain constructs used. Our ulp1-C204 protein is shorter than the one used in the above studies (C218), and we showed it has reduced activity compared with full-length Ulp1 (Fig. 2). We have tested the ulp1-NΔ403 (C218)–expressing allele in our strains and confirmed its dominant-lethal effect (unpublished data). A simple explanation would be that ulp1-C218 has a higher specific activity than ulp1-C204, so any loss of constraints on its activity in vivo could cause a growth defect. Supporting this view, stronger expression of ulp1-C204 is also dominant lethal (Fig. 4). Panse et al. (2003) restricted their functional analysis to suppression of this (dominant) lethality. In contrast, the ability of ulp1-C204 and several of our other ulp1 alleles to support growth allowed us to evaluate multiple potential functions of the Ulp1 N domain, which revealed that this domain has additional roles beyond NPC docking and that NPC docking of Ulp1 is not necessarily essential.
Evolutionary diversification of Ulps
Deletion of the NH2-terminal domain of Ulp1 causes a remarkable switch in its in vivo substrate specificity: many normal Ulp1 targets are no longer processed efficiently, whereas the majority of Ulp2 substrates are now cleaved by the truncated Ulp1 protein (Figs. 2, 6, and 7). At higher concentrations, the Ulp1 UD can cleave essentially all detectable SUMO conjugates in yeast cells. It is easy to imagine an “Ur-Ulp” of broad specificity that, through a process of gene duplication and divergence, gave rise to multiple enzymes with more restricted and independently regulable functions. Some of these might even have evolved to act on Ubls other than SUMO.
S. cerevisiae, with only two SUMO isopeptidases of the Ulp class, can be converted to something more like the presumed primitive condition simply by removing one of the two enzymes, Ulp2, and reprogramming the other by removal of its NH2-terminal regulatory domain. In the example of the mammalian SENP2 gene, multiple specificities can be generated from a single gene by alternative mRNA splicing, with distinct subcellular locations for the product of each splice variant. Adding to this complexity in mammals is the presence of multiple additional members of the Ulp/SENP family. Thus, we can expect even greater functional specialization and diversification of Ulps in these organisms. It will be a challenge to find exact parallels between the functions of these proteins and their orthologs in different model eukaryotes such as yeast. Nevertheless, analysis of the yeast SUMO isopeptidases should continue to shed light on how the substrate specificity of this important group of enzymes is controlled.
Materials and methods
Strains and plasmids
Media for yeast and bacterial growth and genetic procedures for manipulating yeast and bacteria were described previously (Ausubel et al., 1989; Guthrie and Fink, 1991). Yeast strains used in this study were MHY500 (a his3-Δ200 leu2–3,112 ura3–52 lys2–801 trp1–1), MHY1380 (α his3-Δ200 leu2–3,112 ura3–52 lys2–801 trp1–1 ulp2Δ::HIS3), and MHY1321 (a his3-Δ200 leu2–3,112 ura3–52 lys2–801 trp1–1 ulp1Δ::HIS3/ YCp50-ULP1) (Chen et al., 1993; Li and Hochstrasser, 1999, 2000). The E. coli strain JM101 was used for GST fusion protein expression and recombinant DNA cloning (Ausubel et al., 1989).
All of the ULP1 deletions were derived from YCplac22-ULP1 (Li and Hochstrasser, 1999) by homologous recombination in yeast. The ULP1 gene in the pACYC184 vector was used for PCR amplification with a primer that annealed to the 3′ end of the ULP1 ORF and a bridging primer that contained sequences which fused elements bridging the sequence to be deleted. The PCR products were cotransformed into wild-type MHY500 cells with Kpn21, Xba1-cut YCplac22-ULP1, and Trp+ colonies were selected. Recombinant plasmids from the Trp+ colonies were recovered in E. coli, and the segments derived from PCR amplification were sequenced to verify that the correct deletion was introduced. This series of ulp1 deletions encoded fusions of the NH2-terminal 19 residues of Ulp1 to sequences COOH-terminal to the desired deletion endpoint. We created Ulp1 deletions C478 and C204 with only the initiator methionine and found no differences in their ability to complement ulp1Δ lethality or in their levels of expression.
DNA encoding the MYC9 epitope was incorporated into the YCplac22-ULP1 and ulp1 -C478, -C459, -C449, -C275, -C204 and -C173 plasmids by inserting a Kpn21, EcoRV-cut fragment from YRTAG310-ULP1-MYC9 into the recipient plasmids after treating them sequentially with EcoRI, Klenow polymerase, and Kpn21. YCplac22ulp1-N417, and -N172 were constructed by in vivo recombination. MYC9-tagged ulp1-N417, -N349, and -N172 were constructed by in vivo recombination by cotransforming Kpn21, XhoI-digested and gel-purified YCplac22-ULP1-MYC9 plasmid together with PCR-generated DNA fragments that deleted the desired COOH-terminal region.
Plasmids for expression of GST-Ulp1 protein derivatives in E. coli were made with the vector pGEX-KG and for expression in yeast with pEMBL-GST (Baldari et al., 1987). For the latter, transcription was induced either by growth in galactose or by contransforming with pGal4-ER-VP16 plasmid (Louvion et al., 1993), which allows induction of GAL-driven gene expression in glucose-containing media by addition of β-estradiol to 100 nM (final concentration). Plasmids pEMBL-GST-ULP1 and pEMBL-GST-ulp1C580S were made by subcloning XhoI-BamHI fragments from pGEX-ULP1 and pGEX-ulp1C580S (Li and Hochstrasser, 1999), respectively, into BamHI, SalI-cut vector. Other GST fusion alleles were generated by PCR and cloning into the pGEX-KG and pEMBL-GST vectors.
Immunofluorescence and Western immunoblot analyses
Immunofluorescence experiments were executed as described (Li and Hochstrasser, 2000). The cells were incubated with a 1:100 dilution of anti-MYC mAb (9E10; Covance), and secondary Oregon green–conjugated anti–mouse IgG (Molecular Probes) was also diluted 100-fold in 1% BSA-PBS. A drop of DAPI-containing mounting solution was applied to the air-dried cells, and the samples were sealed under a cover slide.
Western immunoblot analysis was performed as described previously (Li and Hochstrasser, 1999). Cells were harvested at OD600 ∼1, and extracts were prepared by the alkaline lysis procedure. PVDF (Whatman) membranes were bound with a 1:3,000 dilution of a partially purified anti-Smt3 polyclonal antibody (Li and Hochstrasser, 1999) or a 1:3,000 dilution of an anti-MYC mAb (9E10; Covance). Membranes were stripped and reprobed with an anti-PGK (1:4,000 dilution) mAb (Molecular Probes) to evaluate protein loading.
Recombinant protein purification and enzyme assays
GST fusion proteins were expressed in E. coli JM101 by addition of IPTG to 1 mM for 3 h at 37°C. The fusion proteins were purified on glutathione-agarose (Sigma-Aldrich) and eluted with glutathione. The purified recombinant proteins were dialyzed against 50 mM Tris-HCl, pH 8.0, 100 mM NaCl, 1 mM dithiothreitol, and 30% glycerol. Purification and radiolabeling of the substrate His6-Ub-Smt3-HA for the in vitro cleavage reactions was described previously (Li and Hochstrasser, 2000).
Cleavage of 35S-labeled His6-Ub-Smt3-HA was performed in reaction buffer containing 50 mM Tris-HCl, pH 7.5, 150 mM NaCl, and 1 mM dithiothreitol and stopped by addition of SDS gel sample buffer and heating to 100°C for 3 min. The samples were separated by 12.5% SDS-PAGE, and cleavage was quantitated using a Storm 860 Imager and ImageQuant software (Molecular Dynamics). For kinetic measurements of processing, concentrations of GST-Ulp1 and GST–Ulp1-C204 were 0.93 and 4.1 nM, respectively. Substrate was present in 102–103 molar excess over enzyme. Duplicate measurements at each reaction time point were made.
Pulse–chase analysis
Pulse–chase analysis was done as described previously (Chen et al., 1993). Briefly, mid-log phase cultures were pulse labeled with 35S-Translabel (ICN) for 5 min at 30°C and chased in buffer containing cycloheximide (0.5 mg/ml) and excess unlabeled methionine (10 mM). At various time points, aliquots of cells were taken and heated at 100°C in a 1% SDS buffer. Immunoprecipitation was performed after addition of 1 ml Triton lysis buffer (150 mM NaCl, 50 mM Hepes, pH 7.5, 5 mM EDTA, 1% Triton X-100) to 0.1 ml of SDS-lysed cells, using a 1:100 dilution of anti-HA mAb (16E12; Covance). The antigen–antibody complex was precipitated with protein G–agarose (Repligen).
Acknowledgments
We thank Alaron Lewis, Dave Schwartz, and Mary Kroetz for comments on the manuscript.
This work was supported by National Institutes of Health grant GM53756.
S.-J. Li's present address is Celera SSF, 180 Kimball Way, South San Francisco, CA 94080.
Footnotes
Abbreviations used in this paper: HU, hydroxyurea; NPC, nuclear pore complex; Ubl, ubiquitin-like protein; UD, Ulp domain; Ulp, Ubl-specific protease.
References
- Ausubel, F.M., R. Brent, R.E. Kingston, D.D. Moore, J.G. Seidman, J.A. Smith, and K. Struhl. 1989. Current Protocols in Molecular Biology. John Wiley and Sons, New York.
- Bachant, J., A. Alcasabas, Y. Blat, N. Kleckner, and S.J. Elledge. 2002. The SUMO-1 isopeptidase Smt4 is linked to centromeric cohesion through SUMO-1 modification of DNA topoisomerase II. Mol. Cell. 9:1169–1182. [DOI] [PubMed] [Google Scholar]
- Baldari, C., J.A. Murray, P. Ghiara, G. Cesareni, and C.L. Galeotti. 1987. A novel leader peptide which allows efficient secretion of a fragment of human interleukin 1 beta in Saccharomyces cerevisiae. EMBO J. 6:229–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer, P., A. Arndt, S. Metzger, R. Mahajan, F. Melchior, R. Jaenicke, and J. Becker. 1998. Structure determination of the small ubiquitin-related modifier SUMO-1. J. Mol. Biol. 280:275–286. [DOI] [PubMed] [Google Scholar]
- Best, J.L., S. Ganiatsas, S. Agarwal, A. Changou, P. Salomoni, O. Shirihai, P.B. Meluh, P.P. Pandolfi, and L.I. Zon. 2002. SUMO-1 Protease-1 regulates gene transcription through PML. Mol. Cell. 10:843–855. [DOI] [PubMed] [Google Scholar]
- Chen, P., P. Johnson, T. Sommer, S. Jentsch, and M. Hochstrasser. 1993. Multiple ubiquitin-conjugating enzymes participate in the in vivo degradation of the yeast MATα2 repressor. Cell. 74:357–369. [DOI] [PubMed] [Google Scholar]
- Chung, C.H., and S.H. Baek. 1999. Deubiquitinating enzymes: their diversity and emerging roles. Biochem. Biophys. Res. Commun. 266:633–640. [DOI] [PubMed] [Google Scholar]
- Ding, J., W.J. McGrath, R.M. Sweet, and W.F. Mangel. 1996. Crystal structure of the human adenovirus proteinase with its 11 amino acid cofactor. EMBO J. 15:1778–1783. [PMC free article] [PubMed] [Google Scholar]
- Gong, L., S. Millas, G.G. Maul, and E.T. Yeh. 2000. Differential regulation of sentrinized proteins by a novel sentrin-specific protease. J. Biol. Chem. 275:3355–3359. [DOI] [PubMed] [Google Scholar]
- Guthrie, C., and G.R. Fink. 1991. Guide to Yeast Genetics and Molecular Biology. Academic Press, San Diego, CA.
- Hang, J., and M. Dasso. 2002. Association of the human SUMO-1 protease SENP2 with the nuclear pore. J. Biol. Chem. 277:19961–19966. [DOI] [PubMed] [Google Scholar]
- Hochstrasser, M. 1996. Ubiquitin-dependent protein degradation. Annu. Rev. Genet. 30:405–439. [DOI] [PubMed] [Google Scholar]
- Hochstrasser, M. 2000. Evolution and function of ubiquitin-like protein-conjugation systems. Nat. Cell Biol. 2:E153–E157. [DOI] [PubMed] [Google Scholar]
- Hochstrasser, M. 2001. SP-RING for SUMO: new functions bloom for a ubiquitin-like protein. Cell. 107:5–8. [DOI] [PubMed] [Google Scholar]
- Hoege, C., B. Pfander, G.L. Moldovan, G. Pyrowolakis, and S. Jentsch. 2002. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature. 419:135–141. [DOI] [PubMed] [Google Scholar]
- Jentsch, S., and G. Pyrowolakis. 2000. Ubiquitin and its kin: how close are the family ties? Trends Cell Biol. 10:335–342. [DOI] [PubMed] [Google Scholar]
- Johnson, E.S., and G. Blobel. 1999. Cell cycle–regulated attachment of the ubiquitin-related protein SUMO to the yeast septins. J. Cell Biol. 147:981–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, E.S., and A.A. Gupta. 2001. An E3-like factor that promotes SUMO conjugation to the yeast septins. Cell. 106:735–744. [DOI] [PubMed] [Google Scholar]
- Kim, K.I., S.H. Baek, and C.H. Chung. 2002. Versatile protein tag, SUMO: its enzymology and biological function. J. Cell. Physiol. 191:257–268. [DOI] [PubMed] [Google Scholar]
- Li, S.-J., and M. Hochstrasser. 1999. A new protease required for cell-cycle progression in yeast. Nature. 398:246–251. [DOI] [PubMed] [Google Scholar]
- Li, S.J., and M. Hochstrasser. 2000. The yeast ULP2 (SMT4) gene encodes a novel protease specific for the ubiquitin-like Smt3 protein. Mol. Cell. Biol. 20:2367–2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, H., A. Keriel, C.R. Morales, N. Bedard, Q. Zhao, P. Hingamp, S. Lefrancois, L. Combaret, and S.S. Wing. 2000. Divergent N-terminal sequences target an inducible testis deubiquitinating enzyme to distinct subcellular structures. Mol. Cell. Biol. 20:6568–6578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louvion, J.-F., B. Havaux-Copf, and D. Picard. 1993. Fusion of GAL4-VP16 to a steroid-binding domain provides a tool for gratuitous induction of galactose-responsive genes in yeast. Gene. 131:129–134. [DOI] [PubMed] [Google Scholar]
- Melchior, F. 2000. SUMO–nonclassical ubiquitin. Annu. Rev. Cell Dev. Biol. 16:591–626. [DOI] [PubMed] [Google Scholar]
- Mossessova, E., and C.D. Lima. 2000. Ulp1-SUMO crystal structure and genetic analysis reveal conserved interactions and a regulatory element essential for cell growth in yeast. Mol. Cell. 5:865–876. [DOI] [PubMed] [Google Scholar]
- Nishida, T., F. Kaneko, M. Kitagawa, and H. Yasuda. 2001. Characterization of a novel mammalian SUMO-1/Smt3-specific isopeptidase, a homologue of rat axam, which is an axin-binding protein promoting beta-catenin degradation. J. Biol. Chem. 276:39060–39066. [DOI] [PubMed] [Google Scholar]
- Panse, V.G., B. Kuster, T. Gerstberger, and E. Hurt. 2003. Unconventional tethering of Ulp1 to the transport channel of the nuclear pore complex by karyopherins. Nat. Cell Biol. 5:21–27. [DOI] [PubMed] [Google Scholar]
- Park, K.C., J.H. Kim, E.-J. Choi, S.W. Min, S. Rhee, S.H. Baek, S.S. Chung, O. Bang, D. Park, T. Chiba, et al. 2002. Antagonistic regulation of myogenesis by two deubiquitinating enzymes, UBP45 and UBP69. Proc. Natl. Acad. Sci. USA. 99:9733–9738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickart, C.M. 2001. Mechanisms underlying ubiquitination. Annu. Rev. Biochem. 70:503–533. [DOI] [PubMed] [Google Scholar]
- Saitoh, H., and J. Hinchey. 2000. Functional heterogeneity of small ubiquitin-related protein modifiers SUMO-1 versus SUMO-2/3. J. Biol. Chem. 275:6252–6258. [DOI] [PubMed] [Google Scholar]
- Schwienhorst, I., E.S. Johnson, and R.J. Dohmen. 2000. SUMO conjugation and deconjugation. Mol. Gen. Genet. 263:771–786. [DOI] [PubMed] [Google Scholar]
- Seufert, W., B. Futcher, and S. Jentsch. 1995. A ubiquitin-conjugating enzyme involved in the degradation of both S- and M-phase cyclins. Nature. 373:78–81. [DOI] [PubMed] [Google Scholar]
- Stade, K., F. Vogel, I. Schwienhorst, B. Meusser, C. Volkwein, B. Nentwig, R.J. Dohmen, and T. Sommer. 2002. A lack of SUMO conjugation affects cNLS-dependent nuclear protein import in yeast. J. Biol. Chem. 277:49554–49561. [DOI] [PubMed] [Google Scholar]
- Stephens, R.S., S. Kalman, C. Lammel, J. Fan, R. Marathe, L. Aravind, W. Mitchell, L. Olinger, R.L. Tatusov, Q. Zhao, et al. 1998. Genome sequence of an obligate intracellular pathogen of humans: Chlamydia trachomatis. Science. 282:754–759. [DOI] [PubMed] [Google Scholar]
- Strunnikov, A.V., L. Aravind, and E.V. Koonin. 2001. Saccharomyces cerevisiae SMT4 encodes an evolutionarily conserved protease with a role in chromosome condensation regulation. Genetics. 158:95–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi, Y., M. Iwase, M. Konishi, M. Tanaka, A. Toh-e, and Y. Kikuchi. 1999. Smt3, a SUMO-1 homolog, is conjugated to Cdc3, a component of septin rings at the mother-bud neck in budding yeast. Biochem. Biophys. Res. Commun. 259:582–587. [DOI] [PubMed] [Google Scholar]
- Takahashi, Y., J. Mizoi, E.A. Toh, and Y. Kikuchi. 2000. Yeast Ulp1, an Smt3-specific protease, associates with nucleoporins. J. Biochem. 128:723–725. [DOI] [PubMed] [Google Scholar]
- Weissman, A.M. 2001. Themes and variations on ubiquitylation. Nat. Rev. Mol. Cell Biol. 2:169–178. [DOI] [PubMed] [Google Scholar]
- Wilkinson, K.D., and M. Hochstrasser. 1998. The deubiquitinating enzymes. Ubiquitin and the Biology of the Cell. J.M. Peters, J.R. Harris, and D. Finley, editors. Plenum Press, New York. 99–125.
- Zhang, H., H. Saitoh, and M.J. Matunis. 2002. Enzymes of the SUMO modification pathway localize to filaments of the nuclear pore complex. Mol. Cell. Biol. 22:6498–6508. [DOI] [PMC free article] [PubMed] [Google Scholar]