

Abstract
Cell survival depends on proper propagation of protective signals through intracellular signaling intermediates. We report here that calponin homology domain–containing integrin-linked kinase (ILK)–binding protein (CH-ILKBP), a widely expressed adaptor protein localized at plasma membrane-actin junctions, is essential for transmission of survival signals. Cells that are depleted of CH-ILKBP undergo extensive apoptosis despite the presence of cell–extracellular matrix contacts and soluble growth factors. The activating phosphorylation of protein kinase B (PKB/Akt), a key regulator of apoptosis, is impaired in the absence of CH-ILKBP. Importantly, loss of CH-ILKBP prevents the membrane translocation of PKB/Akt. Furthermore, forced membrane targeting of PKB/Akt bypasses the requirement of CH-ILKBP for the activating phosphorylation of PKB/Akt, suggesting that CH-ILKBP is required for the membrane translocation but not the subsequent phosphorylation of PKB/Akt. Finally, we show that loss of CH-ILKBP is also required for the full activation of extracellular signal–regulated kinase (ERK)1/2. However, restoration of the PKB/Akt activation is sufficient for protection of cells from apoptosis induced by the depletion of CH-ILKBP despite the persistent suppression of the ERK1/2 activation. Thus, CH-ILKBP is an important component of the prosurvival signaling pathway functioning primarily by facilitating the membrane translocation of PKB/Akt and consequently the activation of PKB/Akt in response to extracellular survival signals.
Keywords: apoptosis; CH-ILKBP; protein kinase B/Akt; membrane translocation; ILK
Introduction
Cell survival is a fundamental process that requires cellular interactions with appropriate extracellular survival factors (e.g., growth factors and extracellular matrix) and proper propagation of the survival signals through intracellular signaling pathway. Recent studies have established protein kinase B (PKB)*/Akt as a key signaling intermediate in the survival pathway (for reviews see Downward, 1998; Brazil and Hemmings, 2001; Lawlor and Alessi, 2001; Cantley, 2002). PKB/Akt activation is a multistep process involving coordinated actions of several catalytic and noncatalytic molecules. An early and obligatory step in the activation of PKB/Akt is its translocation from cytosol to the plasma membrane, often near or at cell–cell or cell–matrix contacts (Watton and Downward, 1999). The membrane-bound PKB/Akt is then phosphorylated at Ser473 near the COOH terminus and Thr308 in the activation loop by upstream protein kinases (Alessi et al., 1996), which leads to full activation of PKB/Akt. Activated PKB/Akt, in turn, phosphorylates several key apoptosis mediators, resulting in protection of cells from apoptosis. Therefore, identification of factors that participate in the cellular control of PKB/Akt activation is essential for understanding the mechanism by which cells control apoptosis.
Recent development of specific and efficient gene silencing in mammalian cells by RNA interference (RNAi) has allowed us to critically evaluate the role of proteins in cell survival and the mechanism whereby they function. Calponin homology domain–containing integrin-linked kinase (ILK)–binding protein (CH-ILKBP; Tu et al., 2001) (also known as actopaxin [Nikolopoulos and Turner, 2000] or α-parvin [Olski et al., 2001]) is a member of the CH-ILKBP/actopaxin/affixin/parvin protein family (Wu and Dedhar, 2001) that interacts with ILK, paxillin, and actin and clusters at the plasma membrane-actin junctions. In this study, we show that CH-ILKBP is an important antiapoptotic protein. Furthermore, we provide evidence showing that CH-ILKBP functions in cell survival by facilitating the membrane translocation of PKB/Akt and consequently the activation of PKB/Akt in response to extracellular survival signals.
Results and discussion
CH-ILKBP is crucial for cell survival
We employed RNAi to deplete CH-ILKBP in human cells. Transfection of HeLa cells with a CH-ILKBP small interfering RNA (siRNA) (Fig. 1 A, lane 1) but not an irrelevant control RNA (Fig. 1 A, lane 2) resulted in a near total loss of CH-ILKBP. Probing the same samples with an antiactin antibody showed that the level of actin was not altered (Fig. 1 B). Thus, the CH-ILKBP siRNA effectively suppresses the expression of CH-ILKBP without globally altering protein expression. CH-ILKBP forms a ternary complex with ILK and PINCH-1 in cells (Tu et al., 2001). Consistent with this, depletion of CH-ILKBP modestly reduced the levels of ILK (Fig. 1 C) and PINCH-1 (Fig. 1 D). The suppression of CH-ILKBP expression in the CH-ILKBP siRNA transfectants (Fig. 1 E) but not the control siRNA transfectants (Fig. 1 F) was confirmed by immunofluorescent staining. Staining of the cells with an antibody to paxillin (a marker of focal adhesions) showed that focal adhesions were formed in the CH-ILKBP–deficient cells (Fig. 1 G) and the control cells (Fig. 1 H). Moreover, staining of the cells with an anti-ILK antibody showed that a substantial amount of ILK (or proteins that share a common epitope with ILK) was clustered at focal adhesions (Fig. 1 I) despite the suppression of CH-ILKBP expression. To further analyze this, we transfected HeLa cells expressing GFP-ILK or GFP–PINCH-1 with CH-ILKBP siRNA and the control RNA, respectively. GFP-ILK (Fig. 1 L) and GFP–PINCH-1 (Fig. 1 N) localized to cell–matrix adhesions in both the CH-ILKBP siRNA transfectants (Fig. 1, K and M) and the control transfectants (unpublished data). Although depletion of CH-ILKBP prevented neither the formation of cell–matrix adhesions nor the localization of ILK and PINCH-1 to the adhesion sites, it induced a dramatic increase of apoptosis as revealed by the appearance of a large percentage of TUNEL-positive cells (Fig. 2, A and B). Analyses of the activity of caspase-3, a key mediator of apoptosis, showed that its activity was markedly increased in the absence of CH-ILKBP (Fig. 2 C). These results suggest that (a) CH-ILKBP is essential for cell survival albeit it is not absolutely required for the formation of cell–matrix adhesions and (b) CH-ILKBP acts in the cell survival pathway upstream of caspase-3.
Figure 1.

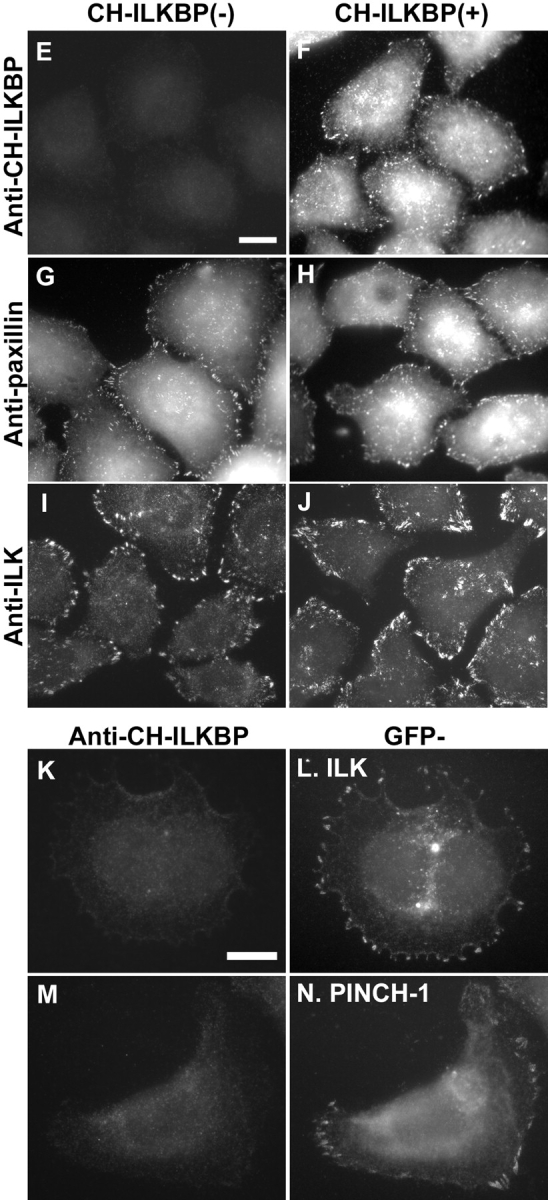
RNAi suppression of CH-ILKBP expression. (A–D) HeLa cells (lane 3), CH-ILKBP siRNA (lane 1), or the control RNA (lane 2) transfectants were analyzed by Western blotting with anti–CH-ILKBP mAb 3B5 (A, 10 μg proteins/lane), polyclonal antiactin antibody (B, 1 μg proteins/lane), anti-ILK mAb 65.1 (C, 10 μg proteins/lane), and polyclonal anti–PINCH-1 antibody (D, 10 μg proteins/lane). As we showed previously (Li et al., 1999), an additional protein band with an apparent molecular mass of ∼85 kD, which likely represents a protein that shares an epitope with ILK, was also detected by the anti-ILK mAb (C). (E–J) The CH-ILKBP siRNA (E, G, and I) and the control siRNA (F, H, and J) transfectants were stained with mAbs recognizing CH-ILKBP (E and F), paxillin (G and H), and ILK (I and J), respectively. Bar, 25 μm. (K–N) HeLa cells expressing GFP-ILK or GFP–PINCH-1 were transfected with CH-ILKBP siRNA and stained with anti–CH-ILKBP mAb 3B5 and a Rhodamine redTM–conjugated anti–mouse IgG antibody. Cells were observed under a fluorescence microscope with rhodamine (K and M) and GFP (L and N) filters. Bar, 15 μm.
Figure 2.
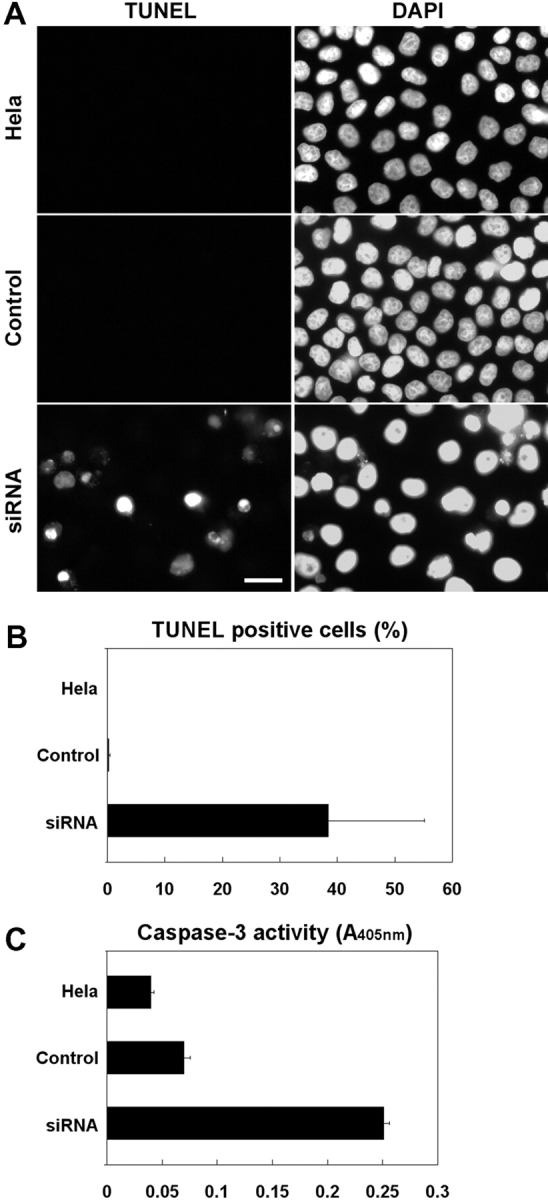
CH-ILKBP is crucial for cell survival. (A) HeLa cells, the control transfectants, and the CH-ILKBP siRNA transfectants were fixed and then incubated in the TUNEL reaction mixture, washed, and stained with DAPI. Apoptotic (TUNEL-positive) cells were detected by fluorescence microscopy. (B) The percentages of TUNEL-positive cells were calculated by counting at least 800 cells from five randomly selected fields. Data represent means ± SD. (C) The activities of caspase-3 in HeLa cells and the control and CH-ILKBP siRNA transfectants were quantified. Data represent means ± SD from two independent experiments.
CH-ILKBP regulates PKB/Akt phosphorylation
To gain insight into the mechanism by which CH-ILKBP functions in cell survival, we analyzed the effect of loss of CH-ILKBP on the activating phosphorylation of PKB/Akt, a key membrane-proximal survival signaling intermediate that is activated in response to growth factors and cell–matrix adhesion. Stimulation of HeLa cells with serum (Fig. 3 A, lane 6) or insulin-like growth factor (IGF)-1 (Fig. 3 B, lane 6) induced phosphorylation of PKB/Akt at both Ser473 and Thr308. However, the activating phosphorylation of PKB/Akt was impaired in the CH-ILKBP–deficient cells (Fig. 3, A and B, lane 4 compared with 6) but not the control cells (Fig. 3, A and B, lane 5 compared with 6). In parallel experiments, a similar amount of PKB/Akt protein was detected in the CH-ILKBP–deficient cells and the control cells (Fig. 3, A and B, lanes 1–6). Loss of CH-ILKBP did not alter the protein level or phosphorylation of phosphoinositide-dependent protein kinase 1 (PDK1) (Fig. 3, A and B), an upstream Ser/Thr kinase responsible for the Thr308 phosphorylation of membrane-bound PKB/Akt. Collectively, these results suggest that CH-ILKBP is critically involved in the cellular phosphorylation of PKB/Akt without altering the protein level or phosphorylation of PDK1.
Figure 3.
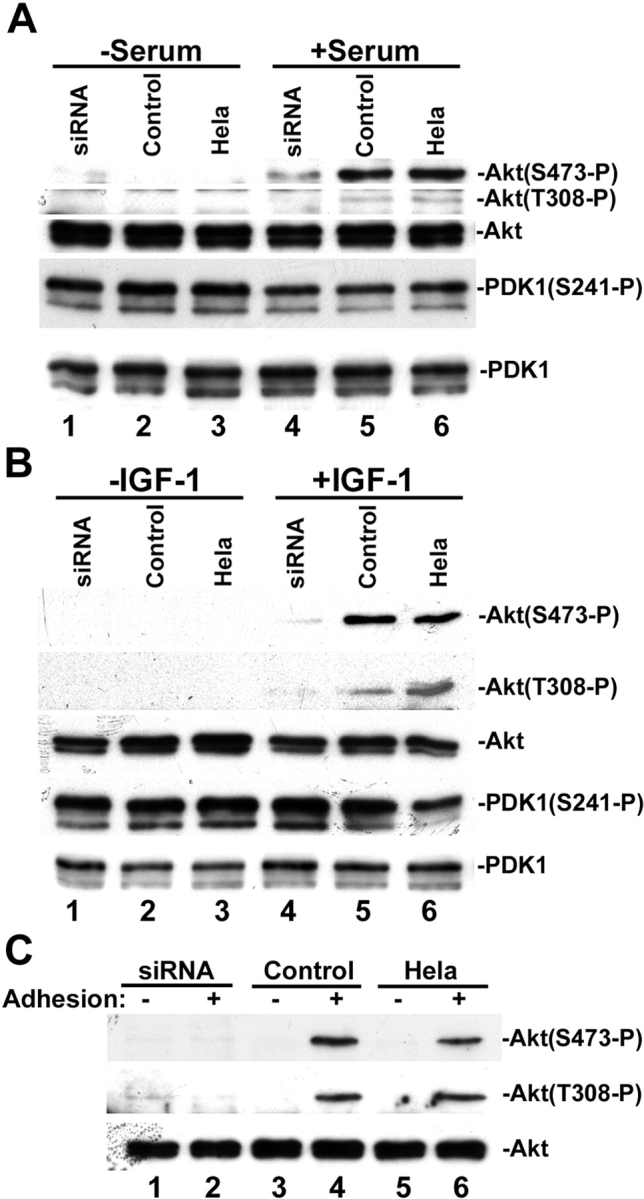
CH-ILKBP regulates PKB/Akt phosphorylation. (A and B) The CH-ILKBP siRNA transfectants (lanes 1 and 4), the control RNA transfectants (lanes 2 and 5), and the parental HeLa cells (lanes 3 and 6) were serum starved (lanes 1–3) and stimulated with 10% FCS (A, lanes 4–6) or 2 ng/ml IGF-1 (B, lanes 4–6) for 10 min. The cell lysates (10 μg/lane) were analyzed by Western blotting with antibodies recognizing Akt, phospho-Akt(Ser473), phospho-Akt(Thr308), phosphor-PDK1(Ser241), and PDK1, respectively. (C) The CH-ILKBP siRNA transfectants (lanes 1 and 2), the control RNA transfectants (lanes 3 and 4), and HeLa cells (lanes 5 and 6) were kept in suspension for 2 h and then either harvested (lanes 1, 3, and 5) or allowed to adhere on laminin-coated plates for 30 min (lanes 2, 4, and 6). The cell lysates (15 μg/lane) were analyzed by Western blotting with antibodies recognizing Akt, phospho-Akt(Ser473), and phospho-Akt(Thr308), respectively.
PKB/Akt can be activated not only by soluble survival factors but also by integrin-mediated cell–matrix adhesion (Khwaja et al., 1997; King et al., 1997; Gu et al., 2002). To test whether CH-ILKBP is required for cell adhesion-induced activating phosphorylation of PKB/Akt, we placed the CH-ILKBP–deficient cells and CH-ILKBP–expressing cells on laminin. Adhesion of the CH-ILKBP–expressing cells to laminin induced phosphorylation of PKB/Akt at both Ser473 and Thr308 (Fig. 3 C, lanes 4 and 6). By marked contrast, the phosphorylation of PKB/Akt was impaired in the absence of CH-ILKBP (Fig. 3 C, lane 2). Together, these results suggest that CH-ILKBP is required for both the soluble survival factor– and the matrix adhesion–induced phosphorylation of PKB/Akt.
CH-ILKBP functions in the PKB/Akt activation by facilitating the membrane translocation of PKB/Akt
We next investigated the mechanism by which CH-ILKBP functions in the activating phosphorylation of PKB/Akt. Because membrane translocation of PKB/Akt precedes and is essential for the activating phosphorylation of PKB/Akt (Andjelkovic et al., 1997; Scheid et al., 2002), we tested whether CH-ILKBP is required for the membrane translocation of PKB/Akt. Stimulation of HeLa cells or the control cells with IGF-1 induced translocation of PKB/Akt from cytosol to the membrane (Fig. 4 A, lanes 4 and 6), where it was phosphorylated at Ser473 (Fig. 4 B, lanes 4 and 6) and Thr308 (Fig. 4 C, lanes 4 and 6). By marked contrast, the PKB/Akt membrane translocation (Fig. 4 A, lane 2) and phosphorylation at Ser473 (Fig. 4 B, lane 2) and Thr308 (Fig. 4 C, lane 2) were impaired in the absence of CH-ILKBP. Probing the samples with an anti–CH-ILKBP antibody confirmed that CH-ILKBP was depleted in the membrane and the cytosol fractions of the CH-ILKBP siRNA transfectants (Fig. 4 D, lanes 1 and 2). Loss of CH-ILKBP did not significantly alter the amount of PDK1 associated with the membrane (Fig. 4 E, lanes 1 and 2). These results indicate that CH-ILKBP is required for the membrane translocation of PKB/Akt but not the association of PDK1 with the membrane.
Figure 4.

Loss of CH-ILKBP impairs the membrane translocation of PKB/Akt. (A–E) The CH-ILKBP siRNA-transfectants (lanes 1 and 2), the control transfectants (lanes 3 and 4), and the parental HeLa cells (lanes 5 and 6) were serum starved (lanes 1, 3, and 5) or stimulated with 2 ng/ml IGF-1 for 5 min (lanes 2, 4, and 6). Samples from the cytosol (10 μg proteins/lane) and the membrane (corresponding to 20 μg cytosol proteins/lane) fractions were analyzed by Western blotting with antibodies recognizing Akt (A), phospho-Akt(Ser473) (B), phospho-Akt(Thr308) (C), CH-ILKBP (D), and PDK1 (E), respectively. (F) HeLa cells were transfected with the pUSEamp(+)/myr-Akt1 vector encoding myristoylated PKB/Akt (lanes 1–4) or a pUSEamp(+) vector lacking Akt sequence as a control (lanes 5–8). The myristoylated PKB/Akt and the control vector transfectants were then transfected with the CH-ILKBP siRNA (lanes 1, 2, 5, and 6) or the control RNA (lanes 3, 4, 7, and 8). The cell lysates (10 μg/lane) were analyzed by Western blotting with antibodies recognizing Akt, phospho-Akt(Ser473), phospho-Akt(Thr308), and CH-ILKBP, respectively.
We next sought to confirm that CH-ILKBP participates in the activation of PKB/Akt by facilitating the membrane translocation of PKB/Akt. We reasoned that if facilitating the membrane translocation of PKB/Akt is the primary function of CH-ILKBP in the activation of PKB/Akt, loss of CH-ILKBP should not impair phosphorylation of membrane-bound PKB/Akt. To test this, we expressed myristoylated PKB/Akt in HeLa cells (Fig. 4 F, lanes 1–4). The myristoylated PKB/Akt, which is constitutively bound to the membrane (Andjelkovic et al., 1997), was phosphorylated at Ser473 and Thr308 in the absence (Fig. 4 F, lanes 1 and 2) and the presence (Fig. 4 F, lanes 3 and 4) of CH-ILKBP. In parallel experiments, stimulation with IGF-1 induced phosphorylation of wild-type PKB/Akt at Ser473 and Thr308 in the presence (Fig. 4 F, lane 8) but not the absence (Fig. 4 F, lane 6) of CH-ILKBP. Thus, although CH-ILKBP is required for the Ser473 and Thr308 phosphorylation of wild-type PKB/Akt, it is not required for the Ser473 and Thr308 phosphorylation of the membrane-bound PKB/Akt, confirming that CH-ILKBP functions in the cellular activation of PKB/Akt by facilitating the membrane translocation but not the phosphorylation reaction of PKB/Akt.
PKB/Akt is the primary target of CH-ILKBP in cell survival
Stimulation of cells with serum induced phosphorylation of not only PKB/Akt but also ERK1/2 and p38 MAPK (Fig. 5 A, lanes 5 and 6). Loss of CH-ILKBP modestly inhibited serum induced phosphorylation of ERK1/2 (Fig. 5 A, lane 4 compared with 5 and 6) but not that of p38 MAPK (Fig. 5 A, lanes 4–6). A modest inhibitory effect on IGF-1–induced ERK1/2 phosphorylation has also been observed (Fig. 5 B, lane 4). To test whether PKB/Akt is the primary target of CH-ILKBP in cell survival, we analyzed the effect of expression of the constitutively active PKB/Akt on apoptosis induced by the loss of CH-ILKBP. Expression of myristoylated PKB/Akt, which overrode the inhibition on the membrane translocation, and consequently the activating phosphorylation of PKB/Akt induced by the loss of CH-ILKBP (Fig. 4 F), did not release the modest inhibition of ERK1/2 phosphorylation (Fig. 5 C, lane 2 compared with 4). However, it did rescue cells from apoptosis induced by the loss of CH-ILKBP (Fig. 5, D and E). In control experiments, expression of the constitutively active PKB/Akt did not release the siRNA-mediated suppression of CH-ILKBP expression (Fig. 5 F). Thus, PKB/Akt, and specifically the membrane translocation of PKB/Akt, is the primary target of CH-ILKBP in protection of cells from apoptosis.
Figure 5.

PKB/Akt is the primary target of CH-ILKBP in cell survival. (A and B) CH-ILKBP modulates ERK phosphorylation. The CH-ILKBP siRNA transfectants (lanes 1 and 4), the control transfectants (lanes 2 and 5), and HeLa cells (lanes 3 and 6) were serum starved (lanes 1–3) and then stimulated with 10% FCS (A, lanes 4–6) or 2 ng/ml IGF-1 (B, lanes 4–6) for 10 min. The cell lysates (10 μg/lane) were analyzed by Western blotting with antibodies recognizing Erk1/2 (p44/42), phosphor-Erk1/2(Thr202/Tyr204) (phosphor-p44/42), and phosphor-p38 MAPK (Thr180/Tyr182), respectively. (C) The myristoylated PKB/Akt-expressing cells (see Fig. 4 F) were transfected with the CH-ILKBP siRNA (lanes 1 and 2) or the control RNA (lanes 3 and 4). The cells were serum starved (lanes 1 and 3) and then stimulated with 2 ng/ml IGF-1 (lanes 2 and 4) for 10 min. The cell lysates (10 μg protein/lane) were analyzed by Western blotting with antibodies recognizing Erk1/2 and phosphor-Erk1/2(Thr202/Tyr204), respectively. (D–F) Expression of membrane-bound PKB/Akt bypasses the requirement of CH-ILKBP for cell survival. Apoptotic cells were detected using TUNEL (D), and the percentages of the TUNEL-positive cells (E) were calculated as in the legend to Fig. 2. Vector, HeLa cells transfected with a pUSEamp(+) vector lacking Akt sequence and the control RNA; CH-ILKBP(−) + Vector, HeLa cells transfected with a pUSEamp(+) vector lacking Akt sequence and the CH-ILKBP siRNA; CH-ILKBP(−) + myr-Akt, HeLa cells transfected with the pUSEamp(+)/myr-Akt1 vector encoding myristoylated PKB/Akt and the CH-ILKBP siRNA. (F) The lysates (10 μg/lane) from the transfectants were analyzed by Western blotting with anti–CH-ILKBP mAb 3B5.
The results presented above have established CH-ILKBP as an important antiapoptotic protein, since depletion of CH-ILKBP results in a dramatic increase of apoptosis. In addition, they shed light on the mechanism by which CH-ILKBP functions in protection of cells from apoptosis. CH-ILKBP is clearly required for the activating phosphorylation of PKB/Akt in response to extracellular survival factors. Although CH-ILKBP is also involved in the modulation of phosphorylation of Erk1/2, which likely plays a role in the regulation of other cellular processes such as proliferation or differentiation, the primary function of CH-ILKBP in the survival pathway is to regulate the activation of PKB/Akt, since expression of constitutively active PKB/Akt is sufficient for protection of cells from apoptosis induced by the loss of CH-ILKBP despite the persistent suppression of the ERK1/2 phosphorylation.
How does CH-ILKBP regulate the activation of PKB/Akt? PKB/Akt activation is a complex process involving at least two major steps, namely the membrane translocation and the subsequent phosphorylation of PKB/Akt at Ser473 and Thr308. Therefore, the activation of PKB/Akt could be regulated at either the membrane translocation or the phosphorylation steps. Our findings that (a) loss of CH-ILKBP impairs the membrane translocation of PKB/Akt and (b) forced targeting of PKB/Akt to the membrane restores its phosphorylation at both Ser473 and Thr308 suggest that CH-ILKBP is required for the membrane translocation but not the subsequent phosphorylation reaction of PKB/Akt.
Cell matrix adhesion is crucial for cell survival (Meredith et al., 1993; Frisch and Screaton, 2001; Lee and Juliano, 2002; Miranti and Brugge, 2002). Watton and Downward (1999) have shown that during PKB/Akt activation, PKB/Akt is translocated to sites near or at cell matrix or cell–cell contacts. The finding that CH-ILKBP is required for the membrane translocation of PKB/Akt raises an interesting possibility that the close vicinity of cell matrix contacts and the PKB/Akt membrane translocation sites may reflect a need for a local event involving certain cell matrix adhesion components (e.g., CH-ILKBP) during the membrane translocation process. It is interesting to note that although human CH-ILKBP (Tu et al., 2001) and affixin (Yamaji et al., 2001) are encoded by two different genes, they share significant sequence similarity, particularly in the CH2 domains that mediate the ILK binding. Overexpression of the ILK-binding CH2 fragments of CH-ILKBP/actopaxin or affixin in mammalian cells altered the dynamics of cell shape change but did not block the eventual establishment of cell matrix adhesions (Nikolopoulos and Turner, 2000; Tu et al., 2001; Yamaji et al., 2001), possibly due to the presence of other linkages between extracellular matrix and the actin cytoskeleton. In this study, we have found that cell matrix adhesion structures were formed in the absence of CH-ILKBP (Fig. 1). Thus, the impairment of PKB/Akt membrane translocation induced by the depletion of CH-ILKBP is not caused by a lack of cell matrix contacts but instead by a compositional change of the cell matrix contacts.
Because ILK is involved in the activation of PKB/Akt (Delcommenne et al., 1998; Lynch et al., 1999), a simple mechanism would be that loss of CH-ILKBP impairs the proper localization of ILK and thereby inhibits the activation of PKB/Akt. Indeed, mutations within the COOH-terminal domain of ILK that disrupt the binding of the CH2 domains of CH-ILKBP/actopaxin abolish the localization of ILK to cell matrix adhesions (Nikolopoulos and Turner, 2002; Zhang et al., 2002). However, depletion of CH-ILKBP does not appear to prevent the localization of ILK to cell matrix adhesions, suggesting that other proteins binding to the same ILK site (e.g., other members of the CH-ILKBP/actopaxin/affixin/parvin family) could play a similar role supporting the localization of ILK to the adhesion sites. Although there likely exist multiple ILK-binding proteins that can support the localization of ILK to the adhesion sites, CH-ILKBP is indispensable for the PKB/Akt membrane translocation and cell survival. Thus, although our data do not necessarily rule out a role of CH-ILKBP in ILK localization, they do not support a model in which CH-ILKBP functions in the PKB/Akt activation by merely facilitating the localization of ILK to cell matrix adhesion sites.
The mechanism by which ILK functions in the activation of PKB/Akt is complex and is an area under intensive research. Overexpression of ILK, but not that of an ILK point mutant (GH31R) in which Glu359 in the COOH-terminal domain was substituted with Lys, enhanced the Ser473 phosphorylation of PKB/Akt (Delcommenne et al., 1998). In addition, Delcommenne et al. (1998) have found that ILK can phosphorylate PKB/Akt at Ser473 in vitro and therefore proposed that ILK regulates PKB/Akt activation by direct phosphorylation of Ser473. Lynch et al. (1999) have confirmed that overexpression of ILK promotes the Ser473 phosphorylation of PKB/Akt, but they proposed that ILK regulates the Ser473 phosphorylation of PKB/Akt by an indirect mechanism, since ILK mutants bearing mutations at sites that are predicted to be essential for kinase activity were also able to promote the Ser473 phosphorylation. The finding that ILK possesses protein kinase activity has been confirmed by several reports, and the promotion of the PKB/Akt activation by ILK has been repeatedly observed (for review see Wu and Dedhar, 2001). However, biochemical purification and characterization of the Ser473 kinase suggest that the Ser473 kinase is distinct from ILK (Hill et al., 2002). The findings presented in this paper raise an interesting possibility, which is not necessarily mutually exclusive with the model involving ILK kinase activity, that ILK and CH-ILKBP work in concert in facilitating the membrane translocation and consequently the activation of PKB/Akt. This possibility is supported by recent findings that the dominant-negative ILK GH31R mutant is incapable of binding to CH-ILKBP (Nikolopoulos and Turner, 2002). In further supporting of this, we have found that overexpression of an ILK-binding CH-ILKBP COOH-terminal fragment inhibited the activation of PKB/Akt (unpublished data). Clearly, future studies are required to further test this possibility.
Apoptosis is a fundamental process that is tightly regulated in organogenesis and in cell and tissue homeostasis. Dysregulation of apoptosis is a major cause of human diseases such as cancers. Therefore, the work presented in this report not only provides new information on the regulation of PKB/Akt membrane translocation and apoptosis but also suggest that CH-ILKBP, given its crucial role in the cellular activation of PKB/Akt, could serve as a useful target in the therapeutic intervention of human diseases involving abnormal apoptosis.
Materials and methods
Antibodies and other reagents
Rabbit antibodies against AKT, phospho-Akt(Ser473), phospho-Akt(Thr308), PDK1, phosphor-PDK1(Ser241), p44/42 MAPK (Erk1/2), phosphor-p44/42 MAPK (Thr202/Tyr204), and phosphor-p38 MAPK (Thr180/Tyr182) were from Cell Signaling Technology, Inc. Mouse antipaxillin mAb (clone 349) was from BD Transduction Laboratories. IGF-1, FBS, and cell culture media were from Invitrogen.
RNA interference
The sequences of CH-ILKBP siRNA were selected based on a method described previously (Elbashir et al., 2002). The targeted sequence that effectively mediates the silencing of CH-ILKBP expression is 5′-AAUGAGGUGCGAACAAUGGUG-3′ (sense sequence). The 21-nucleotide synthetic siRNA duplex was prepared by Dharmacon Research. HeLa cells were transfected with the CH-ILKBP siRNA or a 21-nucleotide irrelevant RNA duplex as a control using Oligofectamine (Invitrogen). Depletion of CH-ILKBP was confirmed by Western blotting.
Immunofluorescent staining
Immunofluorescent staining was performed as described (Li et al., 1999; Tu et al., 2001). Briefly, cells were plated on fibronectin-coated coverslips and cultured for 24 h. The cells were then fixed and stained with anti–CH-ILKBP (Tu et al., 2001), anti-ILK (Li et al., 1999), and antipaxillin mAbs. The mouse primary antibodies were detected with a Rhodamine redTM–conjugated anti–mouse IgG antibody (Jackson ImmunoResearch Laboratories).
DNA constructs and transfection
The DNA construct (pUSEamp(+)/myr-Akt1) encoding myristoylated Akt was from Upstate Biotechnology. HeLa cells were transfected with pUSEamp(+)/myr-Akt1 or a pUSEamp(+) vector lacking Akt sequence as a control using LipofectAmine PLUS (Invitrogen). To analyze subcellular localization of ILK and PINCH-1, HeLa cells were transfected with pEGFP-ILK and pEGFP–PINCH-1 expression vectors as described (Zhang et al., 2002). 1 d after DNA transfection, the cells were transfected with the CH-ILKBP siRNA or a 21-nucleotide irrelevant RNA duplex using Oligofectamine (Invitrogen). The transfectants were plated on fibronectin-coated 24 h after siRNA transfection and cultured for an additional 24 h. The cells were fixed and analyzed by immunofluorescent staining as described above.
Cell adhesion
Cells were starved in serum-free DME for 18–24 h and then harvested with 0.05% trypsin-EDTA. Cells were washed with serum-free DME containing 1% BSA, resuspended in the same medium, and kept in suspension for 2 h at 37°C. The cells were either immediately collected and lysed with 1% Triton X-100 in PBS containing protease inhibitors (lysis buffer) or allowed to adhere on laminin-coated dishes for 30 min and then lazed. The cell lysates were analyzed by Western blotting.
Serum and IGF-1 stimulation
Cells were starved in serum-free DME for 18–24 h and then stimulated with DME containing FBS or IGF-1. At the end of stimulation, cells were either immediately lysed with the lysis buffer and analyzed by Western blotting or fractionated as described below.
Cell fractionation
Cytosol and membrane fractions were prepared as described (Scheid et al., 2002). Briefly, cells were suspended in hypotonic lysis buffer (25 mM Tris, pH 7.4, 5 mM EDTA, 1 mM dithiothreitol, 1 mM Na3VO4, 2.5 mM Na4P2O7, and protease inhibitors) and disrupted by five passages through a 27-gauge needle. Large cell debris was pelleted by centrifugation at 2,000 g for 5 min at 4°C. The supernatants were centrifuged at 100,000 g for 20 min at 4°C. The resulting supernatant (cytosol) was collected, and the pellet was resuspended in lysis buffer containing 1% Triton X-100. The lysate was again centrifuged at 100,000 g for 20 min at 4°C, and the supernatant (membrane) was collected. The distributions of proteins in the cytosol and membrane fractions were analyzed by Western blotting.
Apoptosis
Cells (as specified in each experiment) were cultured in DME supplemented with 10% FBS. Apoptosis was analyzed 48 h after RNA transfection using terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL) and caspase-3 assays. The TUNEL assay was performed using an In Situ Cell Death Detection kit (Roche). The caspase-3 activities were measured using a colorimetric caspase-3 assay kit from Calbiochem.
Acknowledgments
This work was supported by National Institutes of Health grants GM65188 and DK54639 to C. Wu.
Footnotes
Abbreviations used in this paper: CH-ILKBP, calponin homology domain–containing ILK-binding protein; ERK, extracellular signal–regulated kinase; IGF, insulin-like growth factor; ILK, integrin-linked kinase; PKB, protein kinase B; RNAi, RNA interference; siRNA, small interfering RNA.
References
- Alessi, D.R., M. Andjelkovic, B. Caudwell, P. Cron, N. Morrice, P. Cohen, and B.A. Hemmings. 1996. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 15:6541–6551. [PMC free article] [PubMed] [Google Scholar]
- Andjelkovic, M., D.R. Alessi, R. Meier, A. Fernandez, N.J. Lamb, M. Frech, P. Cron, P. Cohen, J.M. Lucocq, and B.A. Hemmings. 1997. Role of translocation in the activation and function of protein kinase B. J. Biol. Chem. 272:31515–31524. [DOI] [PubMed] [Google Scholar]
- Brazil, D.P., and B.A. Hemmings. 2001. Ten years of protein kinase B signalling: a hard Akt to follow. Trends Biochem. Sci. 26:657–664. [DOI] [PubMed] [Google Scholar]
- Cantley, L.C. 2002. The phosphoinositide 3-kinase pathway. Science. 296:1655–1657. [DOI] [PubMed] [Google Scholar]
- Delcommenne, M., C. Tan, V. Gray, L. Rue, J. Woodgett, and S. Dedhar. 1998. Phosphoinositide-3-OH kinase-dependent regulation of glycogen synthase kinase 3 and protein kinase B/AKT by the integrin-linked kinase. Proc. Natl. Acad. Sci. USA. 95:11211–11216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downward, J. 1998. Mechanisms and consequences of activation of protein kinase B/Akt. Curr. Opin. Cell Biol. 10:262–267. [DOI] [PubMed] [Google Scholar]
- Elbashir, S.M., J. Harborth, K. Weber, and T. Tuschl. 2002. Analysis of gene function in somatic mammalian cells using small interfering RNAs. Methods. 26:199–213. [DOI] [PubMed] [Google Scholar]
- Frisch, S.M., and R.A. Screaton. 2001. Anoikis mechanisms. Curr. Opin. Cell Biol. 13:555–562. [DOI] [PubMed] [Google Scholar]
- Gu, J., A. Fujibayashi, K.M. Yamada, and K. Sekiguchi. 2002. Laminin-10/11 and fibronectin differentially prevent apoptosis induced by serum removal via phosphatidylinositol 3-kinase/Akt- and MEK1/ERK-dependent pathways. J. Biol. Chem. 277:19922–19928. [DOI] [PubMed] [Google Scholar]
- Hill, M.M., J. Feng, and B.A. Hemmings. 2002. Identification of a plasma membrane Raft-associated PKB Ser473 kinase activity that is distinct from ILK and PDK1. Curr. Biol. 12:1251–1255. [DOI] [PubMed] [Google Scholar]
- Khwaja, A., P. Rodriguez-Viciana, S. Wennstrom, P.H. Warne, and J. Downward. 1997. Matrix adhesion and Ras transformation both activate a phosphoinositide 3-OH kinase and protein kinase B/Akt cellular survival pathway. EMBO J. 16:2783–2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King, W.G., M.D. Mattaliano, T.O. Chan, P.N. Tsichlis, and J.S. Brugge. 1997. Phosphatidylinositol 3-kinase is required for integrin-stimulated AKT and Raf-1/mitogen-activated protein kinase pathway activation. Mol. Cell. Biol. 17:4406–4418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawlor, M.A., and D.R. Alessi. 2001. PKB/Akt: a key mediator of cell proliferation, survival and insulin responses? J. Cell Sci. 114:2903–2910. [DOI] [PubMed] [Google Scholar]
- Lee, J.W., and R.L. Juliano. 2002. The alpha5beta1 integrin selectively enhances epidermal growth factor signaling to the phosphatidylinositol-3-kinase/Akt pathway in intestinal epithelial cells. Biochim. Biophys. Acta. 1542:23–31. [DOI] [PubMed] [Google Scholar]
- Li, F., Y. Zhang, and C. Wu. 1999. Integrin-linked kinase is localized to cell-matrix focal adhesions but not cell-cell adhesion sites and the focal adhesion localization of integrin-linked kinase is regulated by the PINCH-binding ANK repeats. J. Cell Sci. 112:4589–4599. [DOI] [PubMed] [Google Scholar]
- Lynch, D.K., C.A. Ellis, P.A. Edwards, and I. Hiles. 1999. Integrin-linked kinase regulates phosphorylation of serine 473 of protein kinase B by an indirect mechanism. Oncogene. 18:8024–8032. [DOI] [PubMed] [Google Scholar]
- Meredith, J.E., Jr., B. Fazeli, and M.A. Schwartz. 1993. The extracellular matrix as a cell survival factor. Mol. Biol. Cell. 4:953–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranti, C.K., and J.S. Brugge. 2002. Sensing the environment: a historical perspective on integrin signal transduction. Nat. Cell Biol. 4:E83–E90. [DOI] [PubMed] [Google Scholar]
- Nikolopoulos, S.N., and C.E. Turner. 2000. Actopaxin, a new focal adhesion protein that binds paxillin LD motifs and actin and regulates cell adhesion. J. Cell Biol. 151:1435–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolopoulos, S.N., and C.E. Turner. 2002. Molecular dissection of actopaxin-integrin-linked kinase-paxillin interactions and their role in subcellular localization. J. Biol. Chem. 277:1568–1575. [DOI] [PubMed] [Google Scholar]
- Olski, T.M., A.A. Noegel, and E. Korenbaum. 2001. Parvin, a 42 kDa focal adhesion protein, related to the alpha-actinin superfamily. J. Cell Sci. 114:525–538. [DOI] [PubMed] [Google Scholar]
- Scheid, M.P., P.A. Marignani, and J.R. Woodgett. 2002. Multiple phosphoinositide 3-kinase-dependent steps in activation of protein kinase B. Mol. Cell. Biol. 22:6247–6260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu, Y., Y. Huang, Z. Zhang, Y. Hua, and C. Wu. 2001. A new focal adhesion protein that interacts with integrin-linked kinase and regulates cell adhesion and spreading. J. Cell Biol. 153:585–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watton, S.J., and J. Downward. 1999. Akt/PKB localisation and 3′ phosphoinositide generation at sites of epithelial cell-matrix and cell-cell interaction. Curr. Biol. 9:433–436. [DOI] [PubMed] [Google Scholar]
- Wu, C., and S. Dedhar. 2001. Integrin-linked kinase (ILK) and its interactors: a new paradigm for the coupling of extracellular matrix to actin cytoskeleton and signaling complexes. J. Cell Biol. 155:505–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaji, S., A. Suzuki, Y. Sugiyama, Y. Koide, M. Yoshida, H. Kanamori, H. Mohri, S. Ohno, and Y. Ishigatsubo. 2001. A novel integrin-linked kinase-binding protein, affixin, is involved in the early stage of cell–substrate interaction. J. Cell Biol. 153:1251–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y., K. Chen, Y. Tu, A. Velyvis, Y. Yang, J. Qin, and C. Wu. 2002. Assembly of the PINCH-ILK-CH-ILKBP complex precedes and is essential for localization of each component to cell-matrix adhesion sites. J. Cell Sci. 115:4777–4786. [DOI] [PubMed] [Google Scholar]