

Abstract
Parathyroid hormone (PTH)-related protein (PTHrP)-knockout mice die at birth with a chondrodystrophic phenotype characterized by premature chondrocyte differentiation and accelerated bone formation, whereas overexpression of PTHrP in the chondrocytes of transgenic mice produces a delay in chondrocyte maturation and endochondral ossification. Replacement of PTHrP expression in the chondrocytes of PTHrP-knockout mice using a procollagen II-driven transgene results in the correction of the lethal skeletal abnormalities and generates animals that are effectively PTHrP-null in all sites other than cartilage. These rescued PTHrP-knockout mice survive to at least 6 months of age but are small in stature and display a number of developmental defects, including cranial chondrodystrophy and a failure of tooth eruption. Teeth appear to develop normally but become trapped by the surrounding bone and undergo progressive impaction. Localization of PTHrP mRNA during normal tooth development by in situ hybridization reveals increasing levels of expression in the enamel epithelium before the formation of the eruption pathway. The type I PTH/PTHrP receptor is expressed in both the adjacent dental mesenchyme and in the alveolar bone. The replacement of PTHrP expression in the enamel epithelium with a keratin 14-driven transgene corrects the defect in bone resorption and restores the normal program of tooth eruption. PTHrP therefore represents an essential signal in the formation of the eruption pathway.
Keywords: gene targeting/transgenic mice/differentiation/bone resorption/osteoclasts
Although parathyroid hormone (PTH)-related protein (PTHrP) was originally discovered by virtue of its elaboration from tumors associated with the syndrome of humoral hypercalcemia of malignancy, this may represent the only circumstance in which sufficient quantities of the peptide are released into the general circulation to exert systemic effects (1). The PTH and PTHrP genes represent the sequelae of an ancient gene duplication; structural similarity and sequence homology at the amino termini of the respective mature peptides mediate their binding to and stimulation of the type I PTH/PTHrP receptor with equal affinity (2, 3). Activation of this G protein-coupled receptor on osteoblasts and renal tubule cells is responsible for the classical effects of PTH on calcium and phosphate metabolism and, similarly, mediates the paraneoplastic effects of PTHrP. PTHrP and its mRNA are typically expressed in a focal pattern within surface epithelia, whereas the type I receptor is diffusely distributed in the adjacent mesenchyme (4, 5). In skeletal growth plates, PTHrP is expressed in the perichondrium and maturing chondrocytes, whereas its receptor is primarily found in the proximal prehypertrophic layer. Thus, PTHrP appears to act locally in a paracrine or autocrine fashion (reviewed in ref. 6).
A number of recent experiments implicate PTHrP as a developmental regulatory molecule. Targeted overexpression of PTHrP in the epidermis causes both delay and failure of hair follicle initiation (7), and overexpression in mammary myoepithelial cells delays and profoundly diminishes the extent of ductal proliferation and branching morphogenesis (8). Disruption of the PTHrP gene results in a lethal skeletal dysplasia characterized by the premature maturation of chondrocytes and accelerated mineralization of bone (9), whereas overexpression of the peptide in the proliferating and prehypertrophic chondrocytes of transgenic mice causes a profound delay in chondrocyte differentiation and endochondral ossification (10). In addition, a constitutively activated mutant type I receptor is the cause of Jansen-type metaphyseal chondrodysplasia in humans, a disease characterized by delayed endochondral bone formation (11). PTHrP thus appears to function as an attenuator of programmed differentiation in the genesis of a number of organ systems.
Because the perinatal lethality in the PTHrP-knockout mice prevents any assessment of potential consequences of the absence of the peptide on postnatal development, we attempted to correct the primary skeletal defects in the knockouts by replacing PTHrP expression in the skeleton with a chondrocyte-targeted transgene. This approach was designed to create a viable animal that is effectively PTHrP-null in all tissues except cartilage.
MATERIALS AND METHODS
The generation and genotyping of PTHrP-knockout mice and of procollagen II (col II)-PTHrP and keratin 14 (K14)-PTHrP transgenic mice have been described (7, 9, 10, 12). Rescued PTHrP-knockout animals were fed a liquid diet (Bioserv, Frenchtown, NJ). Neonatal skeletons were stained with alizarin red S to differentiate calcified tissues (10). Nondecalcified sections of neonatal mandibles were stained with toluidine blue (13). Mandibles from older mice were decalcified in 10% EDTA for 10 days and sections were stained with hematoxylin/eosin. Radiographic analyses of tooth development were carried out with a Hewlett-Packard Faxitron at 27 kV for 40 sec. Tartrate-resistant acid phosphatase activity was detected with reagents from Sigma. Mouse skulls were prepared by incubation with Dermestid beetle larvae (14). K14 localization and in situ hybridization were carried out as described (7, 12).
RESULTS
Generation of Rescued PTHrP-Knockout Mice.
To this end, we took advantage of a transgenic model of PTHrP overexpression targeted to proliferating and prehypertrophic chondrocytes via the murine procollagen (α1) type II promoter (10). However, rather than using a line of transgenic mice with high levels of PTHrP expression associated with a profound delay in endochondral ossification and resultant short-limbed dwarfism (10), we chose instead a line with lower expression levels, in which mineralization was retarded only in the tail vertebrae at birth and appeared to be normal soon thereafter (see below). Expression of the transgene in these animals could be detected only in cartilage; serum calcium levels were normal and circulating PTHrP was undetectable (not shown). These col II-PTHrP transgenics were crossed to PTHrP-null heterozygotes, and resultant offspring carrying both the transgene and a PTHrP null allele were again crossed to PTHrP-null heterozygotes, thereby generating col II-PTHrP transgenic, PTHrP-null homozygous animals.
Skeletal Phenotype.
As shown in Fig. 1, differential staining of calcified tissue in col II-PTHrP transgenic, PTHrP-knockout neonates indicated that the skeletal abnormalities seen in the PTHrP-knockouts are largely corrected. Transgenic knockouts were indistinguishable from wild-type littermates in size, and the limbs were of an appropriate length. Ossification of long bones, pelvis, and vertebrae was not premature, and there was no evidence of inappropriate mineralization of the costal or intersternebral cartilages. Endochondral growth plates also appeared to be normal at birth (data not shown). Mineralization of the endochondral bones of the base of the skull appeared to be normal (Fig. 1), and cranial doming was not evident, although foreshortening of the maxilla and mandible, which form principally by intramembranous ossification (15), appeared to persist. Therefore, the targeting of PTHrP expression to chondrocytes via the col II promoter appeared sufficient to restore the normal program of endochondral differentiation in PTHrP-knockout mice.
Figure 1.
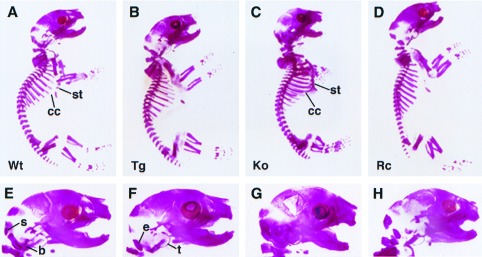
Staining of calcified tissue in neonatal mice with alizarin red S. From left to right are wild-type (A), col II-PTHrP transgenic (B), PTHrP-knockout (C), and rescued PTHrP-knockout (D) phenotypes. The PTHrP-knockout animals (C) display inappropriate ossification of the costal cartilage (cc) and along the length of the sternum (st). These abnormalities appear to be largely corrected in the rescued mice (D), in which ossification is confined to the sternebrae and does not extend to the costal cartilage. A magnified view of the rescued-knockout cranium (H) shows substantial correction of the accelerated mineralization of the endochondral bones of the cranial base seen in the knockout (G); s, supraoccipital; b, basioccipital; e, exoccipital; t, tympanic ring. The tips of the emerging mandibular incisors can be discerned in both the normal (E) and transgenic (F) animals, but not in the PTHrP-knockout (G) or rescued-knockout (H) mice. Wt, wild type; Tg, transgenic; Ko, knockout; Rc, rescued-knockout.
Although rescued PTHrP-knockout mice were born at the appropriate ratio of one in eight, less than half survived past 3 weeks of age. Although indistinguishable from their littermates at birth, these animals failed to thrive and exhibited a 50% reduction in both size and weight by the time of weaning. Because of a lack of functional dentition, rescued animals were unable to eat solid food and were placed on a liquid diet at weaning. With increasing age, rescued animals displayed a progressive short-limbed dwarfism, foreshortening of the maxillary-occipital axis, and doming of the calvarium (Fig. 2), features typical of a classical chondrodystrophy (16). Shallow eye sockets associated with the abnormal craniofacial development were the apparent cause of the noticeable ocular proptosis. Rescued PTHrP-knockout mice have a life span of ≈6 months; the cause of death is presently unknown.
Figure 2.
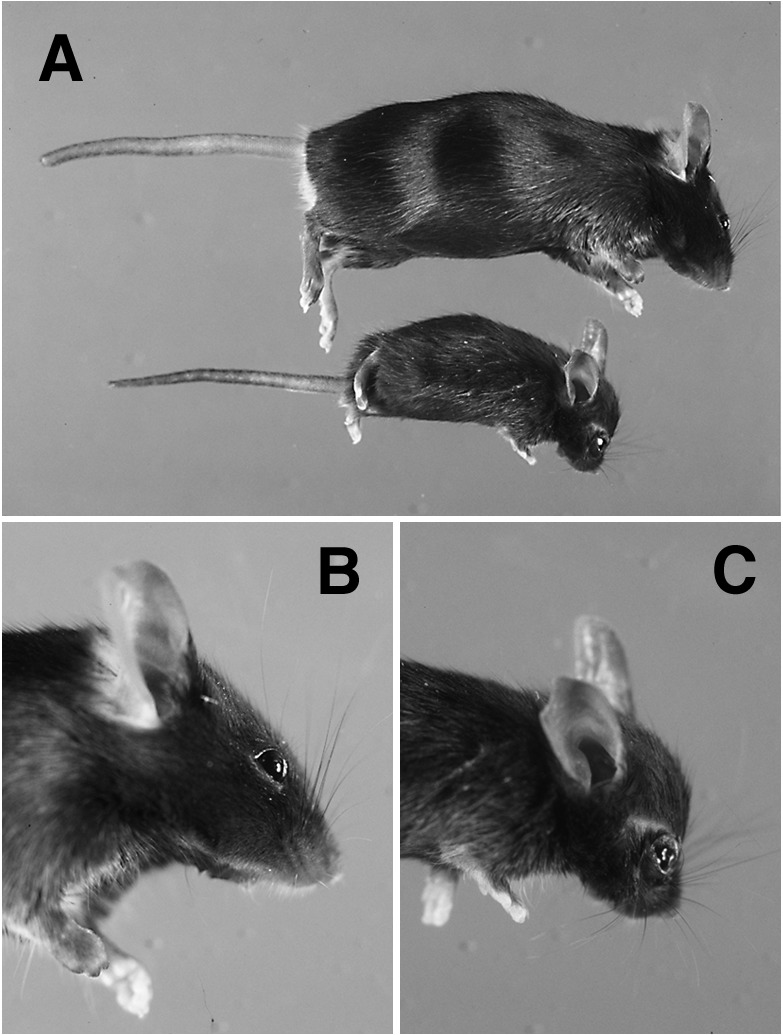
Gross appearance of rescued PTHrP-knockout mice. (A) Relative size of a rescued-knockout mouse at 8 weeks of age (below) as compared with a wild-type littermate (above). Foreshortening of the limbs is apparent by this age. The cranial chondrodystrophy in the rescued-knockout mouse (C), as compared with a normal animal (B), includes doming of the calvarium, frontal bossing, and a flattened snout.
Failure of Tooth Eruption.
As shown in Fig. 3, incisors and molars were absent from both the mandible and maxilla of adult rescued PTHrP-knockout mice, and the bone surfaces overlying expected sites of eruption were intact. Murine incisors normally emerge at ≈1 week of age (17), followed by the molars (M1 through M3) beginning at ≈2 weeks (18). Cranial radiographs taken at 2 weeks of age (Fig. 4) revealed a failure of tooth eruption and root formation in the rescued-knockouts, whereas the increased density of the incisors and molars suggested that they had become trapped and impacted by the surrounding alveolar bone. Histologic examination of mandibles from rescued-knockouts in late gestation showed that tooth morphogenesis was unaffected by the absence of PTHrP (data not shown). By birth, however, the impaction process began to interfere with the late stages of incisor development. Unimpeded growth of alveolar bone in the rescued animal (Fig. 5 A and B) compressed the epithelial layers along the labial surface of the incisor to such a degree that the normally prominent ameloblast layer, which is responsible for enamel deposition (17), was destroyed and enamel was lacking. Inspection of 1-week-old animals (Fig. 5 C and D) revealed impaction in the molar teeth as well. The ameloblast layer was still intact and enamel had been deposited, but the molar crypt was choked with bone and the teeth were visibly distorted. Ultimately, the teeth became irreversibly ankylosed because of the fusion of the dental cementum with the encroaching alveolar bone (data not shown).
Figure 3.
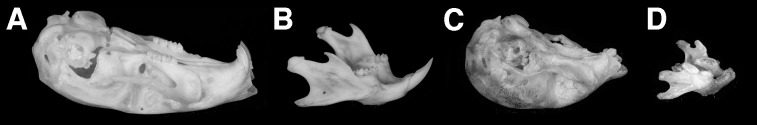
Comparison of skulls from adult wild-type mice (A and B) and rescued PTHrP-knockouts (C and D). External incisors and molars are clearly absent from both the maxilla (compare A and C) and mandible (compare B and D) in these animals. The lack of functional dentition in the rescued-knockout is superimposed on the underlying features of the cranial chondrodystrophy, which includes foreshortening of the maxilla and mandible.
Figure 4.
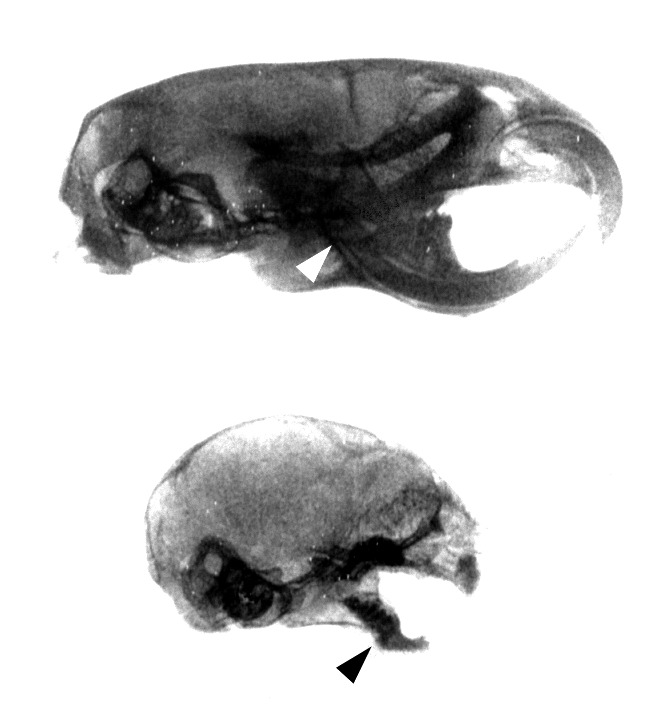
Radiographic analysis of tooth eruption in wild-type littermates (Upper) and rescued PTHrP-knockout mice (Lower) at 2 weeks of age. Normal eruption of incisors and molars (Upper) does not occur in the rescued-knockout mouse (Lower); the radiographic densities apparent in the mandible and maxilla of the rescued-knockouts represent impacted teeth. Also, the root of the lower incisor extends under the molars to M3 in the normal mandible (arrowhead, Upper), whereas the incisor root in the rescued-knockout fails to reach M1 (arrowhead, Lower), a finding typical of osteopetrosis.
Figure 5.
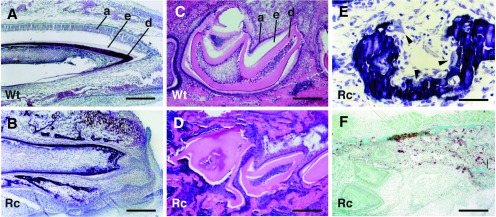
Tooth histology. (A and B) Sagittal, nondecalcified sections of incisors from a rescued PTHrP-knockout neonate (B) and a wild-type littermate (A) stained with toluidine blue. A well developed ameloblast layer (a) is readily apparent on the labial aspect of the normal tooth (A), but is lacking in the mutant (B), which is crowded by the surrounding alveolar bone. The dark dentin layer (d) is intact but the adjacent enamel (e) is absent from the labial surface of the incisor (the space between the ameloblast layer and the enamel in A is a sectioning artifact). (C and D) Mandibles from 1-week-old wild-type (C) and rescued-knockout mice (D) were decalcified and then sectioned sagittally through the molar crypt. Distortion of the teeth in the rescued-knockout animal caused by progressive impaction is readily apparent. The space between the ameloblast (a) and dentin (d) layers defines the area occupied by the enamel (e) that has been removed through decalcification. (E) Magnification of an nondecalcified sagittal section through the region between the incisor and the first molar of a mandible from a rescued-knockout animal. Osteoclasts (arrowheads) can be seen attached to the surfaces of the alveolar bone. (F) Coronal sections of mandibles from neonatal rescued-knockout animals were stained for tartrate-resistant acid phosphatase activity. Tartrate-resistant acid phosphatase-positive cells (stained brown) are present in the alveolar bone immediately adjacent to the lateral aspect of the molars in a pattern indistinguishable from that found in wild-type littermates (not shown). Wt, wild type; Rc, rescued-knockout. [Bar = 64 μm in (A and B); 32 μm (C, D, and F), and 16 μm (E).]
The process of tooth eruption requires the resorption of the bone overlying the direction of tooth movement by activated osteoclasts; a similar process occurs at the base of the tooth to permit extension of the roots (19). The failure to form an eruption pathway in the rescued PTHrP-knockout animals, therefore, indicated a defect in either osteoclast formation or function. Abundant multinucleated osteoclasts were present throughout the mandibular bone of rescued-knockouts and could be observed along the alveolar bone surfaces surrounding both molars and incisors (arrows in Fig. 5E). In addition, tartrate-resistant acid phosphatase activity, a marker of osteoclast differentiation (20), identified mature osteoclasts immediately adjacent to teeth in the pre-eruptive phase of development (Fig. 5F). These findings, therefore, implicated a defect in osteoclast function, rather than an impairment of formation or differentiation, in the failure of eruption in the rescued-knockouts.
Localization of PTHrP and the Type I Receptor.
Murine first molars (M1) emerge through the oral epithelium between the 16th and 17th postnatal day, but the eruption process in these teeth begins by day 10 (18). In situ hybridization of normal molars at day 10 showed a high level of PTHrP mRNA in the coronal epithelial layers of M1, both in the outer enamel epithelium and in the adjacent stellate reticulum (Fig. 6). Type I receptor mRNA was strongly expressed in the alveolar bone surrounding M1 with the exception of the region immediately above the tooth where the bone was already resorbed. Expression of receptor mRNA was lower, but detectable, in the dental mesenchyme, both in the periphery of the pulp and in the follicle layer that lies between the outer enamel epithelium and the surrounding bone (17, 18). The development of the second molar (M2) lags behind that of M1 by several days (18) and so, at day 10, the stellate reticulum in M2 had just begun to acquire high levels of PTHrP mRNA expression (Fig. 6). The bony roof of the molar crypt above M2 was still intact at this point and thus exhibited strong receptor mRNA expression. The third molar (M3) is delayed by more than a week relative to M1 and expressed PTHrP only in the outer enamel epithelium (21). The sequential nature of molar growth thus provides a developmental time course, indicating a dramatic increase in PTHrP mRNA expression in the epithelial layers between the crown of the tooth and the overlying bone before the formation of the eruption pathway.
Figure 6.

In situ hybridization of a normal mandible sectioned sagittally through the molar crypt and hybridized with murine probes for the PTH/PTHrP receptor (A) and PTHrP (B). The receptor is found principally in bone (b), but can also be detected in the dental mesenchyme, both in the dental pulp (dp) and the dental follicle layer (df). PTHrP localizes to the outer enamel epithelium (oe) and the stellate reticulum (sr). Some enamel (e) remains after decalcification; d, dentin. [The scale bar represents 0.12 mm.]
Replacement of PTHrP in the Enamel Epithelium Restores Tooth Eruption.
Although the high level of PTHrP mRNA in the stellate reticulum before tooth eruption strongly suggested a role for PTHrP expression by this epithelial layer in the formation of the eruption pathway, the contribution of other cell types could not be excluded. Osteoblasts, for example, also express PTHrP (22) and are present in the alveolar bone surrounding the tooth. To establish an essential function for epithelially derived PTHrP in the process of tooth eruption, we attempted to replace PTHrP expression in the epithelial component of the developing tooth in PTHrP-null mice. For this purpose, we used a K14-PTHrP transgene that had been previously used to target the peptide to endogenous sites of PTHrP expression in the epithelia of the skin and the mammary gland (8, 9). Several cytokeratins, including K14, are expressed in the epithelial layers surrounding the developing tooth (23). As shown in Fig. 7A, immunohistochemical staining confirmed the presence of K14 expression throughout the layers of the reduced enamel epithelium in normal murine teeth before their eruption. This expression pattern replicates that for PTHrP at this stage of development (21) and thus provided the basis for the attempted phenotypic rescue.
Figure 7.
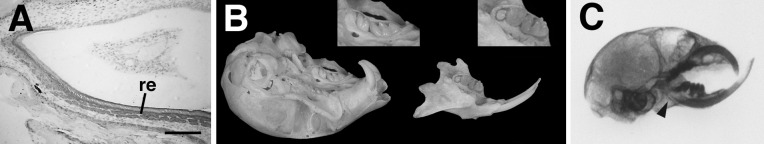
Dental phenotype of doubly rescued PTHrP-knockout mice. (A) Immunohistochemical staining for K14 (dark gray) in a sagittal section from a normal neonatal incisor localizes to the reduced enamel epithelium (re) on the labial surface. (B) Eruption of both incisors and molars (enlarged in Insets) is readily apparent in the maxilla and mandible of an adult doubly rescued animal. (C) A sagittal radiograph of an adult doubly-rescued PTHrP-knockout mouse clearly reveals the malocclusion of the incisors in the rescued-knockout caused by the underlying chondrodystrophy. Note the restoration of root extension in the lower incisor (arrowhead). (Bar = 27 μm.)
Breeding of the K14-PTHrP transgene onto a PTHrP-null background was carried out in a manner analogous to that used above. K14-PTHrP transgenic, PTHrP-null heterozygotes were first derived and then bred to the col II-PTHrP transgenic, PTHrP-null heterozygotes to obtain K14-PTHrP/col II-PTHrP double transgenic, PTHrP-null homozygotes at a frequency of 1 in 16. These doubly transgenic, rescued PTHrP-knockout mice were identical to their col II-PTHrP rescued counterparts with regard to growth rate, body size, life span, normalized neonatal skeleton and progressive chondrodystrophy. However, both incisors and molars were observed to erupt normally and on schedule in doubly rescued animals. Adult mice possessed complete sets of upper and lower dentition, although there was some crowding caused by the underlying chondrodystrophic distortion of the jaws (Fig. 7B). An unusual property of murine incisors, but not molars, is that they continue to grow throughout the animal’s lifetime and are impeded only by occlusion with the opposing teeth (19). As a result of the uncorrected chondrodystrophy in the skulls of doubly rescued mice, the lower, mandibular incisors protruded beyond the upper incisors in the foreshortened maxilla (Fig. 7C), and the resultant malocclusion allowed unchecked growth of the lower incisors. Upper incisors remained partially occluded and consequently were less affected.
DISCUSSION
Models of PTHrP overexpression have implicated this peptide as an attenuator of programmed differentiation in the epidermis, mammary gland, and endochondral bone (7, 8, 10), and disruption of the PTHrP gene has established an essential role for the peptide in prenatal skeletal development (9). The rescue of the PTHrP-knockout mouse has now allowed an assessment of the effects of the peptide’s absence on postnatal development. Rescued PTHrP-knockout animals manifested a number of developmental abnormalities, including dwarfing, craniofacial chondrodystrophy, and a failure of tooth eruption. In addition, these animals displayed a defect in the early branching morphogenesis of the mammary epithelium (12) and abnormal epidermal differentiation (unpublished observations).
Although of normal size at birth, rescued mice exhibited retarded growth by the time of weaning. It is possible that this failure to thrive may simply reflect an impaired ability to nurse caused by the distortion of the jaws, compounded by the absence of erupted incisors. Although a liquid diet proved essential for survival, rescued PTHrP-knockout mice nevertheless failed to achieve a normal growth rate after weaning, and displayed progressive stunting of the limbs. Schipani et al. (24) have also reported the correction of skeletal abnormalities in the PTHrP-knockout mouse, in this case by the chondrocyte-targeted expression of a constitutively-active type I PTH/PTHrP receptor, and noted premature closure of the epiphyseal growth plates in these animals; loss of endochondral growth plate cartilage was observed in the col II-PTHrP rescued-knockout mice as well (data not shown). Thus, whereas transgenic expression of PTHrP (or an activated receptor) targeted to proliferating and prehypertrophic chondrocytes can adequately compensate for the absence of the endogenous peptide during the early formation of the growth plate when these cell types are abundant, it appears to be quantitatively insufficient to maintain the measured rate of differentiation necessary to prevent the eventual disappearance of these cell layers in the mature growth plate. This loss of growth potential in the long bones ultimately results in short-limbed dwarfism.
Reduction or restriction in the size of the base of the skull and subsequent doming of the membranous bones of the cranial vault caused by continued growth of the brain are classical hallmarks of chondrodystrophy (16). In the PTHrP-knockout mouse, the premature ossification of the endochondral bones in the base of the skull is the likely cause of the evident cranial chondrodystrophy (9). Because the col II gene is expressed at high levels in the cartilage precursors of these bones (25), accelerated mineralization of the cranial base was essentially corrected in neonatal rescued PTHrP-knockout mice, and prominent doming of the skull was not apparent at this stage of development. As these animals matured, however, they displayed an increasingly chondrodystrophic cranial phenotype, presumably caused by a loss of growth potential and consequent size restriction in the endochondral bones in the base of the skull. The failure to correct the foreshortening of the maxilla and mandible in the rescued-knockout mice was anticipated because of the lack of col II expression in the cell types involved in the intramembranous ossification process at these sites (15, 25).
Finally, the rescued PTHrP-knockout mouse revealed an absolute requirement for PTHrP in the process of tooth eruption. Teeth appeared to develop normally but failed to escape from the surrounding bone or form roots and eventually became impacted, findings that are typically associated with osteopetrosis, a disorder of impaired osteoclast function. A limiting step in the process of tooth eruption is the resorption of bone overlying the crown to create an eruption pathway (19, 26). Before the scheduled eruption of the first molars in the rodent at 2 weeks of age, the secretion of colony-stimulating factor-1 by the dental follicle promotes an influx of mononuclear stem cells and a concomitant increase in the number of multinucleated osteoclasts in the coronal portion of the bony crypt, peaking at 3 days postnatally (27). Lesions in any of the principal stages of osteoclast development can disable the bone resorption process. For example, teeth fail to erupt in three of the murine models of osteopetrosis, the op/op mutant (which lacks colony-stimulating factor-1) and the c-fos and c-src knockouts, because of defects in osteoclast formation, differentiation, and activation, respectively (28–30). In the rescued PTHrP-knockout mouse, osteoclast formation and differentiation appeared to be normal, thus implicating a functional defect in the failure to create an eruption pathway. In this context then, PTHrP represents an essential signal for osteoclast activation that is distinct from the signal(s) mediating the migration and differentiation of osteoclast precursors. There is no evidence of generalized osteopetrosis, however, in the skeletons of PTHrP-knockout mice (9) or in adult PTHrP-null heterozygotes, which display abnormalities in trabecular bone due to haplotype insufficiency (31). Thus, the defect in bone resorption cannot be intrinsic to the osteoclast but must be caused by a deficiency specific to the microenvironment of the tooth.
Interestingly, osteoclasts surrounding the teeth of rescued PTHrP-knockout animals did not appear to be nonfunctional with regard to the normal turnover of alveolar bone. In contrast to the findings in the c-src-knockout mice (30), the alveolar bone in the rescued animals was not overtly osteopetrotic, and resident osteoclasts were found within resorption lacunae (see Fig. 5E). Under nonpathophysiological conditions, bone turnover is a precisely coordinated process, in which the rates of formation and resorption are balanced, or coupled, to maintain homeostasis of bone mass (32). The formation of the eruption pathway through the overlying bone, however, appears to represent an example of markedly uncoupled resorption, and it is this process that is defective in the rescued PTHrP-knockout mice. Although the capacity of PTH (and PTHrP) to increase osteoclast activity and promote bone resorption is well established (33), osteoclasts do not possess functional PTH/PTHrP receptors, and their activation appears to be mediated indirectly through a paracrine or juxtacrine interaction with osteoblasts or other receptor-bearing cells in bone (20, 32–34). Given the restriction of the defect in bone resorption in the rescued PTHrP-knockout animals to the microenvironment of the tooth, however, PTH/PTHrP receptor-bearing cells in the dental follicular mesenchyme must also be considered as a potential source of a PTHrP-induced activity that augments osteoclastic bone resorption. Such a role for PTHrP would correspond to a true “PTH-like” function for this peptide in vivo, which would be in accordance with the conserved amino terminal domains of the two proteins and their shared use of a single receptor.
Acknowledgments
We thank A. E. Broadus and J. Wysolmerski for insight and advice, and J.-P. Zhang for expert technical assistance. We are extremely grateful to R. Baron and S. Marks, Jr., for sharing their expertise in osteoclast biology and tooth eruption, respectively. This work was supported by National Institutes of Health Grants AR30102, DK48108, and DK07058, and was facilitated by the Molecular Core of the Yale Diabetes Endocrine Research Center (National Institutes of Health Grant DK45735).
ABBREVIATIONS
- PTH
parathyroid hormone
- PTHrP
PTH-related protein
- K14
keratin 14
- col II
procollagen II
References
- 1. Broadus A E, Stewart A F. In: The Parathyroids. Bilezekian J P, Levine M A, Marcus R, editors. New York: Raven; 1994. pp. 259–294. [Google Scholar]
- 2.Jüppner H, Abou-Samra A-B, Freeman M, Kong X F, Schipani E, Richards J, Kolakowski L F, Hock J, Potts J T, Kronenberg H M, et al. Science. 1991;254:1024–1026. doi: 10.1126/science.1658941. [DOI] [PubMed] [Google Scholar]
- 3.Abou-Samra A-B, Jüppner H, Force T, Freeman M W, Kong X-F, Schipani E, Urena P, Richards J, Bonventre J V, Potts J T, Jr, Kronenberg H M, et al. Proc Natl Acad Sci USA. 1992;89:2732–2736. doi: 10.1073/pnas.89.7.2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Campos R V, Asa S L, Drucker D J. Cancer Res. 1991;51:6351–6357. [PubMed] [Google Scholar]
- 5.Lee K, Deeds J D, Segre G V. Endocrinology. 1995;136:453–463. doi: 10.1210/endo.136.2.7835276. [DOI] [PubMed] [Google Scholar]
- 6.Philbrick W M, Wysolmerski J J, Galbraith S, Orloff J J, Yang K H, Vasavada R C, Weir E C, Broadus A E, Stewart A F. Physiol Rev. 1996;76:127–173. doi: 10.1152/physrev.1996.76.1.127. [DOI] [PubMed] [Google Scholar]
- 7.Wysolmerski J J, Broadus A E, Zhou J, Fuchs E, Milstone L M, Philbrick W M. Proc Natl Acad Sci USA. 1994;91:1133–1137. doi: 10.1073/pnas.91.3.1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wysolmerski J J, McCaughern-Carucci J F, Daifotis A G, Broadus A E, Philbrick W M. Development (Cambridge, UK) 1995;121:3539–3547. doi: 10.1242/dev.121.11.3539. [DOI] [PubMed] [Google Scholar]
- 9.Karaplis A C, Luz A, Glowacki J, Bronson R J, Tybulewicz V L J, Kronenberg H M, Mulligan R C. Genes Dev. 1994;8:277–289. doi: 10.1101/gad.8.3.277. [DOI] [PubMed] [Google Scholar]
- 10.Weir E C, Philbrick W M, Amling M, Neff L A, Baron R, Broadus A E. Proc Natl Acad Sci USA. 1996;93:10240–10245. doi: 10.1073/pnas.93.19.10240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schipani E, Kruse K, Jüppner H. Science. 1995;268:98–100. doi: 10.1126/science.7701349. [DOI] [PubMed] [Google Scholar]
- 12.Wysolmerski J J, Philbrick W M, Dunbar M E, Lanske B, Kronenberg H, Karaplis A, Broadus A E. Development (Cambridge, UK) 1998;125:1285–1294. doi: 10.1242/dev.125.7.1285. [DOI] [PubMed] [Google Scholar]
- 13.Baron R A, Vignery A, Neff L, Silverglate A, Santa Marie A. In: Bone Histomorphometry: Techniques and Interpretation. Recker R R, editor. Boca Raton, FL: CRC; 1983. pp. 13–35. [Google Scholar]
- 14.Hefti E, Trechel U, Rufenacht H, Fleisch H. Calcif Tissue Int. 1980;31:44–47. doi: 10.1007/BF02407166. [DOI] [PubMed] [Google Scholar]
- 15.Thorogood P. In: The Skull, Volume 1: Development. Hanken J, Hall B K, editors. Chicago: Univ. of Chicago Press; 1993. pp. 112–152. [Google Scholar]
- 16.Johnson D R. In: The Skull, Volume 1: Development. Hanken J, Hall B K, editors. Chicago: Univ. of Chicago Press; 1993. pp. 248–288. [Google Scholar]
- 17.Hay M F. Arch Oral Biol. 1961;3:86–109. doi: 10.1016/0003-9969(61)90142-x. [DOI] [PubMed] [Google Scholar]
- 18.Cohn S A. Am J Anat. 1957;101:295–319. doi: 10.1002/aja.1001010205. [DOI] [PubMed] [Google Scholar]
- 19.Marks S C, Jr, Schroeder H E. Anat Rec. 1996;245:374–393. doi: 10.1002/(SICI)1097-0185(199606)245:2<374::AID-AR18>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 20.Roodman G D. Endocr Rev. 1996;17:308–332. doi: 10.1210/edrv-17-4-308. , 1996. [DOI] [PubMed] [Google Scholar]
- 21.Beck F, Tucci J, Russell A, Senior P V, Ferguson M W J. Cell Tissue Res. 1995;280:283–290. doi: 10.1007/BF00307800. [DOI] [PubMed] [Google Scholar]
- 22.Suda N, Gillespie M T, Traianedas K, Zhou H, Ho P M N, Hards D K, Allan E H, Martin T J, Moseley J M. J Cell Physiol. 1996;166:94–104. doi: 10.1002/(SICI)1097-4652(199601)166:1<94::AID-JCP11>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 23.Feghali-Assaly M, Sawal M H, Serres G, Forrest N, Ouhayoun F P. J Periodont Res. 1994;29:185–195. doi: 10.1111/j.1600-0765.1994.tb01212.x. [DOI] [PubMed] [Google Scholar]
- 24.Schipani E, Lanske B, Hunzelman J, Luz A, Kovacs C S, Lee K, Pirro A, Kronenberg H, Jüppner H. Proc Natl Acad Sci USA. 1997;94:13689–13694. doi: 10.1073/pnas.94.25.13689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Metsaranta M, Garofolo S, Smith C, Vuorio E. Dev Dyn. 1995;204:202–210. doi: 10.1002/aja.1002040211. [DOI] [PubMed] [Google Scholar]
- 26.Marks S C., Jr Connect Tissue Res. 1995;32:149–157. doi: 10.3109/03008209509013718. [DOI] [PubMed] [Google Scholar]
- 27.Wise G E, Lin F. J Dent Res. 1995;74:303–306. doi: 10.1177/00220345950740010301. [DOI] [PubMed] [Google Scholar]
- 28.Yoshida H, Hayashi S, Kunisada T, Ogawa M, Nishikawa S, Okamura H, Sudo T, Schultz L D, Nishikawa S. Nature (London) 1990;345:442–444. doi: 10.1038/345442a0. [DOI] [PubMed] [Google Scholar]
- 29.Grigoriadis A E, Wang Z-Q, Cecchini M G, Hofstetter W, Felix R, Fleisch H A, Wagner E F. Science. 1994;266:443–448. doi: 10.1126/science.7939685. [DOI] [PubMed] [Google Scholar]
- 30.Lowe C, Yoneda T, Boyce B F, Chen H, Mundy G R, Soriano P. Proc Natl Acad Sci USA. 1993;90:4485–4489. doi: 10.1073/pnas.90.10.4485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Amizuka N, Karaplis A C, Henderson J E, Warshawsky H, Lipman M L, Matsuki Y, Ejiri S, Tanaka M, Izumi N, Ozawa H, Goltzman D. Dev Biology. 1996;175:166–176. doi: 10.1006/dbio.1996.0104. [DOI] [PubMed] [Google Scholar]
- 32.Martin T J, Ng K W, Suda T. In: Metabolic Bone Disease, Part I. Tiegs R D, editor. Philadelphia: Saunders; 1989. pp. 833–858. [Google Scholar]
- 33.Mundy G R, Roodman G D. In: Bone and Mineral Research. Peck W A, editor. Vol. 5. Amsterdam: Elsevier; 1987. pp. 209–280. [Google Scholar]
- 34.Rodan G A, Martin T J. Calcif Tissue Int. 1981;33:349– 351. doi: 10.1007/BF02409454. [DOI] [PubMed] [Google Scholar]