

Abstract
While evidence is accumulating that phosphoinositide signaling plays a crucial role in growth factor and hormone receptor down-regulation, this signaling pathway has also been proposed to regulate endosomal membrane transport and multivesicular endosome biogenesis. Here, we have followed the fate of the down-regulated EGF receptor (EGFR) and bulk transport (fluid phase) markers in the endosomal pathway in vivo and in vitro. We find that bulk transport from early to late endosomes is not affected after inhibition of the phosphatidylinositol-3-phosphate (PI3P) signaling pathway, but that the EGFR then remains trapped in early endosomes. Similarly, we find that hepatocyte growth factor–regulated tyrosine kinase substrate (Hrs) is not directly involved in bulk solute transport, but is required for EGFR sorting. These observations thus show that transport and sorting can be uncoupled in the endosomal pathway. They also show that PI3P signaling does not regulate the core machinery of endosome biogenesis and transport, but controls the sorting of down-regulated receptor molecules in early endosomes via Hrs.
Keywords: EGF receptor; phosphoinositide; FYVE; PHOX and PX; multivesicular body
Introduction
Recent studies have highlighted the role of phosphatidylinositol-3-phosphate (PI3P) signaling in growth factor and hormone receptor down-regulation (Simonsen et al., 2001). Additionally, this signaling pathway has also been proposed to regulate endosomal membrane transport and multivesicular endosome biogenesis (Reaves et al., 1996; Fernandez-Borja et al., 1999; Odorizzi et al., 2000; Futter et al., 2001). In addition to PI 3-kinase, candidate effectors of this PI3P signaling pathway include phosphoinositide-binding proteins, including the PI3P 5-kinase Fab1p (Odorizzi et al., 1998), hepatocyte growth factor–regulated tyrosine kinase substrate (Hrs) in conjunction with other class E proteins (Bishop et al., 2002; Raiborg et al., 2002), and members of the SNX family (Haft et al., 1998; Sato et al., 2001). The precise functions of these proteins in protein trafficking and membrane transport are still unclear. More importantly, it is not known to what extent this signaling pathway as such plays a role in transport, protein sorting, or endosome biogenesis. Here, we report that bulk transport from early to late endosomes, including biogenesis of endosomal intermediates and their subsequent fusion with late endosomes, does not depend on PI3P signaling, but that phosphoinositides tightly control the sorting of down-regulated EGF receptor (EGFR) in early endosomes via Hrs.
Results and discussion
Bulk transport from early to late endosomes was followed with soluble markers that are incorporated nonselectively within the endosomal content, e.g., fluorescent dextran or HRP, whereas the EGFR was used as a reporter for the trafficking of down-regulated receptors. Since solutes may follow more than one endocytic pathways (Nichols and Lippincott-Schwartz, 2001), the fate of endocytosed dextran was first compared with that of down-regulated EGFR after EGF treatment. After 10 min, both markers colocalized in early endosomes (Fig. 1 A), containing the Rab5 effector early endosomal antigen 1 (EEA1) (Simonsen et al., 1998). After subsequent incubation for 45 min (not shown) or 90 min at 37°C, both EGFR and its ligand EGF reached vesicles that contained the late endosomal-specific lipid, lysobisphosphatidic acid (LBPA) (Kobayashi et al., 1998) (not shown) or Lamp1 (Fig. 1 B), a very abundant protein of late endosomes and lysosomes (Aniento et al., 1993a; Griffiths et al., 1988). Similarly, under the same conditions, dextran reached vesicles that contained Lamp1 (not shown) or LBPA (Fig. 1 C). Hence, both solutes and EGFR follow the same pathway to early endosomes and then to late endocytic compartments.
Figure 1.
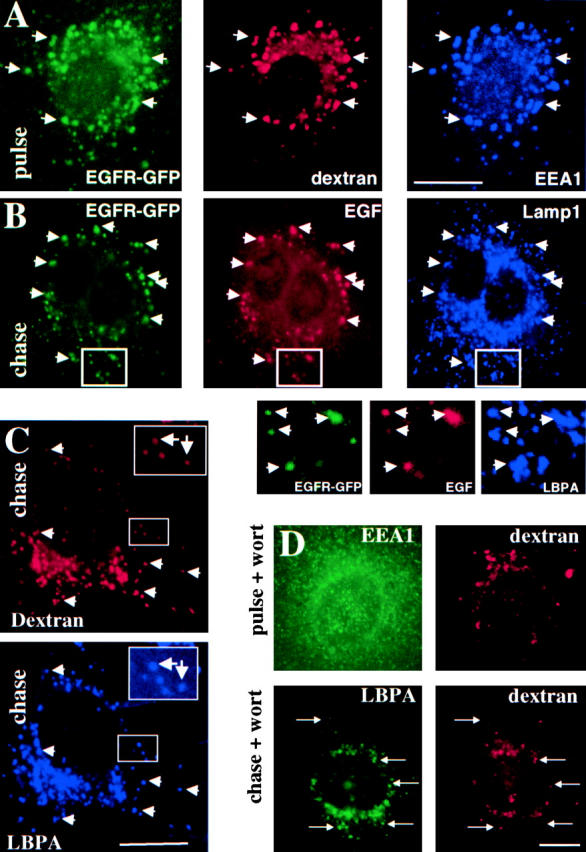
Transport from early to late endosomes after PI3 kinase inhibition. (A) BHK cells expressing EGFR-GFP were preincubated with EGF for 1 h at 4°C and washed; this pretreatment was always used for EGF or EGFR endocytosis. Cells were incubated for 10 min at 37°C with rhodamine-dextran (pulse), labeled with antibodies against the indicated antigens, and analyzed by triple channel fluorescence microscopy. (B) BHK cells overexpressing EGFR-GFP were incubated with EGF-biotin and streptavidin-phycoerythrin for 1 h at 4°C and then for 10 min at 37°C, followed by a 90-min chase in the presence of 0.5 mg/ml leupeptin and analyzed as in described for A; high magnification views of the indicated areas are shown below the micrographs. (C) A pulse of rhodamine dextran was endocytosed as described for A and then chased for 45 min; cells were analyzed as described for A. (D) A pulse of rhodamine dextran was endocytosed as described for A into BHK cells with or without chase (as in C); 100 nM wortmannin (Wort) was present during both the pulse and the chase. Cells were analyzed as described for A. Bars: (A–C) 5 μm; (D) 2.5 μm.
When transport from early to late endosomes is inhibited, HRP is regurgitated into the medium and fails to accumulate intracellularly (Clague et al., 1994; Gu et al., 1997; Mayran et al., 2003). In contrast, when PI3 kinase was inhibited with wortmannin, although HRP internalization was decreased, accumulation was not affected (not shown), suggesting that transport from early to late endosomes does not depend on PI3P signaling. Indeed, PI3 kinase inhibition did not affect transport of dextran to late endosomes containing LBPA (Fig. 1 D, and see quantification in Fig. 3 C) or Lamp1 (see Fig. 3 A). As a control, we confirmed that wortmannin did however lead to the release of EEA1 from early endosomes (Fig. 1 D), and reduced the PI3P content of early endosomal fractions (not shown). To interfere more specifically with PI3P-dependent functions, we used a double FYVE PI3P-binding domain (2xFYVE) that binds PI3P with high specificity (not shown) and inhibits early endosome fusion in vitro (see Fig. 4 B), as expected (Gillooly et al., 2000). When linked to GFP, 2xFYVE colocalized with dextran internalized for 10 min and EEA1 on early endosomes (Fig. 2 A). In agreement with the lack of effect of wortmannin, GFP-2xFYVE did not affect dextran transport to late endosomes (Fig. 2 A, and quantification in Fig. 3 C). Similarly, endocytosed mouse IgGs, a fluid phase marker, were transported to lysosomes and degraded whether or not GFP-2xFYVE was expressed, whereas IgGs accumulated intracellularly when lysosomal degradation was blocked with leupeptin (Fig. 2 B). We thus concluded that PI3P signaling is not involved in bulk endosomal membrane transport.
Figure 3.


PI3P signaling regulates EGFR sorting in early endosomes. (A) Cells expressing EGFR-GFP (pretreated with EGF, as in Fig. 1 A) were incubated for 10 min at 37°C with rhodamine-dextran, followed by a 90-min chase with 100 nM wortmannin, labeled with antibodies against the indicated antigens and analyzed by triple channel fluorescence. (B) Cells transfected with GFP-2xFYVE were incubated with EGF-biotin and streptavidin-phycoerythrin for 1 h at 4°C and then for 10 min at 37°C followed (chase) or not (pulse) by a 90-min chase in the presence of leupeptin, labeled with antibodies against the indicated antigens and analyzed by triple channel fluorescence. (C) Cells expressing GFP-2xFYVE (FYVE), or treated (wort) or not treated (control: ctrl) with wortmannin were labeled with endocytosed EGF-biotin/streptavidin-phycoerythrin (as in B) or dextran (as in A), except that the chase time period was 45 min, and then processed for immunofluorescence using antibodies against the early endosomal (EE) marker EEA1 or the late endosomal (LE) marker LBPA, as indicated. For each condition, the total number of vesicles labeled with EGF or dextran was counted from ≥15 cells in three different experiments (expressed as 100%), as well as the percentage of these vesicles that also contained the EE (EEA1 in control cells; FYVE in FYVE-expressing cells) or the LE endosomal marker LBPA (except with wortmannin, since the drug releases EEA1). (D) After EGF pretreatment (as in Fig. 1 A), cells overexpressing EGFR-GFP were incubated for 10 min at 37°C followed (chase) or not (pulse) by a 35-min chase with wortmannin (wort) and processed for immunofluorescence. (E) The number of vesicles in D that contained both GFP-EGFR and TfR was counted, and is expressed as a percentage of the total number of GFP-EGFR–positive vesicles (C). Bars: (A and B) 2.5 μm; (D) 5 μm.
Figure 4.

PI3P signaling is not necessary for ECV biogenesis. (A) The different transport steps that were studied in vitro are shown with reference to the corresponding panels: homotypic early endosome (EE) fusion (B), ECV biogenesis (C), and ECV fusion with late endosomes (LE) (D). ECVs lose the capacity to fuse with EEs in vitro (E) and acquire the capacity to fuse with LEs (D). (B–F) The homotypic fusion of early endosomes (B), the formation of ECVs (C), and the fusion of ECVs formed in vitro with late (D) or early (E) endosomes were measured in vitro in the absence (control) or presence of 100 nM wortmannin, 3 μg/100 μg endosomal protein GST-2xFYVE or C215S mutant, and ATP, as indicated. Note the relatively high efficiency of ECV fusion with late (D) but not early (E) endosomes, when compared with the efficiency of ECV formation (C). (F) Early endosomes were incubated with or without GST-2xFYVE and analyzed by Western blotting using antibodies against the indicated proteins.
Figure 2.

GFP-2xFYVE does not inhibit bulk transport to late endosomes in vivo. (A) As in Fig. 1 A, a pulse of rhodamine dextran was endocytosed with or without chase in cells expressing a double FYVE domain associated to GFP (GFP-2xFYVE). Cells were analyzed as described in the legend to Fig. 1 A. (B) Cells expressing or not expressing GFP-2xFYVE were incubated overnight with 40 μg/ml mouse IgGs with or without 50 μg/ml leupeptin. Endocytosed IgGs were revealed using anti-mouse antibodies and cells were analyzed by double channel fluorescence microscopy. The total number of labeled vesicles (containing IgGs in red) was counted for each condition in control cells or cells expressing GFP-2xFYVE. Vesicles in ≥20 cells were counted for each condition in three different experiments. Numbers were similar whether GFP-2xFYVE was present or not, and are expressed as a percentage of the total number of vesicles in leupeptin-treated cells, to facilitate direct comparison. Bar, 2.5 μm.
In marked contrast to our observations with solutes, EGFR endocytosed for 45 min (Fig. 3 C) or even for 90 min after EGF addition failed to reach late endocytic compartments containing LBPA (not shown) or Lamp1 (Fig. 3 A) after inhibition of PI3 kinase with wortmannin. Then, EGFR was trapped in early/recycling endosomes containing the transferrin receptor (TfR) (Fig. 3 A and D, and quantification in E). Solute transport was unaffected in the same cells (Fig. 3 A). Similarly, endocytosed EGF was not transported to LBPA-containing late endosomes in cells expressing GFP-2xFYVE, but was trapped within early endosomes, containing both EEA1 (not shown) and GFP-2xFYVE (Fig. 3 B, and quantification in C). In a previous study, the lysosomal degradation of EGFR preinternalized at 20°C was reported to be insensitive to wortmannin (Futter et al., 2001), perhaps because EGFR had been transported beyond the early endosomal sorting checkpoint before drug addition (Griffiths et al., 1988).
These differences between solute and receptor trafficking prompted us to investigate the role of PI3P signaling in transport from early to late endosomes in more detail. To this end, we used a well-established assay that reconstitutes the formation of the transport intermediates (multivesicular bodies or endosomal carrier vesicles [ECVs]) from donor early endosomes (Aniento et al., 1996). In this assay, the content of early endosomes is labeled with the solute HRP, and early endosomes are prepared. After in vitro incubation, newly formed ECVs exhibit a lower density than donor early endosomal membranes and are conveniently separated by floatation in gradients. The efficiency of vesicle formation is then calculated from the amounts of HRP present in donor endosomes and vesicles formed in vitro. We confirmed with an established fusion assay that ECVs formed in vitro were functional (Aniento et al., 1993a; Gruenberg et al., 1989), since they lost the capacity to fuse with early endosomes (Fig. 4 E, and outline in A) in contrast to donor membranes (Fig. 4 B, and outline in A), but acquired the capacity to fuse with late endosomes (Fig. 4 D, and outline in A).
Using this assay, we found that the in vitro biogenesis of ECVs was not affected by wortmannin or by GST-2xFYVE (Fig. 4 C). Nor was the fusion of ECVs with late endosomes (Fig. 4 D). As a control, we confirmed that both wortmannin and GST-2xFYVE, but not the inactive C215S mutant, inhibited the homotypic fusion of early endosomes (Gruenberg et al., 1989) (Fig. 4 B), as expected, since the Rab5 effector EEA1 was released from endosomes by GST-2xFYVE (Fig. 4 F). These observations thus demonstrate that PI3P signaling is not required for ECV biogenesis in vitro and subsequent docking/fusion with late endosomes.
We then used the same assay, but with the EGFR as a marker instead of HRP. EGF-induced endocytosis of the receptor was allowed to proceed for a few minutes to accumulate EGFR in early endosomes. These were then purified (Fig. 5 A) and used as donor membranes. In a highly selective manner, EGFR was incorporated into newly formed ECVs (Fig. 5 C), in contrast to TfR, which remained in donor membranes (Fig. 5 B). Interestingly, incorporation of EGFR into newly formed ECVs was significantly reduced when GST-2xFYVE, but not the inactive C215S mutant, was added to the assay (Fig. 5 C). GST-2xFYVE was recruited by donor membranes (Fig. 5 D) and to a lesser extent by ECVs depleted in EGFR (Fig. 5 C), consistently with the distribution of PI3P on early endosomes and ECVs (Gillooly et al., 2000). Similarly, EGFR incorporation into newly formed ECVs was also inhibited by wortmannin (Fig. 5 C). The selective inhibition of EGFR, but not HRP, incorporation into ECVs by GST-2xFYVE, but not by the C215S mutant, could be reproduced in vitro using donor membranes containing both endocytosed HRP and EGFR (not shown).
Figure 5.

PI3P signaling regulates EGFR sorting. (A) EGF was pulsed for 10 min at 37°C in cells expressing EGFR-GFP (pretreated with EGF, as in Fig. 1 A). Early endosome (EE), late endosome (LE), and heavy membrane (HM) fractions (Aniento et al., 1993a) were then analyzed by Western blotting using anti-GFP antibodies. (B–D) The in vitro formation of ECVs was measured (as in Fig. 4 C) using donor early endosomes from cells expressing EGFR-GFP (as in A) in the presence or absence of ATP, wortmannin (Wort), GST-2xFYVE, or C215S mutant. Fractions containing donor early endosomes (EE) and vesicles formed in vitro were analyzed by Western blotting using antibodies against the indicated antigens. In B and D, 50 and 10% of the donor membranes (EE), respectively, were analyzed. Scanning of the lanes showed that, within the 30-min incubation of the assay, ∼10% of the EGFR present in untreated donor membranes was incorporated into newly formed ECVs. This value is somewhat lower than EGF transport to late endosomes in vivo (∼60% after 45 min; Fig. 3 C), in part because one round of vesicle formation only is reconstituted in vitro (Aniento et al., 1993a).
In our in vitro assay, EGFR and HRP were incorporated into newly formed ECVs with a similar efficiency (Fig. S3, available online at http://www.jcb.org/cgi/content/full/jcb.200303018/DC1). Similarly, in vivo values for EGFR and dextran were also in the same range. Clearly, however, sorting and transport in vitro occurred with a somewhat reduced efficiency (∼30%), when compared with the in vivo situation, as expected. More importantly, EGFR sorting was inhibited to the same extent by wortmannin and 2xFYVE in vivo as well as in vitro, whereas these treatments did not affect solute transport. Altogether, these data demonstrate that EGFR sorting into ECVs, but not solute transport, is inhibited when interfering with PI3P signaling.
Wortmannin was previously reported to inhibit MVB morphogenesis by interfering with the membrane invagination process, thereby causing a vacuolation of early and late endosomes (Reaves et al., 1996; Fernandez-Borja et al., 1999; Futter et al., 2001). Under our experimental conditions, however, wortmannin did not affect the appearance of endosomes (Fig. 3), nor the amounts and distribution of LBPA, a marker of late endosome invaginations (Kobayashi et al., 1998) (Figs. S1 and S2, available online at http://www.jcb.org/cgi/content/full/jcb.200303018/DC1).
Hrs is likely to mediate the PI3P-dependent sorting of EGFR into ECVs. Indeed, Hrs contains a FYVE domain, is wortmannin sensitive and is involved in EGF down-regulation, presumably by linking ubiquitinated EGFR with endosomal clathrin (Bishop et al., 2002; Lloyd et al., 2002; Raiborg et al., 2002). Hrs overexpression was previously found either to inhibit (Bean et al., 2000) or not inhibit (Raiborg et al., 2001) the recycling pathway, and to interfere with EGFR and dextran transport from early to late endosomes (Raiborg et al., 2001). After low Hrs-myc overexpression to limit problems associated with high overexpression, EGFR remained trapped in early endosomes containing Hrs-myc, and failed to reach late endosomes (Fig. 6 B, and quantification in A), in agreement with Raiborg et al. (2001). Dextran, however, did not accumulate in Hrs-myc–labeled early endosomes and reached late endosomes containing LBPA. This apparent discrepancy with Raiborg et al. (2001), like perhaps the differential effects of Hrs on the recycling pathway (Bean et al., 2000; Raiborg et al., 2001), may well reflect differences in the levels of Hrs overexpression.
Figure 6.
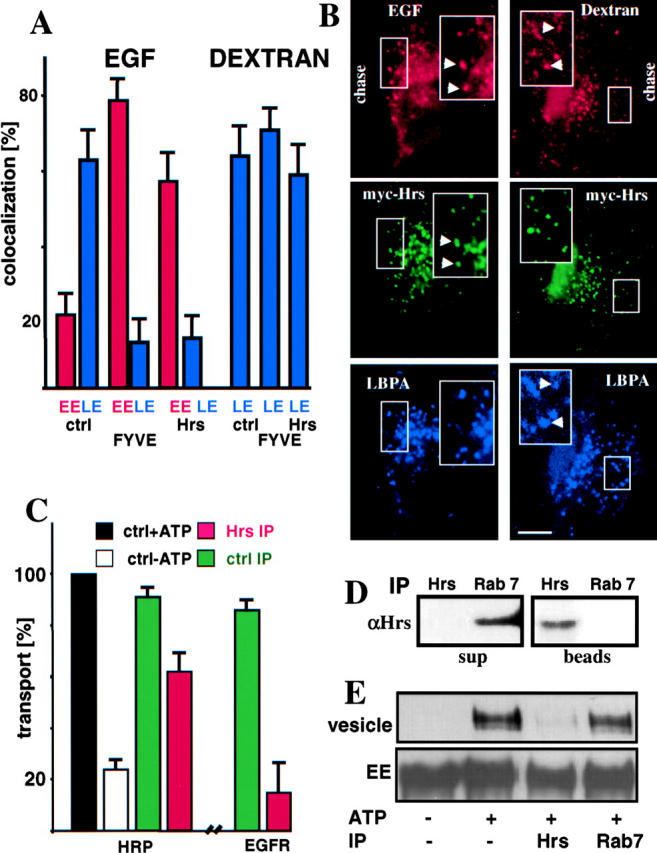
Hrs regulates EGFR sorting, but not bulk transport to late endosomes. (A and B) As in Fig. 3 A and B, control cells (ctrl) or cells expressing either GFP-2xFYVE (FYVE) or myc-Hrs were labeled with EGF-biotin/streptavidin-phycoerythrin or dextran, except that the marker was endocytosed for 45 min followed by a 60-min chase, and analyzed by triple channel fluorescence microscopy (B). In A, the colocalization of dextran or EGF with the early endosomal marker (EE, GFP-2xFYVE or myc-Hrs) or the late endosomal marker (LE, LBPA) was quantified as in Fig. 3 C. (C) ECV formation in vitro was measured with HRP as a marker, as in Fig. 4, using cytosol depleted of Hrs by immunoprecipitation (Hrs IP), or mock-treated with a control anti-Rab7 antibody (ctrl IP). (D) These cytosols (after Hrs immunoprecipitation or control immunoprecipitation, as in C) were analyzed by SDS electrophoresis and Western blotting with anti-Hrs (αHrs) antibodies. (E) The incorporation of GFP-EGFR into ECVs was followed as in Fig. 5, using Hrs-depleted or mock-treated (Rab7) cytosol (as in C). GFP-EGFR was analyzed in donor membranes (EE; 50% of the donor membranes) and vesicles (whole fraction) formed in vitro (vesicles) by Western blotting with anti-GFP antibodies. The lanes were scanned, and the quantification is shown in C, as a percentage of the total amounts of GFP-EGFR in the vesicle fraction of controls. Bar, 2.5 μm.
We therefore used our in vitro transport assay to further investigate the role of Hrs in ECV biogenesis. Fig. 6 D shows that cytosol depleted of Hrs by immunoprecipitation failed to support EGFR incorporation into newly formed ECVs (Fig. 6 E, and quantification in C). Cytosol depleted of Rab7 (not shown), used as a control, was without effect (Fig. 6, C and E). In contrast, the in vitro biogenesis of ECVs labeled with HRP was only marginally affected by Hrs immunodepletion (Fig. 6 C). This agrees with the findings that, despite defective membrane invagination, solute transport occurs along the degradation pathway of hrs mutant Drosophila cells at a rate similar to wild-type cells (Lloyd et al., 2002). Altogether, our in vivo and in vitro data indicate that Hrs, like PI3P signaling, regulates EGFR, but not solute, trafficking along the degradation pathway, strongly suggesting that Hrs mediates PI3P-dependent EGFR sorting into ECVs.
In conclusion, our data show that bulk transport from early to late endosomes in vivo and biogenesis of functional endosomal intermediates in vitro are not affected, when interfering with the PI3P signaling pathway or with Hrs functions. In marked contrast, EGFR then remains trapped within early/recycling endosomes, and fails to be transported to late endosomes in vivo or to be incorporated into newly formed endosomes in vitro. Similarly, the ubiquitination machinery, which regulates EGFR sorting, does not play a role in solute transport along the degradation pathway (van Kerkhof et al., 2001). Our data thus show that membrane transport and receptor sorting can be uncoupled in the endosomal pathway. They also demonstrate that, although PI3P signaling does not regulate the core machinery of endosome transport, it controls the sorting of down-regulated receptor molecules in early endosomes, presumably via Hrs.
Materials and methods
Reagents
Wortmannin, EGF, and mouse IgG were from Sigma-Aldrich; fugene and leupeptin were from Roche Diagnostic; anti-TfR antibody was from Zymed Laboratories; anti-EEA1 antibody was from BD Bioscience; EGF-biotin and 10-kD rhodamine-dextran were from Molecular Probes; and rhodamine-labeled or AMCA-labeled anti–mouse IgGs were from Jackson ImmunoResearch Laboratories. pGEX-2xFYVE and pGEX-2xFYVEC215S have been described (Gillooly et al., 2000), as well as anti-LBPA antibodies (Kobayashi et al., 1998), and anti-Hrs antibodies and Hrs-myc (Raiborg et al., 2001). EGFR-GFP was from Alexander Sorkin (University of Colorado, Denver, Colorado).
In vivo experiments
When indicated, BHK-21 (Gruenberg et al., 1989) and HeLa cells (Rojo et al., 1997) were transfected with fugene 36 h before the experiment. To label early or late endosomes, cells were incubated for 10 min at 37°C with 3 mg/ml rhodamine-dextran (pulse) or further incubated without the marker (chase) for the indicated time period, with or without 100 nM wortmannin (for long incubations, the drug was always added freshly after 30 min). Alternatively, cells were first incubated for 1 h at 4°C with EGF-biotin and streptavidin-phycoerythrin. The marker was endocytosed with the same pulse–chase protocole as mentioned here. For quantification, cells expressing or not expressing GFP-2xFYVE were separately incubated with EGF-biotin/streptavidin-phycoerythrin (as described here), or with rhodamine-dextran for 10 min, followed by a 45- or 60-min chase, and processed for immunofluorescence. When indicated, cells were incubated for 16 h at 37°C with 40 µg/ml mouse IgG with or without 50 µg/ml leupeptin. An EGFR-GFP cell line was obtained after transfection of BHK cells using a lentiviral vector (Trono, 2002) with a construct encoding the EGFR fused to GFP (Carter and Sorkin, 1998) under the control of a tetracycline promoter. The clonal population was obtained by limited dilution after a first selection by FACS.
Transport assays
Early and late endosomes were separated by floatation in a step sucrose gradient (Aniento et al., 1993a; Gorvel et al., 1991), and recovered at d ≈ 1.1031g/cm3 (35%/25% sucrose interface) and d ≈ 1.0772g/cm3 (25%/8.5% sucrose), respectively. Cytosol was prepared from rat liver (Aniento et al., 1993b), aliquoted, frozen, and stored in liquid nitrogen. When indicated, the cytosol was depleted of Hrs by immunoprecipitation, or mock-treated with anti-Rab7 as a control (Aniento et al., 1993a). The homotypic fusion activity of early endosomes was measured after mixing in the assay two early endosome populations, which had been separately labeled with endocytosed biotinylated HRP and avidin, respectively (Aniento et al., 1993a; Gruenberg et al., 1989). The mixture was incubated with ATP and cytosol for 45 min at 37°C with or without PI3 kinase inhibitors or 3 μg recombinant protein/100 μg endosomal protein. The avidin–biotinylated HRP (bHRP) complex formed upon fusion was immunoprecipitated with anti-avidin antibodies, and the enzymatic activity of bHRP was quantified and expressed as a percentage of the maximal amount of avidin–bHRP complex formed after detergent solubilization (efficiency).
We measured the formation of ECVs from donor early endosomal membranes, using endocytosed HRP as a marker of the endosomal content, as described (Aniento et al., 1996; Gu and Gruenberg, 2000). In the assay, donor early endosomes were incubated for 30 min at 37°C with ATP and cytosol, with or without drugs or recombinant proteins, as above. ECVs formed in vitro were separated by floatation in a sucrose gradient, and the HRP content of both ECVs and donor membranes was quantified. The efficiency of the reaction was calculated as the percentage of total HRP present in ECVs formed in vitro. To measure the fusion activity of ECVs formed in vitro with late endosomes, ECVs were prepared using biotinylated HRP instead of HRP (Aniento et al., 1996; 1993b). The fusion assay was carried out as described above for early endosomes, except that avidin was endocytosed for 5 min followed by a 40-min chase, and then late endosomal fractions were used in the assay. To study transport of EGFR-GFP in vitro, cells were pretreated with 20 μg/ml brefeldin A for 1 h at 37°C, to deplete early endosomes from Golgi markers (Gu et al., 1997; Rojo et al., 1997). Cells were incubated for 1 h at 4°C with 0.5 μg/ml of EGF and 20 μg/ml brefeldin A, and then for 10 min at 37°C, and fractionated as above. In vitro formation of ECVs was as described here, except that cytosol and donor membranes were reduced 10-fold and the volume of the reaction was adjusted. Donor membranes and ECVs formed in vitro were analyzed by SDS gel electrophoresis and Western blotting.
Other methods
The 2x FYVE motif was isolated from the pGEMEGFP 2xFYVE (Gillooly et al., 2000) after an EcoRI-SalI digestion and introduced into the peGFPC2. Recombinant GST-2xFYVE and C215S mutant were produced in bacteria and purified (Cavalli et al., 2001). ELISA of LBPA in fractions was as described using Immulon I 96-well plates (Kobayashi et al., 1998). Quantification of protein was according to Bradford (1976). Western blot analysis was carried out using peroxidase-conjugated sheep anti–mouse or goat anti–rabbit IgG as secondary antibodies and detected by chemiluminescence using the ECL reagent (Amersham Biosciences). Immunofluorescence was as described (Rojo et al., 1997).
Online supplemental material
Figs. S1 and S2 illustrate our observations that LBPA is not affected by PI3-kinase inhibition, and Fig. S3 includes detailed quantification of the efficiency of transport measured in vivo and in vitro. All supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200303018/DC1.
Acknowledgments
We thank Marie-Hélène Beuchat, Monique Beulet, and Marie–Claire Velluz for expert technical assistance, and Gisou van der Goot for critical reading of the manuscript.
This study was supported by grants from the Swiss National Science Foundation and the Human Frontier Science Programme Organization (to J. Gruenberg). A. Petiot was a recipient of Roche Foundation and ARC (Association pour la Recherche sur le Cancer) fellowships.
The online version of this article includes supplemental material.
Abbreviations used in this paper: bHRP, biotinylated HRP; ECV, endosomal carrier vesicle; EEA1, early endosomal antigen 1; EGFR, EGF receptor; Hrs, hepatocyte growth factor–regulated tyrosine kinase substrate; LBPA, lysobisphosphatidic acid; PI3P, phosphatidylinositol-3-phosphate; TfR, transferrin receptor.
References
- Aniento, F., N. Emans, G. Griffiths, and J. Gruenberg. 1993. a. Cytoplasmic dynein-dependent vesicular transport from early to late endosomes. J. Cell Biol. 123:1373–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aniento, F., E. Roche, A. Cuervo, and E. Knecht. 1993. b. Uptake and degradation of glyceraldehyde-3-phosphate dehydrogenase by rat liver lysosomes. J. Biol. Chem. 268:10463–10470. [PubMed] [Google Scholar]
- Aniento, F., F. Gu, R. Parton, and J. Gruenberg. 1996. An endosomal βcop is involved in the pH-dependent formation of transport vesicles destined for late endosomes. J. Cell Biol. 133:29–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bean, A.J., S. Davanger, M.F. Chou, B. Gerhardt, S. Tsujimoto, and Y. Chang. 2000. Hrs-2 regulates receptor-mediated endocytosis via interactions with Eps15. J. Biol. Chem. 275:15271–15278. [DOI] [PubMed] [Google Scholar]
- Bishop, N., A. Horman, and P. Woodman. 2002. Mammalian class E vps proteins recognize ubiquitin and act in the removal of endosomal protein-ubiquitin conjugates. J. Cell Biol. 157:91–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford, M.M. 1976. A rapid and sensitive method for the quantitation of microgram quantitites of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248–254. [DOI] [PubMed] [Google Scholar]
- Carter, R.E., and A. Sorkin. 1998. Endocytosis of functional epidermal growth factor receptor-green fluorescent protein chimera. J. Biol. Chem. 273:35000–35007. [DOI] [PubMed] [Google Scholar]
- Cavalli, V., F. Vilbois, M. Corti, M.J. Marcote, K. Tamura, M. Karin, S. Arkinstall, and J. Gruenberg. 2001. The stress-induced MAP kinase p38 regulates endocytic trafficking via the GDI:Rab5 complex. Mol. Cell. 7:421–432. [DOI] [PubMed] [Google Scholar]
- Clague, M., S. Urbé, F. Aniento, and J. Gruenberg. 1994. Vacuolar ATPase activity is required for endosomal carrier vesicle formation. J. Biol. Chem. 269:21–24. [PubMed] [Google Scholar]
- Fernandez-Borja, M., R. Wubbolts, J. Calafat, H. Janssen, N. Divecha, S. Dusseljee, and J. Neefjes. 1999. Multivesicular body morphogenesis requires phosphatidyl-inositol 3-kinase activity. Curr. Biol. 9:55–58. [DOI] [PubMed] [Google Scholar]
- Futter, C.E., L.M. Collinson, J.M. Backer, and C.R. Hopkins. 2001. Human VPS34 is required for internal vesicle formation within multivesicular endosomes. J. Cell Biol. 155:1251–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillooly, D.J., I.C. Morrow, M. Lindsay, R. Gould, N.J. Bryant, J.M. Gaullier, R.G. Parton, and H. Stenmark. 2000. Localization of phosphatidylinositol 3-phosphate in yeast and mammalian cells. EMBO J. 19:4577–4588 (In Process Citation). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorvel, J.P., P. Chavrier, M. Zerial, and J. Gruenberg. 1991. Rab 5 controls early endosome fusion in vitro. Cell. 64:915–925. [DOI] [PubMed] [Google Scholar]
- Griffiths, G., B. Hoflack, K. Simons, I. Mellman, and S. Kornfeld. 1988. The mannose-6-phosphate receptor and the biogenesis of lysosomes. Cell. 52:329–341. [DOI] [PubMed] [Google Scholar]
- Gruenberg, J., G. Griffiths, and K.E. Howell. 1989. Characterisation of the early endosome and putative endocytic carrier vesicles in vivo and with an assay of vesicle fusion in vitro. J. Cell Biol. 108:1301–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu, F., F. Aniento, R. Parton, and J. Gruenberg. 1997. Functionnal dissection of COP-I subunits in the biogenesis of multivesicular endosomes. J. Cell Biol. 139:1183–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu, F., and J. Gruenberg. 2000. ARF1 regulates pH-dependent COP functions in the early endocytic pathway. J. Biol. Chem. 275:8154–8160. [DOI] [PubMed] [Google Scholar]
- Haft, C.R., M. de la Luz Sierra, V.A. Barr, D.H. Haft, and S.I. Taylor. 1998. Identification of a family of sorting nexin molecules and characterization of their association with receptors. Mol. Cell. Biol. 18:7278–7287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi, T., E. Stang, K.S. Fang, P. de Moerloose, R.G. Parton, and J. Gruenberg. 1998. A lipid associated with the antiphospholipid syndrome regulates endosome structure/function. Nature. 392:193–197. [DOI] [PubMed] [Google Scholar]
- Lloyd, T.E., R. Atkinson, M.N. Wu, Y. Zhou, G. Pennetta, and H.J. Bellen. 2002. Hrs regulates endosome membrane invagination and tyrosine kinase receptor signaling in Drosophila. Cell. 108:261–269. [DOI] [PubMed] [Google Scholar]
- Mayran, M., R.G. Parton, and J. Gruenberg. 2003. Annexin II regulates multivesicular endosome biogenesis in the degradation pathway of animal cells. EMBO J. 22:3242–3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols, B.J., and J. Lippincott-Schwartz. 2001. Endocytosis without clathrin coats. Trends Cell Biol. 11:406–412. [DOI] [PubMed] [Google Scholar]
- Odorizzi, G., M. Babst, and S.D. Emr. 1998. Fab1p PtdIns(3)P 5-kinase function essential for protein sorting in the multivesicular body. Cell. 95:847–858. [DOI] [PubMed] [Google Scholar]
- Odorizzi, G., M. Babst, and S.D. Emr. 2000. Phosphoinositide signaling and the regulation of membrane trafficking in yeast. Trends Biochem. Sci. 25:229–235. [DOI] [PubMed] [Google Scholar]
- Raiborg, C., K.G. Bache, D.J. Gillooly, I.H. Madshus, E. Stang, and H. Stenmark. 2002. Hrs sorts ubiquitinated proteins into clathrin-coated microdomains of early endosomes. Nat. Cell Biol. 4:394–398. [DOI] [PubMed] [Google Scholar]
- Raiborg, C., K.G. Bache, A. Mehlum, E. Stang, and H. Stenmark. 2001. Hrs recruits clathrin to early endosomes. EMBO J. 20:5008–5021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reaves, B.J., N.A. Bright, B.M. Mullock, and J.P. Luzio. 1996. The effect of wortmannin on the localisation of lysosomal type I integral membrane glycoproteins suggests a role for phosphoinositide 3-kinase activity in regulating membrane traffic late in the endocytic pathway. J. Cell Sci. 109:749–762. [DOI] [PubMed] [Google Scholar]
- Rojo, M., R. Peppercok, G. Emery, R. Kellner, E. Stang, R.G. Parton, and J. Gruenberg. 1997. Involvement of the transmembrane protein p23 in biosynthetic protein transport. J. Cell Biol. 139:1119–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato, T.K., M. Overduin, and S.D. Emr. 2001. Location, location, location: membrane targeting directed by PX domains. Science. 294:1881–1885. [DOI] [PubMed] [Google Scholar]
- Simonsen, A., R. Lippe, S. Christoforidis, J.M. Gaullier, A. Brech, J. Callaghan, B.H. Toh, C. Murphy, M. Zerial, and H. Stenmark. 1998. EEA1 links PI(3)K function to Rab5 regulation of endosome fusion. Nature. 394:494–498 (In Process Citation). [DOI] [PubMed] [Google Scholar]
- Simonsen, A., A.E. Wurmser, S.D. Emr, and H. Stenmark. 2001. The role of phosphoinositides in membrane transport. Curr. Opin. Cell Biol. 13:485–492. [DOI] [PubMed] [Google Scholar]
- Trono, D.E. 2002. Lentiviral vectors. Curr. Top. Microbiol. Immunol. 261:211–227. [DOI] [PubMed] [Google Scholar]
- van Kerkhof, P., C.M. Alves dos Santos, M. Sachse, J. Klumperman, G. Bu, and G.J. Strous. 2001. Proteasome inhibitors block a late step in lysosomal transport of selected membrane but not soluble proteins. Mol. Biol. Cell. 12:2556–2566. [DOI] [PMC free article] [PubMed] [Google Scholar]