

Abstract
Using the Cre/loxP system we conditionally inactivated β-catenin in endothelial cells. We found that early phases of vasculogenesis and angiogenesis were not affected in mutant embryos; however, vascular patterning in the head, vitelline, umbilical vessels, and the placenta was altered. In addition, in many regions, the vascular lumen was irregular with the formation of lacunae at bifurcations, vessels were frequently hemorrhagic, and fluid extravasation in the pericardial cavity was observed. Cultured β-catenin −/− endothelial cells showed a different organization of intercellular junctions with a decrease in α-catenin in favor of desmoplakin and marked changes in actin cytoskeleton. These changes paralleled a decrease in cell–cell adhesion strength and an increase in paracellular permeability. We conclude that in vivo, the absence of β-catenin significantly reduces the capacity of endothelial cells to maintain intercellular contacts. This may become more marked when the vessels are exposed to high or turbulent flow, such as at bifurcations or in the beating heart, leading to fluid leakage or hemorrhages.
Keywords: desmoplakin; adherens junctions; angiogenesis; vasculogenesis; vascular permeability
Introduction
Endothelial cells are linked one to another through adhesive junctional structures that include tight and adherens junctions (Takeichi, 1990; Dejana et al., 1995; Gumbiner, 1996, 2000; Tsukita et al., 2001). Adherens junctions are formed by an endothelial-specific member of the cadherin family called VE-cadherin, which forms zipper-like structures along the cell periphery and promotes cell–cell adhesion (Dejana et al., 1999). Furthermore, in lymphatics and some venous endothelium, VE-cadherin takes part in the organization of complexus adhaerentes (Schmelz et al., 1994). In these junctional structures, VE-cadherin is linked to plakoglobin, which through binding to desmoplakin, can mediate the anchorage to vimentin (Franke et al., 1988; Dejana et al., 1995; Valiron et al., 1996; Kowalczyk et al., 1998).
The null mutation of VE-cadherin prevented vascular remodeling and induced degeneration and regression of vascular structures resulting in early fetal death (Carmeliet et al., 1999). These defects may be due to the lack of both adhesive and signaling properties of the protein. VE-cadherin may signal through β-catenin, which binds cadherin cytoplasmic tail and α-catenin, which in turn links the complex to actin cytoskeleton (Aberle et al., 1996; Zhurinsky et al., 2000). However, when β-catenin is released and stabilized in the cytosol, it translocates to the nucleus, associates Tcf transcription factors, and modulates gene transcription (Gumbiner, 1995; Behrens et al., 1996; Miller and Moon, 1996; Zhurinsky et al., 2000; Hurlstone and Clevers, 2002). Therefore, cadherins (and VE-cadherin in particular) may signal in an indirect way by maintaining β-catenin at the membrane and preventing its nuclear translocation.
Furthermore, a large family of growth and differentiation factors called Wnts acts through β-catenin stabilization and transcriptional activity (Peifer and Polakis, 2000). Endothelial cells express some Wnt receptors, denominated frizzled (Li et al., 1999), and bind and respond to Wnt-1 (Wright et al., 1999).
β-Catenin is rather ubiquitous, and inactivation of its gene results in the death of the embryo at gastrulation due to defects in anterior–posterior axis formation and mesoderm organization (Haegel et al., 1995; Huelsken et al., 2000). To study the role of β-catenin in the development of the vascular system, we undertook a conditional gene inactivation strategy using the Cre/loxP system and Tie2-Cre transgenic animals. Tie2 is an endothelial specific receptor, and the promoter has cell specific expression (Schlaeger et al., 1997; Kisanuki et al., 2001).
We found that mutant embryos die in utero within embryonic day 11.5–13.5 (E11.5–13.5) due to alterations of vascular morphogenesis. Vessels have enlarged or irregular lumen, an altered network organization, and are frequently hemorrhagic. At EM, endothelial cells present increased fenestration, and intercellular junctions appear less convoluted. Endothelial cells cultured from β-catenin–null animals have a different organization of adherens junctions: α-catenin is substantially reduced, whereas junctional desmoplakin is markedly increased. It is likely that vascular fragility and the presence of lacunae at bifurcations, observed at times of sustained shear stress, are due to inadequate junctional organization and intercellular adhesion.
Results
β -Catenin gene inactivation in endothelial cells is embryonically lethal
To specifically inactivate the β-catenin gene in endothelial cells, mice containing a floxed β-catenin allele were intercrossed with transgenic mice expressing Cre under the control of Tie2 endothelial-specific promoter (Schlaeger et al., 1997; Brault et al., 2001; Kisanuki et al., 2001). The β-catenin flox mice have been used successfully to inactivate β-catenin in vivo (Brault et al., 2001; Hari et al., 2002; Lickert et al., 2002). The four genotypes obtained from the crossing are shown in Fig. 1 A (see Materials and methods for the mating strategy). The ability of Tie2-Cre to recombine the flox allele specifically in the endothelium was tested by genomic PCR from embryonic yolk sacs. As reported in Fig. 1 A, the flox del allele was detected only in E10.5 animals positive for Tie2-Cre (Fig. 1 A, compare lane 3 and lane 4), suggesting that Cre expression in the endothelial cells of the yolk sac was capable of recombining the loxP sites within β-catenin gene. The presence of the unrecombined flox allele in the same lane is attributable to the nonendothelial cells abounding in the yolk sac used for the genomic PCR. The flox del allele was detected as early as E8.5 (unpublished data). Examination of the offspring indicated that heterozygous (Fig. 1 A, lane 1 and lane 4) were viable and fertile and appeared to be normal. In contrast, no mouse with endothelial β-catenin flox del/flox del (Fig. 1 A, lane 2) was born (Table I). Analyzing the embryos at different times after conception, we found that up to 9.5 d, β-catenin mutants were morphologically indistinguishable from control embryos (either wild types or littermates that had not inherited the complete set of alleles). However, ∼50 and 100% of embryos died within E11.5 and E13.5, respectively (Table I).
Figure 1.

Cre recombinase efficiently recombines the β -catenin flox allele in vivo. (A) Genomic PCR on E10.5 embryos from Tie2-Cre x β-catenin flox/flox crossings for the β-catenin gene and for Tie2-Cre gene. The four lanes represent the four different genotypes obtained: β-catenin flox del/flox, Cre − (+/−, lane 1), β-catenin flox del/flox, Cre + (−/−, lane 2), β-catenin flox/wt, Cre − (+/+, lane 3), and β-catenin flox/wt, Cre + (+/−, lane 4). In this last lane, note the presence of the recombined β-catenin flox del band when the flox allele is in the presence of Tie2-Cre recombinase. (B) Western blot of β-catenin protein in endothelial cell lines established from wt (control) and β-catenin flox/flox del, Tie2-Cre + embryos (−/−). (C-J) Immunofluorescence for anti-PECAM and anti-β-catenin antibodies on serial cryosections from wild-type (+/+, control) and β-catenin flox/flox del, Tie2-Cre + (−/−, mutant) E10.5 embryos. Heart sections show positive staining of the endocardium (e) with PECAM antibodies in both control (C) and β-catenin −/− (E) animals. β-Catenin staining is undetectable in mutant embryos (F). β-Catenin is also absent in the dorsal aorta (G and H, Ao) and in the yolk sac vessels (I and J, arrow) of the mutant embryos. Bar in H refers to C–H, and bar in J refers to I and J.
Table I.
Genotype of progeny from β-catenin flox/flox x Tie2-Cre intercrosses
| Endothelial β
-catenin genotypea
|
Number of viable −/− | |||
|---|---|---|---|---|
| +/+ | +/− | −/− | ||
| E9.5 | 12 (21) | 30 (55) | 13 (23) | 13 (100) |
| E10.5 | 30 (19) | 87 (56) | 39 (25) | 37 (95) |
| E11.5 | 41 (27) | 71 (46) | 42 (27) | 21 (50) |
| E12.5 | 4 (31) | 4 (31) | 5 (38) | 1 (20) |
| E13.5 | 1 (5) | 14 (70) | 5 (25) | 0 |
| P20 | 25 (36) | 44 (64) | 0 | 0 |
E, embryonic day; P, postnatal day.
Numbers are given with percentage in parentheses.
Cre expression in Tie2-Cre animals crossed with ROSA26R was previously found in endothelial and endocardial cells at E8.5 and at E7.5 in a subset of extra embryonic mesodermal components of the yolk sac, considered as endothelial precursors (unpublished data; Kisanuki et al., 2001). No significant Cre expression was found in other tissues (Kisanuki et al., 2001).
Tissues and cell types, but not endothelial cells, were positive for β-catenin staining by immunohistochemistry, indicating that inhibition of β-catenin expression was specific for endothelial cells (a typical staining is reported in Fig. 1, C–J).
To unequivocally prove that the β-catenin gene was fully inactivated in endothelial cells, we isolated and cultured endothelial cell lines from β-catenin +/+ (control) and −/− animals. Three different cell lines have been examined, derived from animals at 9.5 d of development, and none of them expressed detectable amounts of β-catenin by Western blot (Fig. 1 B) or PCR analysis (unpublished data). In some cases, contaminating fibroblasts were observed at early stages of culture of the cells, which presented positive staining for β-catenin at immunofluorescence (unpublished data). However, only cells negative for VE-cadherin were positive for β-catenin, further indicating cell-specific inactivation of β-catenin gene.
Vascular defects in endothelial β-catenin −/− mutants
At E9.5, endothelial β-catenin mutant mice were morphologically indistinguishable from control littermates and showed a similar vascular pattern in the embryo proper as well as in the yolk sac (Fig. 2, A and B; unpublished data). However, at E10.5, the β-catenin mutant mice had pale and less perfused yolk sacs (Fig. 2, C and D). In the yolk sac of mutant embryos, the primary vascular plexus was present and correctly organized, whereas the vitelline vessels presented a smaller diameter (Fig. 2, E and F). The average diameter of the vitelline vessels connecting the embryo to the yolk sac (Fig. 2, E and F, arrows) was 60 ± 30 μm in the β-catenin mutant mice and 145 ± 10 μm in the control. The maximum diameter of the vitelline vessels in the yolk sac (Fig. 2, E and F, arrowheads) was 82 ± 18 μm in the mutant and 170 ± 20 μm in the control (numbers obtained out of five embryos). The number of branches was not significantly different for the same vessel length comparing mutant and control embryos (6.3 ± 1.1 primary branches in the mutant and 5.3 ± 1.5 in the control for 2 mm length of the major vitelline vessel, measured out of three embryos). Histological sections of the yolk sac showed the presence of endodermal (n), endothelial (e), mesothelial (m), and nucleated blood cells (b) in both control (Fig. 2 G) and mutant (Fig. 2 H) mice. Umbilical vessels were often increased in number, presented a markedly smaller diameter, and were abnormally branched or anastomosed, as compared with controls (Fig. 2, I–L).
Figure 2.
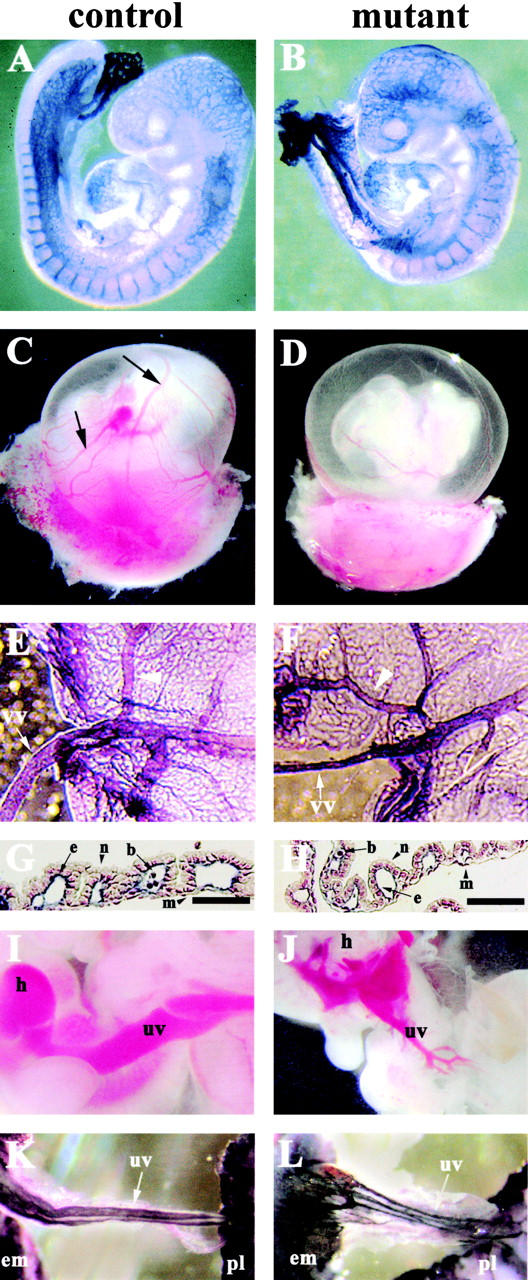
Phenotypic defects in endothelial β -catenin–null embryos. (A and B) E9.5 embryos after whole-mount immunohistochemistry with anti-PECAM antibodies. No major differences can be observed in the vascular phenotype of control (A) and mutant (B) embryos at this stage. (C–J) E10.5 embryos. Although major vitelline vessels are visible in control embryos after dissection (C, arrows), the mutant yolk sac appeares paler (D). In E–H, K, and L, whole-mount staining for PECAM was performed. In E (control) and F (mutant), note the difference in diameter of vitelline (vv) and major vessels (arrowheads) in the yolk sac. In both control (G) and mutant embryos (H), PECAM and nuclear fast red staining of yolk sac sections shows essentially indistinguishable vessels with the presence of endothelial (e), mesothelial (m), blood (b), and endodermal cells (n). (I–L) Umbilical vessels (uv) in 10.5 (I and J) and 11.5 embryos (K and L). In the mutants, umbilical vessels are smaller in diameter, ramified, or anastomosed (J and L) as compared with the control (I and K). h, heart; em, embryo; pl, placenta. Bars, 100 μm.
In the embryo proper, the vascular pattern in the head was defective (Fig. 3, A–D). Whole-mount staining with anti-PECAM antibodies showed an altered development of blood vessels in the primitive neural plexus. Namely, blind ending vessels and the formation of lacunae-like bifurcations were observed in the mutants (Fig. 3, B and D, arrowheads). Furthermore, cephalic vessels in the mutants showed an inconstant diameter and often formed acute turns and branching as compared with the control (Fig. 3, C and D, arrowhead). Transverse histological sections of the head confirmed the irregularity of vessel diameter, as indicated, by the different size of the internal carotid artery, shown in two sequential sections of the mutant embryos (Fig. 3, E and F, arrows).
Figure 3.
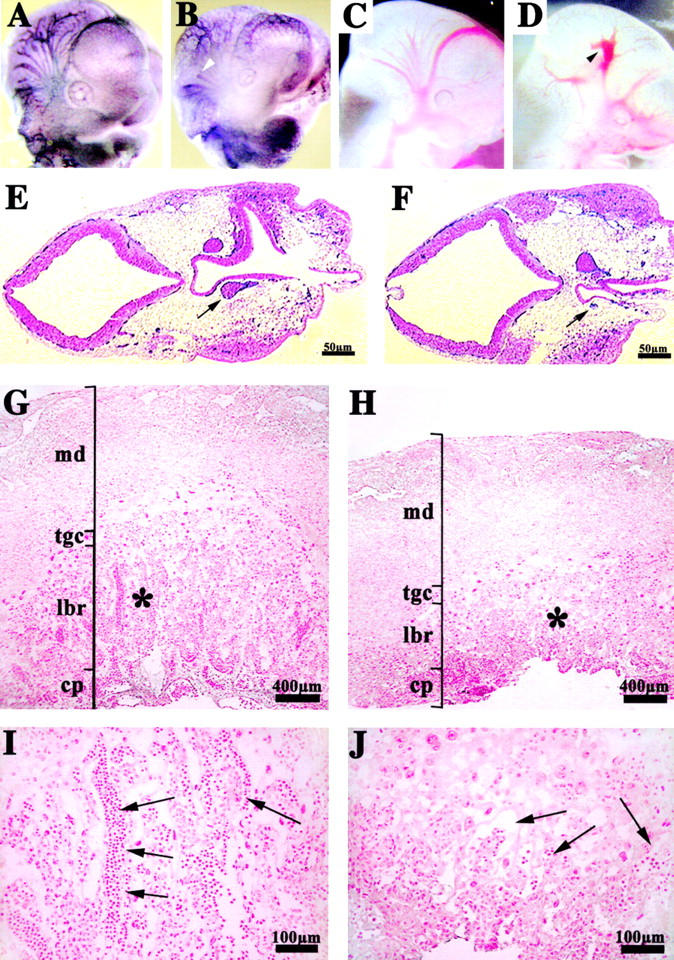
Vascular defects in the placenta and head of endothelial β -catenin–null embryos. Whole-mount immunohistochemistry for anti-PECAM antibody in E10.5 embryos (A and B). In the head, mutant embryos (B and D) compared with controls (A and C) show a less organized vascular network with vessels of irregular diameter and shape. In mutants, blind ending vessels (B, arrowhead) and lacunae-like bifurcations (D, arrowhead) can be often observed. PECAM and nuclear fast red staining in two serial transverse sections, separated by 60 μm, of E10.5 mutant head (E and F). The arrows point to the diameter of the internal carotid artery, which is significantly different in the two sections. Placenta histology of control (G and I) and mutant E10.5 embryos (H and J) shows that, although the maternal decidua (md), the chorionic plate (cp), and the trophoblast giant cells (tgc) are comparable, the labyrinthine layer (lbr) is reduced in mutants. I and J represent higher magnifications of the zones indicated by the asterisks in G and H, respectively. Embryonic blood vessels (arrows), are substantially more abundant in the labyrinthine zone of the control (I), than in the mutant (J).
Placenta tissues presented differences in mutant embryos. Although the chorionic plate and trophoblast giant cells appeared to be normal, the labyrinthine layer was reduced in thickness and less vascularized as compared with control littermates. A reduced number of fetal blood vessels penetrated in the labyrinthine area, and they mainly remained concentrated in the chorionic plate (Fig. 3, G–J, arrows).
About 50% of the embryos presented hemorrhages in different vascular areas such as the head and the dorsal vessels (Fig. 4, B and C, arrowheads). The heart of the mutant embryos was significantly smaller as compared with the wild type, and frequently showed extensive liquid accumulation in the pericardial cavity (Fig. 4, D and E, arrows).
Figure 4.

Hemorrhages and fluid extravasation in β -catenin mutant embryos. E10.5 control (A and D) and mutant embryos (B, C, and E). Arrowheads in B and C indicate regions of extended hemorrhages. In the mutants, we observed fluid accumulation in the pericardial cavity (arrow in E in comparison to controls in D) suggesting increase of permeability of cardiac microvasculature.
β-catenin −/− endocardial and vascular endothelial cells are morphologically altered in vivo
As shown in Fig. 5, in mutant embryos endocardial and vascular endothelial cells were more elongated than control vessels, resulting in a continuously thinner endothelial layer. The absolute minimum thickness of the endothelium instead was not altered (Fig. 5, A and B). Junctional overlapping typical of endothelial cells was impaired and consistently reduced in essentially all the specimens considered (Fig. 5, C and D).
Figure 5.
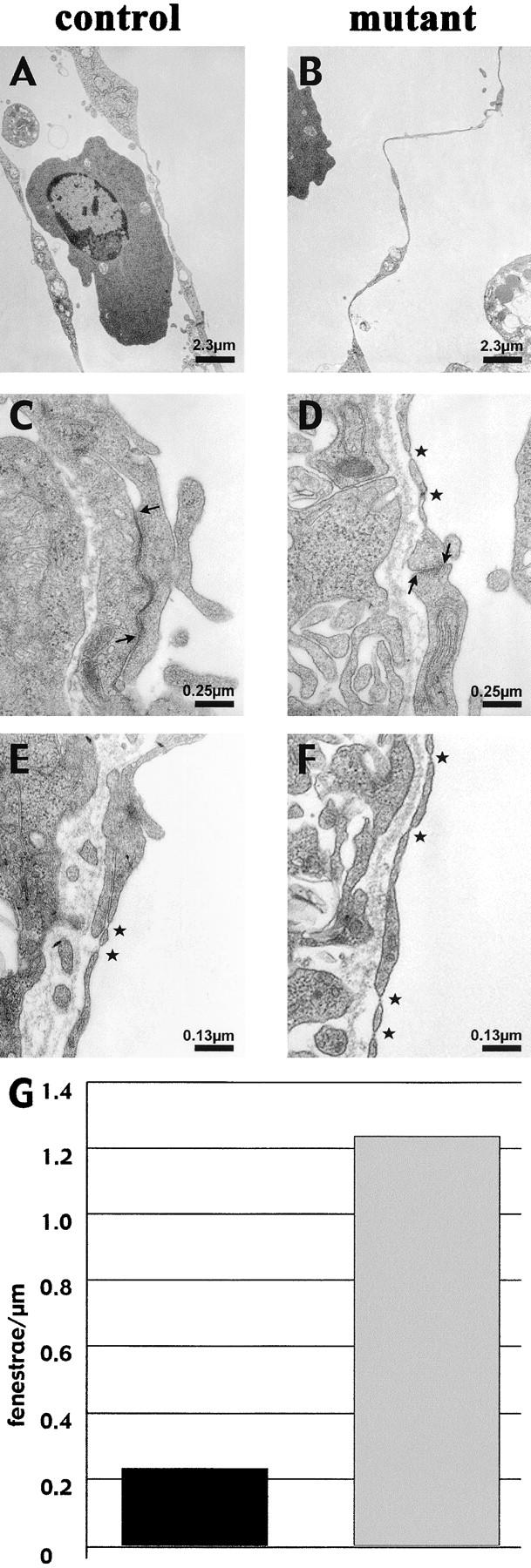
Thinner endocardium and increased number of fenestrations in β -catenin–null embryos. In electron micrographs, the endocardial lining of the heart appear continuously thinner in the null embryos compared with control littermates (compare A with B), whereas the myocardium is equally organized (not depicted). Ultrathin sections of yolk sac vessels of control and mutant embryos show similar features as the heart endocardium, with thinner endothelial walls and less organized junctions in β-catenin–deficient embryos. In the latter, the inter-endothelial junctions show a reduced junctional overlapping (see arrows), as compared with controls (C and D; asterisks point to fenestrations). Furthermore, a higher incidence of fenestrations (asterisks) in the yolk sac vasculature of null embryos can be observed (compare D, E, and F), which are quantified as number of fenestrae per length unit of endothelial wall (G; 10 pictures counted for each genotype).
In addition to the changes in cell and junction morphology, mutant endothelial cells presented a higher degree of fenestration. These structures are discontinuities in the endothelial cell membranes, allowing the passage of small mol wt constituents and which likely derive from a fusion of the apical and basal membrane (Feng et al., 1999). Typical examples are reported in Fig. 5 (E and F), in which yolk sac endothelial cells are analyzed. In control embryos, the yolk sac endothelium was only poorly fenestrated along the vessel wall. In contrast, in mutant endothelium a sixfold increase in the number of fenestrae could be observed (Fig. 5 G).
β-catenin −/− endothelial cells present altered intercellular junctional organization both in vitro and in vivo
To study whether the morphological and functional defects observed in β-catenin −/− endothelial cells in vivo were independent from their tissue context, we isolated and cultured endothelial cells from mutant and wild-type embryos. β-Catenin −/− endothelial cells presented all endothelial markers analyzed (Fig. 6–8; unpublished data), but cell morphology was significantly different. β-Catenin −/− cells were more elongated as compared with control cells (Fig. 6, C and D), and presented thinner and longer bundles of actin filaments, with sensibly less organized anchorage sites at the periphery of the cell (Fig. 6, E and F).
Figure 6.
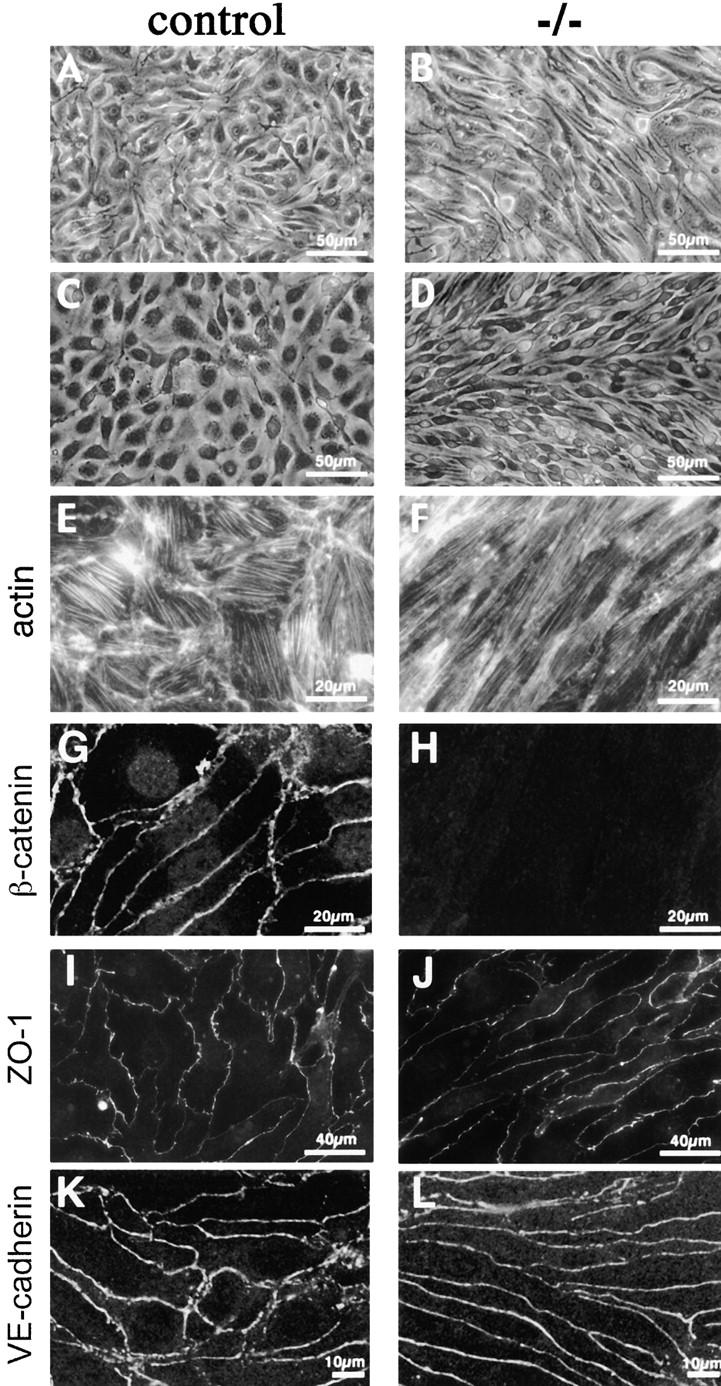
Morphological differences and junctional organization in control and β -catenin −/− endothelial cells in culture. A represents β-catenin flox/flox del (control) endothelial cells, whereas B represents the same cells after infection with an adeno vector expressing Cre recombinase (β-catenin flox del/flox del, or −/−). In C, E, and G, endothelial cells cultured from control embryos are shown, whereas D, F, and H show endothelial cells from β-catenin −/− embryos. Although control endothelial cells exhibit classical cobblestone morphology (A and C), β-catenin −/− cells are more elongated and have a fibroblastoid/mesenchymal morphology (B and D). Phalloidin staining shows more elongated and thinner bundles of actin filaments in β-catenin −/− cells (F), as compared with controls (E). β-Catenin staining is undetectable at intercellular contacts of −/− cells (compare G and H), whereas ZO-1 (I and J) and VE-cadherin (K and L) are correctly organized at intercellular junctions.
Figure 8.

Biochemical characterization of the junctional complex. (A) Association of VE-cadherin with α, β-catenins, and plakoglobin. Endothelial cell extracts from control and β-catenin −/− cells were immunoprecipitated with antibodies to VE-cadherin. The immunoprecipitates (IP) and an equal amount of total protein extracts (Total) from the two cell types were immunoblotted (IB) with the antibodies indicated on the left. Note the decrease in the level of both total and VE-cadherin-bound α-catenin. (B) Increase in desmoplakins level in β-catenin −/− and in control cell lysates. Arrowheads indicate the bands corresponding to desmoplakin I (250 kD) and II (220 kD).
To understand whether changes observed were due to modifications acquired during embryonic development, we cultured endothelial cells from β-catenin flox/flox del Cre − (+/−) animals, which did not present any apparent developmental alterations. These cells were infected with an adeno vector expressing Cre recombinase (Anton and Graham, 1995; Wang et al., 1995) in order to inactivate β-catenin gene in vitro. Western blot analysis confirmed the absence of β-catenin in these cells (Fig. S3, available at http://www.jcb.org/cgi/content/full/jcb.200212157/DC1), and they underwent similar changes in morphology and cytoskeletal organization as β-catenin −/− cells derived from embryos (see Fig. 6, A–D, and Fig. S1).
The +/− cells were cultured and systematically compared with β-catenin +/+ and −/− cells. To exclude the toxicity of Cre recombinase, we also cultured endothelial cells derived from flox/wt Cre + animals (+/−). In all the assays performed, these cell types were indistinguishable from β-catenin +/+ cells.
The expression and distribution of junctional markers was studied by immunofluorescence microscopy. We observed that essentially all the junctional proteins considered (including PECAM, JAM, VE-cadherin, ZO-1, p120, and occludin) were correctly expressed and distributed at intercellular junctions (see Figs. 6–8; unpublished data). The only exceptions were plakoglobin, which presented a slightly brighter staining at intercellular contacts (Fig. 7, A and B), and α-catenin, which was strongly reduced at cell–cell contacts (Fig. 7, C and D). These observations were further confirmed by biochemical analyses. Immunoprecipitation of VE-cadherin followed by Western blot with specific antibodies revealed changes in cadherin–catenin complex composition. α-catenin was strongly reduced (4–5-fold) in the complex and in total amount, whereas plakoglobin was slightly increased (1–1.5-fold; Fig. 8 A). The total level of p120 and VE-cadherin and their level associated to the complex were comparable (Fig. 8 A; unpublished data). Because α-catenin was reduced, plakoglobin could have another partner. Desmoplakin is a preferential binding protein for plakoglobin, but is weakly expressed in vascular endothelium (Franke et al., 1988; Schmelz et al., 1994). We found that desmoplakin was 2–3-fold increased in extracts of β-catenin −/− cells as compared with controls (Fig. 8 B). In addition, desmoplakin was found at intercellular contacts only in β-catenin −/− cells, but not in control cells (Fig. 7, E and F). The same characterization of adherens junctions composition was performed in β-catenin flox endothelial cells treated either with adeno-Cre or the empty vector, and we found essentially the same results (Figs. S1–S3, available at http://www.jcb.org/cgi/content/full/jcb.200212157/DC1).
Figure 7.
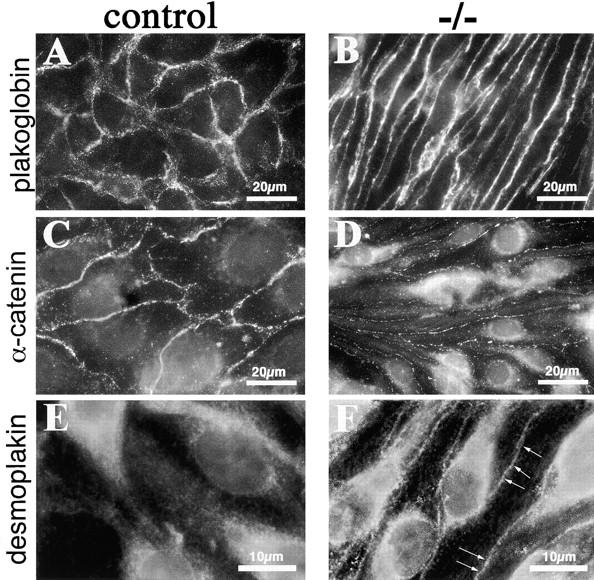
Immunostaining of junctional proteins in β-catenin −/− endothelial cells. Although plakoglobin is correctly organized at cell–cell contacts in β-catenin −/− embryos (compare A and B), α-catenin antibodies give a very faint and discontinuous staining at cell–cell contacts in β-catenin −/−, as compared with control cells (compare C and D). Note the presence of desmoplakin at intracellular junctions only in β-catenin −/− cells (F, arrows), whereas in control cells, the same antibody gives only a faint and diffuse staining (E).
The organization of endothelial cell junctions was also studied in vivo by immunostaining of vascular sections of E10.5 embryos. Consistently with the data obtained on cultured cells, we found that α-catenin was barely visible at interendothelial contacts of mutant embryos as compared with controls (Fig. 9, A–D). Desmoplakin was detectable only in the endothelium of mutant mice, but not in controls (Fig. 9, E to H, arrows). VE-cadherin and plakoglobin were correctly expressed in mutant and control mice (Fig. S4, available at http://www.jcb.org/cgi/content/full/jcb.200212157/DC1). Therefore, endothelial immunostaining in vivo supports the observations made on cultured cells.
Figure 9.
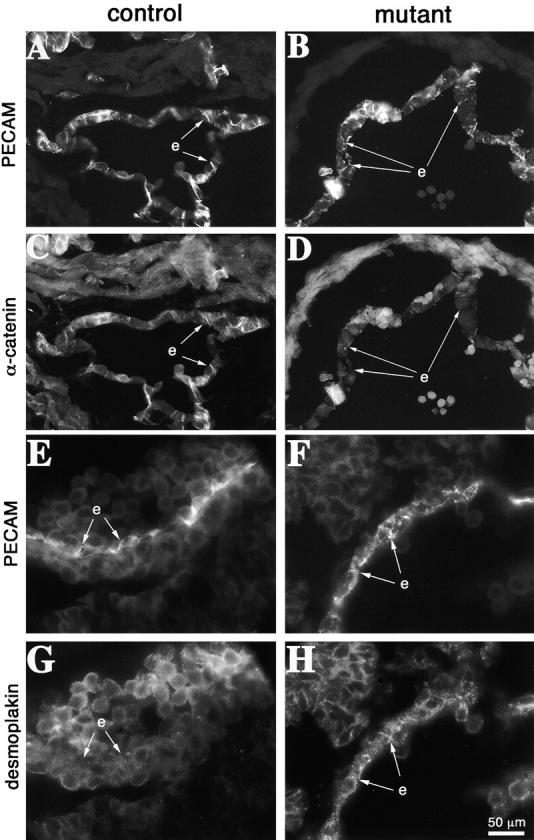
Junctional organization in control and mutant embryos. Cryosections from E10.5 embryos were analyzed for the codistribution of PECAM (A and B) and α-catenin (C and D). PECAM is equally expressed in control (A) and mutant embryos (B), and in some regions it is possible to evidentiate its localization at cell–cell contacts (arrows). In contrast, in mutant embryos α-catenin shows a weaker endothelial staining and colocalization with PECAM at intercellular junctions is rare (compare C and D, arrows). Desmoplakin is detectable in endothelial cells in β-catenin–null embryos and colocalizes with PECAM at cell–cell contacts (F and H). In control embryos desmoplakin staining was negative in endothelial cells (E and G). e, endocardium. Bar in H refers to A–H.
Cell–cell adhesion strength is reduced in absence of β-catenin
When intercellular junctions are affected, cells tend to detach from the monolayer and migrate as single cells (Corada et al., 2002). As reported in Fig. 10 (A–C), single migrating cells were very few in control monolayers, whereas cells lacking β-catenin detached from the monolayer and migrated into the wound much more efficiently. Differences in cell migration velocity or cell proliferation at the cell front among β-catenin +/+ and −/− cells were excluded by the phagokinetic track assay and BrdU incorporation respectively (see supplemental Materials and methods section, Fig. S5; available at http://www.jcb.org/cgi/content/full/jcb.200212157/DC1).
Figure 10.
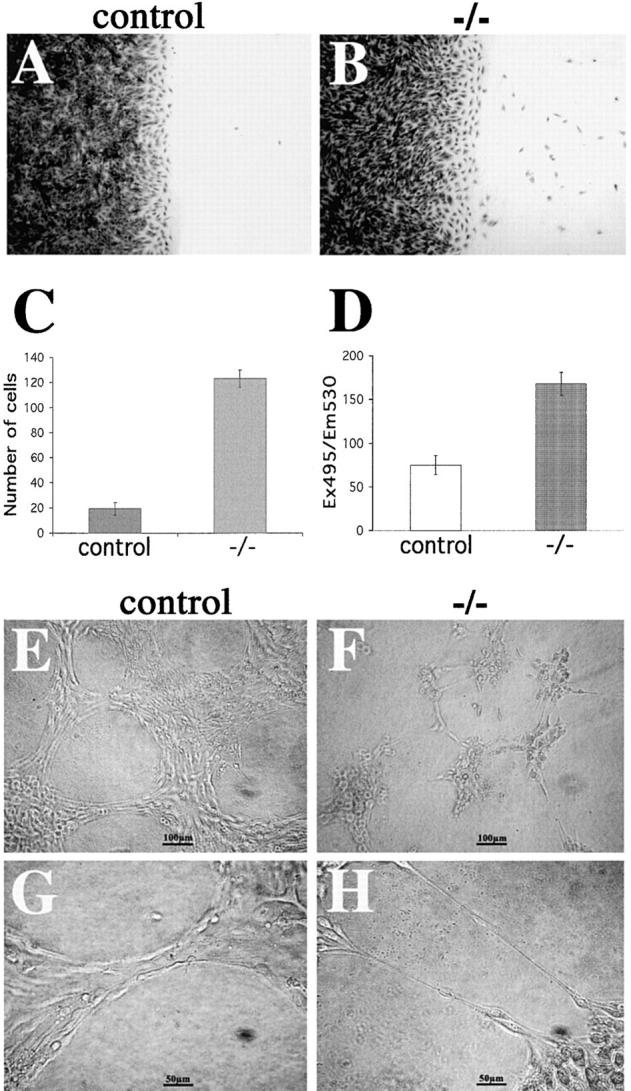
Cell–cell adhesion strength in control and mutant endothelial cells. (A–C) Analysis of endothelial cell migration into a wound. Crystal violet staining of control (A) and β-catenin −/− (B) endothelial cells 8 h after mechanical wounding of the monolayer. C reports the number of migrating single cells into the wound at 8 h. (D) Permeability across endothelial cell monolayers. β-Catenin −/− confluent endothelial cells show a higher passage of FITC-dextran as compared with the control cells. In C and D, columns represent means ± SEM of quadruplicates of a typical experiments out of three performed. (E–H) β-catenin–null endothelial cells (−/−) form only small, disorganized aggregates interconnected with thin, single-cell elongations on Matrigel™ matrix (G and H), whereas control endothelial cells are able to organize into aggregates and bundles and show a typical cordlike structure on Matrigel™ matrix (E and F).
As a second assay, we tested paracellular permeability of endothelial cell monolayers grown on Transwell filters (Corada et al., 2002). As reported in Fig. 10 D, β-catenin −/− cells were more permeable to high mol wt dextran as compared with control cells. Cell density was not significantly different between the two cell lines as measured by crystal violet staining of the cells grown on the filters at d 4 (in a typical experiment, OD 540 nm of β-catenin −/− cells was 1.216 ± 0.13 and β-catenin −/− = 1.495 ± 0.06; values are means of four filters ± SD).
Finally, we tested the mutant cell lines for their capacity to aggregate and form cords in Matrigel™ matrix. As shown in Fig. 10 (E–H), cells lacking β-catenin were unable to organize in aggregates or cords of large diameter, but tended to form only thin filaments connecting small and disorganized cell clumps.
Discussion
In this paper, we report that endothelial cells do not need β-catenin for early phases of vasculogenesis and angiogenesis, but require this molecule for correct vascular patterning and maintenance of vascular integrity at later stages of development. The defects in β-catenin–null animals were localized in specific regions such as the head vascular network, large vitelline and umbilical vessels, and the placenta.
Placental insufficiency and hypoxia might be the cause of death and of some defects of the mutant embryos (such as the smaller diameter of vessels in the yolk sac). However, many of the phenotypic alterations observed (such as the abnormal branching of umbilical vessels, lacunae-like bifurcations, blind ending or inconstant diameter of vessels, localized hemorrhages in different regions of the microvasculature, or changes in junction organization) appear specific because they are not shared by embryos with comparable placental insufficiency (Kozak et al., 1997; for review see Roberts et al., 2001; Kuang et al., 2002; Yang et al., 2003).
A serious concern for the late and localized vascular defects in β-catenin −/− embryos may be a partial, late, nonuniform, or ectopic expression of Tie2-Cre transgene. However, from different evidences this possibility seems unlikely. First, the Tie2 transgene is expressed in hemangioblasts at early (E7.5) stages of development (Kisanuki et al., 2001), and is able to induce recombination of the β-catenin gene in vivo at least at E8.5 (this paper). Therefore, endothelial cells in the progeny should present the recombined gene as proven by histological staining of the endothelium in different vessels in vivo and by isolating and culturing endothelial cell lines from mutant embryos. Staining in vivo and cell cultures were obtained from embryos at E9.5 when no apparent vascular defect was yet detectable. In both cases, β-catenin was undetectable arguing against a partial effect of the recombinase. Furthermore, ectopic expression of the transgene also looks unlikely, as shown by positive staining of the surrounding nonendothelial tissues and by experiments performed using the same animal strains crossed with “indicator” mice expressing a floxed stop codon for Lac Z expression (Kisanuki et al., 2001).
A typical defect of β-catenin–null animal vessels was the abnormal, frequently inconsistent lumen accompanied by the diffuse occurrence of hemorrhages. Changes in endothelial cell morphology and function in the mutants may explain, at least in part, the phenotype. At EM, endothelial cells of β-catenin–null animals were more flatten and elongated. Inter-endothelial junctions looked less elaborate compared with control animals, and the incidence and number of fenestrae were increased. All these changes would argue in favor of more fragile and permeable vessels, which would explain the presence of vascular leakage and hemorrhages.
This possibility is further supported by analyses on cultured β-catenin–null endothelial cells. These cells present differences in morphology and cytoskeletal organization and, most importantly, in the molecular organization of adherens junctions, where α-catenin was markedly decreased. In other experimental systems, similar changes parallels marked junction weakening (Kuroda et al., 1998; Vasioukhin et al., 2000).
In contrast to epithelial cells, endothelial cells do not have desmosomes, neither do they express desmogleins or desmocollins (Franke et al., 1988). However, they have complexus adhaerentes (Schmelz et al., 1994) formed by VE-cadherin linked to plakoglobin or p0071 (Calkins et al., 2002), which through the binding to desmoplakin, mediate the anchorage to vimentin (Valiron et al., 1996; Kowalczyk et al., 1998; Gallicano et al., 2001). Although desmoplakin and vimentin are present in other types of endothelium (Valiron et al., 1996; Kowalczyk et al., 1998), complexus adhaerentes are preferentially observed in lymphatics, where junctions are relatively weak, allowing a dynamic passage of cells and solutes (Schmelz et al., 1994; Stacker et al., 2002). We speculate that in the absence of β-catenin, plakoglobin may bind to α-catenin only to a limited extent while increasing its interaction with desmoplakin (see the scheme in Fig. 11). According to this model, we found that both the level of desmoplakin and its distribution at intercellular junctions were significantly increased in β-catenin −/− cells. These molecular changes lead to an overall decrease of the number of strong, actin-based adherens junctions to weak, more dynamic vimentin based complexus adhaerentes. Consistently, it was recently reported that β-catenin is reduced in lymphatic as compared with vascular endothelium (Petrova et al., 2002).
Figure 11.

Proposed model for the molecular reorganization of endothelial intercellular junctions in the absence of β-catenin. (A) In the presence of β-catenin (β), α-catenin (α) is normally expressed at cell–cell junctions and connects VE-cadherin/β-catenin or VE-cadherin/plakoglobin (PG) complex to actin cytoskeleton. (B) In the absence of β-catenin, the decrease in α-catenin, accompanied by increase in desmoplakin (DP), may lead to a shift from α-catenin and actin based junctions to desmoplakin and vimentin-based complexus adhaerentes.
In vivo, EM showed morphological changes of endothelial cells in different parts of the vasculature and also in regions where vessels were apparently not severely affected, such as in the microvasculature of the yolk sac or at early developmental stages. It is possible that when vessels are exposed to low hemodynamic stress, such as at early stages of development or in regions of low shear, the defects due to the lack of β-catenin remain silent. In contrast, when vessels are exposed to high shear, such as in small arteries, at bifurcations or in the heart, the endothelial cells are stretched and interendothelial junctions may break causing vascular leakage. This would explain the late and localized occurrence of vascular dilations and hemorrhages.
In other cells, such as in the epithelium, inactivation of β-catenin did not modify intercellular cell–cell adhesion strength (Huelsken et al., 2000; Posthaus et al., 2002). However, this may be due to the presence of other strong junctional structures such as desmosomes or more elaborated tight junctions (Milton and Knutson, 1990).
Besides its role as cadherin partner, β-catenin is an important signaling and transcriptional factor. The absence of β-catenin would lead to a block of Wnt signaling in endothelial cells. The role of Wnt in vascular development is still largely obscure. Inactivation of Fzd 5 gene, a receptor of Wnt 5a, Wnt 10b and Wnt 2, is embryonically lethal due to defects in vascular organization in the yolk sac and placenta (Ishikawa et al., 2001). These defects, such as reduction of large vessel lumen in the yolk sac, were, for many aspects, similar to those observed in β-catenin −/− animals in the same organs, suggesting that they are due to a lack of β-catenin transcriptional activity. However, in contrast to this last report, we were unable to detect significant endothelial cell detachment or apoptosis in the vessels of the yolk sac. A possible explanation is that Fzd 5 is expressed also in cells surrounding the endothelium and the apoptotic effect may be the result of an altered interaction of the endothelium with the mesenchyma.
To test the possibility that some of the observed changes in junctional protein expression, such as α-catenin, may be due to β-catenin transcriptional activity, we infected β-catenin −/− endothelial cells with two different constructs with constitutive Lef/β-catenin transcriptional activity (LEFΔN-βCTA and LEFΔN-VP16, a gift from Kris Vleminckx; Vleminckx et al., 1999). We found that although both constructs were able to increase the expression of known target genes for β-catenin signaling, such as cyclin D1, none of them changed α-catenin expression (Fig. S6, available at http://www.jcb.org/cgi/content/full/jcb.200212157/DC1) or cell morphology. In addition, according to the only available paper on the effect of Wnt-1 overexpression and β-catenin transcriptional activity in cultured brain microvascular endothelial cells (Wright et al., 1999), we cocultured endothelial cells with 293 cells transfected with a vector expressing Wnt-1. Also in this case, using different culture conditions, we could not detect changes in adherens junction composition or cell morphology (unpublished data). Overall, these data suggest that the classic Wnt signaling pathway doesn't play a major role in the reported junctional changes.
In a transgenic mouse model with a reporter gene under the control of β-catenin/TCF-responsive elements (Maretto et al., 2003), reporter activity in blood vessels was found from E10.5, but only at distinct sites of neoangiogenesis, such as in microvessels invading the brain parenchyma. This suggests that β-catenin transcriptional activity in vascular development may be transient and expressed only when angiogenesis occurs.
Another possibility to explain the presence of lacunae and irregular vascular lumen may be related to the role of β-catenin in modulating endothelial cell activation by VEGF. In a recent work (Lampugnani et al., 2003), we found that in absence of β-catenin, VEGF receptor-2 signaling to cell proliferation is not inhibited by confluency. This may cause an uncontrolled endothelial cell proliferation and an irregular organization of the vasculature.
In conclusion, inhibition of β-catenin expression in the endothelium leads to the death of embryos in utero due to alterations in vascular morphogenesis. Interestingly, VE-cadherin gene inactivation induced a more dramatic vascular phenotype leading to fetal death at earlier stages of development (Carmeliet et al., 1999). Hence, VE-cadherin plays a complex role in vascular morphogenesis, which seems to a substantial extent independent from β-catenin interaction and signaling.
Materials and methods
Transgenic mice and crossings
The generation of β-catenin flox and flox del mice has been described previously (Brault et al., 2001). The Tie2-Cre transgenic mice were a gift of Dr. Yanagisawa (University of Texas Medical Center, Dallas, TX) (Schlaeger et al., 1997; Kisanuki et al., 2001). In the mating scheme devised, only one flox allele needs to undergo recombination to create endothelial β-catenin −/− mice. In a first cross, homozygous Tie2-Cre transgenic mice were mated with mice heterozygous for the β-catenin flox del allele. 50% of the offspring inheriting both the flox del allele and a Tie2-Cre were then mated with homozygous flox β-catenin mice to obtain embryos with the Tie2-Cre transgene together with one flox and one flox del allele. DNA isolated from yolk sac of embryos and tail biopsies were used for genotyping animals. Different combinations of wild-type, flox and/or flox del β-catenin alleles were identified by PCR using primers RM41, RM42, and RM43 as described in Brault et al. (2001). For the identification of the Tie2-Cre gene, F-Tie2 (5′-GAGGTGTGGCAGGCTTGAGAT-3′) and antisense primer Cre minus (5′-CCAGCAGGCGCACCATTGCCC-3′) generate a 1,200-bp product.
Antibodies
Mouse mAbs were as follows: anti-β-catenin, anti-α-catenin, anti-plakoglobin, anti-p120, and anti-P-tyrosine (PY20, HRP-conjugated) were from Transduction Laboratories. Rat mAbs were from the Dejana laboratory as follows: anti-VE-cadherin clone BV13, (Corada et al., 1999), anti-CD31/PECAM clone MEC7.46, (Vecchi et al., 1994). The polyclonal rabbit anti-ZO-1 was purchased from Zymed Laboratories. The polyclonal rabbit anti-desmoplakin Ch-1 has been described previously (Valiron et al., 1996).
Embryological techniques and immunostaining
For whole-mount staining with anti-PECAM antibodies, embryos were fixed in Dent's fixative (methanol and DMSO, 4:1) to minimize unspecific binding and for permeabilization, and embryos were incubated for 3 h in PBS containing 0.1% Tween 20 and 2% BSA. The anti-PECAM primary antibody was followed by an amplification step with rabbit anti–rat IgG (DakoCytomation) and the secondary antibody rat APAAP (alcaline phosphatase and rat anti-alcaline phosphatase; DakoCytomation) and developed with the NBC/BCIP kit from Promega.
For histological examination, fixed embryos were dehydrated, embedded in paraffin, and sectioned at 5 μm, according to standard procedures. Sections were dewaxed, rehydrated, and counter-stained with Nuclear Fast Red (Vector Laboratories). Immunostaining on PFA-fixed specimens was performed as described previously (Lampugnani et al., 2002). Embryos or cultured cells (see below) were immersion fixed in 4% freshly depolymerized PFA in HBSS (0.1 M Hepes, 0.15 M NaCl, 5 mM KCl, 5 mM glucose, 1 mM CaCl2 and 1 mM MgCl2, pH 7.4) for 1 h on ice. For α-catenin and desmoplakin staining, cultured cells were fixed for 5 min at −20°C in methanol. Cells were subjected to the labeling procedure (see below), whereas the embryos were subsequently transferred to a graded sucrose series of 12, 15, and 18% sucrose in HBSS, embedded in Tissue Tek® O.C.T. (Sakura Finetek), frozen in liquid nitrogen, and stored at −80°C before cryosectioning.
Cryostat sections (8–12 μm thick) were cut using a cryotome (model CM3050S; Leica), mounted on poly-l-lysine–coated glass slides (Menzel-Glaser), and dried. Sections were postfixed in 4% PFA/HBSS on ice and washed in Tris-buffered saline (pH 7.6) containing 1 mM CaCl2 (TBS).
For immunostaining of embryo's sections and for endothelial cells, unspecific binding was minimized by incubation in 5% (wt/vol) skimmed milk for 30 min, 0.3% (vol/vol) Triton X-100 (Serva), and 0.04% (wt/vol) NaN3 in TBS. Antibodies were diluted in TBS containing 1% BSA and incubated over night at 4°C. After several washes in TBS, sections and cells were incubated with secondary antibody for 1 h at RT. To visualize cell nuclei, samples were washed in TBS and counterstained with nuclear dye Hoechst 33258 (Sigma-Aldrich). Specimens were mounted in 90% glycerol containing paraphenylenediamine as an anti-bleaching agent, and fluorescence was observed with an epifluorescence microscope (Leica) and digitally documented with a camera, (SenSys; Roper Scientific). Images were computer processed using Adobe Photoshop® version 6 for Macintosh.
Conventional EM
EM specimens were fixed and embedded as described previously (Liebner et al., 2000). For the quantification of fenestrae, the total length of the vessel wall was measured using Carnoy software, version 2.0 for Macintosh, and the number of fenestrations was counted at the same magnification. For the graphical presentation, the relative incidence of fenestrae/μm was calculated.
Culture of endothelial cells and adeno-Cre infection
Endothelial cells with homozygous null mutation of the β-catenin gene (β-catenin flox del/flox del) were isolated from E9.5 embryos (Carmeliet et al., 1999; Balconi et al., 2000). The endothelial nature of the cells was confirmed by FACS®, Western blot, and immunofluorescence microscopy with antibodies to endothelial markers as described in the Results section.
Cells were routinely cultured in DME with 20% FCS (Hyclone), endothelial cell growth supplement (5 μg/ml, home-made from calf brain), and heparin (100 μg/ml; Sigma-Aldrich) maintenance medium (Balconi et al., 2000) on gelatin-coated tissue culture vessels.
The adeno vector–expressing Cre recombinase was a gift of Frank Graham (McMaster University, Hamilton, Ontario, Canada) and was described previously (Anton and Graham, 1995; Wang et al., 1995). For the infection, 2 plaque-forming units/cell were used. To increase efficiency, a second infection was performed after 9–12 h.
Immunoprecipitation and Western blot analysis
105/cm2 cells were cultured for 4 d in maintenance medium before the experiments, and scraped in lysis buffer (50 mM Tris and 150 mM NaCl, pH 7.4, containing 1% Triton X-100, 1% Nonidet P-40, 1 mM phenylmethylsulfonyl fluoride, 2 mM Ca2+, 15 μg/ml leupeptin, 71 μg/ml phenanthrolyne, and 20 U/ml aprotine [Sigma-Aldrich], 300 μM vanadate, and 600 mM hydrogen peroxide). The insoluble material was removed by centrifugation at 12,000 rpm for 10 min. Alternatively, cultured cells were lysated by boiling in a modified Leamli sample buffer (2% SDS, 20% glycerol, and 125 mM Tris-HCl, pH 6.8). The protein content was measured according to the BCA method (Pierce Chemical Co.). For immunoprecipitation experiments, samples (each containing 900 μg proteins) were precleared with protein G (Amersham Biosciences) and incubated with protein G previously conjugated with the appropriate antibody. After washing, immunoprecipitates or total cell lysates were separated by SDS-PAGE under reducing conditions and were analyzed in immunoblot with specific antibodies.
In vitro functional tests
Paracellular permeability through endothelial cell monolayers and in vitro wounding were measured as described previously (Corada et al., 2002).
Cord formation in Matrigel™ matrix
The formation of tridimensional cord-like structures on Matrigel™ matrix was as described previously (Taraboletti et al., 2002).
Online supplemental material
A concise description of the data presented in each supplemental figure is introduced on citation in the text. Details on the experimental procedure and further comments on the data reported can be found in the online supplemental legends, available at http://www.jcb.org/cgi/content/full/jcb.200212157/DC1.
Acknowledgments
This work was supported by Associazione italiana per la Ricerca sul Cancro, the European Community (QLRT-2001-02059; QLG1-CT-1999-01036; QT-CT-1999-00020); Marie Curie fellow (QLK6-CT-2000-52056); Agenzia Spaziale Italiana, Associazione Duchenne Parent Project, Italian Ministry of Health, Ministry of University and Scientific and Technological Research, Telethon Italy, CNR/MIUR (CNR.02.731.DEJA), MIUR/FIRB (RBNE01MAWA_009, RBNE01F8LT_007), and Cofin 2002 (2001053777_002).
The online version of this article includes supplemental material.
Abbreviation used in this paper: E, embryonic day.
References
- Aberle, H., H. Schwartz, and R. Kemler. 1996. Cadherin-catenin complex: protein interactions and their implications for cadherin function. J. Cell. Biochem. 61:514–523. [DOI] [PubMed] [Google Scholar]
- Anton, M., and F.L. Graham. 1995. Site-specific recombination mediated by an adenovirus vector expressing the Cre recombinase protein: a molecular switch for control of gene expression. J. Virol. 69:4600–4606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balconi, G., R. Spagnuolo, and E. Dejana. 2000. Development of endothelial cell lines from embry Beck onic stem cells: A tool for studying genetically manipulated endothelial cells in vitro. Arterioscler. Thromb. Vasc. Biol. 20:1443–1451. [DOI] [PubMed] [Google Scholar]
- Behrens, J., J.P. von Kries, M. Kühl, L. Bruhn, D. Wedlich, R. Grosschedl, and W. Birchmeier. 1996. Functional interaction of β-catenin with the transcription factor LEF-1. Nature. 382:638–642. [DOI] [PubMed] [Google Scholar]
- Brault, V., R. Moore, S. Kutsch, M. Ishibashi, D.H. Rowitch, A.P. McMahon, L. Sommer, O. Boussadia, and R. Kemler. 2001. Inactivation of the beta-catenin gene by Wnt1-Cre-mediated deletion results in dramatic brain malformation and failure of craniofacial development. Development. 128:1253–1264. [DOI] [PubMed] [Google Scholar]
- Calkins, C.C., B.L. Hoepner, C.M. Law, M.R. Novak, S.V. Setzer, M. Hatzfeld, and A.P. Kowalczyk. 2002. The Armadillo family protein p0071 is a VE-cadherin- and desmoplakin-binding protein. J. Biol. Chem. 278:1774–1783. [DOI] [PubMed] [Google Scholar]
- Carmeliet, P., M.G. Lampugnani, L. Moons, F. Breviario, V. Compernolle, F. Bono, G. Balconi, R. Spagnuolo, B. Oostuyse, M. Dewerchin, et al. 1999. Targeted deficiency or cytosolic truncation of the VE-cadherin gene in mice impairs VEGF-mediated endothelial survival and angiogenesis. Cell. 98:147–157. [DOI] [PubMed] [Google Scholar]
- Corada, M., M. Mariotti, G. Thurston, K. Smith, R. Kunkel, M. Brockhaus, M.G. Lampugnani, I. Martin-Padura, A. Stoppacciaro, L. Ruco, et al. 1999. Vascular endothelial-cadherin is an important determinant of microvascular integrity in vivo. Proc. Natl. Acad. Sci. USA. 96:9815–9820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corada, M., L. Zanetta, F. Orsenigo, F. Breviario, M.G. Lampugnani, S. Bernasconi, F. Liao, D.J. Hicklin, P. Bohlen, and E. Dejana. 2002. A monoclonal antibody to vascular endothelial-cadherin inhibits tumor angiogenesis without side effects on endothelial permeability. Blood. 100:905–911. [DOI] [PubMed] [Google Scholar]
- Dejana, E., M. Corada, and M.G. Lampugnani. 1995. Endothelial cell-to-cell junctions. FASEB J. 9:910–918. [PubMed] [Google Scholar]
- Dejana, E., G. Bazzoni, and M.G. Lampugnani. 1999. Vascular endothelial (VE)-cadherin: only an intercellular glue? Exp. Cell Res. 252:13–19. [DOI] [PubMed] [Google Scholar]
- Feng, D., J.A. Nagy, K. Pyne, I. Hammel, H.F. Dvorak, and A.M. Dvorak. 1999. Pathways of macromolecular extravasation across microvascular endothelium in response to VPF/VEGF and other vasoactive mediators. Microcirculation. 6:23–44. [PubMed] [Google Scholar]
- Franke, W.W., P. Cowin, C. Grund, C. Kuhn, and H.P. Kapprell. 1988. The endothelial junction: the plaque and its components. Endothelial Cell Biology. N. Simionescu and M. Simionescu, editors. Plenum Press, New York. 147–166.
- Gallicano, G.I., C. Bauer, and E. Fuchs. 2001. Rescuing desmoplakin function in extra-embryonic ectoderm reveals the importance of this protein in embryonic heart, neuroepithelium, skin and vasculature. Development. 128:929–941. [DOI] [PubMed] [Google Scholar]
- Gumbiner, B.M. 1995. Signal transduction by β-catenin. Curr. Opin. Cell Biol. 7:634–640. [DOI] [PubMed] [Google Scholar]
- Gumbiner, B.M. 1996. Cell adhesion: the molecular basis of tissue architecture and morphogenesis. Cell. 84:345–357. [DOI] [PubMed] [Google Scholar]
- Gumbiner, B.M. 2000. Regulation of cadherin adhesive activity. J. Cell Biol. 148:399–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haegel, H., L. Larue, M. Ohsugi, L. Fedorov, K. Herrenknecht, and R. Kemler. 1995. Lack of beta-catenin affects mouse development at gastrulation. Development. 121:3529–3537. [DOI] [PubMed] [Google Scholar]
- Hari, L., V. Brault, M. Kleber, H.Y. Lee, F. Ille, R. Leimeroth, C. Paratore, U. Suter, R. Kemler, and L. Sommer. 2002. Lineage-specific requirements of {beta}-catenin in neural crest development. J. Cell Biol. 159:867–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huelsken, J., R. Vogel, V. Brinkmann, B. Erdmann, C. Birchmeier, and W. Birchmeier. 2000. Requirement for beta-catenin in anterior-posterior axis formation in mice. J. Cell Biol. 148:567–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurlstone, A., and H. Clevers. 2002. T-cell factors: turn-ons and turn-offs. EMBO J. 21:2303–2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa, T., Y. Tamai, A.M. Zorn, H. Yoshida, M.F. Seldin, S. Nishikawa, and M.M. Taketo. 2001. Mouse Wnt receptor gene Fzd5 is essential for yolk sac and placental angiogenesis. Development. 128:25–33. [DOI] [PubMed] [Google Scholar]
- Kisanuki, Y.Y., R.E. Hammer, J. Miyazaki, S.C. Williams, J.A. Richardson, and M. Yanagisawa. 2001. Tie2-Cre transgenic mice: a new model for endothelial cell-lineage analysis in vivo. Dev. Biol. 230:230–242. [DOI] [PubMed] [Google Scholar]
- Kowalczyk, A.P., P. Navarro, E. Dejana, E.A. Bornslaeger, K.J. Green, D.S. Kopp, and J.E. Borgwardt. 1998. VE-cadherin and desmoplakin are assembled into dermal microvascular endothelial intercellular junctions: a pivotal role for plakoglobin in the recruitment of desmoplakin to intercellular junctions. J. Cell Sci. 111:3045–3057. [DOI] [PubMed] [Google Scholar]
- Kozak, K.R., B. Abbott, and O. Hankinson. 1997. ARNT-deficient mice and placental differentiation. Dev. Biol. 191:297–305. [DOI] [PubMed] [Google Scholar]
- Kuang, S.Q., L. Liao, H. Zhang, F.A. Pereira, Y. Yuan, F.J. DeMayo, L. Ko, and J. Xu. 2002. Deletion of the cancer-amplified coactivator AIB3 results in defective placentation and embryonic lethality. J. Biol. Chem. 277:45356–45360. [DOI] [PubMed] [Google Scholar]
- Kuroda, S., M. Fukata, M. Nakagawa, K. Fujii, T. Nakamura, T. Ookubo, I. Izawa, T. Nagase, N. Nomura, H. Tani, et al. 1998. Role of IQGAP1, a target of the small GTPases Cdc42 and Rac1, in regulation of E-cadherin-mediated cell-cell adhesion. Science. 281:832–835. [DOI] [PubMed] [Google Scholar]
- Lampugnani, M.G., A. Zanetti, F. Breviario, G. Balconi, F. Orsenigo, M. Corada, R. Spagnuolo, M. Betson, V. Braga, and E. Dejana. 2002. VE-cadherin regulates endothelial actin activating Rac and increasing membrane association of Tiam. Mol. Biol. Cell. 13:1175–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampugnani, M., A. Zanetti, M. Corada, T. Takahashi, G. Balconi, F. Breviario, F. Orsenigo, A. Cattelino, R. Kemler, T.O. Daniel, and E. Dejana. 2003. Contact inhibition of VEGF-induced proliferation requires vascular endothelial cadherin, beta-catenin, and the phosphatase DEP-1/CD148. J. Cell Biol. 161:793–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, L., H. Yuan, C.D. Weaver, J. Mao, G.H. Farr, III, D.J. Sussman, J. Jonkers, D. Kimelman, and D. Wu. 1999. Axin and Frat1 interact with dvl and GSK, bridging Dvl to GSK in Wnt-mediated regulation of LEF-1. EMBO J. 18:4233–4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lickert, H., S. Kutsch, B. Kanzler, Y. Tamai, M.M. Taketo, and R. Kemler. 2002. Formation of multiple hearts in mice following deletion of beta-catenin in the embryonic endoderm. Dev. Cell. 3:171–181. [DOI] [PubMed] [Google Scholar]
- Liebner, S., H. Gerhardt, and H. Wolburg. 2000. Differential expression of endothelial beta-catenin and plakoglobin during development and maturation of the blood-brain and blood-retina barrier in the chicken. Dev. Dyn. 217:86–98. [DOI] [PubMed] [Google Scholar]
- Maretto, S., M. Cordenonsi, S. Dupont, P. Braghetta, V. Broccoli, A.B. Hassan, D. Volpin, G.M. Bressan, and S. Piccolo. 2003. Mapping Wnt/beta-catenin signaling during mouse development and in colorectal tumors. Proc. Natl. Acad. Sci. USA. 100:3299–3304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, J.R., and R.T. Moon. 1996. Signal transduction through β-catenin and specification of cell fate during embryogenesis. Genes Dev. 10:2527–2539. [DOI] [PubMed] [Google Scholar]
- Milton, S.G., and V.P. Knutson. 1990. Comparison of the function of the tight junctions of endothelial cells and epithelial cells in regulating the movement of electrolytes and macromolecules across the cell monolayer. J. Cell. Physiol. 144:498–504. [DOI] [PubMed] [Google Scholar]
- Peifer, M., and P. Polakis. 2000. Wnt signaling in oncogenesis and embryogenesis–a look outside the nucleus. Science. 287:1606–1609. [DOI] [PubMed] [Google Scholar]
- Petrova, T.V., T. Makinen, T.P. Makela, J. Saarela, I. Virtanen, R.E. Ferrell, D.N. Finegold, D. Kerjaschki, S. Yla-Herttuala, and K. Alitalo. 2002. Lymphatic endothelial reprogramming of vascular endothelial cells by the Prox-1 homeobox transcription factor. EMBO J. 21:4593–4599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posthaus, H., L. Williamson, D. Baumann, R. Kemler, R. Caldelari, M.M. Suter, H. Schwarz, and E. Muller. 2002. beta-Catenin is not required for proliferation and differentiation of epidermal mouse keratinocytes. J. Cell Sci. 115:4587–4595. [DOI] [PubMed] [Google Scholar]
- Roberts, A.W., L. Robb, S. Rakar, L. Hartley, L. Cluse, N.A. Nicola, D. Metcalf, D.J. Hilton, and W.S. Alexander. 2001. Placental defects and embryonic lethality in mice lacking suppressor of cytokine signaling 3. Proc. Natl. Acad. Sci. USA. 98:9324–9329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlaeger, T.M., S. Bartunkova, J.A. Lawitts, G. Teichmann, W. Risau, U. Deutsch, and T.N. Sato. 1997. Uniform vascular-endothelial-cell-specific gene expression in both embryonic and adult transgenic mice. Proc. Natl. Acad. Sci. USA. 94:3058–3063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmelz, M., R. Moll, C. Kuhn, and W.W. Franke. 1994. Complexus adhaerentes, a new group of desmoplakin-containing junctions in endothelial cells: II. Different types of lymphatic vessels. Differentiation. 57:97–117. [DOI] [PubMed] [Google Scholar]
- Stacker, S.A., M.G. Achen, L. Jussila, M.E. Baldwin, and K. Alitalo. 2002. Lymphangiogenesis and cancer metastasis. Nat. Rev. Cancer. 2:573–583. [DOI] [PubMed] [Google Scholar]
- Takeichi, M. 1990. Cadherins: a molecular family important in selective cell-cell adhesion. Annu. Rev. Biochem. 59:237–252. [DOI] [PubMed] [Google Scholar]
- Taraboletti, G., S. D'Ascenzo, P. Borsotti, R. Giavazzi, A. Pavan, and V. Dolo. 2002. Shedding of the matrix metalloproteinases MMP-2, MMP-9, and MT1-MMP as membrane vesicle-associated components by endothelial cells. Am. J. Pathol. 160:673–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukita, S., M. Furuse, and M. Itoh. 2001. Multifunctional strands in tight junctions. Nat. Rev. Mol. Cell Biol. 2:285–293. [DOI] [PubMed] [Google Scholar]
- Valiron, O., V. Chevrier, Y. Usson, F. Breviario, D. Job, and E. Dejana. 1996. Desmoplakin expression and organization at human umbilical vein endothelial cell-to-cell junctions. J. Cell Sci. 109:2141–2149. [DOI] [PubMed] [Google Scholar]
- Vasioukhin, V., C. Bauer, M. Yin, and E. Fuchs. 2000. Directed actin polymerization is the driving force for epithelial cell-cell adhesion. Cell. 100:209–219. [DOI] [PubMed] [Google Scholar]
- Vecchi, A., C. Garlanda, M.G. Lampugnani, M. Resnati, C. Matteucci, A. Stoppacciaro, H. Schnurch, W. Risau, L. Ruco, A. Mantovani, et al. 1994. Monoclonal antibodies specific for endothelial cells of mouse blood vessels. Their application in the identification of adult and embryonic endothelium. Eur. J. Cell Biol. 63:247–254. [PubMed] [Google Scholar]
- Vleminckx, K., R. Kemler, and A. Hecht. 1999. The C-terminal transactivation domain of beta-catenin is necessary and sufficient for signaling by the LEF-1/beta-catenin complex in xenopus laevis. Mech. Dev. 81:65–74. [DOI] [PubMed] [Google Scholar]
- Wang, P., M. Anton, F.L. Graham, and S. Bacchetti. 1995. High frequency recombination between loxP sites in human chromosomes mediated by an adenovirus vector expressing Cre recombinase. Somat. Cell Mol. Genet. 21:429–441. [DOI] [PubMed] [Google Scholar]
- Wright, M., M. Aikawa, W. Szeto, and J. Papkoff. 1999. Identification of a Wnt-responsive signal transduction pathway in primary endothelial cells. Biochem. Biophys. Res. Commun. 263:384–388. [DOI] [PubMed] [Google Scholar]
- Yang, Z.Z., O. Tschopp, M. Hemmings-Mieszczak, J. Feng, D. Brodbeck, E. Perentes, and B.A. Hemmings. 2003. PKBalpha regulates placental development and fetal growth. J. Biol. Chem. In press. [DOI] [PubMed] [Google Scholar]
- Zhurinsky, J., M. Shtutman, and A. Ben-Ze'ev. 2000. Plakoglobin and beta-catenin: protein interactions, regulation and biological roles. J. Cell Sci. 113:3127–3139. [DOI] [PubMed] [Google Scholar]