

Abstract
During puberty, mouse mammary epithelial ducts invade the stromal mammary fat pad in a wave of branching morphogenesis to form a complex ductal tree. Using pharmacologic and genetic approaches, we find that mammary gland branching morphogenesis requires transient matrix metalloproteinase (MMP) activity for invasion and branch point selection. MMP-2, but not MMP-9, facilitates terminal end bud invasion by inhibiting epithelial cell apoptosis at the start of puberty. Unexpectedly, MMP-2 also represses precocious lateral branching during mid-puberty. In contrast, MMP-3 induces secondary and tertiary lateral branching of ducts during mid-puberty and early pregnancy. Nevertheless, the mammary gland is able to develop lactational competence in MMP mutant mice. Thus, specific MMPs refine the mammary branching pattern by distinct mechanisms during mammary gland branching morphogenesis.
Keywords: apoptosis; matrix metalloproteinases; stromal–epithelial interaction; terminal end bud; tissue inhibitor of metalloproteinases
Introduction
The mammary gland is changed during puberty from a small, simply branched, relatively quiescent epithelial tissue in the corner of the mammary stromal fat pad into a dynamic tissue, in which ducts undergo dichotomous and lateral branching and invade and fill the fat pad. In mice, this hormone-dependent burst of mammary gland branching morphogenesis begins with the formation of bulbous terminal end buds (TEBs) at the invading front of epithelial ducts at ∼3 wk old. Several distinct mechanisms regulate branching morphogenesis of the mammary gland. TEBs are driven forward, invade the fat pad and undergo dichotomous branching through bifurcation. Behind TEBs, mature ducts sprout laterally to form secondary branches. In contrast to the invasion of TEBs, where immature epithelium invades directly into the adipose tissue of the fat pad, lateral branches must invade through a barrier of myoepithelial cells, basement membrane (BM) and stromal ECM that surrounds mature ducts (Wiseman and Werb, 2002). The invading epithelium must communicate with the stroma to coordinate these events; however, the mechanisms by which this cross talk regulates mammary gland branching morphogenesis are poorly understood (Affolter et al., 2003). Matrix metalloproteinases (MMPs) are stromal factors that are ideally positioned to regulate stromal–epithelial cross talk (Sternlicht and Werb, 2001). MMPs could regulate mammary gland branching morphogenesis by clearing a path for invading ducts by degrading ECM barriers and permitting ductal penetration into the mammary fat pad. MMPs also influence cell signaling. They can change the extracellular microenvironment and thereby alter stromal–epithelial signaling. In addition, cleavage of growth factors, cytokines and cell–cell adhesion proteins by MMPs can affect their activities. MMPs can also release factors that are sequestered in the ECM, thus, making them bioavailable. In three-dimensional cultures of mouse mammary epithelial cells, MMP activity is necessary for growth factor–induced branching, and recombinant MMP-3 (stromelysin-1) is sufficient to induce branching, in the absence of an added growth factor (Simian et al., 2001). Similarly, an autoactivating MMP-3 transgene, targeted to the mouse mammary gland, accelerates branching morphogenesis, inducing supernumerary ductal branching and precocious appearance of lobular alveoli (Sympson et al., 1994; Witty et al., 1995). In contrast, introduction of exogenous tissue inhibitor of metalloproteinases-1 (TIMP-1) into pubertal mammary gland, via a pellet, retards ductal invasion (Fata et al., 1999). Therefore, MMPs may regulate branching morphogenesis in the mammary gland by influencing stromal–epithelial cross talk. However, how specific endogenous MMPs contribute to the different aspects of elaboration of the ductal tree has not been determined.
Here, we modified MMP activity broadly in pubertal female mice using either a small molecule inhibitor of MMPs, GM6001, or by expressing TIMP-1, an endogenous inhibitor of MMP activity, as a transgene or by deleting endogenous TIMP-1. Then, we used genetic analysis in mice to elucidate specific roles for MMP-2 (gelatinase A) and MMP-3 (stromelysin-1).
Results
MMPs have distinct expression patterns in the pubertal mammary gland
First, we determined where MMPs were expressed in the mammary gland by in situ hybridization on sections of pubertal mouse mammary tissue (Fig. 1). Four MMPs were expressed in distinct locations in the mammary gland during puberty. MMP-2 mRNA was concentrated primarily in the periductal stroma and was weakly expressed by adipose tissue. Surprisingly, its expression was reduced at sites of initiating buds or side branches (Fig. 1 D). The mRNA for MMP-14, the principal activator of MMP-2, (Will et al., 1996) overlapped MMP-2 expression only in part. MMP-14 was mainly stromal, but also present in the epithelium, with no reduction at branch initiation sites; instead it was highly concentrated within and around TEBs. MMP-3 was exclusively stromal and located in periductal stroma and the adipose tissue, with no difference at sites of initiating branches. MMP-9 was expressed at low levels throughout the gland in the epithelium and the stroma. There were spots of concentrated MMP-9 (gelatinase B) mRNA that correspond to macrophages.
Figure 1.
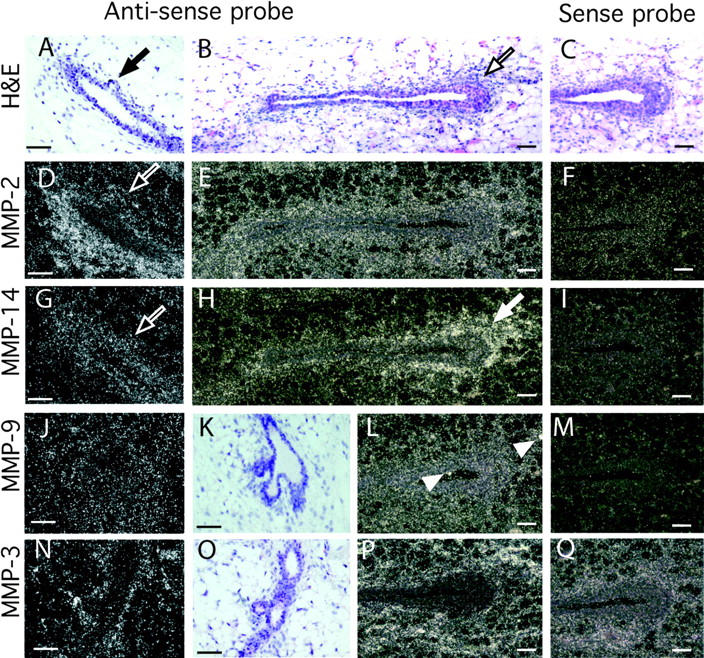
Localization of MMPs-2, -3, -9, and -14 mRNA within the mammary gland. Mammary glands were taken at 50 d old and sectioned. (A–C, K, and O) Hematoxylin and eosin counterstain of mammary gland sections in G–J and N, respectively. Note the initiating lateral branch in A (black arrow) and TEB in B (black outlined arrow). In situ hybridization analysis was performed with the following antisense and sense probes (as indicated): (D–F) MMP-2, (G–I) MMP-14, (J, L, and M) MMP-9, and (N, P, and Q) MMP-3. Note the reduction in MMP-2, but not MMP-14 mRNA, at the initiating lateral branch in the adjacent sections D and G (white outlined arrows); the localization of MMP-14 around the TEB in H (white arrow) and the spots of MMP-9 expression probably localized in macrophages in L (white arrow heads). Bars, 50 μm.
Inhibiting mammary epithelial branching morphogenesis MMP activity by GM6001 inhibits ductal invasion, but induces precocious lateral budding
To determine if MMP activity plays a role in branching morphogenesis during pubertal mammary development, we compared mammary glands from mice treated from 3.5 wk old with a broad spectrum MMP inhibitor to controls treated with the vehicle. GM6001 works well in inhibiting MMP function in vivo (Alexander et al., 1996; Kheradmand et al., 2002), and inhibits all MMPs tested with Ki values of <100 nM (Grobelny et al., 1992; Gijbels et al., 1994). Ducts of mice treated with GM6001 showed much less invasion than controls (Fig. 2, A, B, and H), and stopped about where they were at the beginning of the treatment (not depicted). An unexpected observation in the GM6001-treated mice was that the mammary ducts displayed supernumerary budding along the primary ducts compared with the controls (Fig. 2 B, inset). These buds appeared to be lateral branches that were initiated, but failed to elongate and invade into the mammary fat pad.
Figure 2.
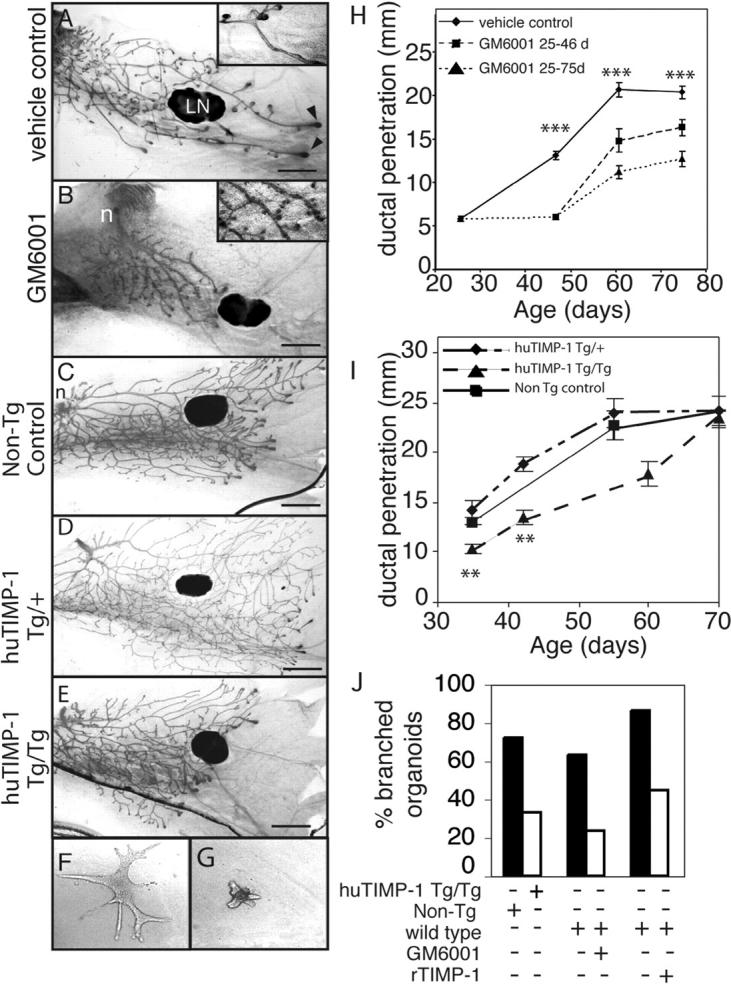
MMP inhibition disrupts mammary gland branching morphogenesis. (A and B) Mammary gland whole mounts from 6.5-wk-old mice treated daily with (A) vehicle or (B) GM6001 from 3.5 wk old. Note the extra ductal budding in the GM6001 treated mammary glands (inset). LN, lymph node; arrowheads, TEBs. Bar, 1 mm. (C–E) Mammary gland whole mounts from (C) nontransgenic control mice and (D) mice hemizygous and (E) homozygous for the β-actin human TIMP-1 transgene at 35 d old. n, nipple. Bar, 1 mm. (F and G) Primary mammary organoids grown for 7 d in the presence of EGF in collagen gels from (F) nontransgenic control mice or (G) huTIMP-1 transgenic mice. (H and I) Penetration of mammary ducts into fat pad of control mice, mice continually treated with GM6001 until sacrifice and mice that were treated with GM6001 from 3.5- to 6.5-wk-old using 8–12 mammary glands per data point (t test compared vehicle-control with mice continually treated with GM6001; H) and (I) nontransgenic control, Tg/+ and Tg/Tg hu-TIMP-1 transgenic mice using 4–12 mammary glands per data point. Data are mean ± SEM. t test compared Tg/+ with Tg/Tg. (J) The percentage of branched organoids in response to 7-d treatment with EGF. Organoids were derived from hu-TIMP-1 transgenic mice, nontransgenic controls, or wild-type mice and grown in absence or presence of GM6001 or recombinant TIMP-1 as indicated. In all panels: ***, P < 0.0005; **, P < 0.005, unpaired, two-tailed t test.
The GM6001-treated mammary glands had fewer TEBs than controls. In many of the GM6001-treated mice, some primary ducts had no TEBs at their ends. Moreover, the TEBs that formed were generally smaller than those of controls. At 6.5 wk old, 1/11 treated mice had no TEBs. By 8.5 wk, the number with no TEBs had risen to 33% and by 10.5 wk, 100% of the GM6001-treated mice had no TEBs. The control mice were normal. In all cases, serum estrogen levels were normal and there was still space in the fat pad (unpublished data).
To determine if the complete inhibition of ductal invasion by GM6001 was reversible, we treated mice between 3.5 and 6.5 wk and then stopped the treatment. We found that, after 2 and 4 wk of relief from GM6001 treatment, the mammary ducts partially recovered (Fig. 2 H). This observation demonstrates that GM6001 did not produce irreversible damage, and that the ductal tree still had the potential to recommence growth and invasion. GM6001 treatment was stopped at 6.5 wk followed by 4 wk of recovery, 80% of the mice had TEBs and space in the fat pad, indicating that their ducts were still actively invading into the fat pad.
Transgenic overexpression of TIMP-1 attenuates mammary epithelial branching morphogenesis
As a second approach, we inhibited activity of most MMPs using transgenic mice that overexpress human TIMP-1 driven by an β-actin promoter (huTIMP-1 mice). We found that the mammary ducts of mice that expressed huTIMP-1 transgenes from both alleles (Tg/Tg) showed decreased invasion, taking 2 wk longer to reach the edges of the mammary fat pad compared with wild-type controls (Fig. 2 I). Thus, exogenous TIMP-1 retards, but unlike GM6001, does not completely block ductal invasion. Interestingly, the primary mammary ducts of mice with only one transgene (Tg/+) grew to the same extent as the wild-type glands (Fig. 2 I), suggesting that there is a threshold for TIMP-1 to inhibit ductal invasion. TIMP-1 Tg/Tg mice showed only a slight effect on secondary branches, compared with GM6001-treated mice (Fig. 2, C–E).
Inhibiting MMP activity in organotypic cultures attenuates mammary epithelial branching
The in vivo experiments with GM6001 and the huTIMP-1 transgene inhibited MMPs systemically, and not just in the mammary gland. To determine if MMP inhibition acts locally on mammary cells, we isolated mammary organoids (Simian et al., 2001) from wild-type or Tg/Tg β-actin huTIMP-1 mice and cultured them in collagen gels. Upon addition of EGF or keratinocyte growth factor (FGF-7), wild-type organoids extended several branches that contained lumens. However, few huTIMP-1 organoids formed branches, and, in those that did, the branches were shorter and fewer in number (Fig. 2, F, G, and J; not depicted). Recombinant TIMP-1 and GM6001 added to organoid cultures also inhibited branching in the same manner (Fig. 2 J). Together, with our previous studies (Simian et al., 2001), these experiments indicate that branching of mammary cells is directly responsive to MMP inhibition.
TIMP-1 knockout mice have altered TEB morphology
If inhibition of MMP activity attenuates mammary ductal invasion, then the absence of TIMP-1 could reduce MMP action and change mammary morphogenesis. TIMP-1 mRNA is up-regulated during mouse mammary gland pubertal development (Fata et al., 1999), but we found no change in primary duct elongation in TIMP-1 −/− mice compared with controls, and only small increases in the number of branch points (Fig. 3 D). This is not surprising because at least three TIMP family members are expressed in the mammary gland. However, TEBs were much larger in TIMP-1 −/− mice than their wild-type littermates (Fig. 3 B), indicating that TIMP-1 contributes to maintenance of TEB morphology.
Figure 3.

TIMP-1 is not necessary for ductal invasion or branching in the mammary gland. (A and B) Whole mounts of mammary glands of 42-d-old TIMP-1 +/+ (A) and TIMP-1 −/− mice (B). Note the enlarged TEBs of TIMP-1 −/− mice (inset). Bars, 1 mm. (C and D) Penetration of mammary ducts into fat pad (C) and number of branch points beyond the lymph node at 42 d old (D) of TIMP-1 +/+ and TIMP-1 −/− mice. Data are mean (C) ± SEM or (D) ± SD using four to eight mammary glands per data point.
Together, with the results of GM6001, these data indicate that MMP activity regulates aspects of branching morphogenesis in the mammary gland. Therefore, we investigated which MMPs are involved.
MMP-2 regulates ductal invasion
MMP-2 is expressed in the stromal compartment around mammary ducts during branching morphogenesis and is present in an activated form (Fig. 1; Fata et al., 1999). MMP-2 influences branching in the pseudoglandular stage of lung development (Kheradmand et al., 2002), making it a good candidate for regulating mammary branching. Although MMP-2 −/− mice show attenuated tumor growth (Itoh et al., 1997, 1998), the females are able to nurture their young. We compared mammary glands of age- and estrus-matched MMP-2 −/−, MMP-2 +/−, and wild-type mice. Because there was no discernable heterozygous phenotype, we used the MMP-2 +/− mice as controls. There was no difference in the morphology of MMP-2 −/− mammary glands compared with littermate controls at 20 d old, indicating that MMP-2 is not needed before pubertal development. However, we found retarded invasion of the mammary ducts of MMP-2 −/− mice into the stromal fat pad at 30 d (Fig. 4, A, B, and I). This corroborates our in situ data showing that mRNA for the activator of MMP-2, namely MMP-14, is concentrated right at the invasive front of ducts (Fig. 1). This corresponds to the site of highest MMP activity as seen by in situ zymography (unpublished data). This phenotype was most evident in early puberty at 30 d old and the difference was significant relative to the fat pad length. However, the retardation was transient, and by 50 d, the difference was no longer significant (Fig. 4, C, D, and I). Therefore, MMP-2 regulates only the initial events in ductal invasion after puberty.
Figure 4.

MMP-2 −/− mice have reduced ductal invasion and increased lateral branching. (A–D) Whole mounts of mammary glands of an (A and C) MMP-2 +/− and (B and D) MMP-2 −/− mice (A and B) 30 d old and (C and D) 50 d in estrus. Note supernumerary branching in D (inset). Example of ramified branch (arrows) and unramified branch or bud (arrowheads). Bars, 1 mm. (E–H) Sections through TEBs in mammary glands from 30-d-old (E and G) MMP-2 +/− and (F and H) MMP-2 −/− mice. (E and F) Immunohistochemistry for Ln-1. (G and H) TUNEL assay. Apoptotic cells are red. Bars, 25 μm. (I–L) Penetration of mammary ducts of (I) MMP-2 +/− and MMP-2 −/− and (J) MMP-9 +/− and MMP-9 −/− mice, (K) number of branches per millimeter, and (L) number of unramified branches per millimeter at 50 d from primary mammary ducts of MMP-2 +/− and −/− mice. Data are mean ± SEM (I, K, and L) using 8–16 or (H) 4–8 mammary glands per data point. (M and N) Percentage of BrdU (M) or TUNEL (N) positive cells within TEBs of MMP-2 −/− and MMP-2 +/− 30-d-old mice. Data are mean percent per TEB ± SD. ***, P < 0.0005; **, P < 0.005; *, P < 0.05, unpaired, two-tailed t test.
Then, we sought the mechanism by which MMP-2 might regulate ductal penetration. TEBs formed properly and in a timely manner in MMP-2 −/− mice, indicating that puberty was not delayed, and the glands responded to ovarian hormones. MMP-2 has many ECM substrates (Sternlicht and Werb, 2001), but we did not observe a build up of BM proteins laminin-1 (Ln-1; Fig. 4, C and D) and collagen IV (Coll IV), or interstitial ECM proteins (fibrillar collagens, tenascin-C, fibronectin, and vitronectin) surrounding TEBs or periductally (not depicted). TEBs increase in number by bifurcation or dichotomous branching (Wiseman and Werb, 2002). However, there were no differences in the number of TEBs in the mammary glands of MMP-2 −/− mice compared with controls (unpublished data).
Alternatively, MMP-2 could regulate cell proliferation and/or apoptosis, both of which occur at high rates within TEBs, to provide cells for ductal extension and for hollowing out the ductal lumen, respectively (Humphreys et al., 1996). We found no overt defects in TEB structure, size, association with the stroma, or cell proliferation in MMP-2 −/− mice at 25 and 30 d (Fig. 4 and not depicted). However, the TEBs of MMP-2 −/− mice had almost twice the level of apoptosis and activated caspase-3 compared with controls (Fig. 4, G, H, and N; and not depicted). Thus, MMP-2 supports epithelial cell survival, and in its absence there may be too few cells available to form the growing duct. These data also suggest that the mechanism of MMP-2 regulation of ductal morphogenesis and invasion of TEBs occurs largely through path finding by a pushing action from the increasing mass of proliferating cells, rather than through a path clearing action of the MMP.
MMP-2 represses lateral budding
MMPs have long been thought to facilitate cell migration by degrading ECM. However, we made the unexpected observation that MMP-2 mRNA was down-regulated along the primary ducts at sites where new lateral branches initiate (Fig. 1 D). This raises the question of whether MMP-2 has a negative role in lateral branching. We determined the number of branch points arising from primary ducts as a function of ductal length (to account for the difference in lengths). In the absence of MMP-2, mammary ducts had more lateral branches than controls (Fig. 4 K). The branching defect was confined to lateral branching and did not affect TEB bifurcation. Interestingly, the increase was confined to secondary buds and branches arising from the primary ducts (Fig. 4 L). There was no difference in tertiary branching (i.e., the frequency of ramified secondary branches; unpublished data). Therefore the absence of MMP-2 fosters the premature initiation of buds and small branches. Importantly, this supernumerary branching only began ∼40 d old, about the time that the primary ducts mature, and was most evident ∼50 d. The loss of MMP-2 accelerated the rate at which the secondary branches appeared, but by the time the mammary gland matured at 70 d old, the MMP-2–null mice displayed a normal ductal tree. This suggests that MMP-2 regulates the rate at which branches appear, but does not affect the selection of branch sites. Thus, MMP-2 has two roles: during early puberty it promotes TEB invasion and supports cell survival, and later in puberty it represses the rate of lateral branching. In contrast, MMP-9 has no obvious role in mammary gland branching morphogenesis. Mice deficient for MMP-9, a close relative of MMP-2, which is present and active (Fig. 1; Fata et al., 1999), yielded no differences in ductal length or branching (Fig. 4 J and not depicted).
MMP-3 regulates secondary duct formation during mammary branching morphogenesis
To initiate a new branch from a mature duct, it is necessary to degrade the BM and stromal ECM underlying epithelial cells that are primed for proliferation and invasion. Our previous data suggest that MMP-3 degrades BM components, sheds the extracellular domain of E-cadherin and regulates differentiation of the adipogenic stroma in the mammary gland (Sympson et al., 1994; Alexander et al., 1996; Thomasset et al., 1998). MMP-3 also facilitates contraction of dermal fibroblasts during wound healing (Bullard et al., 1999).
MMP-3 −/− mice showed a loss of function phenotype (Fig. 5, A and B) that was the mirror image of the gain of function phenotype that we described previously in WAP-MMP-3 transgenic mice (Fig. 5, I and J; Sympson et al., 1994). The heterozygote and wild-type mice were indistinguishable. In contrast to MMP-2 −/− mice, the ductal tree of mammary glands from MMP-3 −/− mice was much sparser, yet there were no differences in primary ductal invasion compared with controls (Fig. 5, A, B, and L). Both the frequency of branches and the total number of branch points were greatly reduced in MMP-3 −/− mice (Fig. 5 M and not depicted). However, there was no difference in the number of TEBs (not depicted) between MMP-3 −/− glands and controls, indicating that MMP-3 was needed for lateral branching, rather than dichotomous branching through TEB bifurcation. Furthermore, ramified secondary branches were absent (Fig. 5 N), implying that MMP-3 regulates both secondary and tertiary branching. This phenotype was transient and most evident around 50 d old (Fig. 5, M and N). By 70 d old, the mammary ductal tree in MMP-3 −/− mice was indistinguishable from controls, indicating that other factors can compensate for MMPs after 50 d.
Figure 5.

MMP-3 is required for lateral branching in the mammary gland. (A–H) Whole mounts of mammary glands of (A, C, E, and G) MMP-3 +/− and (B, D, F, and H) MMP-3 −/− mice at (A and B) 42 d old in estrus, (C and D) 6 d of pregnancy, (E and F) 9 d of pregnancy, and (G and H) 13 d of pregnancy. (I and J) Whole mounts of mammary glands of 70-d-old virgin (I) nontransgenic controls or (J) WAP-MMP-3 transgenic mice. Bars, 1 mm. (K) Wild-type mammary gland stained for Ln-1. Note reduction in Ln-1 where lateral branches are budding (arrows). Bar, 25 μm. (L–N) Penetration of ducts (L), number of total branches per millimeter (M), and number of ramified secondary branches per millimeter (N) from primary ducts of MMP-3 −/− and MMP-3 +/− mammary glands. Data are mean ± SEM using 4–12 mammary glands per data point. ***, P < 0.0005; **, P < 0.005; *P, < 0.05, unpaired, two-tailed t test.
At the onset of pregnancy, another phase of lateral branching occurs from the mature mammary ducts and continues until mid-pregnancy when alveoli form on the expanded ductal tree. Upon analysis, the ductal trees of mammary glands from pregnant MMP-3 −/− mice were much sparser than controls at 6 and 9 d of pregnancy (Fig. 5, C–F). Again, the phenotype was transient and by the 13 d of pregnancy, there were no gross differences between knockouts and controls (Fig. 5, G and H). Thus, MMP-3 induces secondary and tertiary lateral branching midway through puberty and again in early pregnancy. Indeed, mammary glands of MMP-3 −/− mice function and their pups have no problems feeding (Lund et al., 2000).
MMP-2 and -3 have opposite effects on branching morphogenesis during puberty. This raises the question of what phenotype MMP-2; MMP-3 double null mice would show in their mammary glands. We examined 11 mammary glands from MMP-2 −/−; MMP-3 −/− mice. These mammary glands resembled wild-type mammary glands, albeit with slightly delayed elongation of secondary branches (unpublished data). These data suggest that these two MMPs function in the epithelial microenvironment in different locations in a network of interacting pathways designed to give the final branching pattern.
We found that the mammary glands of WAP-MMP-3 transgenic mice in which an autoactivating MMP-3 is targeted to the mammary gland by the whey acidic protein promoter show supernumerary lateral branching (Fig. 5, I and J), in keeping with our earlier observations (Sympson et al., 1994), but no change in the rate of primary ductal elongation (not depicted). Consistently, the WAP-MMP-3 transgenic mice had supernumerary secondary and tertiary branches (Sympson et al., 1994) that were greater in number than those seen during the precocious branching seen in MMP-2 −/− mice, approaching the density of branching seen in early pregnancy (Fig. 5, C and E).
For lateral branches to initiate, the sites must first be determined. Then, the epithelial cells must proliferate, migrate, and invade through periductal stroma and ECM that surround mature ducts. MMP-3 cleaves BM components including Ln-1, nidogen, and Coll IV in the mammary gland (Alexander et al., 1996). By immunohistochemistry, we observed that MMP-3 −/− and wild-type mammary glands had similar levels of Ln-1 and Coll IV in the BM surrounding mature ducts. However, both Ln-1 and Coll IV were specifically degraded at sites of branch formation (Fig. 5 K and not depicted). Although we could not locate the points where branches failed to form, there were fewer sites of ECM degradation along the ductal length in MMP-3 −/− glands, which is consistent with the reduced number of branch points (Fig. 5, M and N). These data indicate that, unlike down-regulation of MMP-2 (Fig. 4 K), which allows a more rapid emergence of side branches that eventually total the normal number, MMP-3 affects the selection of branch sites by itself, and in excess, can trigger branch formation from stem and progenitor cells that lie dormant along the ducts.
These data suggest that MMP-3 has three roles in epithelial morphogenesis: it participates in the selection and spacing of the lateral branches; it facilitates lateral branching by degrading BM components and allowing epithelial cells to invade into the stromal spaces; and it regulates the phenotype of stromal fibroblasts/preadipocytes, which in turn affect the epithelium.
Discussion
Factors that initiate and control the outgrowth of individual branches and reiteration of the branching process are just being elucidated in the mammary gland. Here, we have shown that two MMPs have distinct functions in the correct execution of these steps. It is significant that MMPs are made almost exclusively in the mesenchymal compartment. As such, they are critical mediators of the epithelial–mesenchymal cross talk and the transient epithelial to mesenchymal transitions needed for a branch to form (Affolter et al., 2003). Intriguingly, MMPs may directly regulate migratory activity by cleaving ECM molecules like laminin-5, turning it into a motogen (Koshikawa et al., 2000). Our studies in vivo and other studies suggest that programmed cell death contributes to the morphology of branching organs, particularly in formation of a lumen (Humphreys et al., 1996; Blatchford et al., 1999; Debnath et al., 2002), and that MMPs mediate these processes (Boudreau et al., 1995). At least part of the function of the MMPs is to activate TGF-β, which inhibits lateral branching (Bottinger et al., 1997; Yu et al., 2001; Ewan et al., 2002).
Our results show that, in mice, there are distinct mechanisms and phases of mammary gland branching morphogenesis, both positive and negative, which are regulated by MMPs. During early puberty, MMP-2 supports the invasion of TEBs into the stromal fat pad, by protecting against excessive apoptosis within TEBs. Later in puberty, MMP-2 acts on the mature primary duct to repress excessive secondary lateral budding and branching. In contrast, MMP-3 acts on both primary and secondary ducts to induce secondary and tertiary branch formation. Thus, the MMP-3 loss of function analysis parallel the gain of function phenotypes, characterized by a significant delay in secondary and tertiary branching, is precisely the opposite of the gain of function phenotype seen in mammary-targeted MMP-3 transgenic mice, in which supernumerary lateral branching and eventual tumor formation occur (Sympson et al., 1994; Sternlicht et al., 1999).
MMP inhibitors regulate mammary morphogenesis
The demonstration that an exogenous huTIMP-1 transgene inhibits mammary ductal invasion suggests that these effects are due to the inhibition of a TIMP-1–inhibitable MMP. In addition, the implantation of TIMP-1 pellets has a similar effect locally, yet these pellets may result in unphysiologically high levels of TIMP-1 (Talhouk et al., 1992; Alexander et al., 1996) or produce an inflammatory response to surgery (Fata et al., 1999). Moreover, TIMP-1 can exert growth promoting activity independent of its MMP inhibitory activity (Baker et al., 2002). Nevertheless, we saw similar effects on mammary branching morphogenesis, both in vivo and in vitro, with a synthetic MMP inhibitor, GM6001. Thus, we conclude that, in the mammary gland, TIMP-1 acts by inhibiting the proteolytic activity of MMPs, and that MMP activity is required for normal mammary gland branching morphogenesis. GM6001 also induced the regression of TEBs, yet this was not obvious in the absence of MMP-2 or -3 nor in TIMP-1–overexpressing transgenic mice, suggesting that an unidentified MMP plays a role in TEB maintenance. TIMP-1 and GM6001 have different inhibitory activities against specific MMPs. TIMP-1 does not inhibit MMP-14 or MMP-19 at physiological concentrations, and is less effective at inhibiting MMP-2 than MMP-3. On the other hand, high concentrations of GM6001 may inhibit ADAM-TS metalloproteinases. The response of the ducts to MMP inhibition was also dose dependent. A double dose of the huTIMP-1 transgene was needed to suppress normal ductal development.
That the loss of TIMP-1 only produced a mild gain of function phenotype is not surprising because there are four TIMP family members, each of which is expressed in the mammary epithelium and adipogenic stroma (Fata et al., 1999; Alexander et al., 2001), as well as other endogenous MMP inhibitors (Welm et al., 2002). Thus, MMP-mediated effects on the mammary gland are still controlled, despite the absence of TIMP-1. Surprisingly however, the mammary phenotype in the TIMP-1–deficient mice was less pronounced than that seen in mice with only a 50% decrease in their normal TIMP-1 levels due to an antisense TIMP-1 transgene. Consistent with the notion that MMPs influence ductal elongation and branching, these partially TIMP-1–deficient mice had longer ducts and supernumerary branching compared with controls and no difference in TEB size (Fata et al., 1999). Thus, their more pronounced phenotype may be due to compensatory responses, the antisense repression of additional TIMPs, or strain-specific differences in sensitivity to MMP inhibition.
Invasion of TEBs requires MMP-2 activity
MMP-2 regulates the initial invasion of TEBs into the fat pad. MMP-2 has been implicated in the induction of apoptosis through destruction of ECM, leading to altered adhesion (anoikis; Lund et al., 1996; Roberts et al., 2002) or by allowing infiltration of toxic immune cells (Wielockx et al., 2001). In contrast, MMP-2 promotes cell survival in the TEB, which is a site of both proliferation and cell death. Thus, MMP-2 may release survival factors sequestered by binding proteins or the ECM. However, TEBs are multilayered and the apoptotic cells are found close to the lumen (Fig. 6; Humphreys et al., 1996), which suggests that these cells are not dying by anoikis. Thus, other potential substrates, such as insulin-like growth factor binding proteins, which are MMP-2 substrates (Fowlkes et al., 1999) that can be inhibited from signaling in vivo by MMP inhibitors (Martin et al., 1999), may be responsible for the survival-promoting effects of MMP-2. The enlarged TEBs of the TIMP-1–deficient mammary glands may also be related to a reduction in apoptosis due to increased MMP-2 activity, leading to an overabundance of cells. MMP-2 presumably allows sufficient cells to accumulate for ductal extension to ensue. Although it is likely that cell migration is important for branching and the invasion of the epithelial cell layer into the fat pad, our results suggest that branches may be pushed outwards by cell division. This does not preclude the possibility that they are also pulled out by migratory cells.
Figure 6.

Model for different phases of mammary gland branching morphogenesis. Before puberty, the mammary epithelial is small and simply branched. In response to the release of estrogen (E) and growth hormone (GH), at ∼3 wk old TEBs form. MMP-2 is then involved in inducing TEBs to invade and the ducts begin to fill the fat pad by branching dichotomously through bifurcation. At ∼6–8 wk old, the mammary ducts branch laterally. This process is suppressed by MMP-2 and induced by MMP-3 and may be related to changes in the response of the gland to progesterone (P) and prolactin (PRL). The fat pad is filled with ducts at ∼10 wk old and is relatively quiescent until pregnancy, when there is another wave of lateral branching that is regulated by MMP-3, P, and PRL before the formation of lobular alveoli.
MMP-2 may have functions that facilitate TEB invasion in addition to protection from apoptosis. Although we did not detect a build up of Ln-1 and Coll IV at the invading front of the TEBs, there may be a build up of other ECM components. MMP-2 produces a bioactive fragment from γ2 chain of laminin-5 that induces breast epithelial cells to migrate in vitro (Giannelli et al., 1997). MMP-2 is important in angiogenesis (Itoh et al., 1998; Kato et al., 2001) and may regulate blood vessel formation for the newly formed mammary epithelium.
MMP-14 is an activator of MMP-2 (Will et al., 1996; for review see Sternlicht and Werb, 2001). The localization of MMP-14 (a membrane-bound MMP) at the invading front of TEBs, together with the requirement for MMP-2 in TEB invasion, strongly suggests that MMP-14 anchors MMP-2 activity at this invading front. Moreover, TIMP-2 is part of the MMP-14 complex that activates MMP-2, and in contrast to TIMP-1, implantation of TIMP-2 Elvax pellets increases ductal invasion locally (Fata, J.E., and R. Khokha, personal communication). Thus, MMP-14 and TIMP-2 may localize and activate MMP-2 at the invasive front of TEBs and so assist the invasion of primary ducts.
MMPs differentially regulate lateral branching
We predicted that an MMP capable of BM degradation would be expressed at branch initiation points. Instead, we found that expression of MMP-2 is specifically down-regulated at branch points. Because supernumerary branches appeared in MMP-2 −/− mice and in mice treated with GM6001, the selection of active sites for branching may be regulated by MMP-2, such that that potential branch sites that still express MMP-2 are inhibited. TGFβ is activated by MMP-2 (Yu and Stamenkovic, 2000) and is a potential effector of MMP-2–mediated branch inhibition, because TGFβ signaling represses lateral branching and proliferation in the mammary gland (Kordon et al., 1995; Joseph et al., 1999). Importantly, regulation of epithelial invasion or morphogenesis by MMP-2 depends very much on context, because at different sites and phases, MMP-2 promotes (at the invasion front of TEBs) and inhibits (from mature ducts) epithelial invasion. Thus, MMP-2 differentially regulates epithelial morphogenesis depending on its microenvironment.
In contrast to MMP-2, our data from knock out and transgenic mice demonstrate that MMP-3 induces secondary and tertiary ducts in the mammary gland both midway through puberty and again at pregnancy. The extrusion of lateral branches from mature ducts requires that the initiating cells break though BM and a thick layer of interstitial ECM (Wiseman and Werb, 2002). Indeed Ln-1 protein levels are reduced at sites of initiating branches (Fig. 5 K). Thus, MMP-3 may break down the physical BM barrier to allow lateral branching. Interestingly, Ln-1 is produced by myoepithelial cells and stabilizes the polarity of epithelial cells (Gudjonsson et al., 2002). Because epithelial cells depolarize when initiating new branches (O'Brien et al., 2002). MMP-3 may degrade Ln-1 to allow epithelial depolarization and migration at bud initiation sites. MMP-3 also cleaves the ectodomain of E-cadherin, which inhibits E-cadherin function and could induce invasion of breast epithelial cells (Lochter et al., 1997; Noe et al., 2001).
The requirement for MMP activity for ductal elongation and lateral branching was transient in all cases except treatment with GM6001. This suggests that MMPs facilitate developmental processes, possibly by increasing bioavailability of signaling factors or loosening the ECM barrier, but are not absolutely required for these events. Alternatively, there may be compensation by other proteases. The delays observed may be because the compensatory protease is more inefficient than the original MMP, but can eventually complete the task. This may be why the effects of GM6001 were not transient: there was no MMP activity to compensate.
Mammary gland branching morphogenesis occurs in distinct phases
The mammary gland is patterned by distinct mechanisms: ductal invasion, bifurcation of TEBs, and lateral branching from mature ducts. MMPs regulate at least two of these mechanisms, and yet the requirement for MMPs becomes apparent at different ages. MMP-2 was required for invasion at 3–4 wk old, whereas MMP-2 and -3 were needed to regulate lateral branching after 6 wk old. It is interesting that the hormonal pathways (prolactin and progesterone) that control tertiary branching in the mammary gland (Hovey et al., 2002) are also up-regulated at this time. Expression of prolactin and progesterone receptor mRNAs increase in the mammary gland at 6 and 8 wk old, respectively (Hovey et al., 2001) and progesterone receptor protein is redistributed between 6 and 12 wk old (Seagroves et al., 2000). This suggests that midway through puberty, ducts undergo a maturation phase, regulated by MMPs and hormones, which manifests as the ability to sprout and ramify lateral ducts. Thus, we propose that mammary gland branching morphogenesis occurs in distinct phases (Fig. 6). First, at around 3 wk old, TEBs are formed in response to estrogen and growth hormone, and ducts begin to invade in an MMP-2–dependent manner. Then, ∼6 wk old and in response to prolactin and progesterone, ducts mature and sprout lateral branches, which is dependent on both MMP-2 and -3. Then during early pregnancy, another round of MMP-3–dependent lateral branching occurs, which also requires progesterone and prolactin (Hovey et al., 2002).
Unlike the Drosophila trachea, branch outgrowth and elongation in the mammary gland are associated with cell division, and, thus, the former must somehow be coupled to the latter. Stem cells capable of repeated cycles of growth are embedded in the ductwork (Welm et al., 2002). This phenomenon appears to occur in Drosophila, where tracheoblasts repopulate the tracheal system after each larval state (Sato and Kornberg, 2002).
Our work separates branching morphogenesis into phenotypically recognizable stages each regulated by distinct MMPs. This prototype may facilitate mapping the factors that have already been found to regulate these different branching mechanisms into a regulatory network, which, so far, has been unfeasible (Hennighausen and Robinson, 2001).
Much of what we have learned from the way MMPs regulate mammary branching morphogenesis can be applied to increasing our understanding of how MMPs regulate epithelial morphogenesis and invasion in human diseases, such as metastatic cancer. We have found that their action depends greatly on context, and different MMPs can have opposing effects. Our challenge is now to elucidate the detailed mechanisms underlying these biologic events.
Materials and methods
Mice
Mice used for the GM6001 experiment were Balb/C. GM6001 (3-[N-hydroxycarbamoyl]-[2R]-isobutylpropionyl-l-tryptophan methylamide) was synthesized according to Grobelny et al. (1992) and administered daily i.p. at 100 mg/kg body weight as a 20 mg/ml slurry in 4% carboxymethylcellulose in 0.9% PBS from 3.5 wk old until indicated. Controls were treated with a daily injection of 4% carboxymethylcellulose. We analyzed 8–12 treated and control mice for each time point.
The transgenic mice expressing a human TIMP-1 transgene under the control of the β-actin promoter have been described previously (Alexander et al., 1996) and were on a CD-1 background. Wild-type mice were nontransgenic siblings, Tg/+ transgenics were mice resulting from transgenic × wild-type crosses and Tg/Tg transgenic mice were mice resulting from transgenic × transgenic crosses, in which both parents had only had transgenic offspring when mated with a wild-type partner. We analyzed 33 Tg/Tg mice and 40 controls. Mice carrying a targeted null mutation of the TIMP-1 gene were on the Balb/C background and have been described previously (Soloway et al., 1996) and were compared with heterozygous littermates. We analyzed 14 TIMP-1 −/− mice and 11 littermate controls.
Mice carrying a targeted null mutation of MMP-2 have been reported previously (Itoh et al., 1997) and then backcrossed into the FVB/n background for over five generations and compared with heterozygous littermates. We analyzed 42 MMP-2 −/− mice and 45 littermate controls. Mice carrying a targeted null mutation in the MMP-3 gene have been described previously (Mudgett et al., 1998) and were on a mixed out bred background and were compared with heterozygous littermates. We analyzed 36 MMP-3 −/− mice and 22 littermate controls. Transgenic WAP-MMP-3 mice have an auto-activating rat MMP-3 transgene targeted to the mammary epithelium by the mouse WAP gene promoter, they were on the CD-1 background and have been described previously (Sympson et al., 1994) and were compared with wild-type mice from transgenic crosses. These samples were provided by C. Sympson (Lawrence Berkeley Laboratory, Berkeley, CA). We analyzed 25 WAP-MMP-3 mice and 22 controls.
Mammary gland whole mount preparation and morphometric analysis
Female mice were killed during estrus as determined by vaginal smear (Rugh, 1990). The No. 4 (inguinal) mammary glands were whole mounted onto glass slides, stained with alum carmine, and cleared of fat as described previously (Sympson et al., 1994). Whole mounts were photographed at 8× on a stereo microscope (model MZFL111; Leica) equipped with a digital camera (model DXM1200; Nikon) and accompanying software (ACT-1; Nikon) and images transferred to Adobe Photoshop. NIH Image software was used for morphometric analysis. Ductal penetration was the average of the mean length of straight lines from the nipple to the ends of the three longest ducts of each mammary gland. The number of branches per millimeter of duct was the average of the mean number of unbranched and ramified branches on three of the longest primary ducts as a function of the actual length of those ducts. The number of branch points was the mean number of branch points beyond a vertical line through the center of the lymph node of each mammary gland.
Immunohistochemical analysis
Antigen retrieval for rabbit polyclonal antilaminin (1:500; Caltag) was digestion with 0.4% pepsin (Sigma-Aldrich), pH 5.2, for 90 min at 37°C. Endogenous biotin binding and peroxidase activities were blocked with Avidin-Biotin blocking kit (Vector Laboratories) and 3% H2O2, respectively. Primary antibody localization was with biotinylated anti–rabbit IgG (1:250; Sigma-Aldrich), then amplification with ABC reagent (Vector Laboratories). For chromogenic visualization of antigen, Fast DAB (Sigma-Aldrich) was the substrate for HRP with hematoxylin (Zymed Laboratories) counterstain. For fluorescent visualization, tyramide reagent (NEN Life Science Products) was followed by visualization with Alexa 488–streptavidin (1:350; Molecular Probes). Brightfield, fluorescence, and darkfield images were obtained with a microscope (model DMR HC; Leica) using 10×/0.30 and 20×/0.50 HC Plan Fluotar and 40×/0.75 Plan Apochromat air objectives. Color digital images were captured to Adobe Photoshop using a cooled CCD digital camera (model SPOT 100; Diagnostic Instruments) and accompanying SPOT software. To ensure consistent exposure of adjacent sense and antisense in situ hybridization sections, darkfield images were captured using manual R/G/B and gain settings of 0.25/0.4/3 and 16, respectively. Otherwise, automated exposure settings were used after white balance for all other images.
Analysis of cell proliferation and apoptosis within TEBs
TEBs were defined by their morphology (including an open lumen) and their position. For cell proliferation assays, 30-d-old MMP-2 +/− and −/− littermates were injected i.p. with 2 mg/mouse BrdU (Sigma-Aldrich) 2 h before harvest. PFA-fixed No. 4 mammary glands were paraffin embedded and sectioned for the cell proliferation and apoptosis assays. Cells that incorporated BrdU were localized using the BrdU staining kit (Zymed Laboratories). We counted the number of BrdU positive cells as a percentage of total number of cells within each TEB. At least 10 TEBs from three mice (i.e., 2–4 × 103 cells) were counted for each point. To analyze apoptosis, the ApoptagRed kit (Intergen; based on the TUNEL assay) was used and mounted in DAPI containing Vectorshield mounting medium (Vector Laboratories). We defined apoptotic cells as the red stained cells and determined their number as a percentage of the total number of cells within a TEB. Two to three adjacent sections through over 20 TEBs from three mice (i.e., over 104 cells) for each point were assayed.
In situ hybridization analysis of mRNA
The probes and the techniques used for the in situ hybridization have been described previously (Lund et al., 1996, 1999).
Preparation of primary mammary organoids
Primary mammary epithelial organoids consist of epithelial, myoepithelial cells, and some periductal stromal cells. They were prepared from 10-wk-old virgin mice by manual and enzymatic digestion, embedded in type I collagen gels and cultured as described previously (Simian et al., 2001). EGF (Collaborative Research) was used at a final concentration of 50 ng/ml. GM6001 was a gift from R. Galardy (Glycomed Corp, Alameda, CA) and dissolved at 100 mM in dimethylsulfoxide and used at a final concentration of 10 μM. TIMP-1 was used at 150 nM. It was purified from conditioned medium of confluent cultures of baby hamster kidney cells stably transfected with human TIMP-1 (University Technologies International, Inc.), and maintained in DME supplemented with 0.2% lactalbumin hydrolysate and penicillin/streptomycin according to (Howard et al., 1991).
Acknowledgments
We thank Ying Yu, Lidiya Korets, and Bernard Thompson for excellent assistance in maintaining the mouse colony and Joey Hansell and Mari Sciabica for technical assistance.
This work was supported by grants from the National Cancer Institute (CA57621 and CA58207), the U.S. Department of Defense Breast Cancer program (DAMD17-99-1-9113, DAMD17-99-1-9103), the Human Frontiers Science Program (RG0051/1999-M), and a Child Health Research grant from the Charles H. Hood Foundation, MA.
Caroline M. Alexander's present address is McArdle Laboratory for Cancer Research, University of Wisconsin Medical School, Madison, WI 53706-1599.
Abbreviations used in this paper: BM, basement membrane; Ln, laminin; MMP, matrix metalloproteinase; TEB, terminal end bud; TIMP, tissue inhibitor of metalloproteinases.
References
- Affolter, M., S. Bellusci, N. Itoh, B. Shilo, J.P. Thiery, and Z. Werb. 2003. Tube or not tube: remodeling epithelial tissues by branching morphogenesis. Dev. Cell. 4:11–18. [DOI] [PubMed] [Google Scholar]
- Alexander, C.M., E.W. Howard, M.J. Bissell, and Z. Werb. 1996. Rescue of mammary epithelial cell apoptosis and entactin degradation by a tissue inhibitor of metalloproteinases-1 transgene. J. Cell Biol. 135:1669–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, C.M., S. Selvarajan, J. Mudgett, and Z. Werb. 2001. Stromelysin-1 regulates adipogenesis during mammary gland involution. J. Cell Biol. 152:693–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker, A.H., D.R. Edwards, and G. Murphy. 2002. Metalloproteinase inhibitors: biological actions and therapeutic opportunities. J. Cell Sci. 115:3719–3727. [DOI] [PubMed] [Google Scholar]
- Blatchford, D.R., L.H. Quarrie, E. Tonner, C. McCarthy, D.J. Flint, and C.J. Wilde. 1999. Influence of microenvironment on mammary epithelial cell survival in primary culture. J. Cell. Physiol. 181:304–311. [DOI] [PubMed] [Google Scholar]
- Bottinger, E.P., J.L. Jakubczak, D.C. Haines, K. Bagnall, and L.M. Wakefield. 1997. Transgenic mice overexpressing a dominant-negative mutant type II transforming growth factor beta receptor show enhanced tumorigenesis in the mammary gland and lung in response to the carcinogen 7,12-dimethylbenz-[a]-anthracene. Cancer Res. 57:5564–5570. [PubMed] [Google Scholar]
- Boudreau, N., C.J. Sympson, Z. Werb, and M.J. Bissell. 1995. Suppression of ICE and apoptosis in mammary epithelial cells by extracellular matrix. Science. 267:891–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullard, K.M., L. Lund, J.S. Mudgett, T.N. Mellin, T.K. Hunt, B. Murphy, J. Ronan, Z. Werb, and M.J. Banda. 1999. Impaired wound contraction in stromelysin-1-deficient mice. Ann. Surg. 230:260–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debnath, J., K.R. Mills, N.L. Collins, M.J. Reginato, S.K. Muthuswamy, and J.S. Brugge. 2002. The role of apoptosis in creating and maintaining luminal space within normal and oncogene-expressing mammary acini. Cell. 111:29–40. [DOI] [PubMed] [Google Scholar]
- Ewan, K.B., G. Shyamala, S.A. Ravani, Y. Tang, R.J. Akhurst, L. Wakefield, and M.H. Barcellos-Hoff. 2002. Latent TGF-β activation in mammary gland: regulation by ovarian hormones affects ductal and alveolar proliferation. Am. J. Pathol. 160:2081–2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fata, J.E., K.J. Leco, R.A. Moorehead, D.C. Martin, and R. Khokha. 1999. Timp-1 is important for epithelial proliferation and branching morphogenesis during mouse mammary development. Dev. Biol. 211:238–254. [DOI] [PubMed] [Google Scholar]
- Fowlkes, J.L., D.M. Serra, H. Nagase, and K.M. Thrailkill. 1999. MMPs are IGFBP-degrading proteinases: implications for cell proliferation and tissue growth. Ann. NY Acad. Sci. 878:696–699. [DOI] [PubMed] [Google Scholar]
- Giannelli, G., J. Falk-Marzillier, O. Schiraldi, W.G. Stetler-Stevenson, and V. Quaranta. 1997. Induction of cell migration by matrix metalloprotease-2 cleavage of laminin-5. Science. 277:225–228. [DOI] [PubMed] [Google Scholar]
- Gijbels, K., R.E. Galardy, and L. Steinman. 1994. Reversal of experimental autoimmune encephalomyelitis with a hydroxamate inhibitor of matrix metalloproteases. J. Clin. Invest. 94:2177–2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grobelny, D., L. Poncz, and R.E. Galardy. 1992. Inhibition of human skin fibroblast collagenase, thermolysin, and Pseudomonas aeruginosa elastase by peptide hydroxamic acids. Biochemistry. 31:7152–7154. [DOI] [PubMed] [Google Scholar]
- Gudjonsson, T., L. Ronnov-Jessen, R. Villadsen, F. Rank, M.J. Bissell, and O.W. Petersen. 2002. Normal and tumor-derived myoepithelial cells differ in their ability to interact with luminal breast epithelial cells for polarity and basement membrane deposition. J. Cell Sci. 115:39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennighausen, L., and G.W. Robinson. 2001. Signaling pathways in mammary gland development. Dev. Cell. 1:467–475. [DOI] [PubMed] [Google Scholar]
- Hovey, R.C., J.F. Trott, E. Ginsburg, A. Goldhar, M.M. Sasaki, S.J. Fountain, K. Sundararajan, and B.K. Vonderhaar. 2001. Transcriptional and spatiotemporal regulation of prolactin receptor mRNA and cooperativity with progesterone receptor function during ductal branch growth in the mammary gland. Dev. Dyn. 222:192–205. [DOI] [PubMed] [Google Scholar]
- Hovey, R.C., J.F. Trott, and B.K. Vonderhaar. 2002. Establishing a framework for the functional mammary gland: from endocrinology to morphology. J. Mammary Gland Biol. Neoplasia. 7:17–38. [DOI] [PubMed] [Google Scholar]
- Howard, E.W., E.C. Bullen, and M.J. Banda. 1991. Regulation of the autoactivation of human 72-kDa progelatinase by tissue inhibitor of metalloproteinases-2. J. Biol. Chem. 266:13064–13069. [PubMed] [Google Scholar]
- Humphreys, R.C., M. Krajewska, S. Krnacik, R. Jaeger, H. Weiher, S. Krajewski, J.C. Reed, and J.M. Rosen. 1996. Apoptosis in the terminal endbud of the murine mammary gland: a mechanism of ductal morphogenesis. Development. 122:4013–4022. [DOI] [PubMed] [Google Scholar]
- Itoh, T., T. Ikeda, H. Gomi, S. Nakao, T. Suzuki, and S. Itohara. 1997. Unaltered secretion of beta-amyloid precursor protein in gelatinase A (matrix metalloproteinase 2)-deficient mice. J. Biol. Chem. 272:22389–22392. [DOI] [PubMed] [Google Scholar]
- Itoh, T., M. Tanioka, H. Yoshida, T. Yoshioka, H. Nishimoto, and S. Itohara. 1998. Reduced angiogenesis and tumor progression in gelatinase A-deficient mice. Cancer Res. 58:1048–1051. [PubMed] [Google Scholar]
- Joseph, H., A.E. Gorska, P. Sohn, H.L. Moses, and R. Serra. 1999. Overexpression of a kinase-deficient transforming growth factor-beta type II receptor in mouse mammary stroma results in increased epithelial branching. Mol. Biol. Cell. 10:1221–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato, T., T. Kure, J.H. Chang, E.E. Gabison, T. Itoh, S. Itohara, and D.T. Azar. 2001. Diminished corneal angiogenesis in gelatinase A-deficient mice. FEBS Lett. 508:187–190. [DOI] [PubMed] [Google Scholar]
- Kheradmand, F., K. Rishi, and Z. Werb. 2002. Signaling through the EGF receptor controls lung morphogenesis in part by regulating MT1-MMP-mediated activation of gelatinase A/MMP2. J. Cell Sci. 115:839–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kordon, E.C., R.A. McKnight, C. Jhappan, L. Hennighausen, G. Merlino, and G.H. Smith. 1995. Ectopic TGF beta 1 expression in the secretory mammary epithelium induces early senescence of the epithelial stem cell population. Dev. Biol. 168:47–61. [DOI] [PubMed] [Google Scholar]
- Koshikawa, N., G. Giannelli, V. Cirulli, K. Miyazaki, and V. Quaranta. 2000. Role of cell surface metalloprotease MT1-MMP in epithelial cell migration over laminin-5. J. Cell Biol. 148:615–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lochter, A., S. Galosy, J. Muschler, N. Freedman, Z. Werb, and M.J. Bissell. 1997. Matrix metalloproteinase stromelysin-1 triggers a cascade of molecular alterations that leads to stable epithelial-to-mesenchymal conversion and a premalignant phenotype in mammary epithelial cells. J. Cell Biol. 139:1861–1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund, L.R., J. Romer, N. Thomasset, H. Solberg, C. Pyke, M.J. Bissell, K. Dano, and Z. Werb. 1996. Two distinct phases of apoptosis in mammary gland involution: proteinase-independent and -dependent pathways. Development. 122:181–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund, L.R., J. Romer, T.H. Bugge, B.S. Nielsen, T.L. Frandsen, J.L. Degen, R.W. Stephens, and K. Dano. 1999. Functional overlap between two classes of matrix-degrading proteases in wound healing. EMBO J. 18:4645–4656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund, L.R., S.F. Bjorn, M.D. Sternlicht, B.S. Nielsen, H. Solberg, P.A. Usher, R. Osterby, I.J. Christensen, R.W. Stephens, T.H. Bugge, et al. 2000. Lactational competence and involution of the mouse mammary gland require plasminogen. Development. 127:4481–4492. [DOI] [PubMed] [Google Scholar]
- Martin, D.C., J.L. Fowlkes, B. Babic, and R. Khokha. 1999. Insulin-like growth factor II signaling in neoplastic proliferation is blocked by transgenic expression of the metalloproteinase inhibitor TIMP-1. J. Cell Biol. 146:881–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mudgett, J.S., N.I. Hutchinson, N.A. Chartrain, A.J. Forsyth, J. McDonnell, I.I. Singer, E.K. Bayne, J. Flanagan, D. Kawka, C.F. Shen, et al. 1998. Susceptibility of stromelysin 1-deficient mice to collagen-induced arthritis and cartilage destruction. Arthritis Rheum. 41:110–121. [DOI] [PubMed] [Google Scholar]
- Noe, V., B. Fingleton, K. Jacobs, H.C. Crawford, S. Vermeulen, W. Steelant, E. Bruyneel, L.M. Matrisian, and M. Mareel. 2001. Release of an invasion promoter E-cadherin fragment by matrilysin and stromelysin-1. J. Cell Sci. 114:111–118. [DOI] [PubMed] [Google Scholar]
- O'Brien, L.E., M.M. Zegers, and K.E. Mostov. 2002. Opinion: Building epithelial architecture: insights from three-dimensional culture models. Nat. Rev. Mol. Cell Biol. 3:531–537. [DOI] [PubMed] [Google Scholar]
- Roberts, L.M., J.A. Visser, and H.A. Ingraham. 2002. Involvement of a matrix metalloproteinase in MIS-induced cell death during urogenital development. Development. 129:1487–1496. [DOI] [PubMed] [Google Scholar]
- Rugh, R. 1990. The Mouse and its Reproduction and Development. Oxford University Press, Oxford, UK. 430 pp.
- Sato, M., and T.B. Kornberg. 2002. FGF is an essential mitogen and chemoattractant for the air sacs of the Drosophila tracheal system. Dev. Cell. 3:195–207. [DOI] [PubMed] [Google Scholar]
- Seagroves, T.N., J.P. Lydon, R.C. Hovey, B.K. Vonderhaar, and J.M. Rosen. 2000. C/EBPbeta (CCAAT/enhancer binding protein) controls cell fate determination during mammary gland development. Mol. Endocrinol. 14:359–368. [DOI] [PubMed] [Google Scholar]
- Simian, M., Y. Hirai, M. Navre, Z. Werb, A. Lochter, and M.J. Bissell. 2001. The interplay of matrix metalloproteinases, morphogens and growth factors is necessary for branching of mammary epithelial cells. Development. 128:3117–3131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soloway, P.D., C.M. Alexander, Z. Werb, and R. Jaenisch. 1996. Targeted mutagenesis of Timp-1 reveals that lung tumor invasion is influenced by Timp-1 genotype of the tumor but not by that of the host. Oncogene. 13:2307–2314. [PubMed] [Google Scholar]
- Sternlicht, M.D., and Z. Werb. 2001. How matrix metalloproteinases regulate cell behavior. Annu. Rev. Cell Dev. Biol. 17:463–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternlicht, M.D., A. Lochter, C.J. Sympson, B. Huey, J.P. Rougier, J.W. Gray, D. Pinkel, M.J. Bissell, and Z. Werb. 1999. The stromal proteinase MMP3/stromelysin-1 promotes mammary carcinogenesis. Cell. 98:137–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sympson, C.J., R.S. Talhouk, C.M. Alexander, J.R. Chin, S.M. Clift, M.J. Bissell, and Z. Werb. 1994. Targeted expression of stromelysin-1 in mammary gland provides evidence for a role of proteinases in branching morphogenesis and the requirement for an intact basement membrane for tissue-specific gene expression. J. Cell Biol. 125:681–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talhouk, R.S., M.J. Bissell, and Z. Werb. 1992. Coordinated expression of extracellular matrix-degrading proteinases and their inhibitors regulates mammary epithelial function during involution. J. Cell Biol. 118:1271–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomasset, N., A. Lochter, C.J. Sympson, L.R. Lund, D.R. Williams, O. Behrendtsen, Z. Werb, and M.J. Bissell. 1998. Expression of autoactivated stromelysin-1 in mammary glands of transgenic mice leads to a reactive stroma during early development. Am. J. Pathol. 153:457–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welm, B., J. Mott, and Z. Werb. 2002. Developmental Biology: vasculogenesis is a wreck without RECK. Curr. Biol. 12:R209–R211. [DOI] [PubMed] [Google Scholar]
- Wielockx, B., K. Lannoy, S.D. Shapiro, T. Itoh, S. Itohara, J. Vandekerckhove, and C. Libert. 2001. Inhibition of matrix metalloproteinases blocks lethal hepatitis and apoptosis induced by tumor necrosis factor and allows safe antitumor therapy. Nat. Med. 7:1202–1208. [DOI] [PubMed] [Google Scholar]
- Will, H., S.J. Atkinson, G.S. Butler, B. Smith, and G. Murphy. 1996. The soluble catalytic domain of membrane type 1 matrix metalloproteinase cleaves the propeptide of progelatinase A and initiates autoproteolytic activation. Regulation by TIMP-2 and TIMP-3. J. Biol. Chem. 271:17119–17123. [DOI] [PubMed] [Google Scholar]
- Wiseman, B.S., and Z. Werb. 2002. Stromal effects on mammary gland development and breast cancer. Science. 296:1046–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witty, J.P., J.H. Wright, and L.M. Matrisian. 1995. Matrix metalloproteinases are expressed during ductal and alveolar mammary morphogenesis, and misregulation of stromelysin-1 in transgenic mice induces unscheduled alveolar development. Mol. Biol. Cell. 6:1287–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, Q., and I. Stamenkovic. 2000. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes Dev. 14:163–176. [PMC free article] [PubMed] [Google Scholar]
- Yu, Q., Y. Geng, and P. Sicinski. 2001. Specific protection against breast cancers by cyclin D1 ablation. Nature. 411:1017–1021. [DOI] [PubMed] [Google Scholar]