

Abstract
Glial cell line–derived neurotrophic factor (GDNF) and hepatocyte growth factor (HGF) are multifunctional signaling molecules in embryogenesis. HGF binds to and activates Met receptor tyrosine kinase. The signaling receptor complex for GDNF typically includes both GDNF family receptor α1 (GFRα1) and Ret receptor tyrosine kinase. GDNF can also signal independently of Ret via GFRα1, although the mechanism has remained unclear. We now show that GDNF partially restores ureteric branching morphogenesis in ret-deficient mice with severe renal hypodysplasia. The mechanism of Ret-independent effect of GDNF was therefore studied by the MDCK cell model. In MDCK cells expressing GFRα1 but no Ret, GDNF stimulates branching but not chemotactic migration, whereas both branching and chemotaxis are promoted by GDNF in the cells coexpressing Ret and GFRα1, mimicking HGF/Met responses in wild-type MDCK cells. Indeed, GDNF induces Met phosphorylation in several ret-deficient/GFRα1-positive and GFRα1/Ret-coexpressing cell lines. However, GDNF does not immunoprecipite Met, making a direct interaction between GDNF and Met highly improbable. Met activation is mediated by Src family kinases. The GDNF-induced branching of MDCK cells requires Src activation, whereas the HGF-induced branching does not. Our data show a mechanism for the GDNF-induced branching morphogenesis in non-Ret signaling.
Keywords: GDNF; branching morphogenesis; Met; Src; GFRα1
Introduction
Glial cell line–derived neurotrophic factor (GDNF)* regulates ureteric branching in kidney morphogenesis, spermatogenesis, and survival and differentiation of several neuronal populations (Airaksinen et al., 1999; Sariola and Saarma, 1999; Baloh et al., 2000; Meng et al., 2000). The receptor complex for GDNF consists of Ret receptor tyrosine kinase and glycosylphosphatidylinositol (GPI)-linked GDNF family receptor α1 (GFRα1) (Airaksinen et al., 1999). In the embryonic kidney, GDNF is expressed by the metanephric mesenchyme and is repressed by epithelial conversion of the mesenchymal cells (Hellmich et al., 1996; Suvanto et al., 1996). GDNF-releasing beads stimulate ureteric branching in cultured kidneys and promote outgrowth of ectopic ureteric buds from the nephric duct (Sainio et al., 1997). Neutralizing antibodies to GDNF inhibit ureteric branching in kidney culture (Vega et al., 1996).
ret is initially expressed along the nephric duct and the ureteric bud (Pachnis et al., 1993). The receptor becomes restricted to the growing tips of the bud as its branching progresses. GFRα1 is expressed by both ureteric bud and pretubular nephrogenic mesenchyme (Sainio et al., 1997). Targeted disruption of ret, gdnf, or gfrα1 genes results in severe renal hypodysplasia or aplasia (Schuchardt et al., 1994; Pichel et al., 1996; Sanchez et al., 1996; Cacalano et al., 1998), confirming the critical role of GDNF/Ret signaling in the ureteric branching.
Since GFRα1 lacks an intracellular domain, it was initially considered as only a ligand-binding receptor for GDNF. When complexed with two molecules of GDNF, a GFRα1 dimer induces dimerization of Ret, its recruitment to lipid rafts, and transphosphorylation of the tyrosine kinase domains. Lipid rafts are cell membrane domains of sphingolipids and cholesterol packed into moving platforms within the lipid bilayer (Harder et al., 1998). The raft microdomains serve as signaling compartments of the cell membrane, which concentrate raft-specific signaling molecules (Simons and Toomre, 2000). Ret is also activated in trans by GDNF via soluble or matrix-bound GFRα1 (Paratcha et al., 2001). Moreover, GDNF signaling via Ret is different in and outside the lipid rafts (for review see Saarma, 2001).
GDNF can also signal via GFRα1 in a Ret-independent manner (Poteryaev et al., 1999; Trupp et al., 1999). In primary sensory neurones isolated from ret-deficient mice and in a Ret-negative neuroblastoma cell line, GDNF activates Src-type kinases (Poteryaev et al., 1999; Trupp et al., 1999).
There is also indirect evidence that GFRα1 might have Ret-independent roles. First, GDNF binds to GFRα1 in the absence of Ret (Jing et al., 1996). Second, ret and gfrα1 expression patterns do not overlap in many tissues (Sainio et al., 1997; Golden et al., 1999). However, nothing is known about the mechanism and possible biological significance of Ret-independent signaling via GFRα1.
The MDCK dog kidney epithelial cells have been extensively used for studying the molecular mechanisms of branching morphogenesis. Hepatocyte growth factor (HGF), the ligand for Met receptor tyrosine kinase (Naldini et al., 1991), induces scattering, chemotactic movements, and tubule formation of MDCK cells (Stoker et al., 1987; Montesano et al., 1991). In the presence of soluble GFRα1, ret-transfected MDCK cells respond to GDNF like the wild-type MDCK cells respond to HGF (Tang et al., 1998). In vivo, HGF is required for the early development of liver, limb muscles, and placenta, and it is involved in liver regeneration (Birchmeier and Gherardi, 1998). In organ culture, HGF regulates ureteric bud branching and modulates epithelial differentiation of metanephric mesenchymal cells (Karp et al., 1994; Woolf et al., 1995; Sainio et al., 1997).
We now approached the role and mechanism of GFRα1 in branching morphogenesis. These studies were prompted by our observation that the ureteric branching morphogenesis of ret-deficient mice was partially restored in organ culture when GDNF was added to the culture medium. Therefore, we created MDCK clones stably expressing GFRα1 alone or both Ret and GFRα1. When stimulated by GDNF, the GFRα1-expressing, ret-deficient cells formed branching tubules in collagen matrix, but they were completely incapable of responding chemotactically to GDNF. In both GFRα1- and Ret/GFRα1-expressing MDCK cell lines, GDNF induced Met phosphorylation, but the ligand did not directly interact with Met. Pharmacological inhibition of Src-type kinases and transfection experiments with dominant-negative (DN) or activated c-Src showed that Src kinase activity is required for the GDNF-induced activation of Met and tubulogenesis of MDCK cells.
Results
Exogenous GDNF partially restores the renal phenotype of ret-deficient mice
To analyze the possible role of Ret-independent, GFRα1-mediated signaling in ureteric budding and branching during nephrogenesis, we tested the ability of exogenous GDNF to induce ureteric budding or sustain its branching in ret-deficient mice. Embryonic day (E)11 ret −/− urogenital blocks including kidney rudiments were cultured for 4 d without or with 50 ng/ml of GDNF (Fig. 1) . As expected, the ureteric buds of ret −/− mice did not branch or branched rudimentarily in the control media (Schuchardt et al., 1996). When the culture medium was supplemented with GDNF, the number of ureteric bud tips in the hypodysplastic kidneys of ret-deficient mice was increased but not to the level seen in wild-type kidneys (Fig. 1, B and D). Exogenous GDNF increased the number of ureteric bud tips in ret−/−, ret +/−, and wild-type kidney explants (Fig. 1 E). However, with or without exogenous GDNF the number of ret−/− urogenital explants completely lacking a ureteric bud remained the same (Table I). Thus, Ret-independent signaling by GDNF has an apparent role in the ureteric branching but may be less significant in the primary bud formation.
Figure 1.
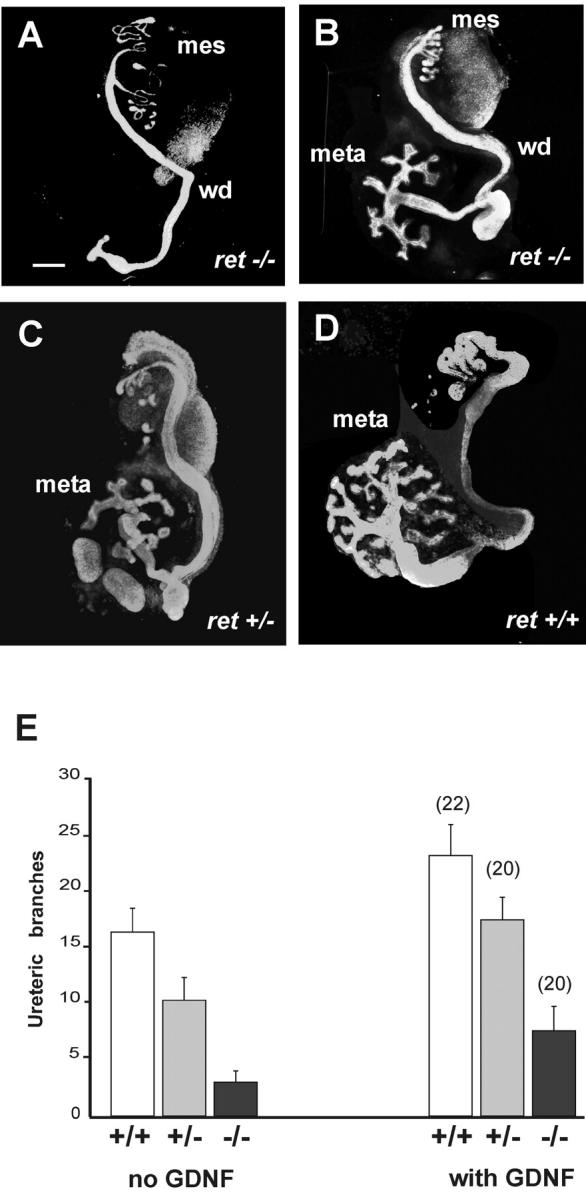
Exogenous GDNF partially restores ureteric branching of ret-deficient kidneys. (A–D) Urogenital block explants including the Wolffian duct (wd), mesonephros (meso), and metanephros (meta) from E11 ret −/− (A and B), ret +/− (C), and ret +/+ (D) mouse embryos. The urogenital blocks from each embryo were separately cultured for 4 d, one side without GDNF (A, C and D) and the other one with 50 ng/ml of GDNF (B). The explants were fixed and immunolabeled as whole mounts with pan-cytokeratin antibodies. Bar, 200 μm. (E) The number of ureteric branches of ret −/−, ret +/−, and ret +/+ kidneys with or without GDNF supplementation. The results represent the means ± SEM. GDNF significantly increases ureteric branch number in ret −/− explants compared with the control media (P < 0.01).
Table I.
Ureteric bud branching in ret −/− , ret +/− , and ret +/+ urogenital explants with or without GDNF supplementation a
| No budding | <5 tips | >5 tips | |||||
|---|---|---|---|---|---|---|---|
| Control | GDNFb | Control | GDNF | Control | GDNF | Number of explants | |
| ret−/− | 7 | 7 | 9 | 4 | 4 | 9 | 20 |
| ret+/− | 0 | 0 | 8 | 4 | 12 | 16 | 20 |
| ret+/+ | 0 | 0 | 4 | 1 | 18 | 21 | 22 |
The results from the experiment shown in Fig. 1 E.
Urgenital blocks from each embryo were seperated by microdissection and cultured for 4 d. One side served as a control, the other one was cultured with 50 ng/ml of GDNF.
GDNF induces branching of gfrα1-transfected/ret-deficient MDCK cells
To study the possible mechanism and mode of action of GDNF in GFRα1 and GFRα1/Ret signaling, MDCK cells were transfected with expression vectors encoding the human ret and rat gfrα1, or gfrα1 only with or without fused GFP. Multiple clones expressing GFRα1 with or without fused GFP and clones expressing Ret together with GFRα1 were identified by RT-PCR and Western blotting. The clonal cell lines expressing GFRα1 with (N3) or without GFP (N14) showed similar biological responses to GDNF (Fig. 2 and unpublished data).
Figure 2.

GDNF induces branching of GFRα1-expressing MDCK cells in three-dimensional collagen gel. (A) Ret/GFRα1- and GFRα1-expressing MDCK cells were grown in collagen gel with GDNF (100 ng/ml), and wild-type MDCK cells were grown in collagen gel with HGF (50 ng/ml). BSA (100 ng/ml) was used as a negative control. Bar, 100 μm. (B) GDNF induces branching of GFRα1 and Ret/GFRα1 cells but not wild-type MDCK cells, which only respond to HGF. Persephin (PSPN; 100 ng/ml) does not induce branching of any MDCK cell line tested. From the total number of cysts in the field, the percentage of cysts with long branches was calculated. Only the branches with the length of more than two cyst diameters were counted. (C) Dose dependency of the GDNF-induced branching of GFRα1- and Ret/GFRα1-expressing MDCK cells. GDNF concentrations are marked per ml. Results are reported as fold of branching cysts over the noninduced control. Means ± SEM of five to eight counted fields are shown. The results are representative of five (A and B) and three (C) independent experiments. (B and C) GDNF significantly increases branching in GFRα1- and Ret/GFRα1-expressing MDCK and HGF increases branching of wild-type MDCK (B) compared with the control media (P < 0.001).
The possible endogenous ret expression by MDCK cells was excluded by Northern blot and RT-PCR. The canine ret cDNA was first cloned from adult dog testis. RT-PCR with canine-specific ret primers and Northern blot showed that ret is not expressed by MDCK cells. Furthermore, in MDCK clones stably expressing gfrα1, no induced ret expression was detected either by RT-PCR with the canine ret primers or Western blotting (unpublished data).
Wild-type MDCK cells form fluid-filled cystic structures in three-dimensional collagen gels. After HGF application, the cells start forming branching tubules (Montesano et al., 1991). When cultured in 100 ng/ml of GDNF, both GFRα1- and Ret/GFRα1-MDCK cells formed branching tubules, whereas wild-type (Fig. 2, A and B) and mock-transfected cells did not (unpublished data). Persephin is the ligand for GFRα4 (Enokido et al., 1998; Thompson et al., 1998) and does not interact with GFRα1. It did not evoke branching of GFRα1- and Ret/GFRα1-expressing MDCK cells (Fig. 2 B). GDNF also induced branching tubulogenesis in wild-type MDCK cells transduced with an adenovirus expressing GFRα1 (unpublished data).
The branching response of the cells expressing GFRα1 alone was highly sensitive to GDNF, since already 0.1 pg/ml of GDNF evoked tubulogenesis. In contrast, the cells coexpressing Ret and GFRα1 started to branch only at 0.1 ng/ml of GDNF (Fig. 2 C). Two GDNF preparations synthesized by different methods by two different manufacturers were tested (see Materials and methods), and both products evoked branching of GFRα1-expressing MDCK cells already at low concentrations (0.1 pg/ml).
GFRα1-expressing cells do not respond chemotactically to GDNF in the absence of Ret
Guided migration of cells toward a chemoattractant is referred to as chemotaxis, whereas enhanced cellular motility is called chemokinesis. Chemotaxis can be tested in the Boyden dual chamber assay by adding the test substance to the lower chamber only, and chemokinesis can be tested by adding the test substance to both upper and lower chambers.
Similar to the HGF-induced chemotaxis of the wild-type MDCK cells (Stoker et al., 1987), the chemotactic migration of ret-transfected MDCK cells is stimulated by GDNF in the presence of soluble GFRα1 (Tang et al., 1998). Accordingly, GDNF was chemotactic to MDCK cells transfected with both ret and the GPI-anchored, membrane-bound form of gfrα1. In contrast, the GFRα1-expressing, ret-deficient MDCK cells did not show a chemotactic response to GDNF under the same conditions (Fig. 3 A).
Figure 3.
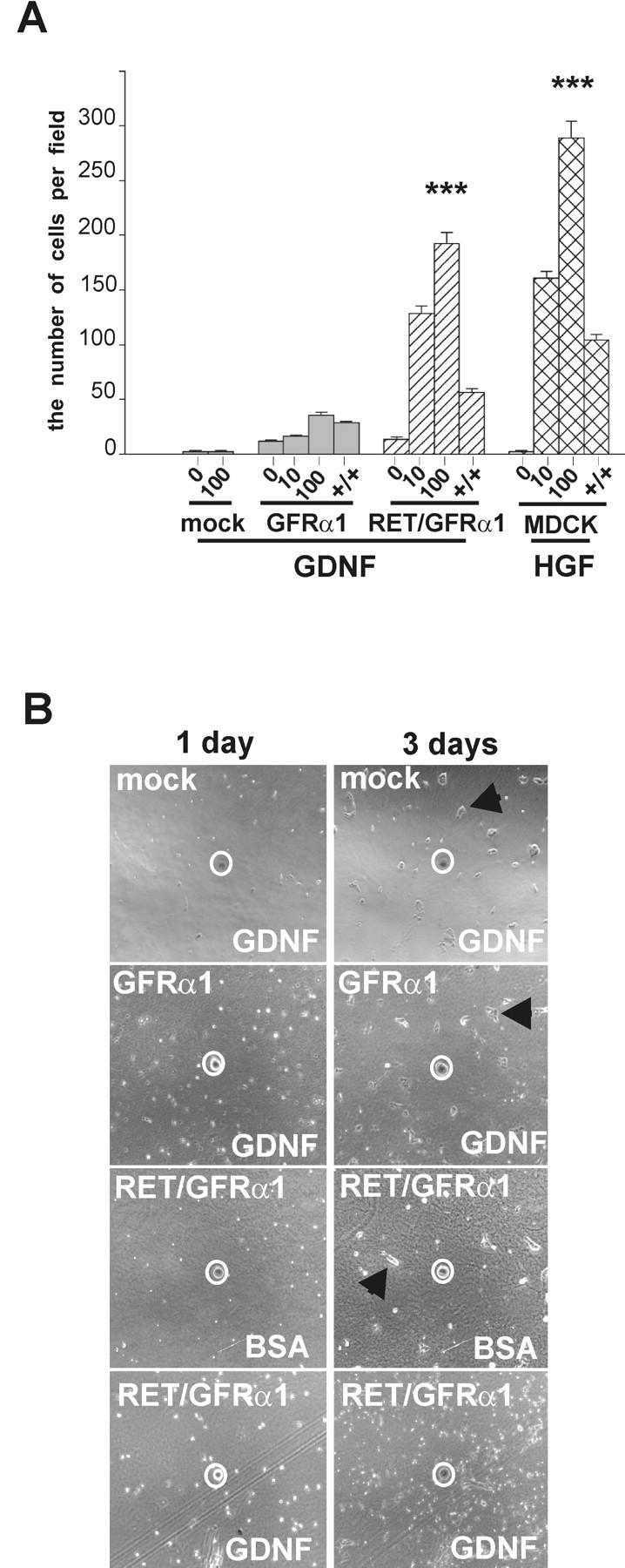
GFRα1-expressing, ret -deficient MDCK cells do not show a chemotactic response to GDNF. (A) In the Boyden chamber chemotaxis assay, the mock-transfected, GFRα1, and Ret/GFRα1 cells were exposed to GDNF (10 and 100 ng/ml), and wild-type MDCK were exposed to HGF (10 and 100 ng/ml). The number of cells was counted as described in Materials and methods. +/+, 100 ng/ml of GDNF or 50 ng/ml of HGF were added to both chambers to assay chemokinesis. The results represent the means ± SEM (n = 3). ***P < 0.001. (B) Chemoattraction assay on collagen matrix. Only Ret/GFRα1-expressing cells migrate toward GDNF-soaked beads. BSA-soaked agarose beads were used as negative control. Beads are marked by a white circle. Note that mock, GFRα1-expressing cells with GDNF-soaked beads and Ret/GFRα1-expressing cells with BSA-soaked bead form clusters of adherent cells (marked with arrowhead) after 3 d, whereas the Ret/GFRα1-expressing cells migrating toward the GDNF bead are scattered.
GDNF was added to both chambers to test its possible chemokinetic effects. The migration of Ret/GFRα1-expressing MDCK cells was reduced threefold compared with their maximal chemotactic response. Similar reduction was observed with HGF in wild-type MDCK cells. In contrast, the migration of GFRα1-expressing cells was only marginally reduced when GDNF was applied to both chambers (Fig. 3 A). Thus, GDNF is chemotactic to Ret/GFRα1-expressing cells and weakly chemokinetic but not chemotactic to GFRα1-expressing, ret-deficient cells.
In another chemotaxis assay (Tang et al. 1998), ret/gfrα1-, gfrα1-, and mock-transfected MDCK cells were seeded on culture dishes coated by type I collagen. Agarose beads were soaked in GDNF (10 ng/μl) or 1% BSA, a bead was placed on top of collagen gel, and the cells were monitored for 3 d. Ret/GFRα1-expressing cells actively migrated toward GDNF-releasing beads but not to those soaked in BSA (Fig. 3 B). gfrα1- and mock-transfected cells were not attracted by the GDNF- or BSA-releasing beads (Fig. 3 B).
GDNF activates Met in both Ret-dependent and -independent signaling
Since both GDNF and HGF promoted branching of MDCK cells and Met is the only receptor known to promote tubule formation in these cells (Santos et al., 1993), we suggested that GDNF may induce Met phosphorylation. In 15 min, GDNF indeed evoked Met phosphorylation in GFRα1- and Ret/GFRα1-expressing MDCK cells but not in wild-type MDCK cells. Saturation was reached at 0.1 pg/ml (Fig. 4, A and B) . The same concentration of GDNF also induced rapid Met phosphorylation in human neuroblastoma SHEP cells (unpublished data), which express GFRα1 but no Ret (Poteryaev et al., 1999). GDNF activated Met in GFRα1- and Ret/GFRα1-expressing MDCK cells already in 15 min (Fig. 4, A and B), and the activation lasted at least 2 h (unpublished data). In the mock-transfected MDCK cells, only HGF phosphorylated Met (Fig. 4 C). In Ret/GFRα1-expressing MDCK cells, Ret was phosphorylated already at 0.1 pg/ml of GDNF, and saturation was reached at 10 ng/ml (Fig. 4 D).
Figure 4.

GDNF induces phosphorylation of Met. (A and B) Dose-dependent phosphorylation of Met by GDNF in GFRα1- and Ret/GFRα1-expressing MDCK cells. Met was activated in 15 min after GDNF application. The bottom panels show the reprobing of the same filter with anti-Met antibodies. The numbers below the lanes indicate the fold of induction of Met tyrosine kinase. (C) Phosphorylation of Met in mock-transfected MDCK cells. Concentrations of GDNF and HGF are given in ng/ml. 30 μg of total proteins were incubated with 10 μl of immobilized phosphotyrosine mAbs, and immunocomplexes were washed and analyzed as described in Materials and methods. (D) Dose-dependent activation of Ret by GDNF in Ret/GFRα1-expressing MDCK cells. The bottom panel shows the reprobing of the same filter with anti-Ret antibodies. The numbers below the lanes indicate the fold of induction of Ret tyrosine kinase. IP, immunoprecipitation; WB, Western blotting; P-tyr, phosphotyrosine. The results are representative of three independent experiments.
In a series of cross-linking immunoprecipitation experiments, we tested whether GDNF activates Met directly or indirectly. Binding of 125I-GDNF to GFRα1-expressing MDCK, SHEP, or COS7 cells and NIH 3T3 cells transiently transfected with gfrα1 was followed by chemical cross-linking and immunoprecipitation with anti-Met antibodies. No high molecular weight complexes were revealed, and in total lysates the bands represent different complexes of 125I-GDNF (monomers or dimers) and the dimers of GFRα1 (Fig. 5) . Different cross-linkers, such as EDC with sulfo-NHS, BS3, DSS, and DSP, were tested, and the result remained the same (Fig. 5 and unpublished data). A direct association of the GDNF receptor complex and Met was not detected in Ret/GFRα1-expressing MDCK cells either (unpublished data).
Figure 5.
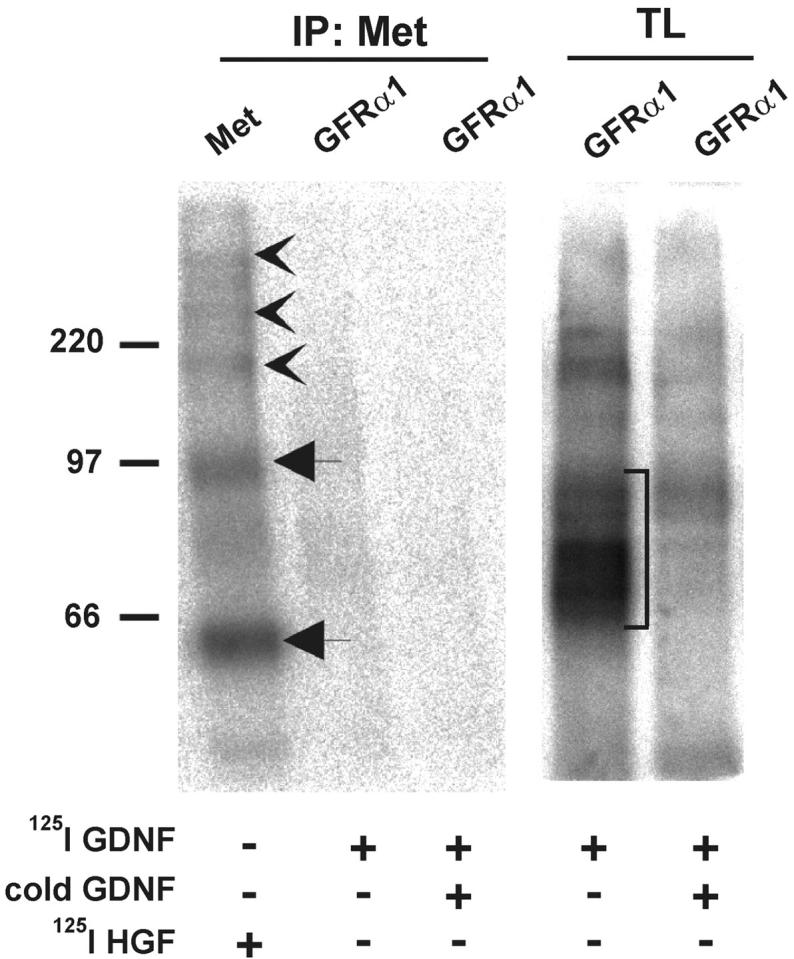
GFRα1 does not complex with Met. Binding of 125I-GDNF to COS7 cells transfected with gfrα1 and 125I-HGF to wild-type COS7 followed by cross-linking with EDC together with sulfo-NHS. Immunoprecipitates with anti-Met antibodies (IP:Met) were analyzed by SDS-PAGE under reducing conditions. In total lysates (TL), different complexes of 125I-GDNF (monomers or dimers) and the dimers of GFRα1 are marked with a square bracket. 125I-HGF α subunit and proHGF are marked by arrows. 125I-HGF–Met complexes are indicated by arrowheads. The results are representative of five independent experiments.
Cross-linking of 125I-HGF in COS7 cells followed by immunoprecipitation with anti-Met antibodies was used as a positive control. It resulted in ∼200-, 250-, and 340-kD complexes under reducing conditions (Fig. 5). They represent different combinations of the Met β-subunit (140 kD), Met-αβ heterodimer (190 kD), and HGF complexes (60–90 kD).
GDNF-induced phosphorylation of Met is mediated by Src family kinases
Already 0.1 pg/ml of GDNF saturated Src phosphorylation at Tyr418, demonstrating Src activation in both GFRα1- and Ret/GFRα1-expressing MDCK cells (Fig. 6 A). 1 μM concentration of the Src-type kinase inhibitor PP2 inhibited GDNF-induced Met activation in both GFRα1- and Ret/GFRα1-expressing MDCK cells but not in the HGF-induced Met phosphorylation in these cells (unpublished data). Accordingly, PP2 strongly reduced the GDNF-dependent phosphorylation of Met in SHEP cells endogenously expressing GFRα1 but not Ret (Fig. 6 B) and did not affect HGF-induced Met phosphorylation (Fig. 6 B).
Figure 6.

GDNF-induced activation of Met requires Src kinase. (A) Dose-dependent Src kinase activation by GDNF in GFRα1- and Ret/GFRα1-expressing MDCK cells. The activation of Src-type kinases was observed after 15 min. The concentrations of GDNF are marked. The bottom panel shows mock-transfected MDCK cells induced with 50 ng/ml of HGF and 100 ng/ml of GDNF. The numbers below the lane indicate the fold of increase in phosphorylation of Tyr418 of Src. The bottom panels show the reprobing of the same filter with anti-Src antibodies. The results are representative of three independent experiments. (B) SHEP cells were grown with GDNF (10 ng/ml and 5 pg/ml) or HGF (10 ng/ml) in the presence of PP2 (1 and 10 μM). To exclude a possible cytotoxic effect of the solvent, DMSO was added to the controls. The bottom panel shows the reprobing of the same filter with anti-Met antibodies. Bottom picture demonstrates wild-type MDCK cells induced with 50 ng/ml of HGF and 100 ng/ml of GDNF. Numbers below the lane indicate the fold of induction of Met tyrosine kinase. The results are representative of three independent experiments.
We used adenoviruses to introduce DN c-Src or activated c-Src to GFRα1- and Ret/GFRα1-expressing and wild-type MDCK cells. Both constructs contained gfp under a separate promoter, which enabled us to monitor the infection efficiency. It was close to 100% in all experiments. Adenoviruses with gfp alone were used as a control. In accordance with the results with PP2, DN c-Src efficiently blocked Src phosphorylation and Met activation induced by GDNF but not that induced by HGF (Fig. 7 A). Expression of activated c-Src resulted in GDNF-independent phosphorylation of Met, which could not be further increased by GDNF. In contrast, HGF enhanced Met phosphorylation in wild-type MDCK-expressing activated c-Src (Fig. 7 A).
Figure 7.

GDNF-induced branching tubulogenesis of GFRα1- and Ret/GFRα1-expressing cells require c-Src kinase. GFRα1- and Ret/GFRα1-expressing and wild-type MDCK cells were infected with adenovirus constructs containing DN c-Src, activated c-Src, or adeno-GFP. (A) GDNF-induced Met activation depends on c-Src kinase. 1 d after the adenovirus infection, GFRα1- and Ret/GFRα1-expressing MDCK cells were induced with GDNF (50 ng/ml) and wild-type MDCK also with HGF (50 ng/ml). Aliquots of total cell lysates were immunoblotted with anti-Y418 Src, and the rest of lysates were immunoprecipitated with anti-Met antibodies and immunoblotted using antiphosphotyrosine antibodies. The results are representative of two independent experiments. (B) After infection cells were put in collagen gel culture, GFRα1- and Ret/GFRα1-expressing cells were grown with or without GDNF (50 ng/ml), wild-type MDCK with or without HGF (50 ng/ml). After 3 d, the cells were fixed and counted as described in Materials and methods. The results are representative of three independent experiments.
GFRα1-, Ret/GFRα1-, and wild-type MDCK cells transduced with adenoviruses expressing DN c-Src, activated c-Src, or GFP were grown in collagen gel to analyze branching responses. DN c-Src suppressed GDNF- but not HGF-induced branching tubulogenesis (Fig. 7 B). Activated c-Src evoked branching in a ligand-independent manner in all tested MDCK lines. In the presence of activated c-Src, HGF but not GDNF further increased the number of branching cysts (Fig. 7 B).
Discussion
We demonstrate the first Ret-independent morphological responses to GDNF. First, it partially restores the ureteric branching of ret-deficient hypodysplastic kidneys when applied to the culture medium. Second, GDNF induces branching but not chemotactic migration of MDCK cells expressing GFRα1 but not Ret. Because Met is the only receptor to promote branching of wild-type MDCK cells, we tested whether GDNF activates Met in non-Ret signaling. Indeed, in MDCK cells and several other cell types, GDNF binding to GFRα1 activates Met indirectly via Src family kinases. Src activation is essential for both Met activation and branching morphogenesis. These data underline the role of GFRα1 in the ureteric branching morphogenesis and provide biochemical and biological evidence for a novel signaling mechanism for GDNF.
The development of the mammalian permanent kidney or metanephros requires reciprocal inductive interactions between the metanephric mesenchyme and the ureteric bud (Kuure et al., 2000). GDNF is an essential mesenchymal signal for ureteric budding and branching (Sariola and Saarma, 1999), and it has been assumed to signal during kidney morphogenesis via the GFRα1 and Ret complex, because GDNF-soaked beads fail to induce ectopic buds from Wolffian ducts of ret-deficient mice (Sainio et al., 1997) and wild-type metanephric mesenchymes cocultured with ret-deficient ureteric buds do not restore branching (Schuchardt et al., 1996). It is notable that the renal phenotype of the mice lacking Ret is variable ranging from total aplasia to hypodysplasia. The metanephric development is initiated in 61% of ret-deficient embryos (Schuchardt et al., 1996). We now managed to partially restore branching of ret-deficient hypodysplastic kidney rudiments by exogenous supplementation of GDNF but failed to decrease with GDNF the number of kidney explants with complete renal aplasia. The data suggest that Ret-independent signaling via GFRα1 rather sustains the ureteric branching than initiates bud formation from the Wolffian duct. However, the proper orientation of the tips of the ureteric buds within the nephrogenic mesechyme might be critically controlled by Ret activity, since it is only in the presence of Ret that the GFRα1-expressing MDCK cells react chemotactically to GDNF.
The molecular mechanisms of chemokinesis, chemotaxis, and tubulogenesis are at least partially different. In HGF signaling, Grb2 and PLCγ are crucial for tubulogenesis but not important for cell scattering (Royal et al., 1997; Gual et al., 2000). A downstream target of PI3-K, p70, is required for MDCK cell motility and dissociation but not for tubulogenesis (Royal et al., 1997). In contrast to the GDNF-induced Ret activation, the GFRα1-mediated Met activation promotes tubulogenesis but only weak chemokinetic motility of MDCK cells. In the presence of Ret, Met activation by GDNF probably involves different downstream adaptors than in GFRα1-mediated, non-Ret signaling and therefore evokes different cell responses.
GFRα1, a GPI-linked receptor, does not have an intracellular domain. In the absence of Ret, GFRα1 apparently employs other transmembrane molecule(s) for signal transduction. The similarity of the GDNF- and HGF-induced branching responses of MDCK cells prompted us to study the possible interplay of GDNF and Met. Indeed, GDNF evokes Met phosphorylation in both GFRα1- and Ret/GFRα1-expressing cells but not in wild-type MDCK cells. Met activation by GDNF is not restricted to a particular cell line or cell type, since it takes place in SHEP cells endogenously expressing GFRα1, in gfrα1-transfected COS7 cells, and NIH 3T3 fibroblasts. However, the cross-linking immunoprecipitation experiments using several different chemical cross-linkers and several cell lines failed to detect any GDNF complexes with Met, making a direct binding of GFRα1 and Met highly improbable.
Src-type kinases were putative candidates to mediate the GDNF signaling from GFRα1 to Met because they are associated with the lipid rafts like GFRα1 (Harder et al., 1998), they are activated in Ret-independent signaling by GDNF (Poteryaev et al., 1999; Trupp et al., 1999), c-Src kinase is associated with Met after receptor activation (Rahimi et al., 1998), and integrin-mediated activation of Ron receptor tyrosine kinase, homologous to Met, requires c-Src (Danilkovitch-Miagkova et al., 2000). Indeed, inhibition of Src-type kinases by PP2 prevents phosphorylation of Met by GDNF but not by HGF. Moreover, expression of DN c-Src blocks GDNF-induced Met phosphorylation and branching tubulogenesis in GFRα1- and Ret/GFRα1-expressing MDCK cells, but it does not affect HGF-induced branching in wild-type MDCK cells. These findings are in agreement with the results reported by Rahimi et al. (1998). They demonstrated that DN c-Src does not alter the phosphorylation level of Met in a mouse mammary carcinoma cell line SP1. Thus, Src family kinases are upstream to Met in the GDNF-induced activation but downstream to Met in the HGF-induced activation. In MDCK cells, c-Src is apparently involved in non-Ret signaling, but the role of other Src kinases remains open.
Already low concentrations of GDNF induce phosphorylation of Met in both GFRα1- and Ret/GFRα1-expressing MDCK cells. Intriguingly, Met is saturated by GDNF at 4 fM (0.1 pg/ml), whereas HGF saturates Met at 0.5 nM (Villa-Moruzzi et al., 1993). The phenomenon of femtomolar concentrations causing activation of a signaling pathway and biological response is not unique. Femtomolar levels of GABA neurotransmitter stimulate migration of a subpopulation of cortical neurons (Behar et al., 1996, 1998). Similarly, the delta opioid peptide [D-Ala2,D-Leu5]enkephalin promotes PC12 cell survival via the MEK-ERK pathway at femtomolar concentration (Hayashi et al., 2002).
Different doses of GDNF and the receptor context define the cellular responses to the ligand. Low doses of GDNF induce branching but not chemotaxis of GFRα1-expressing, ret-deficient MDCK cells. When Ret is present, GDNF induces both branching and chemotaxis but only at a high concentration. Thus, RET obviously negatively controls branching at low doses of GDNF. The GFRα1–Ret complex may be less stable than the GFRα1 complex, which may increase the GDNF binding sites in the absence of Ret and initiate the branching response at low GDNF doses. It is also possible that the GFRα1–Ret complex is internalized without ligand faster than the GFRα1 complex. On the other hand, it is apparent that Ret is essential for the chemotactic response to GDNF.
Kidney development is normally initiated in Met- and HGF-deficient mice (Birchmeier and Gherardi, 1998). These animals die around E13–15 due to severe malformations in placental and liver morphogenesis (Schmidt et al., 1995). In kidney culture, the antibodies neutralizing HGF disrupt kidney development suggesting a role of HGF/Met signaling in kidney morphogenesis (Woolf et al., 1995). On the other hand, GDNF/Ret/GFRα1 signaling plays a crucial role in kidney differentiation both in vivo and in vitro (Schuchardt et al., 1994; Pichel et al., 1996; Sanchez et al., 1996; Cacalano et al., 1998). The in vivo contribution of GDNF/Met signaling in kidney morphogenesis should be further elucidated by Ret/Met- or conditional Met-deficient mice.
Both met and ret are protooncogenes. Met is up-regulated in several different cancer forms (Giordano et al., 2000), and hgf and met are frequently overexpressed in breast carcinomas (Tuck et al., 1996; Ghoussoub et al., 1998). Activated Ret upregulates met in normal human thyrocytes (Ivan et al., 1997). Mutations in met have been found in the familial papillary renal cancer and in few cases of sporadic papillary renal cancer (Schmidt et al., 1997; Zhuang et al., 1998). Oncogenic ret mutations cause multiple endocrine neoplasia type 2A and 2B syndromes, familial medullary thyroid cancer, and pheochromocytomas (Pasini et al., 1996; Edery et al., 1997). c-Src, which is activated after Met and Ret phosphorylation, is highly expressed in human breast cancer (Ottenhoff-Kalff et al., 1992) and is activated in SP1 carcinoma cells (Rahimi et al., 1996, 1998). The sustained activation of c-Src stimulates expression of HGF in carcinoma cells, which may lead to invasiveness and metastasis (Hung and Elliott, 2001). Different cell lines expressing oncogenic forms of Ret possess high Src kinase activity levels (Melillo et al., 1999). The interplay between Met, Ret, and Src kinases might also be crucial in carcinogenesis, since GDNF induces Met phosphorylation in Neuro-2A neuroblastoma cells.
During recent years, evidence for cross-talk between heterologous receptor tyrosine kinases and signaling pathways has rapidly emerged. A neuromodulator, adenosine, acting through the A2A receptors activates Trk neurotrophin receptors in the absence of their ligands (Lee and Chao, 2001). Binding of nerve growth factor to TrkA promotes phosphorylation of Ret in a GDNF-independent manner (Tsui-Pierchala et al., 2002). Met is also activated by factors other than HGF. The Listeria surface protein InIB binds to and phosphorylates Met (Shen et al., 2000), and epidermal growth factor receptor activates Met in transformed cells (Bergstrom et al., 2000; Jo et al., 2000). Src family kinases are one of the mediators between receptor complexes (Danilkovitch-Miagkova et al., 2000; Lee and Chao, 2001). Obviously, a horizontal activation mechanism of a receptor tyrosine kinase by heterologous ligand/receptor systems may be more common than assumed. The horizontal activation of Met by GDNF via Src demonstrates a synergy of two signaling systems, which should be taken in consideration when the biological and pathological effects of Met, GFRα1, or Ret are studied. It remains to be resolved whether other GDNF family ligands using different GFRαs for ligand binding can activate Met or other receptor tyrosine kinases.
Materials and methods
Cell culture and transfections
Early passage MDCK cells were provided by Dr. E. Lehtonen (Haartman Institute, University of Helsinki). Cells were cultured in MEM with 10% FCS. Human SHEP neuroblastoma cells and Neuro-2A were cultured in RPMI 1640 with 10% FCS. NIH 3T3 and COS7 cells were cultured in DME with 10% FCS and transiently transfected with pcDNA3-GFRα1/GFP using FuGene 6TM reagent (Roche). For creation of stable lines, MDCK cells were transfected in equal portions with pcDNA3-Ret, pcDNA3-GFRα1, and pcDNA3-GFRα1/GFP using FuGene 6TM reagent and selected with 400 μg/ml G418 (GIBCO BRL, Life Technologies). After 2 wk of selection, multiple clones were collected and the expression of ret and/or gfrα1 was verified by RT-PCR and Western blotting. Ret/GFRα1 (N7 and N17), GFRα1 (N14), and GFRα1-GFP (N2 and N3) clones which showed high level of exogenous protein expression according to the Western blot were used for further analyses.
GFP-GFRα1 fusion expression plasmid construction
The entire GFP coding sequence except first methionine was amplified by PCR with primers: 5′-aattgctagcgtgagcaagggcgaggagc-3′; 5′-aattgctagcttacttgtacagctcgtcc-3′. The primers contained NheI restriction sites flanking the GFP sequence. The GFRα1 full coding sequence cloned into pcDNA3 expression vector was subjected to “inverse PCR.” The “sense” GFRα1 primer with NheI (5′-aattgctagcgaccgtctggactgtgtgaaag-3′) was designed to anneal to the beginning of mature GFRα1 sequence, whereas the “antisense” primer with NheI (5′-tatagctagctccaccactcacctcggcgg-3′) annealed to the end of signal leader peptide of GFRα1 precursor. The resulting PCR products were digested with NheI and ligated. The expression construct therefore is the NH2-terminal fusion of mature GFRα1 to GFP preceded by in-frame signal peptide of GFRα1 with starting methionine, which targets the fusion to the extracellular protein pathway. The membrane localization of the fusion was checked by confocal microscopy in transiently transfected SHEP and Neuro-2a cells (unpublished data).
Construction of Src mutants recombinant adenoviruses
Wild-type and DN c-Src cDNA were a gift from Dr. Joan Brugge (Harvard Medical School, Boston, MA). Activated c-Src was obtained by introducing a tyrosine to phenylalanine mutation at position 527 using a mutated PCR primer: 5′-GCTCTAGACTATAGGTTCTCCCCGGGCTGGAACTGTGGCTAGTGGAC-3′. The adenoviruses were generated using the pAdEasy recombination system as described in He et al. (1998). Briefly, the Src mutants were cloned in the shuttle vector pAdTrack, recombined in bacteria with the adenoviral vector pAdEasy, linearized, transfected, and amplified in 293A cells. The virus particles were purified on CsCl gradients, dialyzed, and titered in 293A cells. For the infection of GFRα1- and Ret/GFRα1-expressing and wild-type MDCK cells we used 5 pfu/cell of Adeno-DN c-Src, Adeno-activated c-Src and Adeno-GFP diluted in serum-free DME with 15 mM Hepes, pH 7.4.
Cloning of ret cDNA from dog testes
Total RNA was isolated with Trizol reagent (GIBCO BRL, Life Technologies) from autopsy samples of adult dog testes (Veterinary Hospital, University of Helsinki). Reverse transcription reaction was performed using Superscript II RT (GIBCO BRL, Life Technologies). PCR was run for 40 cycles with primers for human c-ret 5′-AGACGTGGTACCTGCATCAGG-3′ and 5′-CGTTGAAGTGGAGCAAGAGG-3′. The PCR product was cloned into pGEM-T vector (Promega) and sequenced (sequence data available from GenBank/EMBL/DDBJ under accession no. AF364316).
Primers from nucleotides 29–49 and 225–244 of GenBank/EMBL/DDBJ sequence no. AF364316 were used in RT-PCR analysis for canine ret expression. For Northern blot, 30 μg of total RNA per lane was separated in 1.2% formaldehyde-agarose gel and transferred by capillary blotting onto Hybond-N membrane (Amersham Biosciences) according to the manufacturer's instructions. Blot was hybridized with [32P]dCTP-labeled canine ret probe (sequence data available from GenBank/EMBL/DDBJ under accession no. AF364316) and washed in stringent conditions (Sambrook et al., 1989).
Western blotting and immunoprecipitation
To analyze Src activation, subconfluent GFRα1- and Ret/GFRα1-expressing MDCK cell cultures were starved for 24 h before induction into serum-free MEM. After a 10-min incubation at 37°C with 50 ng/ml GDNF (Cephalon Inc. or R&D Systems) or 50 ng/ml HGF (Sigma-Aldrich), cells were lysed in lysis buffer supplemented with 1 mM Na-orthovanadate and analyzed on Western blots as described (Lindahl et al., 2001). Blots were probed with the indicated antibodies and developed with ECL reagents (Amersham Biosciences). The following antibodies were used: anti-Y418 Src and anti-Src (BioSource International). Phospho-specific antibody to Tyr418 detects activated form of p60Src (Abram and Courtneidge, 2000).
To detect Met activation, GFRα1- and Ret/GFRα1-expressing MDCK, mock-transfected MDCK, or SHEP cells were starved overnight in MEM or RPMI 1640 with 1% FCS accordingly and in serum-free medium for 2 h prior the induction. After a 15-min incubation at 37°C with indicated concentrations of GDNF or 50 ng/ml, HGF cells were lysed as described. Cleared cell lysates were incubated with anti-Met antibodies (Santa Cruz Biotechnologies, Inc.) overnight at 4°C. Immunoprecipitates were collected with protein A–Sepharose (Amersham Biosciences), washed, separated by SDS-PAGE, and transferred to Hybond-ECL membranes. Membranes were immunoblotted with antiphosphotyrosine antibodies (Upstate Group Inc.) or anti-Met. The same procedure was repeated to detect Ret phosphorylation in Ret/GFRα1-expressing MDCK, only incubation time with GDNF was changed for 2 h. Anti-Ret antibodies (Santa Cruz Biotechnologies, Inc.) were used for immunoprecipitation and immunoblotting.
Alternatively, 30 μg of total proteins were incubated overnight at 4°C with 10 μl immobilized phosphotyrosine mAbs (Cell Signaling, NEB). Immunocomplexes were washed and analyzed as described. Densitometry and quantifications were done using TINA 2.0 program.
For the inhibition of Met and Src activation, SHEP or GFRα1- and Ret/GFRα1-expressing MDCK cells were starved as described above. 1 or 10 μM of PP2 (Calbiochem) was added 30 min before induction by GDNF or HGF. The solvent DMSO was added to the positive controls together with GDNF or HGF.
125I-labeled GDNF and HGF binding, chemical cross-linking
GDNF and HGF were enzymatically iodinated with [125I]NaI (Amersham Biosciences) with lactoperoxidase to a specific activity of 100,000 cpm/ng as described (Lindahl et al., 2001). COS7 cells were transfected with pcDNA3-GFRα1/GFP 2 d prior the assay. 2 nM of 125I-GDNF or 1 nM of 125I-HGF were allowed to bind to cell monolayers for 1–2 h on ice in binding buffer (DME/15 mM Hepes, pH 7.5; 0.2% BSA), washed, and chemically cross-linked for 30 min at RT using BS3, DSS, DSP, or EDC with sulfo-NHS (Pierce Chemical Co.). The nonspecific binding of GDNF was estimated by the amount of 125I-GDNF binding to cells in the presence of 300 nM unlabeled GDNF. Cells were washed, lysed, and immunoprecipitated with anti-Met antibodies as described. Gels were dried and analyzed by phosphorimaging in a BAS Reader 1800 (Fuji).
Cell migration and chemotaxis assays
5 × 104 GFRα1- and Ret/GFRα1-expressing and mock transfected MDCK cells were suspended in 300 μl of MEM with 10% FCS and seeded into 24-well cell culture inserts with the filters (Boyden chambers) (pore size 8 μm; Falcon). The assay was done as described (Tang et al., 1998). Briefly, GDNF or HGF was added to the top or both the bottom and top chambers at the marked concentrations. After a 48-h incubation, nonmigrated cells on the upper surface of the filters were scrapped. Membranes with the cells on the bottom surface were washed with PBS, fixed by 3% glutaraldehyde in PBS, stained with May-Grünwald Giemsa (MGG) solution, dehydrated, and mounted on the slides. Cells in eight fields of each membrane were counted at the magnification 100× under the light microscope. The average and standard error of the mean were calculated. Significance of the differences was estimated by t test.
3.5-cm dishes were coated with collagen I solution, and 20,000 cells were seeded on top of it. GDNF-soaked agarose beads, prepared as described (Sainio et al., 1997), were put on the gel before it solidified. Cells around the beads were photographed daily.
Branching tubule formation assay in collagen gel
Trypsinised cells were mixed 1:3 with collagen type I solution and plated. MEM with 10% FCS was overlaid on the gels with or without GDNF (Cephalon Inc. or R&D Systems) or 50 ng/ml HGF. Cells in collagen were cultured for 3 d; GDNF-containing medium was changed daily. For quantification, cells were cultured for 3 d, fixed by 3% glutaraldehyde in PBS, and counted under a light microscope.
To avoid the effect of possible contamination of GDNF preparation, GDNF from two different sources were tested. One GDNF product was expressed in baculovirus-infected insect cells (Cephalon Inc.), and the another one was expressed in mouse myeloma cell line NSO (R&D Systems).
Kidney cultures
Kidney rudiments were isolated from NMRI mouse embryos at E11 (the vaginal plug day was designated as E0) and cultured on Nuclepore filters (pore size 1 μm) on top of metal grids. The embryos from the breeding of ret +/− mice were genotyped by PCR. The medium (DME with 10% FCS) was supplemented in some dishes with 50 ng/ml GDNF (R&D Systems). After 4 d of culture, the kidney rudiments were fixed in ice-cold methanol and immunohistochemically stained as whole-mounts as described (Sainio et al., 1997). Anti–pan-cytokeratin antibodies (Sigma-Aldrich) were used as primary, and the secondary antibodies were TRITC-anti–mouse IgG (Sigma-Aldrich).
Acknowledgments
We thank Dr. Eero Lehtonen (Haartman Institute, University of Helsinki) for MDCK cells, Dr. Marc Billaud (Centre National de Recherche Scientifique, Lyon, France) for pcDNA3-Ret plasmid, Dr. Carlos Ibanez (Karolinska Institute, Stockholm, Sweden) for pcDNA3-GFRα1 plasmid, Dr. Bert Vogelstein (The Howard Hughes Medical Institute, Baltimore, MD) for Adeno-GFP, and Dr. Joan Brugge (Harvard Medical School) for DN Src construct, Madis Jacobson (University of Tartu, Tartu, Estonia) for his help with chemotaxis assay, Jack Leo for helping in Northern blotting, Marion Andlin and Agnes Viherä for technical assistance, and Matti Airaksinen, Tiina Immonen, Maxim Moshnyakov, and Kirmo Wartiovaara for critical reading of the manuscript. This work was supported by the Academy of Finland, The Centre for International Mobility (CIMO) scholarship, TEKES, the National Technology Agency Organization, HUS (the Hospital District of Helsinki and Uusimaa) Research Funding, Center of Excellence funding by the Academy of Finland, Sigrid Juselius grants to M. Saarma and H. Sariola (Biocentrum Helsinki fellows), and by grants from the National Cancer Institute of Canada to A. Angers-Loustau, and D. Kaplan.
Footnotes
Abbreviations used in this paper: DN, dominant-negative; E, embryonic day; GDNF, glial cell line–derived neurotrophic factor; GFRα1, GDNF family receptor α1; GPI, glycosylphosphatidylinositol; HGF, hepatocyte growth factor.
References
- Abram, C.L., and S.A. Courtneidge. 2000. Src family tyrosine kinases and growth factor signaling. Exp. Cell Res. 254:1–13. [DOI] [PubMed] [Google Scholar]
- Airaksinen, M.S., A. Titievsky, and M. Saarma. 1999. GDNF family neurotrophic factor signaling: four masters, one servant? Mol. Cell. Neurosci. 13:313–325. [DOI] [PubMed] [Google Scholar]
- Baloh, R.H., H. Enomoto, E.M. Johnson, Jr., and J. Milbrandt. 2000. The GDNF family ligands and receptors—implications for neural development. Curr. Opin. Neurobiol. 10:103–110. [DOI] [PubMed] [Google Scholar]
- Behar, T.N., Y.X. Li, H.T. Tran, W. Ma, V. Dunlap, C. Scott, and J.L. Barker. 1996. GABA stimulates chemotaxis and chemokinesis of embryonic cortical neurons via calcium-dependent mechanisms. J. Neurosci. 16:1808–1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behar, T.N., A.E. Schaffner, C.A. Scott, C. O'Connell, and J.L. Barker. 1998. Differential response of cortical plate and ventricular zone cells to GABA as a migration stimulus. J. Neurosci. 18:6378–6387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergstrom, J.D., B. Westermark, and N.E. Heldin. 2000. Epidermal growth factor receptor signaling activates met in human anaplastic thyroid carcinoma cells. Exp. Cell Res. 259:293–299. [DOI] [PubMed] [Google Scholar]
- Birchmeier, C., and E. Gherardi. 1998. Developmental roles of HGF/SF and its receptor, the c-Met tyrosine kinase. Trends Cell Biol. 8:404–410. [DOI] [PubMed] [Google Scholar]
- Cacalano, G., I. Farinas, L.C. Wang, K. Hagler, A. Forgie, M. Moore, M. Armanini, H. Phillips, A.M. Ryan, L.F. Reichardt, M., Hynes, A.M. Davies, and A. Rosenthal. 1998. GFRα1 is an essential receptor component for GDNF in the developing nervous system and kidney. Neuron. 21:53–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danilkovitch-Miagkova, A., D. Angeloni, A. Skeel, S. Donley, M. Lerman, and E.J. Leonard. 2000. Integrin-mediated RON growth factor receptor phosphorylation requires tyrosine kinase activity of both the receptor and c-Src. J. Biol. Chem. 275:14783–14786. [DOI] [PubMed] [Google Scholar]
- Edery, P., C. Eng, A. Munnich, and S. Lyonnet. 1997. RET in human development and oncogenesis. Bioessays. 19:389–395. [DOI] [PubMed] [Google Scholar]
- Enokido, Y., F. de Sauvage, J.A. Hongo, N. Ninkina, A. Rosenthal, V.L. Buchman, and A.M. Davies. 1998. GFRα-4 and the tyrosine kinase Ret form a functional receptor complex for persephin. Curr. Biol. 8:1019–1022. [DOI] [PubMed] [Google Scholar]
- Ghoussoub, R.A.D., D.A. Dillon, T. D'Aquila, B.E. Rimm, E.R. Fearon, and D.L. Rimm. 1998. Expression of c-met is a strong independent prognostic factor in breast carcinoma. Cancer. 82:1513–1520. [DOI] [PubMed] [Google Scholar]
- Giordano, S., A. Maffe, T.A. Williams, S. Artigiani, P. Gual, A. Bardelli, C. Basilico, P. Michieli, and P.M. Comoglio. 2000. Different point mutations in the met oncogene elicit distinct biological properties. FASEB J. 14:399–406. [DOI] [PubMed] [Google Scholar]
- Golden, J.P., J.A. De Maro, P.A. Osborne, J. Milbrant, and E.M. Johnson, Jr. 1999. Expression of neurturin, GDNF, and GDNF family receptor mRNA in the developing and mature mouse. Exp. Neurol. 158:504–528. [DOI] [PubMed] [Google Scholar]
- Gual, P., S. Giordano, T.A. Williams, S. Rocchi, E. Van Obberghen, and P.M. Comoglio. 2000. Sustained recruitment of phospholipase C-gamma to Gab1 is required for HGF-induced branching tubulogenesis. Oncogene. 19:1509–1518. [DOI] [PubMed] [Google Scholar]
- Harder, T., P. Scheiffele, P. Verkade, and K. Simons. 1998. Lipid domain structure of the plasma membrane revealed by patching of membrane components. J. Cell Biol. 141:929–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi, T., L.I. Tsao, and T.P. Su. 2002. Antiapoptotic and cytotoxic properties of delta opioid peptide [D-Ala(2),D-Leu(5)]enkephalin in PC12 cells. Synapse. 43:86–94. [DOI] [PubMed] [Google Scholar]
- He, T.C., S. Zhou, L.T. da Costa, J. Yu, K.W. Kinzler, and B. Vogelstein. 1998. AQ simplified system for generating recombinant adenoviruses. Proc. Natl. Acad. Sci. USA. 95:2509–2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellmich, H.L., L. Kos, E.S. Cho, K.A. Mahon, and A. Zimmer. 1996. Embryonic expression of glial cell-line derived neurotrophic factor (GDNF) suggests multiple developmental roles in neural differentiation and epithelial-mesenchymal interactions. Mech. Dev. 54:95–105. [DOI] [PubMed] [Google Scholar]
- Hung, W., and B. Elliott. 2001. Co-operative effect of c-Src tyrosine kinase and Stat3 in activation of hepatocyte growth factor expression in mammary carcinoma cells. J. Biol. Chem. 276:12395–12403. [DOI] [PubMed] [Google Scholar]
- Ivan, M., J.A. Bond, M. Prat, P.M. Comoglio, and D. Wynford-Thomas. 1997. Activated ras and ret oncogenes induce over-expression of c-met (hepatocyte growth factor receptor) in human thyroid epithelial cells. Oncogene. 14:2417–2423. [DOI] [PubMed] [Google Scholar]
- Jing, S., D. Wen, Y. Yu, P.L. Holst, Y. Luo, M. Fang, R. Tamir, L. Antonio, Z. Hu, R. Cupples, et al. 1996. GDNF-induced activation of the ret protein tyrosine kinase is mediated by GDNFR-alpha, a novel receptor for GDNF. Cell. 85:1113–1124. [DOI] [PubMed] [Google Scholar]
- Jo, M., D.B. Stolz, J.E. Esplen, K. Dorko, G.K. Michalopoulos, and S.C. Strom. 2000. Cross-talk between epidermal growth factor receptor and c-Met signal pathways in transformed cells. J. Biol. Chem. 275:8806–8811. [DOI] [PubMed] [Google Scholar]
- Karp, S.L., A. Ortiz-Arduan, S. Li, and E.G. Neilson. 1994. Epithelial differentiation of metanephric mesenchymal cells after stimulation with hepatocyte growth factor or embryonic spinal cord. Proc. Natl. Acad. Sci. USA. 91:5286–5290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuure, S., R. Vuolteenaho, and S. Vainio. 2000. Kidney morphogenesis: cellular and molecular regulation. Mech. Dev. 92:31–45. [DOI] [PubMed] [Google Scholar]
- Lee, F.S., and M.V. Chao. 2001. Activation of Trk neurotrophin receptors in the absence of neurotrophins. Proc. Natl. Acad. Sci. USA. 98:3555–3560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindahl, M., D. Poteryaev, Y. Liying, U. Arumäe, T. Timmusk, I. Bongarzone, A. Aiello, M.A. Pierotti, M.S. Airaksinen, and M. Saarma. 2001. Human GFRα4 is the receptor for persephin, and is selectively expressed in normal and malignant thyroid medullary cells. J. Biol. Chem. 276:9344–9351. [DOI] [PubMed] [Google Scholar]
- Melillo, R.M., M.V. Barone, G. Lupoli, A.M. Cirafici, F. Carlomagno, R. Visconti, B. Matoskova, P.P. Di Fiore, G. Vecchio, A. Fusco, and M. Santoro. 1999. Ret-mediated mitogenesis requires Src kinase activity. Cancer Res. 59:1120–1126. [PubMed] [Google Scholar]
- Meng, X., M. Lindahl, M.E. Hyvönen, M. Parvinen, D.G. de Rooij, M.W. Hess, A. Raatikainen-Ahokas, K. Sainio, H. Rauvala, M. Lakso, et al. 2000. Regulation of cell fate decision of undifferentiated spermatogonia by GDNF. Science. 287:1489–1493. [DOI] [PubMed] [Google Scholar]
- Montesano, R., K. Matsumoto, T. Nakamura, and L. Orci. 1991. Identification of a fibroblast-derived epithelial morphogen as hepatocyte growth factor. Cell. 67:901–908. [DOI] [PubMed] [Google Scholar]
- Naldini, L., E. Vigna, R.P. Narsimhan, G. Gaudino, R. Zarnegar, G.K. Michalopoulos, and P.M. Comoglio. 1991. Hepatocyte growth factor (HGF) stimulates the tyrosine kinase activity of the receptor encoded by the proto-oncogene c-MET. Oncogene. 6:501–504. [PubMed] [Google Scholar]
- Ottenhoff-Kalff, A.E., G.A.U. Rijksen, A. Hennipman, A.A. Michels, and G.E. Staal. 1992. Characterization of protein tyrosine kinases from human breast cancer: involvement of the c-src oncogene product. Cancer Res. 52:4773–4778. [PubMed] [Google Scholar]
- Pachnis, V., B. Mankoo, and F. Costantini. 1993. Expression of the c-ret proto-oncogene during mouse embryogenesis. Development. 119:1005–1017. [DOI] [PubMed] [Google Scholar]
- Paratcha, G., F. Ledda, L. Baars, M. Coulpier, V. Besset, J. Anders, R. Scott, and C.F. Ibáñez. 2001. Released GFRα1 potentiates downstream signaling, neuronal survival, and differentiation via a novel mechanism of recruitment of c-Ret to lipid rafts. Neuron. 29:171–184. [DOI] [PubMed] [Google Scholar]
- Pasini, B., I. Ceccherini, and G. Romeo. 1996. RET mutations in human disease. Trends Genet. 12:138–144. [DOI] [PubMed] [Google Scholar]
- Pichel, J.G., L. Shen, H.Z. Sheng, A.C. Granholm, J. Drago, A. Grinberg, E.J. Lee, S.P. Huang, M. Saarma, B.J. Hoffer, et al. 1996. Defects in interic innervation and kidney development in mice lacking GDNF. Nature. 382:73–76. [DOI] [PubMed] [Google Scholar]
- Poteryaev, D., A. Titievsky, Y.F. Sun, J. Thomas-Crusells, M. Lindahl, M. Billaud, U. Aurumäe, and M. Saarma. 1999. GDNF triggers a novel RET-independent Src kinase family-coupled signaling via a GPI-linked GDNF receptor α1. FEBS Lett. 463:63–66. [DOI] [PubMed] [Google Scholar]
- Rahimi, N., E. Tremblay, L. McAdam, M. Park, R. Schwall, and B.E. Elliott. 1996. Identification of a hepatocyte growth factor autocrine loop in a murine mammary carcinoma. Cell Growth Differ. 7:263–270. [PubMed] [Google Scholar]
- Rahimi, N., W. Hung, E. Tremblay, R. Saulnier, and B. Elliott. 1998. c-Src kinase activity is required for hepatocyte growth factor-induced motility and anchorage-independent growth of mammary carcinoma cells. J. Biol. Chem. 273:33714–33721. [DOI] [PubMed] [Google Scholar]
- Royal, I., T.M. Fournier, and M. Park. 1997. Differential requirement of Grb2 and PI3-kinase in HGF/SF-induced cell motility and tubulogenesis. J. Cell. Physiol. 173:196–201. [DOI] [PubMed] [Google Scholar]
- Saarma, M. 2001. GDNF recruits the signaling crew into lipid rafts. Trends Neurosci. 24:427–429. [DOI] [PubMed] [Google Scholar]
- Sainio, K., P. Suvanto, J. Davies, J. Wartiovaara, K. Wartiovaara, M. Saarma, U. Aurumäe, X. Meng, M. Lindahl, V. Pachnis, and H. Sariola. 1997. Glial-cell-line-derived neurotrophic factor is required for bud initiation from ureteric epithelium. Development. 124:4077–4087. [DOI] [PubMed] [Google Scholar]
- Sambrook, J., E.F. Fritsch, and T. Maniatis. 1989. Molecular Cloning: A Laboratory Manual. 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Sanchez, M.P., I. Silos-Santiago, J. Frizen, B. He, S.A. Lira, and M. Barbacid. 1996. Renal agenesis and the absence of enteric neurons in mice lacking GDNF. Nature. 382:70–73. [DOI] [PubMed] [Google Scholar]
- Santos, O.F., L.A. Moura, E.M. Rosen, and S.K. Nigam. 1993. Modulation of HGF-induced tubulogenesis and branching by multiple phosphorylation mechanisms. Dev. Biol. 159:535–548. [DOI] [PubMed] [Google Scholar]
- Sariola, H., and M. Saarma. 1999. GDNF and its receptors in the regulation of the ureteric branching. Int. J. Dev. Biol. 43:413–418. [PubMed] [Google Scholar]
- Schmidt, C., F. Bladt, S. Goedecke, V. Brinkmann, W. Zschiesche, M. Sharpe, E. Gherardi, and C. Birchmeier. 1995. Scatter factor/hepatocyte growth factor is essential for liver development. Nature. 373:699–702. [DOI] [PubMed] [Google Scholar]
- Schmidt, L., F.M. Duh, F. Chen, T. Kishida, G. Glenn, P. Choyke, S.W. Scherer, Z. Zhuang, I. Lubensky, M. Dean, et al. 1997. Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. Nat. Genet. 16:68–73. [DOI] [PubMed] [Google Scholar]
- Schuchardt, A., V. D'Agati, L. Larsson-Blomberg, F. Costantini, and V. Pachnis. 1994. Defects in the kidney and enteric nervous system of mice lacking the tyrosine kinase receptor Ret. Nature. 367:380–383. [DOI] [PubMed] [Google Scholar]
- Schuchardt, A., V. D'Agati, V. Pachnis, and F. Costantini. 1996. Renal agenesis and hypodysplasia in ret-k-mutant mice result from defects in ureteric bud development. Development. 122:1919–1929. [DOI] [PubMed] [Google Scholar]
- Shen, Y., M. Naujokas, M. Park, and K. Ireton. 2000. InIB-dependent internalization of Listeria is mediated by the Met receptor tyrosine kinase. Cell. 103:501–510. [DOI] [PubMed] [Google Scholar]
- Simons, K., and D. Toomre. 2000. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 1:31–39. [DOI] [PubMed] [Google Scholar]
- Stoker, M., E. Gherardi, M. Perryman, and J. Gray. 1987. Scatter factor is a fibroblast-derived modulator of epithelial cell mobility. Nature. 327:239–242. [DOI] [PubMed] [Google Scholar]
- Suvanto, P., J.O. Hiltunen, U. Arumäe, M. Moshnyakov, H. Sariola, K. Sainio, and M. Saarma. 1996. Localization of glial cell line-derived neurotrophic factor (GDNF) mRNA in embryonic rat by in situ hybridization. Eur. J. Neurosci. 8:816–822. [DOI] [PubMed] [Google Scholar]
- Tang, M.-J., D. Worley, M. Sanicola, and G. Dressler. 1998. The RET-glial cell-derived neurotrophic factor (GDNF) pathway stimulates migration and chemoattraction of epithelial cells. J. Cell Biol. 142:1337–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson, J., E. Doxakis, L.G. Pinon, P. Strachan, A. Buj-Bello, S. Wyatt, V.L. Buchman, and A.M. Davies. 1998. GFRα-4, a new GDNF family receptor. Mol. Cell. Neurosci. 11:117–126. [DOI] [PubMed] [Google Scholar]
- Trupp, M., R. Scott, S.R. Whittemore, and C.F. Ibáñez. 1999. Ret-dependent and -independent mechanisms of glial cell line-derived neurotrophic factor signaling in neuronal cells. J. Biol. Chem. 274:20885–20894. [DOI] [PubMed] [Google Scholar]
- Tsui-Pierchala, B.A., J. Milbrandt, and E.M. Johnson, Jr. 2002. NGF utilizes c-Ret via a novel GFL-independent, inter-RTK signaling mechanism to maintain the trophic status of mature sympathetic neurons. Neuron. 33:261–273. [DOI] [PubMed] [Google Scholar]
- Tuck, A.B., M. Park, E.E. Sterns, A. Boag, and B.E. Elliott. 1996. Coexpression of hepatocyte growth factor and receptor (Met) in human breast carcinoma. Am. J. Pathol. 148:225–232. [PMC free article] [PubMed] [Google Scholar]
- Vega, Q.C., C.A. Worby, M.S. Lechner, J.E. Dixon, and G.R. Dressler. 1996. Glial cell line-derived neurotrophic factor activates the receptor tyrosine kinase RET and promotes kidney morphogenesis. Proc. Natl. Acad. Sci. USA. 93:10657–10661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villa-Moruzzi, E., S. Lapi, M. Prat, G. Gaudino, and P.M. Comoglio. 1993. A protein tyrosine phosphatase activity associated with the hepatocyte growth factor/scatter factor receptor. J. Biol. Chem. 268:18176–18180. [PubMed] [Google Scholar]
- Woolf, A.S., M. Kolatsi-Joannou, P. Hardman, E. Andermarcher, C. Moorby, L.G. Fine, P.S. Jat, M.D. Noble, and E. Gherardi. 1995. Roles of hepatocyte growth factor/scatter factor and the met receptor in the early development of the metanephros. J. Cell Biol. 128:171–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang, Z., W.S. Park, S. Pack, L. Schmidt, A.O. Vortmeyer, E. Pak, T. Pham, R.J. Weil, S. Candidus, I.A. Lubensky, et al. 1998. Trisomy 7-harbouring non-random duplication of the mutant MET allele in hereditary papillary renal carcinomas. Nat. Genet. 20:66–69. [DOI] [PubMed] [Google Scholar]