

Abstract
Dendritic spines in the central nervous system undergo rapid actin-based shape changes, making actin regulators potential modulators of spine morphology and synapse formation. Although several potential regulators and effectors for actin organization have been identified, the mechanisms by which these molecules assemble and localize are not understood. Here we show that the G protein–coupled receptor kinase–interacting protein (GIT)1 serves such a function by targeting actin regulators and locally modulating Rac activity at synapses. In cultured hippocampal neurons, GIT1 is enriched in both pre- and postsynaptic terminals and targeted to these sites by a novel domain. Disruption of the synaptic localization of GIT1 by a dominant-negative mutant results in numerous dendritic protrusions and a significant decrease in the number of synapses and normal mushroom-shaped spines. The phenotype results from mislocalized GIT1 and its binding partner PIX, an exchange factor for Rac. In addition, constitutively active Rac shows a phenotype similar to the GIT1 mutant, whereas dominant-negative Rac inhibits the dendritic protrusion formation induced by mislocalized GIT1. These results demonstrate a novel function for GIT1 as a key regulator of spine morphology and synapse formation and point to a potential mechanism by which mutations in Rho family signaling leads to decreased neuronal connectivity and cognitive defects in nonsyndromic mental retardation.
Keywords: synapse formation; GIT1; PIX; Rac; spine morphology
Introduction
Synapses are specialized adhesive sites that mediate communication between neurons. Structurally, they are composed of a presynaptic terminal and a postsynaptic region. The postsynaptic side can take the form of dendritic spines, which are actin-rich protrusions on dendrites that receive excitatory synaptic inputs. The formation and maintenance of spines and synapses require precise targeting and coordinated activation of structural and signaling molecules (Nakayama and Luo, 2000; Rao and Craig, 2000; Sheng and Kim, 2002). Although many synaptic proteins have been identified, less is known about the signaling pathways that regulate the formation and pruning of spines and synapses. Imaging studies on living neurons have revealed that dendritic spines are highly dynamic structures, which undergo rapid actin-based shape changes (Fischer et al., 1998). This raises the exciting possibility that actin dynamics provide a cellular basis for synaptic plasticity (Matus, 2000), which underlies cognitive functions such as learning and memory.
Organization of the actin cytoskeleton is regulated by members of the Rho family of small GTPases, including Rho, Rac, and Cdc42, that cycle between an inactive (GDP-bound) and an active (GTP-bound) state (Hall, 1998; Ridley, 2001). The activation of these molecules is tightly controlled by guanine nucleotide exchange factors (GEFs),* which facilitate the exchange of GDP for GTP, and GTPase-activating proteins (GAPs) that promote GTP hydrolysis. Recent studies have shown that regulation of Rho family signaling plays a key role in normal cognitive functions. Three proteins that interact directly with Rho GTPases are mutated in patients with nonsyndromic mental retardation (MR). These proteins are oligophrenin1, a GAP for Rho-GTPases, αPIX, a GEF for Rac and Cdc42, and p21-activated kinase (PAK)3, a Rac and Cdc42 effector (Allen et al., 1998; Billuart et al., 1998; Kutsche et al., 2000; Barnes and Milgram, 2002; Ramakers, 2002). It is not yet known how alterations in Rho signaling result in MR. An attractive hypothesis is that abnormalities in the organization of the actin cytoskeleton give rise to decreased neuronal connectivity and thus impaired cognitive function.
Global activation or shutdown of Rho family signaling usually leads to defects in cellular functions. Thus, one emerging theme is that these proteins are likely to be activated in specific locations within the cell. This could be accomplished by spatial targeting of actin regulators. Adaptor proteins that bind GEFs, GAPs, and their effectors, such as the G protein–coupled receptor kinase–interacting protein (GIT)1, could potentially function in this capacity by concentrating these molecules at sites of actin organization. In epithelial cells and fibroblasts, GIT1 localizes to distinct subcellular compartments, including adhesions, membrane protrusions, and cytoplasmic complexes. GIT1 regulates migration and protrusive activity by assembling and targeting multiprotein signaling complexes, which contain important actin regulators including PIX, a Rac GEF, and PAK, a Rac effector, between the subcellular compartments (Di Cesare et al., 2000; de Curtis, 2001; Manabe et al., 2002).
In epithelial cells and fibroblasts, the function and localization of GIT1 is mediated by a series of domains including the NH2-terminal ADP-ribosylation factor (ARF)–GAP domain that regulates receptor endocytosis (Premont et al., 1998; Claing et al., 2000), ankyrin repeats, Spa2 homology domain (SHD)1 that binds PIX (Zhao et al., 2000), and a COOH-terminal paxillin-binding domain (West et al., 2001; Manabe et al., 2002). In fibroblasts, the COOH-terminal 140 residues of GIT1 (cGIT1), which contain the paxillin binding domain, target it to adhesions, whereas the central domain that contains the ankyrin repeats and the PIX-binding domain is necessary for localization to the cytoplasmic complexes (Manabe et al., 2002).
In this study, we show that GIT1 is enriched in synapses on cultured hippocampal neurons. GIT1 is targeted to the synapse by a novel domain that differs from its adhesion targeting in fibroblasts. Disruption of the postsynaptic localization of GIT1 by a dominant interfering GIT1 mutant that competes for the synaptic binding site results in altered spine morphology and a significant decrease in the density of synapses. These effects result, at least in part, from the mislocalization of PIX. Recruitment of PIX to synapses by GIT1 and the GEF activity of PIX are both necessary for the formation and stabilization of synapse-bearing spines. These results suggest that spatially localized, regulated Rac activity is essential for this process. Thus, GIT1 serves as a key regulator of spine morphology and synapse formation through assembling and targeting multimolecular complexes to synapses where they locally regulate actin dynamics.
Results
GIT1 localizes to synapses in cultured hippocampal neurons
Previous studies showed that GIT1 is highly expressed in the brain both at the mRNA and protein levels (Premont et al., 1998; Zhao et al., 2000). To determine whether GIT1 is expressed in neurons, we performed RT-PCR with hippocampal total RNA using GIT1-specific primers. The sequence of the PCR product was identical to the previously described rat GIT1 sequence, showing the presence of the GIT1 mRNA in these neurons. To confirm the protein expression of GIT1, we subjected a lysate from hippocampal neurons to immunoblot analysis using a GIT1 antibody. We detected a specific band at ∼95 kD, which is the expected mobility of GIT1 (Fig. 1 A). This confirms the presence of GIT1 in the neurons at the protein level since our antibody is specific for GIT1; however, we cannot exclude the possibility that other GIT family members may also be present.
Figure 1.


GIT1 is expressed in cultured hippocampal neurons and enriched in synapses. (A) Western blot of a lysate from day 10 cultured hippocampal neurons. The blot was probed with a GIT1 antibody. A specific band at ∼95 kD confirms the presence of the GIT1 protein in these neurons. (B) Hippocampal neurons at 2–3 wk in culture were double immunostained for endogenous GIT1 (left column) and various synaptic proteins (right column). GIT1 colocalizes with the presynaptic marker SV2 (top). GIT1 shows colocalization with PSD-95 in some puncta (arrowheads), but some GIT1 puncta, especially those on the cell body, do not overlap with PSD-95 (middle, arrows). These puncta show colocalization with the inhibitory synapse marker GAD-6 (bottom). Enlargements of the boxed regions are shown in insets at the bottom right of each panel. Bar, 20 μm. (C) Hippocampal neurons were transfected with either GFP-GIT1 (top) or GIT1-FLAG (bottom) and immunostained for the presynaptic marker synapsin1 at 3 wk in culture. Both GFP-GIT1 and GIT1-FLAG colocalize with synapsin1 in dendritic spines and shafts (arrows). Bar, 20 μm. (D) Hippocampal neurons were transfected with GFP-GIT1 and immunostained for the appropriate synaptic markers at 2–3 wk in culture. The GIT1 clusters on the dendrites (Dendritic) almost completely merge with the postsynaptic marker PSD-95 and are in close apposition to the presynaptic marker synapsin1 (Overlay). The GIT1 clusters on the axons (Axonal) completely merge with the presynaptic marker SV2 and are in close apposition to the postsynaptic marker PSD-95 (Overlay). Note the colocalization of GIT1 clusters with the synaptic markers (arrowheads). Enlargements of individual synapses are shown in the right column. GFP-GIT1 is pseudocolored green, and the synaptic markers are pseudocolored red. Bar, 2 μm.
To examine the subcellular distribution of GIT1 in hippocampal neurons, we immunostained low density cultures with a GIT1 antibody (Manabe et al., 2002). In mature neurons (3 wk in culture), GIT1 accumulated in puncta along neuronal processes, indicating that it might be synaptic. To confirm this, we coimmunostained for GIT1 and the synaptic vesicle protein, SV2. GIT1 clusters colocalized with SV2 staining (Fig. 1 B, top), showing that GIT1 is enriched in synapses. Since brain synapses are categorized as excitatory or inhibitory according to the postsynaptic responses (Craig and Boudin, 2001), we asked whether GIT1 localized in one or both types of synapses. To determine whether GIT1 is present in excitatory synapses, double labeling of GIT1 with the postsynaptic density protein PSD-95 was performed (Fig. 1 B, middle). GIT1 puncta colocalized with PSD-95 puncta in some of the excitatory synapses (Fig. 1 B, arrowheads), although some GIT1 puncta were clearly PSD-95 negative (Fig. 1 B, arrows), suggesting that GIT1 is not restricted to excitatory synapses. Consistent with this, GIT1 colocalized with the GABAergic synaptic marker GAD-6 in clusters on the cell body and dendritic shafts (Fig. 1 B, bottom), indicating that it is also present in inhibitory synapses. Although most of the PSD-95–negative GIT1 puncta correspond to inhibitory synapses, some of the puncta might represent the GIT1 cytoplasmic complexes, which were described previously in non-neuronal cells (Manabe et al., 2002).
The localization of GIT1 to synapses was confirmed by transfecting full-length GFP-tagged and FLAG-tagged GIT1 into hippocampal neurons. In previous studies using fibroblasts, these constructs localized indistinguishably from endogenous GIT1 (Manabe et al., 2002). In hippocampal neurons, the localization of both constructs was also identical to the endogenous GIT1. Both GFP-GIT1 and GIT1-FLAG colocalize in clusters with synapsin1, which, like SV2, labels presynaptic terminals and serves as a synaptic marker (Fig. 1 C). The GIT1 clusters in the dendrites were in close apposition to the presynaptic marker synapsin1 and almost completely merged with the postsynaptic marker PSD-95 (Fig. 1 D). This shows that GIT1 is postsynaptic. We also observed GIT1 in clusters along the axons of the transfected neurons. These clusters completely merged with the presynaptic marker SV2 and were in close apposition to PSD-95 (Fig. 1 D), indicating that GIT1 is not only postsynaptic but also presynaptic. Consistent with this, Kim et al. (2003) recently showed an interaction of GIT1 with the presynaptic protein Piccolo.
Synaptic targeting of GIT1
To identify the region that targets GIT1 to synapses, we expressed various domains of GIT1 as GFP fusion proteins in hippocampal neurons (Fig. 2 A). Their synaptic localization was assayed by GFP fluorescence and coimmunostaining with synaptic markers. We began by examining the localization of cGIT1, since it contains the binding site for paxillin and is responsible for the adhesion targeting of GIT1 in fibroblasts (Manabe et al., 2002). However, in the neurons, cGIT1 did not concentrate in synapses but distributed diffusely throughout the cytoplasm of the soma and processes (Fig. 2 D). Thus, the domain that targets GIT1 to synapses is different than the adhesion localization domain in fibroblasts. As expected, GIT1Δc, which lacks the COOH-terminal paxillin binding domain, localized in synapses (Fig. 2 B). To further localize the targeting domain within GIT1Δc, we first examined whether the NH2-terminal ARF-GAP domain is necessary for synaptic targeting. A central domain deletion mutant, GIT1ΔCD, which contains the ARF-GAP domain and the paxillin binding domain, did not accumulate in synapses (Fig. 2 D). Another mutant that comprises the central domain, CD-GIT1, localized in synapses (Fig. 2 B). Therefore, neither the ARF-GAP nor the paxillin binding domain is necessary for synaptic targeting, pointing to the central domain of GIT1 as the targeting domain.
Figure 2.



Synaptic targeting of GIT1. (A) Schematic diagram of the full-length and deletion constructs of GIT1 (all GFP-tagged at the NH2 terminus except N-SLD and C-SLD, which are FLAG-tagged at the NH2 terminus). The indicated domains are as follows: ARF-GAP domain (ARF-GAP), ankyrin repeats (Ank), Spa2 homology domain 1(SHD1), and paxillin binding site (paxillin). The fusion proteins with synaptic localization are indicated with “+”. N-SLD shows weak localization to synapses which is indicated with “+/−”. (B) GIT1 fusion proteins that show specific localization to synapses. Hippocampal neurons were transfected with the various GIT1 fusion proteins (left column) and stained for synaptic markers (middle column). GIT1Δc and CD-GIT1 were coimmunostained for SV2. CDΔAnk was coimmunostained for PSD-95. CDΔAS/SLD was coimmunostained for synapsin1. Overlays are shown in the right column. The GIT1 constructs were pseudocolored green, and the synaptic markers were pseudocolored red. Bar, 2 μm. (C) FLAG–N-SLD– expressing neurons were coimmunostained for FLAG and synapsin1. N-SLD shows partial localization to synapses (arrows). The overlaid picture is shown in the right column. N-SLD was pseudocolored green, and synapsin1 was pseudocolored red. Bar, 2 μm. (D) GIT1 fusion proteins that fail to localize to synapses. Hippocampal neurons expressing the GFP-tagged GIT1 deletion constructs (left column) were stained for SV2 (middle column). The GIT1 constructs lacking SLD (cGIT1, GIT1ΔCD, nGIT1, and GIT1ΔSLD) do not show synaptic localization. Deletion of the NH2-terminal 32 aa of SLD (SLDΔ32) dramatically reduces localization to the synapse. For localization of C-SLD, neurons expressing FLAG–C-SLD were coimmunostained for FLAG and synapsin1. C-SLD shows a diffuse labeling pattern. The overlays are shown in the right column. The GIT1 constructs were pseudocolored green, and the synaptic markers were pseudocolored red. Bar, 2 μm.
The central domain contains ankyrin repeats, SHD1 (PIX binding domain), and a COOH-terminal flanking region. To further characterize the targeting domain, we constructed two more deletion mutants: CDΔAnk (with the ankyrin repeats deleted from CD-GIT1) and CDΔAS (with both the ankyrin repeats and SHD1 deleted from CD-GIT1) (Zhao et al., 2000). Both mutants localized efficiently to synapses (Fig. 2 B), indicating that neither the ankyrin repeats nor SHD1 is necessary for synaptic targeting. This was further confirmed by the lack of synaptic localization with nGIT1, which only contains the ARF-GAP, ankyrin repeats, and SHD1 (Fig. 2, A and D).
The smallest localizing domain that we identified (CDΔAS) is located between SHD1 and the paxillin binding domain. Since it is responsible for the synaptic targeting of GIT1, we named it SLD for synaptic localization domain. To test if the intact SLD is necessary for synaptic targeting, we prepared a mutant in which the NH2-terminal 32 aa in SLD were deleted (SLDΔ32). This mutant showed significantly decreased localization in synapses (Fig. 2 D), suggesting that these 32 aa contribute to efficient synaptic localization. We also prepared two smaller FLAG-tagged constructs, N-SLD and C-SLD, which contain the NH2-terminal and COOH-terminal half of SLD, respectively. Subcellular localization of these constructs was determined by FLAG and synapsin coimmunostaining. N-SLD showed partial localization to synapses (Fig. 2 C), whereas C-SLD localized diffusely in the cytoplasm (Fig. 2 D). These results suggest that the localization domain resides at least in part in the NH2-terminal half of SLD; however, the COOH terminus of SLD may also contribute to efficient synaptic localization. Finally, an internal deletion mutant with the SLD deleted (GIT1ΔSLD) did not localize to synapses (Fig. 2 D), further confirming that SLD is the localizing domain.
Disruption of the synaptic localization of GIT1 affects spine morphology and synapse formation
To examine the function of GIT1 in synapses, we took a dominant-interfering approach and expressed the smallest localizing construct, SLD, in the neurons. We hypothesized that this construct would prevent GIT1 from localizing to synapses by competing for the synaptic binding sites. This approach has been successful in fibroblasts where the adhesion targeting domain of GIT1 effectively prevents endogenous GIT1 from localizing to adhesions (Manabe et al., 2002). To test if this approach is also effective in reducing the synaptic localization of GIT1, we coexpressed GFP-SLD and GIT1 in the neurons. Indeed, ectopic expression of SLD in neurons showed a dose-dependent effect. In neurons expressing relatively low levels of SLD (<2-fold relative to endogenous GIT1), the synaptic localization of GIT1 was reduced (Fig. 3 A). However, no apparent changes in spine morphology were observed. In neurons expressing high levels of SLD (fivefold relative to endogenous GIT1), GIT1 was distributed diffusely, the number of normal, mushroom-shaped spines was significantly decreased, and the number of long, thin dendritic protrusions was dramatically increased (Fig. 3, B and C). In addition, in the SLD-expressing neurons, the linear density of synapses (number of synapses per 100-μm dendrite) decreased significantly compared with neurons expressing comparable levels of GIT1, nGIT1, and CD-GIT1 (Fig. 3, B and D). When the synaptic density was quantified by another method (the number of synapses per unit area [μm2]), a similar decrease was observed in the SLD-expressing neurons. This effect on synaptic density was observed when SLD was expressed at day 4–7 in culture and the number of synapses quantified at day 14. When we expressed SLD at day 10 in culture and quantified at day 14, the effect on synaptic density is less dramatic. This suggests that SLD affects synapse formation; however, we cannot exclude the possibility that it is also affecting the maintenance of synapses. The effect of SLD on synaptic density is unlikely to be due to neurite outgrowth defects since it does not affect hippocampal neurite extension on poly-l-lysine on which we grow these neurons (unpublished data). Thus, these data suggest that perturbing GIT1 localization results in defects in spine morphology and synapse formation.
Figure 3.

Overexpression of the SLD from GIT1 regulates spine morphology and synaptic density. (A) Hippocampal neurons were cotransfected with GFP-SLD and GIT1-FLAG and stained for synapsin1. Note the localization of GFP-SLD to the synapses in relatively low expressing cells causes a decreased localization of GIT1 in the synapses (arrows). Bar, 20 μm. (B) Hippocampal neurons were transfected with various GIT1 constructs at 1 wk in culture and stained for SV2 at 2 wk in culture. Note the increase in dendritic protrusions (left column) and the decrease in synaptic density (right column) in neurons expressing high levels of GFP-SLD. Bar, 20 μm. (C) Quantification of the number of spines and dendritic protrusions in hippocampal neurons transfected with either GFP-GIT1 or GFP-SLD. 80–100 dendrites from independent transfections were quantified for each construct. The definitions of spines and dendritic protrusions are provided in Materials and methods. (D) Quantification of synaptic density in hippocampal neurons transfected with GIT1, nGIT1, CD-GIT1, or SLD. 85–110 dendrites from independent transfections were quantified for each construct (as described in Materials and methods). The difference between SLD and other GIT1 constructs was statistically significant as determined by Student's t test (*P < 0.0001). Note that even though nGIT1 contains SHD1, it has a decreased affinity for PIX, as assayed by coimmunoprecipitation, when compared with wild-type GIT1 (Zhao et al., 2000; unpublished data).
PIX is targeted to the synapse by GIT1
How does GIT1 affect synapse formation? Since CD-GIT1, which contains the PIX binding domain in addition to SLD, behaved like wild-type GIT1, it is likely that the PIX binding domain is a functional domain. To test this hypothesis, we constructed a GIT1 mutant that has the PIX binding domain deleted (GIT1ΔSHD). As expected, GIT1ΔSHD localized to synapses when expressed at relatively low levels. Neurons expressing high levels of GIT1ΔSHD showed a phenotype similar to the SLD-expressing neurons. The cells had numerous dendritic protrusions (Fig. 4 A) with a decrease in spine and synaptic density (Fig. 4, B and C). This suggests that PIX is acting downstream of GIT1 to affect spine morphology and synapse formation.
Figure 4.
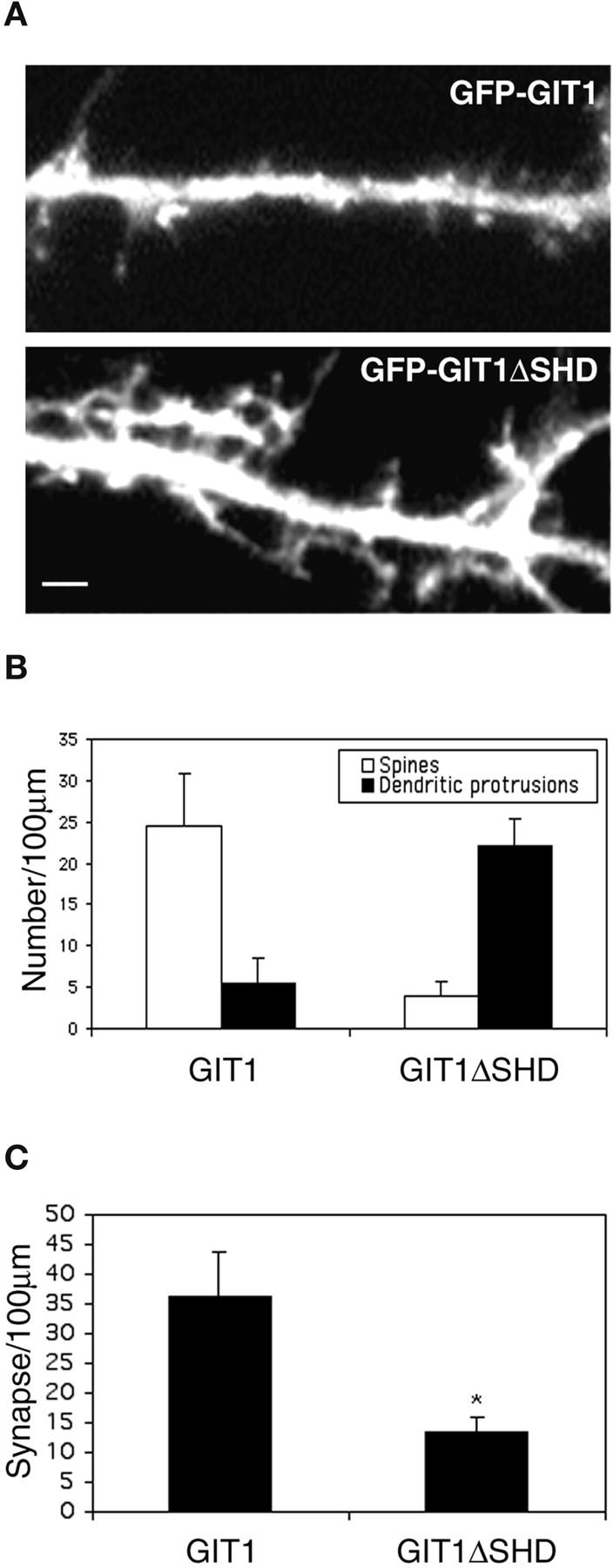
GIT1ΔSHD-expressing neurons show a phenotype similar to SLD-expressing neurons. (A) Hippocampal neurons were transfected with either GFP-GIT1 or GFP-GIT1ΔSHD at day 7 in culture and imaged at day 14 in culture. Note the increase in dendritic protrusions in GIT1ΔSHD-expressing neurons. Bar, 2 μm. (B) Quantification of the number of spines and dendritic protrusions in GIT1- and GIT1ΔSHD-expressing neurons. (C) Quantification of the synaptic density in GIT1- and GIT1ΔSHD-expressing neurons. The difference between GIT1 and GIT1ΔSHD was statistically significant as determined by Student's t test (*P < 0.0001).
To confirm the protein expression of PIX in hippocampal neurons, we performed immunoblot analysis on a hippocampal lysate using a βPIX antibody. We detected a band at ∼78 kD, which is the expected mobility of βPIX (Koh et al., 2001) (unpublished data), indicating that endogenous βPIX is present in hippocampal neurons. In addition to the 78-kD band, another band at ∼55 kD could be detected, which is the expected mobility of an alternative spliced isoform, p50Cool. To see if PIX also localizes to synapses, we transfected HA-tagged βPIX into the neurons and looked at its localization by coimmunostaining with synapsin1. PIX and synapsin1 colocalized in clusters along the processes, suggesting that PIX is synaptic as well (Fig. 5 A). Furthermore, immunostaining of endogenous PIX in hippocampal neurons with a polyclonal βPIX antibody showed that endogenous PIX also localized to synapses (unpublished data). This is consistent with the localization of dPIX, the Drosophila homologue of mammalian PIX, to the postsynaptic density of Drosophila neuromuscular junctions (Parnas et al., 2001).
Figure 5.
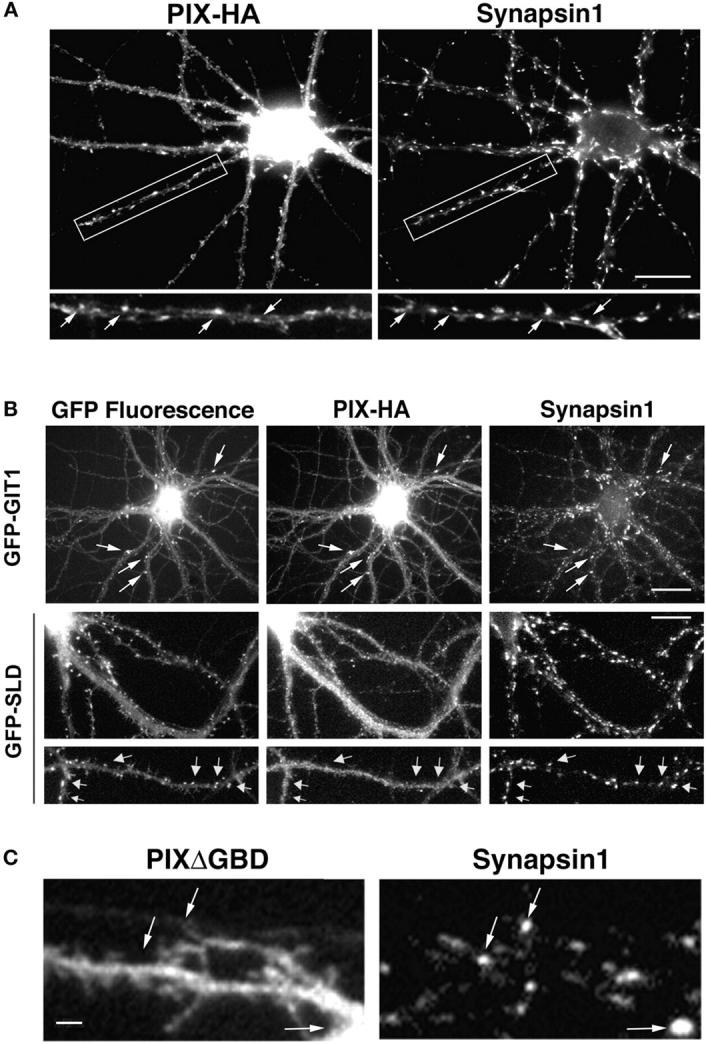
PIX is targeted to the synapses by GIT1. (A) HA-tagged βPIX localizes to the synapses. Hippocampal neurons were transfected with βPIX-HA and stained for HA and synapsin1. Arrows indicate PIX puncta that colocalize with synapsin1 puncta. Bar, 20 μm. (B) The localization of PIX to synapses is inhibited by coexpression of GFP-SLD. Hippocampal neurons were cotransfected with either GFP-GIT1 and PIX-HA or GFP-SLD and PIX-HA. They were fixed and stained for HA and synapsin1. Note the localization of PIX in the synapses when coexpressed with GIT1 (arrows, top) and the decreased localization of PIX to synapses when coexpressed with SLD (arrows, bottom). Bar, 20 μm. (C) A GIT1 binding-deficient PIX mutant (PIXΔGBD) does not localized to synapses. Hippocampal neurons were transfected with HA-PIXΔGBD and coimmunostained for HA and synapsin1. Note the lack of localization to synapses with PIXΔGBD (arrows). Bar, 2 μm.
To determine whether PIX is targeted to synapses by GIT1, we coexpressed GFP-tagged GIT1 constructs and HA-PIX in the neurons. Full-length GIT1 and PIX colocalized in synapses as shown by coimmunostaining with synapsin1 (Fig. 5 B, top). However, in neurons coexpressing GFP-SLD and HA-PIX, PIX showed a diffuse staining pattern with no specific accumulation in synapses (Fig. 5 B, middle and bottom), indicating SLD inhibits PIX localization in synapses. This suggests that PIX is targeted to synapses by GIT1. Furthermore, since SLD inhibits the localization of both GIT1 and PIX, the phenotype of SLD likely results from mistargeting of the GIT1–PIX complex.
To further confirm that PIX is targeted to the synapse by GIT1, we constructed a PIX mutant that is deficient in GIT1 binding (PIXΔGBD) (Koh et al., 2001). Localization of this PIX mutant in hippocampal neurons was examined by transfecting the mutant into the neurons and coimmunostaining with synapsin1. Unlike wild-type PIX, which exhibits synaptic localization, PIXΔGBD showed a diffuse staining pattern with no accumulation in synapses (Fig. 5 C). This shows that GIT1 binding of PIX is necessary for its targeting to synapses.
Effects of PIX mutants on spine morphology and synapse formation
If the SLD phenotype results from mistargeting of the GIT1–PIX complex, increasing the diffuse, nonsynaptic distribution of PIX should give a similar phenotype. To test this hypothesis, we transfected neurons with either wild-type PIX or PIXΔGBD. Wild-type PIX, when expressed at high levels, showed a diffuse labeling pattern. Likewise, PIXΔGBD, which did not localize to synapses, also distributed diffusely. Thus, overexpression of either construct should effectively increase the nonsynaptic distribution of PIX and give a phenotype similar to SLD-expressing neurons. Indeed, in cells expressing high levels of either construct multiple dendritic protrusions were observed with a concomitant decrease in spine and synaptic density (Fig. 6) . This suggests that precise targeting of PIX to synapses by GIT1 is necessary for dendritic spine and synapse formation and that mislocalization of PIX perturbs this process.
Figure 6.

Effects of PIX mutants on spine morphology and synaptic density. (A) Hippocampal neurons were transfected with various PIX constructs at day 7 in culture and imaged at day 14 in culture. Note the increase in dendritic protrusions in PIX- and PIXΔGBD- overexpressing neurons and the decrease in spines and dendritic protrusions in PIX-LL–expressing neurons. “Control” denotes GFP-expressing neurons. Bar, 5 μm. (B) Quantification of the number of spines and dendritic protrusions in various PIX constructs. (C) Quantification of the number of synapses in various PIX constructs. The difference between PIX constructs and the untransfected neurons (control) was statistically significant as determined by the Student's t test (*P < 0.0001).
PIX exhibits GEF activity towards the small GTPases Cdc42 and Rac. To determine if the GEF activity is required for its effects on spine morphology and synapse formation, we examined the effects of PIX-LL, which has mutations (L238R and L239S) in the Dbl homology domain that abolish its GEF activity (Manser et al., 1998). In contrast to neurons expressing wild-type PIX or PIXΔGBD, PIX-LL–expressing neurons showed a significant decrease in both spines and dendritic protrusions (Fig. 6, A and B). This suggests that the GEF activity is necessary for generating the dendritic protrusions in wild-type PIX or PIXΔGBD-overexpressing neurons. Besides the decrease in the number of spines and dendritic protrusions, PIX-LL–expressing neurons also showed a decrease in the number of synapses when compared with the untransfected neurons (Fig. 6, A and C). Thus, the GEF activity of PIX is necessary for both spine morphogenesis and synapse formation. It also points to the small GTPases as the potential downstream effectors of this process.
Effects of Rac mutants on spine morphology and synapse formation
Since active Rac has been reported to induce multiple dendritic protrusions in hippocampal slices (Nakayama et al., 2000), a phenotype like that produced by SLD, altered Rac activation appears a likely mediator of the effects of SLD overexpression. To address this question, we transfected hippocampal neurons with a myc-tagged constitutively active Rac, RacV12, at day 10 in culture. The phenotype was examined 48 h after transfection. RacV12-transfected neurons form numerous dendritic protrusions with a significant decrease in the number of normal spines (Fig. 7, A and C) . This is consistent with the constitutively active Rac phenotype previously reported in hippocampal slices (Nakayama et al., 2000) and in the mouse cerebellar Purkinje cells expressing a RacV12 transgene (Luo et al., 1996). To see if RacV12 has a similar effect on synaptic density as SLD, we immunostained RacV12-transfected neurons with synapsin1 and anti-myc antibodies. RacV12-expressing neurons form significantly fewer synapses than the adjacent untransfected neurons (Fig. 7, A and B; P < 0.0001), suggesting that overactivation of Rac disrupts synapse formation. To further elucidate how Rac activity affects synapse formation, we transfected a myc-tagged dominant-negative version of Rac, RacN17, into the neurons. RacN17-expressing neurons exhibited very smooth dendrites with a drastic reduction in the number of spines compared with the adjacent untransfected neurons. The synaptic density was also significantly reduced (Fig. 7, A–C; P < 0.0001). The effects of Rac mutants on synapse formation are unlikely due to neurite outgrowth defects for two reasons. First, the Rac mutants were transfected at day 10 when the neurites have reached sufficient length for synapses to form. Second, Rac mutants have been shown to affect only axonal growth but not dendritic growth (Luo et al., 1997), whereas the effects of Rac mutants on synaptic density were observed on the dendrites of the transfected neurons.
Figure 7.

Effects of Rac mutants on spine morphology and synaptic density. (A) Effects of Rac mutants on spine morphology and synaptic density. Hippocampal neurons were transfected with myc-tagged RacV12 or RacN17 at day 10 in culture and stained for myc and either rhodamine-conjugated phalloidin (left column) or synapsin1 (right column) at day 12 in culture. Nearby untransfected cells (control) were stained for phalloidin or synapsin1. Note the increase in dendritic protrusions in RacV12-expressing neurons and the decrease in the number of spines in RacN17-expressing neurons. Synaptic density is decreased in both cases. Bar, 2 μm. (B) Quantification of synaptic linear density in neurons transfected with the Rac mutants. Synaptic density is significantly decreased in both RacV12- and RacN17-transfected neurons, *P < 0.0001 (n > 50 for each construct) compared with nearby untransfected neurons (control). (C) Quantification of the number of spines and dendritic protrusions in neurons transfected with the Rac mutants. (D) RacN17 blocks the SLD phenotype. Hippocampal neurons were transfected with either GFP-SLD alone (top) or GFP-SLD and RacN17 (bottom). Note that the dendritic protrusions induced by SLD were completely inhibited by RacN17. Bar, 5 μm.
Rac acts downstream of GIT1
If the dendritic protrusions induced by SLD is mediated by altered Rac activation in the cytoplasm, inhibiting Rac activation should block these protrusions. To test this, we cotransfected GFP-SLD and RacN17 into the neurons. RacN17 completely inhibited the SLD-induced formation of dendritic protrusions (Fig. 7 D). The neurons showed smooth dendrites with very few spines. This suggests that the SLD phenotype is mediated by mislocalized active Rac.
Discussion
Localized changes in the organization and dynamics of the actin cytoskeleton are thought to underlie the formation, maintenance, and plasticity of synaptic connections (Matus, 2000). Although abundant evidence points to the role of Rho family GTPases as pivotal regulators of actin dynamics and organization, the mechanisms that localize their activities to specific sites, like synapses, are not understood. Recent evidence from motile fibroblasts shows that Rac is diffusely distributed throughout the cell, whereas activated Rac is highly localized (Kraynov et al., 2000). In this study, we present evidence that the adapter protein GIT1 serves to localize Rac activity by providing a docking site for PIX, which serves as an exchange factor for Rac and a binding protein for a Rac effector, PAK. Both the recruitment of PIX to synapses and its GEF activity are necessary for the formation and stabilization of synapse-bearing spines (Fig. 8) .
Figure 8.

GIT1 regulates synapse formation. GIT1 is targeted to synapses through the SLD. At the synapse, GIT1, or possibly other related molecules, functions as an adaptor protein recruiting exchanges factors, such as PIX, to synapses where they locally activate Rac. Locally regulated Rac activation is essential for spine morphogenesis and synapse formation. When GIT1/PIX is mislocalized from synapses, Rac is activated outside the synaptic area. Mislocalized active Rac is responsible for the increased dendritic protrusions and decreased synaptic density. Inhibition of the Rac signaling pathway results in a decrease in the density of spines and synapses. The indicated domains of GIT1 are as follows: ARF-GAP domain (ARF-GAP), ankyrin repeats (ANK), Spa2 homology domain 1 (SHD1), synaptic localization domain (SLD), and paxillin binding domain (PAX). The question mark indicates the unknown molecule that targets GIT1 to synapses.
A major observation in our study is that expression of a highly truncated GIT1 mutant, SLD, gives rise to neurons with numerous long and thin dendritic protrusions. Overexpression of PIX, GIT1-binding deficient PIX (PIXΔGBD), or RacV12 recapitulate this phenotype. How do these mutants generate similar phenotypes? When the SLD is overexpressed in neurons, the synaptic localization of endogenous GIT1 and PIX is disrupted, and they become diffusely distributed throughout the neuron. Similarly, overexpression of wild-type PIX or PIXΔGBD also results in increased mislocalization of PIX. We hypothesize that the diffuse distribution of these molecules would produce an increase in Rac activation throughout the neuron. Overexpression of RacV12 produces a similar mislocalization with this molecule diffusely distributed (unpublished data). From these observations, we hypothesize that synapse formation requires properly localized and regulated Rac activation and thus, mislocalized, active Rac induces the formation of multiple dendritic protrusions and disrupts this process in these mutant-transfected neurons.
Interestingly, both RacV12 and RacN17 cause a decrease in synaptic density. One explanation is that synapse formation requires properly localized, critical levels of Rac activation. In the case of RacV12-expressing cells, Rac is constitutively activated and distributed diffusely throughout the processes, which results in aberrant actin organization and an inhibition of synapse formation. In neurons expressing RacN17, the level of active Rac is insufficient to support synapse formation. Another explanation is that cycling of Rac between active and inactive forms is necessary for the formation and/or stabilization of synapses. Expression of either RacV12 or RacN17 blocks the cycling and thus the formation of synapses. The observation that both the constitutively active and dominant-negative mutants of Rac have similar effects has been made in other systems as well. In Drosophila, for example, expression of either mutant results in defects in axonal growth (Luo et al., 1997; Song and Poo, 1999).
Several genes that are involved in the regulation of Rho family signaling are mutated in patients with nonsyndromic MR (Allen et al., 1998; Billuart et al., 1998; Kutsche et al., 2000; Barnes and Milgram, 2002; Ramakers, 2002). However, the mechanisms by which these mutations lead to cognitive defects are not understood. One likely possibility is a decreased neuronal connectivity that results from aberrant actin organization (Marin-Padilla, 1972; Huttenlocher, 1974; Purpura, 1974; Kaufmann and Moser, 2000). Indeed, some children with nonsyndromic MR show abnormalities in dendritic spine morphology in their cerebral cortex, i.e., numerous very long and thin spines and a reduction in the number of stubby and mushroom-shaped spines (Purpura, 1974). In our study, the GIT1, PIX, and Rac mutants that produce mislocalized Rac activity recapitulate this phenotype in cultured neurons. These mutants also cause a decrease in synaptic density. Thus, our results suggest a potential mechanism by which aberrant Rho family signaling can lead to decreased neuronal connectivity and eventually impaired cognitive functions. Interestingly, one of the MR mutants has a large deletion in a PIX isoform, which includes the GIT1 binding domain (Koh et al., 2001) that could result in mislocalized PIX (Kutsche et al., 2000).
Our studies point to several important immediate avenues of investigation. For example, GIT1 is targeted to synapses by a novel site, but the molecule that targets GIT1 to synapses is unknown (Fig. 8). Since a poorly characterized G protein–coupled receptor kinase binding domain resides in the vicinity of the SLD (Premont et al., 2000), G protein–coupled receptors and G protein–coupled receptor kinases may contribute to this process (Pitcher et al., 1998). Further investigation into events downstream of GIT1 is also an important challenge. For example, it would be very interesting to visualize changes in Rac activity during synapse formation and synaptic activity. In addition, since PAK3, a downstream effector of Rac, is mutated in some patients with MR (Allen et al., 1998), it is important to determine the role of PAK3 in this process. Finally, it is possible that other domains of GIT1 besides the SLD also play a role in synaptic activity by interacting with proteins from other pathways. Since GIT1 also localizes to the presynaptic terminals, it is tempting to speculate that it serves additional presynaptic functions.
Materials and methods
Antibodies
Primary antibodies used in this study include GIT1 mAb (1:100; Transduction Labs), GIT1 polyclonal antibody (1:100), which does not cross react with PKL/GIT2 (Manabe et al., 2002), βPIX mAb (1:250; Transduction Labs), SV2 mAb (1:100; University of Iowa hybridoma bank), PSD-95 mAb (6G6, 1:200; Affinity Bioreagents), GAD-6 mAb (1:500; University of Iowa hybridoma bank), synapsin1 polyclonal antibody (1:2,000; Chemicon), HA mAb (12CA5, 1:200; Boehringer), and FLAG mAb (1:400; Stratagene). The polyclonal βPIX antibody was provided by Bo Xiao (The Johns Hopkins University, Baltimore, MD). For immunocytochemistry, the secondary antibodies used (all from ICN) include rhodamine-conjugated anti–mouse IgG antibody (1:500), rhodamine-conjugated anti–rabbit IgG antibody (1:200), FITC-conjugated anti–rabbit IgG antibody (1:500), FITC-conjugated anti–mouse IgG antibody, and 7-amino-4-methyl-coumarin-3 acetic acid ester (AMCA)–conjugated anti–rabbit IgG antibody (1:50). Rhodamine-conjugated phalloidin was obtained from Molecular Probes. For immunoblot analysis, the secondary antibody used was HRP-conjugated anti–mouse IgG antibody from Amersham Biosciences.
Plasmids
Full-length human GIT1 cDNA, which includes the 9 aa insertion (nt 774–800) found in rat GIT1, GIT1-FLAG, GFP-tagged full-length GIT1, GIT1ΔC, cGIT1, and GIT1ΔCD were cloned as described (Manabe et al., 2002). GFP–CD-GIT1 was obtained by subcloning a HindIII (nt 369–1802) fragment into the pEGFP-C3 vector (CLONTECH Laboratories, Inc.). The following GFP-tagged deletion constructs of GIT1 were obtained by subcloning the corresponding GIT1 fragment into the pEGFP-C1 vector (CLONTECH Laboratories, Inc.): CDΔAnk (BglII [nt 774]-HindIII [nt 1802]); SLDΔ32 (FspI [nt 1233]-HindIII [nt 1802]); and nGIT1 ([nt 1]-FspI[nt 1233]). GFP-SLD was obtained by subcloning a PCR fragment (nt 1137-nt 1802) into pEGFP-C1. GFP-GIT1ΔSLD was obtained by subcloning two PCR fragments, nt 1-nt 1163 and nt 1803-nt 2324, into pEGFP-C1. N-SLD and C-SLD were prepared by PCR amplification of nt 1137-nt 1442 and nt 1443-nt 1802, respectively. The PCR products were then subcloned into pcDNA3 vector with a built-in NH2-terminal FLAG tag. GFP-GIT1ΔSHD was obtained by subcloning the appropriate GIT1 fragments (nt1-nt774 and nt1137-nt2324) into pEGFP-C1. HA-tagged mouse βPix was a gift from Chris Turner (SUNY Upstate Medical University, Syracuse, NY). HA-PIX-LL was provided by Lorraine Santy and Jim Casanova (University of Virginia). The L238R, L239S mutations of HA-PIX-LL were introduced by the Quickchange kit (Stratagene) using HA-mouse βPix as the template. HA-PIXΔGBD was generated by subcloning two PCR fragments of βPix (nt1-nt1474 and nt1763-nt2038) into the pEBB-HA vector. Myc-tagged V12-Rac1 and N17-Rac1 were provided by Alan Hall (University College London, London, UK).
Cell culture and transfection
Hippocampal low density cultures were prepared as described previously (Goslin et al., 1998). Neurons were plated at an approximate density of 70 cells/mm2 and were transfected using a modified calcium phosphate precipitation method (Kohrmann et al., 1999). Briefly, for transfection of a 6-cm dish, 6 μg of plasmid DNA was mixed with 120 μl of 250 mM CaCl2 in a polypropylene tube. 120 μl of 2× HBS (274 mM NaCl, 9.5 mM KCl, 15 mM glucose, 42 mM Hepes, 1.4 mM Na2HPO4, pH 7.10–7.15) was then added dropwise to the mixture with aeration. This mixture was added immediately to a 6-cm dish of the neurons with 4 ml of 24-h glia-conditioned medium. When complex formation was observed (typically 30–60 min), the cells were washed twice with HBS (135 mM NaCl, 4 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 10 mM glucose, 20 mM Hepes, pH 7.35), and then glia-conditioned medium with 0.5 mM kynurenic acid was added. Using this method, the transfection efficiency for the hippocampal neurons ranged from 10 to 30%.
Western blot analysis
For Western blot analysis, hippocampal neurons were plated at a density of 280 cells/mm2. At day 10 in culture, neurons were harvested, subjected to SDS-PAGE on a 10% slab, transferred to PVDF, and probed with the appropriate antibodies.
Immunocytochemistry and image analysis
Neurons were fixed in PBS containing 4% PFA with 4% sucrose and permeabilized with 0.2% Triton X-100. Alternatively, they were simultaneously fixed and permeabilized in cold methanol. After blocking with 20% goat serum/PBS, neurons were incubated with the appropriate antibodies in 5% goat serum/PBS. Images were acquired using a cooled CCD camera (Hamamatsu OrcaII) attached to a Nikon TE-300 inverted microscope with a 60× objective (NA1.4; Nikon). To estimate expression levels of the GIT1 constructs, GFP-GIT1–expressing neurons were coimmunostained for total GIT1. The fluorescent intensity levels of GIT1 staining in GFP-GIT1–expressing neurons and untransfected neurons were measured and compared. For low expression the difference is less than 2× and for high expression it is approximately 5×. Neurons expressing high levels of the constructs were chosen for quantification as follows. 85–110 dendrites from independent transfections were randomly selected for each construct. The number of synapses per unit length or per unit area was quantified using NIH Image. We quantified the number of spines and dendritic protrusions by examining 80–100 separate dendrites from independent transfections. Spines are defined as stubby or mushroom-shaped protrusions with associated synapses. Dendritic protrusions are defined as protrusions on the dendrites without associated synapses.
Acknowledgments
We thank Lorraine Santy, Jim Casanova, Chris Turner, Bo Xiao, and Alan Hall for generously providing reagents.
This work was supported by National Institutes of Health grant GM23244. D.J. Webb was supported by National Institutes of Health postdoctoral training grant HD07528-01.
Footnotes
Abbreviations used in this paper: ARF, ADP-ribosylation factor; GAP, GTPase-activating protein; GEF, guanine nucleotide exchange factor; GIT, G protein–coupled receptor kinase–interacting protein; MR, mental retardation; PAK, p21-activated kinase; SHD, Spa2 homology domain; SLD, synaptic localization domain.
References
- Allen, K.M., J.G. Gleeson, S. Bagrodia, M.W. Partington, J.C. MacMillan, R.A. Cerione, J.C. Mulley, and C.A. Walsh. 1998. PAK3 mutation in nonsyndromic X-linked mental retardation. Nat. Genet. 20:25–30. [DOI] [PubMed] [Google Scholar]
- Barnes, A.P., and S.L. Milgram. 2002. Signals from the X: signal transduction and X-linked mental retardation. Int. J. Dev. Neurosci. 20:397–406. [DOI] [PubMed] [Google Scholar]
- Billuart, P., T. Bienvenu, N. Ronce, V. des Portes, M.C. Vinet, R. Zemni, A. Carrie, C. Beldjord, A. Kahn, C. Moraine, and J. Chelly. 1998. Oligophrenin 1 encodes a rho-GAP protein involved in X-linked mental retardation. Pathol. Biol. (Paris). 46:678. [PubMed] [Google Scholar]
- Claing, A., S.J. Perry, M. Achiriloaie, J.K. Walker, J.P. Albanesi, R.J. Lefkowitz, and R.T. Premont. 2000. Multiple endocytic pathways of G protein-coupled receptors delineated by GIT1 sensitivity. Proc. Natl. Acad. Sci. USA. 97:1119–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig, A.M., and H. Boudin. 2001. Molecular heterogeneity of central synapses: afferent and target regulation. Nat. Neurosci. 4:569–578. [DOI] [PubMed] [Google Scholar]
- de Curtis, I. 2001. Cell migration: GAPs between membrane traffic and the cytoskeleton. EMBO Rep. 2:277–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Cesare, A., S. Paris, C. Albertinazzi, S. Dariozzi, J. Andersen, M. Mann, R. Longhi, and I. de Curtis. 2000. p95-APP1 links membrane transport to Rac-mediated reorganization of actin. Nat. Cell Biol. 2:521–530. [DOI] [PubMed] [Google Scholar]
- Fischer, M., S. Kaech, D. Knutti, and A. Matus. 1998. Rapid actin-based plasticity in dendritic spines. Neuron. 20:847–854. [DOI] [PubMed] [Google Scholar]
- Goslin, K., H. Asmussen, and G. Banker. 1998. Rat hippocampal neurons in low-density culture. MIT Press, Cambridge, MA. 339–370.
- Hall, A. 1998. Rho GTPases and the actin cytoskeleton. Science. 279:509–514. [DOI] [PubMed] [Google Scholar]
- Huttenlocher, P.R. 1974. Dendritic development in neocortex of children with mental defect and infantile spasms. Neurology. 24:203–210. [DOI] [PubMed] [Google Scholar]
- Kaufmann, W.E., and H.W. Moser. 2000. Dendritic anomalies in disorders associated with mental retardation. Cereb. Cortex. 10:981–991. [DOI] [PubMed] [Google Scholar]
- Kim, S., J. Ko, H. Shin, J.R. Lee, C. Lim, J.H. Han, W.D. Altrock, C.C. Garner, E.D. Gundelfinger, R.T. Premont, et al. 2003. The GIT family of proteins forms multimers and associates with the presynaptic cytomatrix protein Piccolo. J. Biol. Chem. 278:6291–6300. [DOI] [PubMed] [Google Scholar]
- Koh, C.G., E. Manser, Z.S. Zhao, C.P. Ng, and L. Lim. 2001. Beta1PIX, the PAK-interacting exchange factor, requires localization via a coiled-coil region to promote microvillus-like structures and membrane ruffles. J. Cell Sci. 114:4239–4251. [DOI] [PubMed] [Google Scholar]
- Kohrmann, M., W. Haubensak, I. Hemraj, C. Kaether, V.J. Lessmann, and M.A. Kiebler. 1999. Fast, convenient, and effective method to transiently transfect primary hippocampal neurons. J. Neurosci. Res. 58:831–835. [PubMed] [Google Scholar]
- Kraynov, V.S., C. Chamberlain, G.M. Bokoch, M.A. Schwartz, S. Slabaugh, and K.M. Hahn. 2000. Localized Rac activation dynamics visualized in living cells. Science. 290:333–337. [DOI] [PubMed] [Google Scholar]
- Kutsche, K., H. Yntema, A. Brandt, I. Jantke, H.G. Nothwang, U. Orth, M.G. Boavida, D. David, J. Chelly, J.P. Fryns, et al. 2000. Mutations in ARHGEF6, encoding a guanine nucleotide exchange factor for Rho GTPases, in patients with X-linked mental retardation. Nat. Genet. 26:247–250. [DOI] [PubMed] [Google Scholar]
- Luo, L., T.K. Hensch, L. Ackerman, S. Barbel, L.Y. Jan, and Y.N. Jan. 1996. Differential effects of the Rac GTPase on Purkinje cell axons and dendritic trunks and spines. Nature. 379:837–840. [DOI] [PubMed] [Google Scholar]
- Luo, L., L.Y. Jan, and Y.N. Jan. 1997. Rho family GTP-binding proteins in growth cone signalling. Curr. Opin. Neurobiol. 7:81–86. [DOI] [PubMed] [Google Scholar]
- Manabe, R., M. Kovalenko, D. Webb, and A. Horwitz. 2002. GIT1 functions in a motile, multi-molecular signaling complex that regulates protrusive activity and cell migration. J. Cell Sci. 115:1497–1510. [DOI] [PubMed] [Google Scholar]
- Manser, E., T.H. Loo, C.G. Koh, Z.S. Zhao, X.Q. Chen, L. Tan, I. Tan, T. Leung, and L. Lim. 1998. PAK kinases are directly coupled to the PIX family of nucleotide exchange factors. Mol. Cell. 1:183–192. [DOI] [PubMed] [Google Scholar]
- Marin-Padilla, M. 1972. Structural abnormalities of the cerebral cortex in human chromosomal aberrations: a Golgi study. Brain Res. 44:625–629. [DOI] [PubMed] [Google Scholar]
- Matus, A. 2000. Actin-based plasticity in dendritic spines. Science. 290:754–758. [DOI] [PubMed] [Google Scholar]
- Nakayama, A.Y., and L. Luo. 2000. Intracellular signaling pathways that regulate dendritic spine morphogenesis. Hippocampus. 10:582–586. [DOI] [PubMed] [Google Scholar]
- Nakayama, A.Y., M.B. Harms, and L. Luo. 2000. Small GTPases Rac and Rho in the maintenance of dendritic spines and branches in hippocampal pyramidal neurons. J. Neurosci. 20:5329–5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parnas, D., A.P. Haghighi, R.D. Fetter, S.W. Kim, and C.S. Goodman. 2001. Regulation of postsynaptic structure and protein localization by the Rho-type guanine nucleotide exchange factor dPix. Neuron. 32:415–424. [DOI] [PubMed] [Google Scholar]
- Pitcher, J.A., N.J. Freedman, and R.J. Lefkowitz. 1998. G protein-coupled receptor kinases. Annu. Rev. Biochem. 67:653–692. [DOI] [PubMed] [Google Scholar]
- Premont, R.T., A. Claing, N. Vitale, J.L. Freeman, J.A. Pitcher, W.A. Patton, J. Moss, M. Vaughan, and R.J. Lefkowitz. 1998. beta2-Adrenergic receptor regulation by GIT1, a G protein-coupled receptor kinase-associated ADP ribosylation factor GTPase-activating protein. Proc. Natl. Acad. Sci. USA. 95:14082–14087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Premont, R.T., A. Claing, N. Vitale, S.J. Perry, and R.J. Lefkowitz. 2000. The GIT family of ADP-ribosylation factor GTPase-activating proteins. Functional diversity of GIT2 through alternative splicing. J. Biol. Chem. 275:22373–22380. [DOI] [PubMed] [Google Scholar]
- Purpura, D.P. 1974. Dendritic spine “dysgenesis” and mental retardation. Science. 186:1126–1128. [DOI] [PubMed] [Google Scholar]
- Ramakers, G.J. 2002. Rho proteins, mental retardation and the cellular basis of cognition. Trends Neurosci. 25:191–199. [DOI] [PubMed] [Google Scholar]
- Rao, A., and A.M. Craig. 2000. Signaling between the actin cytoskeleton and the postsynaptic density of dendritic spines. Hippocampus. 10:527–541. [DOI] [PubMed] [Google Scholar]
- Ridley, A.J. 2001. Rho GTPases and cell migration. J. Cell Sci. 114:2713–2722. [DOI] [PubMed] [Google Scholar]
- Sheng, M., and M.J. Kim. 2002. Postsynaptic signaling and plasticity mechanisms. Science. 298:776–780. [DOI] [PubMed] [Google Scholar]
- Song, H.J., and M.M. Poo. 1999. Signal transduction underlying growth cone guidance by diffusible factors. Curr. Opin. Neurobiol. 9:355–363. [DOI] [PubMed] [Google Scholar]
- West, K.A., H. Zhang, M.C. Brown, S.N. Nikolopoulos, M.C. Riedy, A.F. Horwitz, and C.E. Turner. 2001. The LD4 motif of paxillin regulates cell spreading and motility through an interaction with paxillin kinase linker (PKL). J. Cell Biol. 154:161–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, Z.S., E. Manser, T.H. Loo, and L. Lim. 2000. Coupling of PAK-interacting exchange factor PIX to GIT1 promotes focal complex disassembly. Mol. Cell. Biol. 20:6354–6363. [DOI] [PMC free article] [PubMed] [Google Scholar]