

Abstract
The fibronectin (FN)-binding integrins α4β1 and α5β1 confer different cell adhesive properties, particularly with respect to focal adhesion formation and migration. After analyses of α4+/α5+ A375-SM melanoma cell adhesion to fragments of FN that interact selectively with α4β1 and α5β1, we now report two differences in the signals transduced by each receptor that underpin their specific adhesive properties. First, α5β1 and α4β1 have a differential requirement for cell surface proteoglycan engagement for focal adhesion formation and migration; α5β1 requires a proteoglycan coreceptor (syndecan-4), and α4β1 does not. Second, adhesion via α5β1 caused an eightfold increase in protein kinase Cα (PKCα) activation, but only basal PKCα activity was observed after adhesion via α4β1. Pharmacological inhibition of PKCα and transient expression of dominant-negative PKCα, but not dominant-negative PKCδ or PKCζ constructs, suppressed focal adhesion formation and cell migration mediated by α5β1, but had no effect on α4β1. These findings demonstrate that different integrins can signal to induce focal adhesion formation and migration by different mechanisms, and they identify PKCα signaling as central to the functional differences between α4β1 and α5β1.
Keywords: fibronectin; syndecan; PKC; cytoskeleton; vinculin
Introduction
The adhesive interactions of cells with ECM molecules have wide-ranging effects on cellular differentiation and tissue structure in vivo (Bouvard et al., 2001; Hynes, 2002). At the molecular level, these adhesion-dependent signals are mediated by clustering of integrin adhesion receptors, organization of the actin cytoskeleton, and congregation of signaling adaptors and enzymes into specialized morphological structures, including focal adhesions, focal complexes, and fibrillar adhesions (Schoenwaelder and Burridge, 1999; Yamada and Geiger, 1997). The composition of different adhesive contacts is diverse, and therefore suggestive of a varied contribution of these receptors to ECM sensing, intracellular signaling, and cytoskeletal organization, but the key molecular events determining this diversity are unknown. In cultured cells, integrins differentially regulate morphology and cytoarchitecture, fluxes through signaling pathways, and the expression of specific genes (Huhtala et al., 1995; Burridge and Chrzanowska-Wodnicka, 1996; Choquet et al., 1997), but the mechanistically important variations in the signals transduced by different receptors are poorly defined.
Particularly distinctive cellular responses have been observed on substrates recognized by the fibronectin (FN)*-binding integrin α4β1. For example, α4β1 engagement promotes enhanced cell migratory activity while reducing spreading and focal adhesion formation. Elegant studies using integrin chimeras demonstrated that these functional properties were conferred by the α4 cytoplasmic domain (Chan et al., 1992; Kassner et al., 1995), which in turn suggested that this domain either modulates association of cytoskeletal and signaling molecules with its partner β1 subunit differently to other β1-associated α subunits, or that it interacts directly with cytoskeletal and signaling molecules. The latter hypothesis is supported by the fact that α4 can bind directly to paxillin, and that this association contributes to the reduction of spreading and promotion of migration (Liu et al., 1999, Liu and Ginsberg, 2000).
The signaling mechanisms that determine the specific functional properties of other integrins are largely unknown, although work from a number of laboratories has demonstrated that α5β1-dependent focal adhesion formation requires engagement of (and signaling via) a syndecan coreceptor (Woods and Couchman, 2001; Bass and Humphries, 2002). Initially, plating of cells onto the isolated central cell–binding domain (CCBD) and heparin-binding domain (HBD) of FN was shown to promote attachment, but to be insufficient for focal adhesion formation (Izzard et al., 1986; Woods et al., 1986); however, addition of soluble HBD to cells pre-spread on a CCBD fragment triggered vinculin recruitment and actin stress fiber formation (Woods et al., 1986). The site responsible for this activity was narrowed down to a 29-amino acid sequence within the type III repeat 13 of FN (Bloom et al., 1999). Of the four known members of the syndecan family of proteoglycans, only syndecan-4 has been found in focal adhesions (Woods and Couchman, 1994; Baciu and Goetinck, 1995). Treatment of cells with anti-syndecan-4 antibody was also found to trigger focal adhesion formation in cells adherent to the CCBD of FN (Saoncella et al., 1999), and fibroblasts from syndecan-4 knockout mice were unable to respond to the HBD of FN (Ishiguro et al., 2000), implicating syndecan-4 as a key receptor for focal adhesion formation on FN. In serum-starved cells, syndecan-4 was absent from focal adhesions, but was recruited by activation of PKC (Woods and Couchman, 1992; Baciu and Goetinck, 1995). Overexpression of syndecan-4 increased focal adhesions (Echtermeyer et al., 1999), whereas a COOH-terminal truncation mutant acted as a dominant-negative inhibitor of focal adhesion formation (Longley et al., 1999).
Although the role of syndecans in α4β1-mediated adhesion is unknown, it is intriguing that the binding sites for both molecules overlap within FN. The α4β1-binding domain of FN is primarily located in the type III connecting segment (IIICS; Wayner et al., 1989), which is adjacent to the major HBD, the COOH-terminal heparin binding domain of fibronectin (HepII). Three sites each for integrin and heparin binding have been pinpointed within the HepII/IIICS region (Humphries et al., 1986, 1987; McCarthy et al., 1988, 1990; Wayner et al., 1989; Barkalow and Schwarzbauer, 1991; Mould and Humphries, 1991; Mould et al., 1991; Mostafavi-Pour et al., 2001). The overlapping locations of these sites, in part revealed by x-ray crystallography (Sharma et al., 1999), suggest a close coordination between integrin and proteoglycan binding. Here, using melanoma cells that express both α4β1 and α5β1, we have investigated the mechanisms underlying focal adhesion formation and cell migration mediated by engagement of the HepII/IIICS region of FN, and compared this to the CCBD.
Results
Focal adhesion formation mediated by immobilized cell-binding domains of FN
To compare the role of syndecans in focal adhesion formation mediated by the integrins α4β1 and α5β1, A375-SM melanoma cells were plated on defined fragments of FN that interact specifically with each receptor (Fig. 1) . A375-SM cells have previously been shown to express α4β1 and α5β1 (Mould et al., 1990), and we have found by FACS analysis that they also express syndecan-4 (unpublished data). When cells were plated onto a recombinant 50K fragment spanning the CCBD of FN (recognized by α5β1), less than 15% of spread cells formed vinculin-containing focal adhesions (Table I a; Fig. 2 A). Although vinculin was rarely concentrated at sites of attachment, it was notable that these sites did contain α5 (unpublished data) and β1 integrins (Fig. 2 B), suggesting that integrin engagement and clustering had been dissociated from assembly of cytoskeletal structures. It is also notable that talin, α-actinin, focal adhesion kinase, and paxillin were absent from adhesion contacts (unpublished data). When cells were seeded on 50K and the medium was supplemented with a low concentration of soluble heparin-binding fragment of FN (5 μg/ml H/0), there was a significant increase in the percentage of spread cells (from 40–50% to 70–80%), and almost 90% of the spread cells now formed vinculin- and integrin-containing focal adhesions (Table I a; Fig. 2, D–F). The stimulatory effect of H/0 was abolished by co-incubation with heparin (Table I b; Fig. 2, G–I), and could be mimicked by addition of soluble anti-syndecan-4 mAb 5G9 (Fig. 2, J–L). These results confirm the synergistic link between integrin α5β1 and syndecan-4 for focal adhesion formation.
Figure 1.

Schematic diagram of FN and recombinant fragments used in these studies. FN comprises three types of polypeptide module termed types I, II, and III. The CCBD, which binds α5β1, comprises an RGD tripeptide site in III-10 and a synergy site in III-9. The alternatively spliced IIICS, which binds α4β1, contains the peptide sequences LDV (in the NH2-terminal splice segment, IIICS-A) and REDV (in IIICS-C). The main heparin-binding site fragment of FN (HepII) comprises III12–14. The main heparin-binding site in FN (GAG-A) is located in III-13, and additional heparin-binding sites are found in IIICS-B (GAG-B) and III-14 (GAG-C). The three FN fragments used in these studies are 50K (III6–10), H/120 (III12–15, including a full-length 120-amino acid IIICS region), and H/0 (III12–15, without the IIICS).
Table I. Focal adhesion formation on FN fragments.
| aSubstrate | % Spreading without H/0 | % Spreading with H/0 | % Cells with focal adhesions (without H/0) | % Cells with focal adhesions (with H/0) |
|---|---|---|---|---|
| H/120 | 47 ± 5 | 45 ± 5 | 46 ± 3 | 47 ± 5 |
| H/120-GAG-ABC | 48 ± 3 | 50 ± 5 | 52 ± 6 | 56 ± 3 |
| 50K | 52 ± 2 | 75 ± 3 | 14 ± 1 | 86 ± 1 |
| bSubstrate | % Spreading with H/0 | % Spreading with H/0 and heparin | % Cells with focal adhesions (with H/0) | % Cells with focal adhesions (with H/0 and heparin) |
| H/120 | 42 ± 3 | 44 ± 5 | 43 ± 2 | 42 ± 3 |
| H/120-GAG-ABC | 46 ± 4 | 48 ± 3 | 51 ± 4 | 50 ± 2 |
| 50K | 74 ± 2 | 51 ± 4 | 89 ± 1 | 13 ± 2 |
A375-SM cells were seeded onto coverslips coated with 10 μg/ml H/120, H/120-GAG-ABC, or 50K, and the percentage of cells spreading and forming focal adhesions in the presence or absence of 5 μg/ml soluble H/0 was estimated by phase-contrast microscopy and immunocytochemistry, respectively.
The effects of co-incubation of 100 μg/ml heparin and 5 μg/ml soluble H/0 were estimated.
Figure 2.
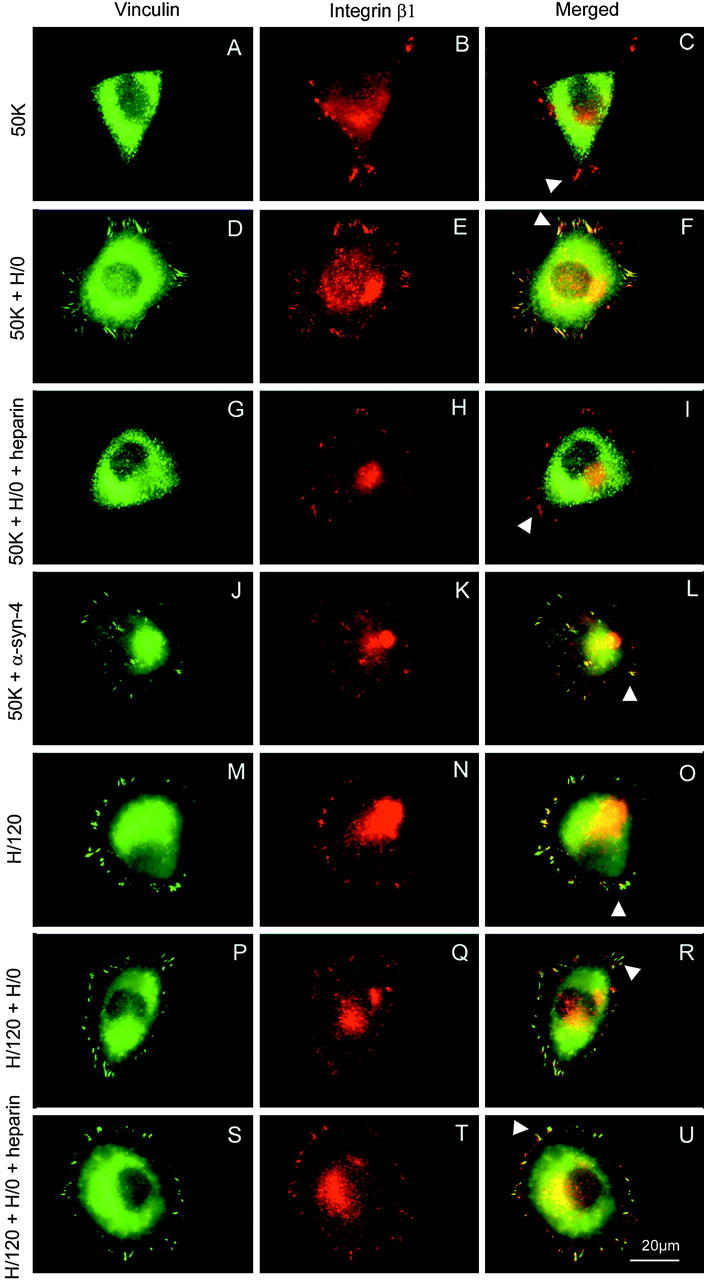
Double immunofluorescence staining for vinculin and integrin β1 in A375-SM cells spread on different adhesive substrates. Cells were incubated for 2 h on 10 μg/ml of each substrate, with or without a further 30 min incubation with 5 μg/ml H/0 or 1:50 anti-syndecan-4. Heparin was added at 100 μg/ml simultaneously with H/0. Cells were fixed, permeabilized, and stained for vinculin (green) and integrin β1 (red). (A–C) Cells spread on 50K alone. The arrowhead in the merged image (C) indicates integrin+/vinculin− clusters. (D–F) Cells prespread on 50K for 2 h followed by addition of H/0. The arrowhead in F indicates colocalization of vinculin and integrin β1. (G–I) Cells prespread on 50K followed by treatment with H/0 in the presence of heparin. The arrowhead in I again indicates integrin+/vinculin− clusters. (J–L) Cells prespread on 50K followed by addition of anti-syndecan-4 antibody. The arrowhead in L indicates colocalization of vinculin and integrin β1. Cells spread on either H/120 (M–O) or H/120 treated with H/0 (P–R) exhibit colocalization of vinculin and integrin β1 (arrowheads in O and R). Addition of heparin and H/0 to cells prespread on H/120 (S–U) had no discernible effect. Bar, 20 μm.
When A375-SM cells were seeded onto the recombinant H/120 fragment of FN, which includes both the heparin/syndecan-binding sites and α4β1 integrin-binding sites in the HepII and IIICS regions, respectively, almost 50% of the spread cells formed focal adhesions (Table I a; Fig. 2, M–O), and exogenous addition of soluble H/0 had no additional stimulatory effect (Table I a; Fig. 2, P–R). Although this result was anticipated, as H/120 includes both integrin- and syndecan-binding sites, it was surprising that addition of 100 μg/ml heparin did not affect focal adhesion formation on H/120 (Table I b; Fig. 2, S–U).
Glycosaminoglycan–binding sites within the HepII/IIICS region are not needed for focal adhesion formation
As one approach to determine whether integrin α4β1 required a syndecan coreceptor to trigger focal adhesion formation, site-directed mutagenesis was performed on H/120 to remove all of its potential GAG-binding sites (found in type III repeats 13 and 14, and the IIICS-B region; Fig. 1). All arginine and lysine residues in these regions were mutated to serine, and the mutant protein (termed H/120-GAG-ABC) was prepared as a GST fusion protein in bacteria. After purification, the protein was analyzed by SDS-PAGE under reducing conditions.
To assess folding of H/120-GAG-ABC, an ELISA assay was performed in which the binding of a panel of HepII/IIICS mAbs was determined. Some of these mAbs, including 9E9 and 16E6, do not Western blot, and therefore recognize conformational epitopes within FN. As shown in Fig. 3 A, all mAbs recognized H/120-GAG-ABC to a similar extent to native H/120 with the exception of 9E9, where binding was reduced by 50%. It is possible that one or more arginine or lysine residues contribute to the epitope for this mAb. The binding of biotinylated heparin to the mutant protein was then investigated. The level of binding to H/120-GAG-ABC was reduced by >98% compared with that of native H/120, and was not significantly different from the background, indicating that all key heparin-binding sites had been removed (unpublished data). Recombinant native and mutated proteins were then tested for their ability to support A375-SM melanoma cell adhesion. The proteins supported attachment in a dose-dependent manner (Fig. 3 B). Attachment to native H/120 variant was maximal at a level of 80%, and a coating concentration of ∼0.7 μg/ml was required for half-maximal attachment. H/120-GAG-ABC showed only slightly lower activity, with a maximal level of cell attachment of ∼65% and a coating concentration of 1.6 μg/ml being required for half-maximal attachment. Finally, H/120-GAG-ABC was unable to trigger vinculin recruitment to focal adhesions when A375-SM cells were prespread on 50K (unpublished data).
Figure 3.
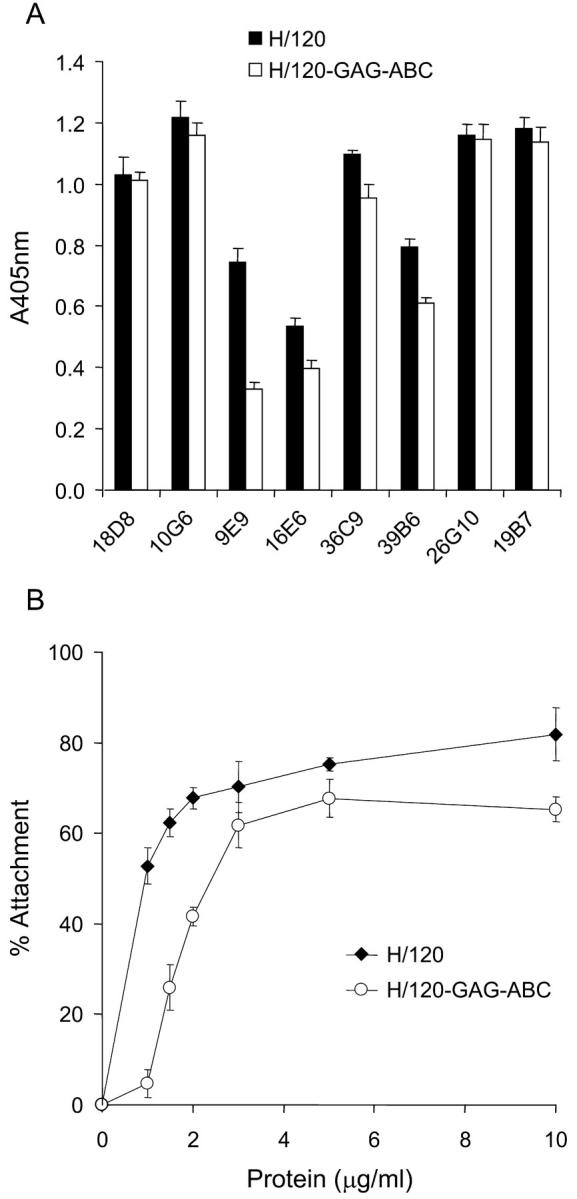
Characterization of recombinant H/120-GAG-ABC. (A) ELISA assay showing the binding of HepII (9E9, 16E6, 36C9, 39B6, 26G10, and 19B7), IIICS-B (18D8) and IIICS-C (10G6) mAbs to immobilized H/120 (solid bars) and its heparin-binding deficient mutant H/120-GAG-ABC (open bars), each coated at 10 μg/ml. (B) Attachment of A375-SM melanoma cells to native H/120 (closed diamonds) and H/120-GAG-ABC (open circles). The level of nonspecific binding, determined from A375-SM melanoma cell attachment to wells coated with BSA alone, was subtracted. Values shown in both panels are mean ± SD of triplicate wells.
The ability of H/120-GAG-ABC to support focal adhesion formation and microfilament polymerization was then assessed by double immunofluorescence microscopy using anti-vinculin antibody and rhodamine-conjugated phalloidin. As shown in Fig. 4 and quantitated in Table I, cells seeded on either H/120 or H/120-GAG-ABC were of a similar shape, and both the number and location of focal adhesions was indistinguishable. Identical results were obtained when A375-SM cells were plated onto either CS1 peptide (as an IgG conjugate) or recombinant VCAM-1-Fc (unpublished data; CS1 is a synthetic peptide containing the major α4β1 binding site within H/120, and VCAM-1 is an alternative α4β1 ligand; neither protein binds heparin). Together, these results indicate that focal adhesion formation mediated by α4β1 does not require simultaneous engagement of a syndecan coreceptor.
Figure 4.

Focal adhesion and actin stress fiber formation in A375-SM melanoma cells seeded on H/120 or H/120-GAG-ABC. Cells were incubated for 2 h on H/120 (A–C) or H/120-GAG-ABC (D–F), fixed and dual-stained for vinculin (A and D) and actin (B and E). Arrows in the merged images (C and F) indicate localization of vinculin at the ends of actin bundles. Bar, 20 μm.
Role of PKCα activation in focal adhesion formation via α4β1 or α5β1
To test the involvement of PKCα in focal adhesion formation, the activation of the enzyme was measured using Western blotting. A375-SM cells adherent to either H/120, H/120-GAG-ABC, 50K, or 50K + soluble H/0 were detergent-extracted, and lysates were blotted with either a pan-anti-PKCα antibody (MC5) or an antiserum that detects activated PKCα (PPA182). Levels of PKCα were quantitated by densitometry. The level of expression of PKCα was the same for all adhesive substrata, but levels of phosphorylation differed (Fig. 5) . Thus, addition of H/0 to cells prespread on 50K potentiated the level of phosphorylation of PKCα eightfold. However, H/120 and H/120-GAG-ABC induced similar levels of PKCα phosphorylation that were indistinguishable from the basal level observed on 50K alone (Fig. 5).
Figure 5.
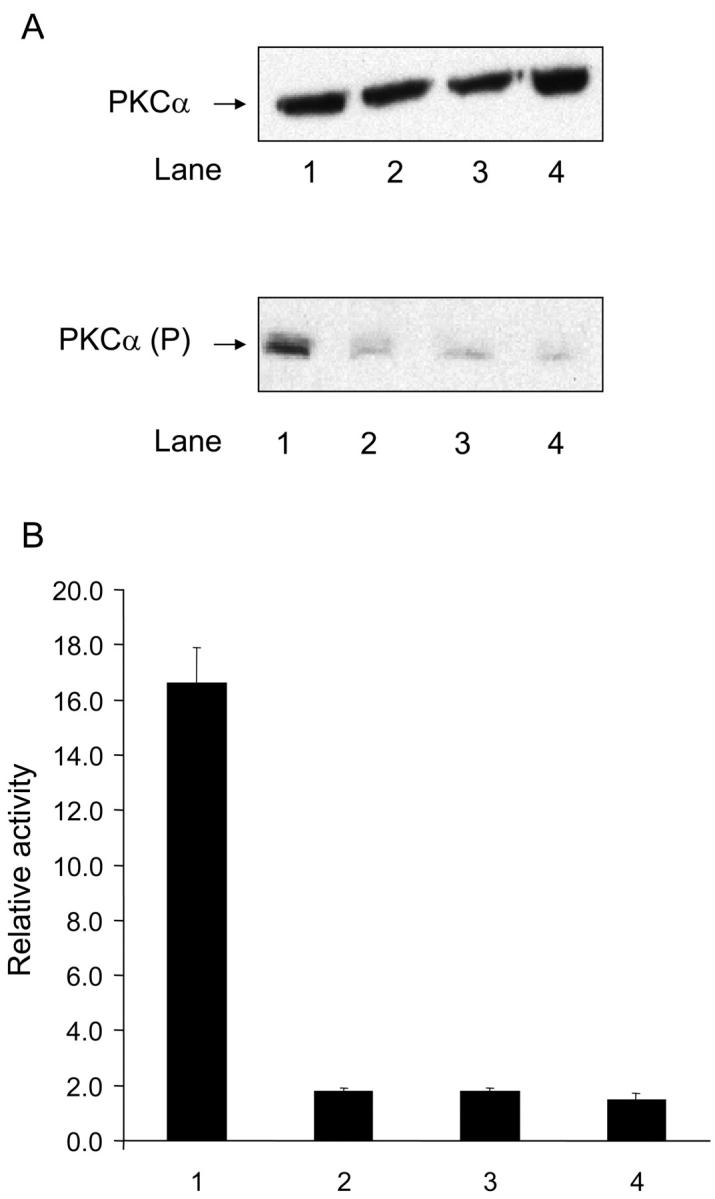
PKCα activation in cells adhering via α5β1 or α4β1. (A) Western blot analysis of total PKCα levels (top; detected with MC5 antibody) or activated PKCα (bottom; detected with PPA182 antibody) in A375-SM cells seeded on 50K+H/0 (lane 1), 50K (lane 2), H/120 (lane 3), and H/120-GAG-ABC (lane 4). (B) Densitometric analysis of A. Results are the mean ± SE of three separate experiments, of which A is representative.
Consistent with these results, pharmacological inhibition of PKCα with bisindolylmaleimide (BIM) blocked vinculin recruitment to focal adhesions induced by H/0 addition to cells seeded on 50K (Fig. 6 A, panels A, D, and G). By contrast, little change was observed in integrin β1 distribution (Fig. 6 A, panels B, E, and H). Vinculin and β1 integrin recruitment induced by H/120 were not substantially affected by BIM (Fig. 6 B; quantitated in Table II), although focal adhesions were not as well organized.
Figure 6.
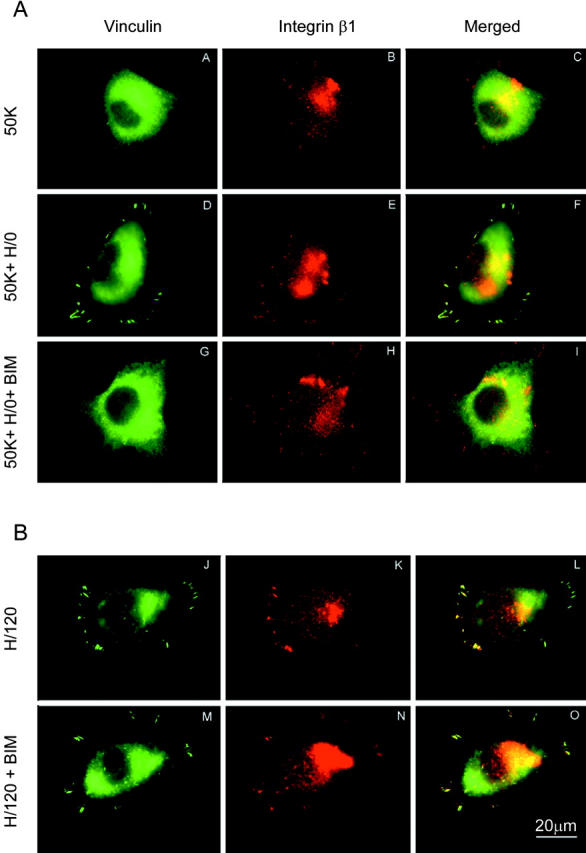
Effects of PKCα inhibition on vinculin and integrin β1 distribution. (A) A375-SM cells were seeded onto 50K (A–C), 50K+H/0 (D–F), or 50K+H/0 in the presence of 10 μg/ml BIM (G–I), and dual-stained for vinculin (A,D, and G) and integrin β1 (B,E, and H). Untreated cells received DMSO as a control. (B) The same experiment was performed for cells seeded onto H/120 without (J–L) or with (M–O) BIM treatment. Bar, 20 μm.
Table II.
Focal adhesion formation in the presence or absence of PKCα inhibitor
| Substrate | % Spreading control | % Spreading (BIM) | % Cells with focal adhesions (control) | % Cells with focal adhesions (BIM) |
|---|---|---|---|---|
| H/120 | 44 ± 5 | 45 ± 4 | 44 ± 4 | 42 ± 2 |
| H/120-GAG-ABC | 48 ± 6 | 51 ± 4 | 56 ± 2 | 52 ± 5 |
| 50K | 54 ± 3 | 51 ± 3 | 15 ± 3 | 12 ± 2 |
| 50K + soluble H/0 | 76 ± 4 | 52 ± 6 | 89 ± 5 | 24 ± 3 |
A375-SM cells were seeded onto coverslips coated with 10 mg/ml native H/120, H/120-GAG-ABC, or 50K (with or without addition of 5 mg/ml soluble H/0), and were co-incubated with 10 mg/ml bisindolylmaleimide (BIM) or DMSO as control. Cell spreading and focal adhesion formation were then estimated by phase-contrast microscopy and immunocytochemistry, respectively.
Role of PKC signaling in A375-SM cell migration mediated by α4β1 and α5β1
To test whether the signaling differences underlying focal adhesion formation mediated by α4β1 and α5β1 were also observed for cell migration, A375-SM cells were plated on recombinant FN fragments, left to form a confluent monolayer overnight, and then scratch-wounded. Under the conditions of this assay, there is a lag phase of ∼6 h during which the cells recover and re-spread, followed by a progressive closure of the wound over the next 24–30 h. Cells failed to migrate when adherent to 50K alone, but treatment with soluble H/0 at the time of wounding stimulated α5β1-dependent migration. When cells were seeded on either H/120 or H/120-GAG-ABC, equivalent rates of α4β1-dependent migration were observed (unpublished data). These findings indicate that α5β1 requires syndecan engagement in order to promote migration, but α4β1 does not.
To examine whether the differential dependence of α4β1 and α5β1 on PKC signaling was manifested during cell movement as well as during focal adhesion formation, the sensitivity of A375-SM cell migration to inhibitors of PKC was tested in a wounding assay. For these studies, the early phase of migration between 6 h and 12 h after wounding was examined. Initially, the effects of the broad spectrum PKC inhibitor, BIM, were tested. As shown in Fig. 7 (and Videos 1–4, available at http://www.jcb.org/cgi/content/full/jcb.200210176/DC1), addition of BIM 6 h after wounding prevented wound closure on a 50K + H/0 substrate, but had no discernible effect on H/120-mediated migration. A DMSO vehicle control showed no inhibition. Thus, the substrate-specific inhibition of focal adhesion formation by BIM was also seen for cell migration.
Figure 7.

Effects of PKCα inhibition on integrin-mediated migration. The migration of A375-SM cells into scratch wounds on either (A) a 50K substrate in the presence of soluble H/0 or (B) an H/120 substrate was examined without treatment (left panels) or in the presence of 10 μg/ml BIM (right panels). Still images were captured at the indicated times after wounding. The edges of the original wound are marked with dashed lines. Bars, 100 μm.
Although BIM exhibits good specificity for PKC, we also attempted to perturb A375-SM migration using a dominant-negative approach. Mutant forms of PKC isoforms, tagged with GFP, were transfected into A375-SM transiently, and then their ability to close wounds was examined. Initially, wild-type pEGFP-PKCα (WT) and the double mutant pEGFP-PKCα-A25E-K368M were tested. A25E is a pseudosubstrate mutation that exposes the DAG/TPA-binding site and catalytic site. K368M is a mutation in the ATP-binding site that leaves the enzyme catalytically inactive. Thus, pEGFP-PKCα-A25E-K368M is inert, but TPA-responsive. On a 50K substrate in the presence of soluble H/0, transfection of pEGFP-PKCα-WT, either with (Fig. 8 A and Video 5) or without (unpublished data) TPA treatment 6 h after wounding, had no discernible effect on cell migration into the wound. When A375-SM cells transfected with dominant-negative pEGFP-PKCα-A25E-K368M were examined in the absence of TPA, migration again occurred normally (Fig. 8 B and Video 6). However, addition of TPA to recruit pEGFP-PKCα-A25E-K368M to the plasma membrane led to an almost complete blockade of cell movement (Fig. 8 C and Video 7). As shown most clearly in the video, GFP-expressing cells remained virtually motionless, exhibiting only occasional membrane ruffling and protrusion. However, nontransfected cells that lacked detectable GFP migrated past them and filled the wound. As an additional control, cells were transfected with pEGFP-PKCα-T497A, a substrate-binding mutant that can also act as a dominant-negative inhibitor. This mutant also prevented A375-SM cell migration on a 50K + H/0 substrate (unpublished data). To probe the contribution of PKCα to cell migration mediated by α4β1, pEGFP-PKCα-A25E-K368M–transfected cells were seeded on H/120 and wounded. In contrast to the findings on 50K + H/0, dominant-negative PKCα had no effect on wound closure (Fig. 8 D and Video 8), again suggesting that α4β1 does not depend on this enzyme.
Figure 8.

Effects of wild-type or dominant-negative PKCα overexpression on integrin-mediated migration. A375-SM cells were transiently transfected with various pEGFP-PKCα constructs and then tested for their ability to migrate into wounds on either a 50K substrate in the presence of soluble H/0 or an H/120 substrate. Still images were captured at the indicated times after wounding, and separate phase, EGFP, and merged images are shown (from left to right). (A) pEGFP-PKCα-WT–transfected cells on 50K + H/0 in the presence of 5 ng/ml TPA; (B) pEGFP-PKCα-A25E-K368M–transfected cells on 50K + H/0 in the absence of TPA; (C) pEGFP-PKCα-A25E-K368M–transfected cells on 50K + H/0 in the presence of 5 ng/ml TPA; (D) pEGFP-PKCα-A25E-K368M–transfected cells on H/120 in the presence of 5 ng/ml TPA. Bars, 50 μm.
Because dominant-negative PKCα might also block the activity of other PKC isoforms by competition for substrates, we performed control experiments with dominant-negative mutants of PKCδ and PKCζ. As shown in Fig. 9 (A and B) and Videos 9 and 10, transfection with either dominant-negative pEGFP-PKCδ-K376M and pEGFP-PKCζ-K281M, with or without TPA treatment, did not reduce A375-SM migration on 50K + H/0. This suggests that the dominant-negative effects of pEGFP-PKCα-A25E-K368M are specific to the α isoform. Interestingly, transfection of wild-type pEGFP-PKCδ, and treatment with TPA, led to a suppression of migration (Fig. 9 C and Video 11). This result is consistent with the previously reported syndecan-4–dependent inhibitory effect of PKCδ on PKCα (Murakami et al., 2002). Together, these results demonstrate that migration mediated by α4β1 and α5β1 exhibits the same differential dependency on PKCα activity as focal adhesion formation.
Figure 9.
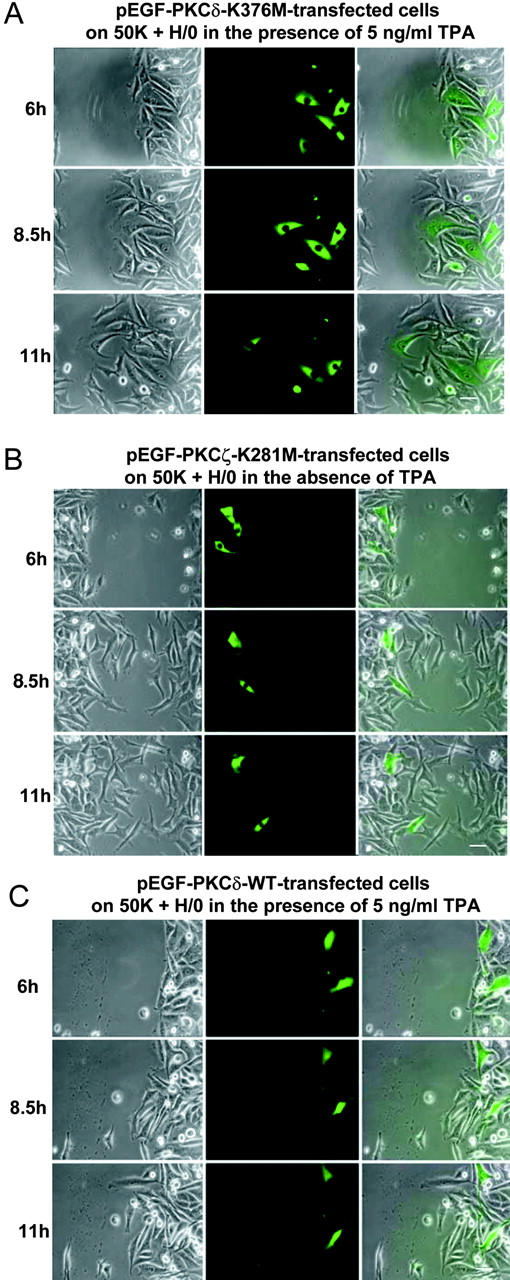
Isoform specificity of the effects of dominant-negative PKCα on α5β1-dependent migration. A375-SM cells were transiently transfected with various pEGFP-PKC constructs and then tested for their ability to migrate into wounds on a 50K substrate in the presence of soluble H/0. Still images were captured at the indicated times after wounding, and separate phase, EGFP, and merged images are shown (from left to right). (A) pEGFP-PKCδ-K376M–transfected cells in the presence of 5 ng/ml TPA; (B) pEGFP-PKCζ-K281M–transfected cells in the absence of TPA; (C) pEGFP-PKCδ-WT in the presence of 5 ng/ml TPA. Bars, 50 μm.
Discussion
In this report, we have compared the mechanisms of focal adhesion formation and cell migration used by A375-SM melanoma cells when adhering to FN via the integrins α4β1 and α5β1. Melanoma cells were selected for these works because they express both integrins, as well as heparan and chondroitin sulfate-containing proteoglycans (Iida et al., 1998). Our major novel findings are as follows: (1) α5β1 and α4β1 have a differential requirement for cell surface proteoglycan engagement for both focal adhesion formation and migration; α5β1 requires a proteoglycan coreceptor (syndecan-4), and α4β1 does not; (2) focal adhesion formation and migration via the two integrins involve activation of different signaling pathways; α5β1/syndecan-4 depends on the catalytic activity of conventional PKC isotypes and signals specifically to activate PKCα, and α4β1 occupancy does not; and (3) transfection of dominant-negative PKC isoforms identify PKCα as the key signaling enzyme responsible for the difference in regulation of α4β1 and α5β1 function. Together, these works demonstrate that the signals controlling integrin-mediated focal adhesion formation and cell translocation are not the same for each receptor, and they define PKCα activation as a key difference in the signaling pathways controlling the function of α4β1 and α5β1.
Initially, we were able to confirm previous reports that cells fail to form focal adhesions, or to migrate, on FN fragments spanning the CCBD unless a heparin-binding fragment is added in soluble form (Izzard et al., 1986; Woods et al., 1986). Using Western blotting to detect activated PKCα and pharmacological or dominant-negative inhibition, we have also confirmed a requirement for PKCα for focal adhesion formation via α5β1/syndecan-4. In contrast to these findings, α4β1-binding ligands stimulated focal adhesion formation without addition of a heparin-binding fragment, and such supplementation did not augment focal adhesion formation. Furthermore, PKCα was not activated, and neither soluble heparin, BIM, nor dominant-negative PKCα blocked focal adhesion formation. These findings indicate that engagement of syndecan-4 with the HBD of FN is not needed for focal adhesion formation via α4β1, and either that separate signaling mechanisms are used by the two integrins to recruit cytoskeletal proteins to focal adhesions, or α4β1 is able to bypass the requirement for syndecan-4–mediated signaling. Given the currently indistinguishable gross composition of α5β1- and α4β1-containing focal adhesions, we favor the latter explanation, and we hypothesize that some of the earliest events in integrin signaling differ between α4β1 and α5β1.
It is now well-established that α4β1 has a number of unusual properties (Chan et al., 1992; Kassner et al., 1995), and it is conceivable that the “gearing” of α4β1 to the cytoskeleton differs from other integrins. The recent finding that the cytoplasmic domain of α4 interacts directly and specifically with the signaling adaptor paxillin (Turner, 2000) may contribute to these different properties (Liu et al., 1999, Liu and Ginsberg, 2000; Han et al., 2001). PIX (Manser et al., 1998) is a nucleotide exchange factor for both Cdc42 and Rac1, and its recruitment by paxillin (through PKL) may potentially enhance membrane ruffling and protrusion through these Rho family GTPases. On the other hand, an integrin such as α5β1, which does not bind paxillin but directly activates PKCα, may stimulate Rho GTPase activities, in a PKC-dependent manner. This hypothesis is consistent with several lines of evidence that Rac1 may act downstream of PKC in the control of cell migration. In melanoma cells, for instance, TPA-induced lamellipodium formation is Rac1-dependent (Ballestrem et al., 2000). By using dominant inhibitory constructs, the activity states of several small GTPases, including Rac1, have been shown to influence TPA-induced disassembly/reassembly of actin stress fibers and focal adhesions in MDCK cells (Imamura et al., 1998). Also, in NIH3T3 cells, growth factor-induced Rac1 activation is sensitive to PKC inhibition, suggesting a hierarchical relationship between the two signaling proteins (Buchanan et al., 2000).
In an attempt to identify the PKC isoform responsible for α5β1-dependent migration, we used a dominant-negative overexpression approach. Overexpression of dominant-negative pEGFP-PKCα (A25E-K368M double mutant) abrogated α5β1-dependent migration in a TPA-dependent manner. Two additional dominant-negative constructs were tested in the same assay, pEGFP-PKCδ-K376M and pEGFP-PKCζ-K281M. As neither was able to perturb α5β1-dependent migration, this provides a clear indication that PKCα plays a specific role. Interestingly, overexpression of wild-type PKCδ did retard migration, a result that is consistent with a recent paper reporting a syndecan-4–dependent inhibitory effect of PKCδ on PKCα (Murakami et al., 2002). PKCδ was shown to phosphorylate the cytoplasmic domain of syndecan-4 and thereby prevent PKCα binding. Therefore, it seems likely that the synergy between integrin α5β1 and syndecan-4 for adhesion and migration relies on the same regulatory pathway.
The downstream targets of PKCα phosphorylation during α5β1-mediated adhesion and migration are not well defined; however, PKCα has been shown to bind directly to syndecan-4 in an interaction mediated by its catalytic domain (Oh et al., 1997a, b; Horowitz and Simons, 1998). In addition, phosphatidylinositol-4,5-bisphosphate (PIP2) appears to make a key contribution to syndecan function through its ability to bind to the variable region of the syndecan-4 cytoplasmic domain (Oh et al., 1998) and to stabilize a “twisted clamp” homodimer conformation discernible by nuclear magnetic resonance (Lee et al., 1998; Shin et al., 2001). This suggests that a ternary complex between syndecan-4, PIP2, and PKCα may form in cells. Integrin ligation has been shown to activate PI-5 kinase, the enzyme that catalyzes the production of PIP2 (McNamee et al., 1993), supporting the close functional link between syndecans and integrins. PIP2 has a wide variety of functions, including the conversion of several cytoskeletal proteins (ERM proteins, vinculin, and talin; Gilmore and Burridge, 1996; Hamada et al., 2000; Martel et al., 2001) from inactive to active forms, and it may be that PIP2 binding to syndecans is a necessary step for the recruitment of these molecules to α5β1-containing focal adhesions. In addition, PKCα has been shown to associate with the ERM protein ezrin (probably subsequent to its release from membrane receptors such as CD44; Legg et al., 2002), phosphorylates its COOH-terminal phosphorylation on the T567 site, and thereby facilitates its conformational activation (Ng et al., 2001; Bretscher et al., 2002; Gautreau et al., 2002). Therefore, α5β1 integrin-derived signals may stimulate ERM activities through both PIP2- and PKC-dependent mechanisms. By contrast, the direct binding of paxillin to α4β1 provides a direct link to vinculin, and therefore talin.
It is also conceivable that the functions of PKCα are required during trafficking of integrin receptors, as studies from our laboratories and others have previously demonstrated close intracellular associations in recycling compartments between integrin β1 and PKCα (Ng et al., 1999a; Podar et al., 2002) and β1 and PKCɛ (Ivaska et al., 2002) by biochemical and fluorescence resonance energy transfer analysis. These findings may be reconciled with the data in this report if either a stable integrin–PKCα interaction at the plasma membrane requires coreceptor function, or if the integrin–PKCα associations in the two compartments differ in their requirement for a coreceptor.
Finally, PKC–integrin interactions are likely to have important implications in growth factor–mediated cell migration. For instance, a constitutive PKCα–β1 integrin complex has been found in multiple myeloma cells and may play a significant role in the development of a VEGF-responsive migratory phenotype (Podar et al., 2002). Furthermore, perturbation of PKC–integrin interaction blocks carcinoma cell chemotaxis (Parsons et al., 2002). This may provide an important mechanism for explaining the well-documented cross-talk between growth factor receptor- and integrin-mediated processes (Wang et al., 1998).
Materials and methods
Antibodies
The mouse mAbs 9E9, 16E6, 39B6, 36C9, 26G10, 10G6, 19B7, and 18D8, which recognize the HepII/IIICS region of human FN, were raised in the Humphries laboratory (Mostafavi-Pour et al., 2001). Mouse mAb 150.9, raised against an NH2-terminal peptide from the human syndecan-4 core protein (Longley et al., 1999), was a gift of Anne Woods (University of Alabama, Birmingham, AL) and mouse monoclonal 5G9 was purchased from Santa Cruz Biotechnology, Inc. Rat anti–human integrin β1 mAb13 was a gift of Ken Yamada (National Institute of Dental and Craniofacial Research, Bethesda, MD). Mouse anti–human integrin α4 mAb HP2/1 was purchased from Serotec. Mouse mAb MC5, which recognizes the v3 region of PKCα, and rabbit pAb PPA182, which recognizes phosphorylated T250 of PKCα, have been reported previously (Ng et al., 1999b). Mouse anti–human vinculin (hVIN-1; 1:400 dilution) was purchased from Sigma-Aldrich.
Cloning and expression of recombinant FN fragments
cDNA clones encoding the H/120 and H/0 variants of the HepII/IIICS region of human FN (Fig. 1) were prepared as described previously (Makarem et al., 1994) with some modifications (Mostafavi-Pour et al., 2001). To create H/120-GAG-ABC, mutations (shown in bold) were introduced into H/120 cDNA by site-directed mutagenesis (Kunkel, 1985) as follows: GAG-A (residues 1697–1704 of FN using Swiss-Prot accession no. PO2751; PPRRARVT to PPSSASVT), GAG-B (two oligonucleotides for residues 1936–1944, HGFRRTTPP to HGFSSTTPP, and residues 1950–1958, IRHRPRPYP to ISHSPSPYP), and GAG-C (two oligonucleotides for residues 1817–1836, KYEKPGSPPREVVPRPRPRPGV to SYESPGSPPSEVVPSPSPGV, and residues 1858–1872, KNNQKSEPLIGRKKT to SNNQSSEPLIGSSST). The 50K CCBD of human FN was produced and purified as described previously (Mould et al., 1997).
Cell attachment
A375-SM cells, a human metastatic melanoma cell line (provided by Josh Fidler, M.D. Anderson Hospital and University of Texas, Houston, TX) were cultured as described previously (Kozlowski et al., 1984). Cell culture reagents were purchased from GIBCO BRL. Cell attachment assays were performed as described previously (Mould et al., 1994; Mostafavi-Pour et al., 2001).
Focal adhesion formation
13-mm diam glass coverslips were derivatized for 30 min with 250 μl 1 mM m-maleimidobenzoyl-N-hydroxysuccinimide ester (Pierce Chemical Co.), washed three times with 1 ml Dulbecco's PBS lacking divalent cations (PBS−), and coated for 1 h at RT with 250 μl of adhesive substrate in PBS−. The solution was aspirated, and 250 μl 10 mg/ml heat-denatured BSA (Humphries et al., 1986) was added to each well for 30 min at RT. The BSA was then aspirated and the coverslips were washed three times with PBS−. In all experiments, A375-SM cells were treated with 25 μg/ml cycloheximide for 2 h to prevent de novo matrix synthesis and were then detached as described previously (Mostafavi-Pour et al., 2001). 10 μg/ml BIM (diluted 1:200 from a DMSO stock; Calbiochem) was added to cells 2 h before detachment. Cells were resuspended to 5 × 104 ml in DME/25 mM Hepes, 0.5 ml aliquots were added to the coverslips, and the cells were incubated for 2 h at 37°C. To test either the effect of soluble heparin-binding fragment (H/0) or clustering of syndecan-4 by anti-syndecan-4 antibody, cells were allowed to spread for 2 h on 50K before the addition of H/0 or antibody for 30 min. Cells were fixed either directly for 20 min at RT with 50 μl 37% (wt/vol) formaldehyde, or unattached cells were removed by two gentle washes with 1 ml PBS−, and remaining cells fixed for 20 min at RT with 250 μl 3% (wt/vol) formaldehyde diluted in PBS−. Coverslips were washed with PBS− and then the formaldehyde was quenched with 0.1 M glycine in PBS− for 20 min at RT. Cells were permeabilized for 4 min at RT with 0.5% (wt/vol) Triton X-100 diluted in PBS−, washed three times with PBS−, and then blocked for 1 h at RT or overnight at 4°C with 3% (wt/vol) BSA in PBS− (blocking buffer). Primary antibodies were diluted in blocking buffer and incubated for 1 h at RT. After washing, antibodies were detected using TRITC-conjugated goat anti–rat IgG (1:100 dilution) and/or FITC-conjugated donkey anti–mouse IgG (1:100 dilution; Jackson ImmunoResearch Laboratories) in blocking buffer for 30 min. F-actin was detected using rhodamine-conjugated phalloidin (1:1,000 dilution; Sigma-Aldrich) in blocking buffer for 30 min. Coverslips were washed three times with PBS− and stained with 250 μl 20 ng/ml DAPI (Sigma-Aldrich) in PBS− for 30 s. Coverslips were mounted face down onto microscope slides using 5 μl Vectashield® (Vector Laboratories), viewed on a microscope (Leica), and immunofluorescence images were taken in the green (FITC) and red (TRITC/rhodamine) channels using a CCD camera. Basic image acquisition and analysis was performed using IPLab software v3.2. Advanced image analysis was performed using Adobe Photoshop® v5.0.
PKC activation
3 × 106 A375-SM cells were resuspended in 300 μl 4× Laemmli SDS sample buffer, with 1 mM sodium vanadate, 1 mM PMSF, 10 μg/ml aprotinin, 10 μg/ml leupeptin, and 5 μg/ml DNase, and were incubated on ice for 45 min. Cell debris was removed by centrifugation at 25,000 g for 30 min at 4°C. Supernatants were analyzed by 4–12% SDS-PAGE using the NuPAGE® Novex Bis-Tris gel system (Invitrogen). Gels were transferred to 0.45 μm nitrocellulose (Schleicher and Schuell) at 10 V (limit 0.5 A) for 30 min. Nonspecific binding sites on nitrocellulose membranes were blocked for 1 h at RT with 3% (wt/vol) BSA in 150 mM NaCl, 10 mM Tris-HCl, pH 7.4, containing 0.1% (wt/vol) Tween-20 (TBS-Tween) as described previously (Ng et al., 1999b; Parekh et al., 2000). Membranes were then incubated for 16 h at 4°C with either MC5 mouse anti–human PKCα diluted 1:500 in 3% (wt/vol) BSA in TBS-Tween for 1 h, or PPA182 rabbit anti-activated human PKCα diluted 1:2,000 in 1% (wt/vol) skimmed milk powder, 1% (wt/vol) BSA, and TBS-Tween. PPA182 was incubated in the presence of 1 μg/ml cognate dephosphorylated peptide to block nonspecific binding of the pAb to the nonphosphorylated form of PKCα (Ng et al., 1999b; Parekh et al., 2000). After three washes with TBS-Tween for 10 min, HRP-conjugated goat anti–mouse (1:1,000 dilution in blocking buffer; Dako) or anti–rabbit IgG (1:4,000 dilution) were added and incubated for 1 h. After three 10-min washes with TBS-Tween, proteins were detected using ECL substrate (NEN Life Science Products).
Wound migration assay
35-mm dishes with 14-mm glass bottoms (MatTek) were coated with either H/120 (10 μg/ml in PBS−) or 50K (50 μg/ml in PBS−) for 1 h, and were blocked with 10 mg/ml heat-denatured BSA (Humphries et al., 1986) for 30 min at RT. A375-SM cells were detached as described previously (Mostafavi-Pour et al., 2001). 2 × 105 cells in complete medium (10% FCS-MEM) were seeded for 16 h at 37°C in a humidified chamber with 5% CO2 until the cells formed a confluent monolayer. 5 μg/ml H/0 was added to the cells at the time of wounding when 50K was used as a substrate. Before wounding, the cell layer was washed two times with PBS−, the medium was replaced, and then the cell monolayer was wounded along the center of the dish using a sterile P10 pipette tip. To test the effect of either anti-α4 antibody (HP2/1), PKC inhibitor (BIM), or PKC activator (TPA) on migration, HP2/1 was added to the medium at a concentration of 10 μg/ml just before wounding, or 10 μg/ml BIM or 5 ng/ml TPA were added to the medium just before videomicroscopy, i.e., 6 h after wounding. The wound width was consistently between 270–300 μm (285 ± 15 μm, n = 30) 1 h after wounding, when the wound had stabilized. Images were taken using a microscope (Axiovert 135; Carl Zeiss MicroImaging, Inc.) equipped with a 20× 0.3 NA objective and a CCD camera (Photometrics Quantix; Roper Scientific). Basic image acquisition and analysis was performed using IPLab software v3.2. The images were processed using Adobe Photoshop® v5.0. Movement was also studied by observing cells using time-lapse video microscopy. The images were taken using a 20× objective at 5-min intervals for 6 h and organized into time-lapse movies using the IPLab image software.
Transfection
To assess the isoform specificity of PKC signaling during integrin-mediated migration, A375-SM cells were transiently transfected with wild-type and dominant-negative pEGFP-PKC constructs. These were as follows: wild-type pEGFP-PKCα, pEGFP-PKCδ and pEGFP-PKCζ, pEGFP-PKCα-A25E (pseudosubstrate site mutation), pEGFP-PKCα-A25E-K368M (pseudosubstrate site and kinase-dead, ATP-binding mutations), pEGFP-PKCα-T497A (kinase-dead, substrate-binding mutant), pEGFP-PKCδ-K376M (kinase-dead, ATP-binding mutant), and pEGFP-PKCζ-K281M (kinase-dead, ATP-binding mutant). pEGFP-PKCα was constructed by subcloning the SacII/StuI fragment from bovine PKCα into pEGFP-C2 (BD Biosciences; CLONTECH Laboratories, Inc.) digested with SacII/SmaI. The various pEGFP-PKCα mutants were constructed using the same strategy using the constructs as described previously (Bornancin and Parker, 1997). The pEGFP-PKCδ and pEGFP-PKCδ-K376M plasmids have been described previously (Srivastava et al., 2002). The pEGFP-PKCζ plasmid was constructed in two steps. A blunted 1.2-kb NdeI/PmeI fragment from pcDNA3.1-PKCζ was ligated into pEGFP-C1 (CLONTECH Laboratories, Inc.) cut with SmaI to make pEGFP-PKMζ. PEGFP-PKCζ was constructed by cutting pEGFP-PKMζ with BspE1/EcoRV and ligating in a fragment from pcDNA3.1-PKCζ cut with NgoMIV/EcoRV. The kinase-dead pEGFP-PKCζ-K281M plasmid was constructed by cutting pEGFP-PKCζ with NotI/XbaI and ligating in the same fragment from pcDNA3-PKCζ-K281M (provided by Anne LeGood, Cancer Research UK Laboratories). All constructs were confirmed by sequencing and expression by Western blot. Transfection of A375-SM cells was conducted in 6-well culture plates (Costar). Cells were detached using 0.05% (wt/vol) trypsin, 0.02% (wt/vol) EDTA in PBS− and seeded at 105 cells/ml in 2 ml growth medium. Cells were transfected the next day using the LipofectAMINE™ 2000 protocol (Invitrogen), when the culture reached 80–90% confluence. Transfected cells were detached (as described under Wound migration assay) and seeded onto 35-mm dishes for wounding assays after 48 h of transfection.
Online supplemental material
Video 1 shows A375-SM melanoma cells on 50K with soluble H/0 in the presence of BIM. Video 2 shows A375-SM melanoma cells on 50K with soluble H/0 in the presence of DMSO. Video 3 shows A375-SM melanoma cells on H/120 in the presence of BIM. Video 4 shows A375-SM melanoma cells on H/120 in the presence of DMSO. Video 5 shows pEGF-PKCα-WT–transfected cells on 50K with soluble H/0 in the presence of 5 ng/ml TPA. Video 6 shows pEGF-PKCαA25E-K368M–transfected cells on 50K with soluble H/0 in the absence of TPA. Video 7 shows pEGF-PKCαA25E-K368M–transfected cells on 50K with soluble H/0 in the presence of 5 ng/ml TPA. Video 8 shows pEGF-PKCαA25E-K368M–transfected cells on H/120 in the presence of 5 ng/ml TPA. Video 9 shows pEGF-PKCδ-K376M–transfected cells on 50K with soluble H/0 in the presence of 5 ng/ml TPA. Video 10 shows pEGF-PKCζ-K281M–transfected cells on 50K with soluble H/0 in the absence of TPA. Video 11 shows pEGF-PKCδ-WT–transfected cells on 50K with soluble H/0 in the presence of 5 ng/ml TPA. Online supplemental material available at http://www.jcb.org/cgi/content/full/jcb.200210176/DC1.
Acknowledgments
We thank Anne Woods, Ken Yamada, Anne LeGood, and Josh Fidler for gifts of antibodies, cDNA clones, and cells.
This work was supported by grants from the Wellcome Trust (045225 to M.J. Humphries), the Iranian Government (to Z. Mostafavi-Pour), the Medical Research Council (to T.T.C. Ng in the form of a Clinician Scientist Grant), and the European Union (to S.J. Parkinson and P.J. Parker).
The online version of this article includes supplemental material.
Footnotes
Abbreviations used in this paper: BIM, bisindolylmaleimide; CCBD, central cell–binding domain; FN, fibronectin; HBD, heparin-binding domain; HepII, COOH-terminal heparin binding domain of fibronectin; IIICS, type III connecting segment; PIP2, phosphatidylinositol-4,5-bisphosphate.
References
- Baciu, P.C., and P.F. Goetinck. 1995. Protein kinase C regulates the recruitment of syndecan-4 into focal contacts. Mol. Biol. Cell. 6:1503–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballestrem, C., B. Wehrle-Haller, B. Hinz, and B.A. Imhof. 2000. Actin-dependent lamellipodia formation and microtubule-dependent tail retraction control-directed cell migration. Mol. Biol. Cell. 11:2999–3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkalow, F.J., and J.E. Schwarzbauer. 1991. Localization of the major heparin-binding site in fibronectin. J. Biol. Chem. 266:7812–7818. [PubMed] [Google Scholar]
- Bass, M.D., and M.J. Humphries. 2002. The cytoplasmic interactions of syndecan-4 orchestrate adhesion receptor and growth factor receptor signalling. Biochem. J. 368:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom, L., K.C. Ingham, and R.O. Hynes. 1999. Fibronectin regulates assembly of actin filaments and focal contacts in cultured cells via the heparin-binding site in repeat III13. Mol. Biol. Cell. 10:1521–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornancin, F., and P.J. Parker. 1997. Phosphorylation of protein kinase C-α on serine 657 controls the accumulation of active enzyme and contributes to its phosphatase-resistant state. J. Biol. Chem. 272:3544–3549. [DOI] [PubMed] [Google Scholar]
- Bouvard, D., C. Brakebusch, E. Gustafsson, A. Aszodi, T. Bengtsson, A. Berna, and R. Fassler. 2001. Functional consequences of integrin gene mutations in mice. Circ. Res. 89:211–223. [DOI] [PubMed] [Google Scholar]
- Bretscher, A., K. Edwards, and R.G. Fehon. 2002. ERM proteins and merlin: integrators at the cell cortex. Nat. Rev. Mol. Cell Biol. 3:586–599. [DOI] [PubMed] [Google Scholar]
- Buchanan, F.G., C.M. Elliot, M. Gibbs, and J.H. Exton. 2000. Translocation of the Rac1 guanine nucleotide exchange factor Tiam1 induced by platelet-derived growth factor and lysophosphatidic acid. J. Biol. Chem. 275:9742–9748. [DOI] [PubMed] [Google Scholar]
- Burridge, K., and M. Chrzanowska-Wodnicka. 1996. Focal adhesions, contractility, and signaling. Annu. Rev. Cell Dev. Biol. 12:463–519. [DOI] [PubMed] [Google Scholar]
- Chan, B.M.C., P.D. Kassner, J.A. Schiro, H.R. Byers, T.S. Kupper, and M.E. Hemler. 1992. Distinct cellular functions mediated by different VLA integrin α subunit cytoplasmic domains. Cell. 68:1051–1060. [DOI] [PubMed] [Google Scholar]
- Choquet, D., D.P. Felsenfeld, and M.P. Sheetz. 1997. Extracellular matrix rigidity causes strengthening of integrin-cytoskeleton linkages. Cell. 88:39–48. [DOI] [PubMed] [Google Scholar]
- Echtermeyer, F., P.C. Baciu, S. Saoncella, Y. Ge, and P.F. Goetinck. 1999. Syndecan-4 core protein is sufficient for the assembly of focal adhesions and stress fibers. J. Cell Sci. 112:3433–3441. [DOI] [PubMed] [Google Scholar]
- Gautreau, A., D. Louvard, and M. Arpin. 2002. ERM proteins and NF2 tumor suppressor: the Yin and Yang of cortical actin organization and cell growth signaling. Curr. Opin. Cell Biol. 14:104–109. [DOI] [PubMed] [Google Scholar]
- Gilmore, A.P., and K. Burridge. 1996. Regulation of vinculin binding to talin and actin by phosphatidylinositol-4,5-bisphosphate. Nature. 381:531–535. [DOI] [PubMed] [Google Scholar]
- Hamada, K., T. Shimizu, T. Matsui, S. Tsukita, S. Tsukita, and T. Hakoshima. 2000. Structural basis of the membrane-targeting and unmasking mechanisms of the radixin FERM domain. EMBO J. 19:4449–4462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han, J., S. Liu, D.M. Rose, D.D. Schlaepfer, H. McDonald, and M.H. Ginsberg. 2001. Phosphorylation of the integrin alpha 4 cytoplasmic domain regulates paxillin binding. J. Biol. Chem. 276:40903–40909. [DOI] [PubMed] [Google Scholar]
- Horowitz, A., and M. Simons. 1998. Phosphorylation of the cytoplasmic tail of syndecan-4 regulates activation of protein kinase Cα. J. Biol. Chem. 273:25548–25551. [DOI] [PubMed] [Google Scholar]
- Huhtala, P., M.J. Humphries, J.B. McCarthy, P.M. Tremble, Z. Werb, and C.H. Damsky. 1995. Cooperative signaling by α5β1 and α4β1 integrins regulates metalloproteinase gene expression in fibroblasts adhering to fibronectin. J. Cell Biol. 129:867–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphries, M.J., S.K. Akiyama, A. Komoriya, K. Olden, and K.M. Yamada. 1986. Identification of an alternatively spliced site in human plasma fibronectin that mediates cell type-specific adhesion. J. Cell Biol. 103:2637–2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphries, M.J., A. Komoriya, S.K. Akiyama, K. Olden, and K.M. Yamada. 1987. Identification of two distinct regions of the type III connecting segment of human plasma fibronectin that promote cell type-specific adhesion. J. Biol. Chem. 262:6886–6892. [PubMed] [Google Scholar]
- Hynes, R.O. 2002. Integrins: bidirectional, allosteric signaling machines. Cell. 110:673–687. [DOI] [PubMed] [Google Scholar]
- Iida, J., A.M. Meijne, T.R. Oegema, T.A. Yednock, N.L. Kovach, L.T. Furcht, and J.B. McCarthy. 1998. A role of chondroitin sulfate glycosaminoglycan binding site in α4β1 integrin-mediated melanoma cell adhesion. J. Biol. Chem. 273:5955–5962. [DOI] [PubMed] [Google Scholar]
- Imamura, H., K. Takaishi, K. Nakano, A. Kodama, H. Oishi, H. Shiozaki, M. Monden, T. Sasaki, and Y. Takai. 1998. Rho and Rab small G proteins coordinately reorganize stress fibers and focal adhesions in MDCK cells. Mol. Biol. Cell. 9:2561–2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiguro, K., K. Kadomatsu, T. Kojima, H. Muramatsu, S. Tsuzuki, E. Nakamura, K. Kusugami, H. Saito, and T. Muramatsu. 2000. Syndecan-4 deficiency impairs focal adhesion formation only under restricted conditions. J. Biol. Chem. 275:5249–5252. [DOI] [PubMed] [Google Scholar]
- Ivaska, J., R.D. Whelan, R. Watson, and P.J. Parker. 2002. PKC epsilon controls the traffic of beta1 integrins in motile cells. EMBO J. 21:3608–3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izzard, C.S., R. Radinsky, and L.A. Culp. 1986. Substratum contacts and cytoskeletal reorganization of BALB/c3T3 cells on a cell-binding fragment and heparin-binding fragments of plasma fibronectin. Exp. Cell Res. 165:320–336. [DOI] [PubMed] [Google Scholar]
- Kassner, P.D., R. Alon, T.A. Springer, and M.E. Hemler. 1995. Specialized functional properties of the integrin α4 cytoplasmic domain. Mol. Biol. Cell. 6:661–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlowski, J.M., I.R. Hart, I.J. Fidler, and N. Hanna. 1984. A human melanoma line heterogeneous with respect to metastatic capacity in athymic nude mice. J. Natl. Cancer Inst. 72:913–917. [PubMed] [Google Scholar]
- Kunkel, T.A. 1985. Rapid and efficient site-specific mutagenesis without phenotypic selection. Proc. Natl. Acad. Sci. USA. 82:488–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, D., E.-S. Oh, A. Woods, J.R. Couchman, and W. Lee. 1998. Solution structure of a syndecan-4 cytoplasmic domain and its interaction with phosphatidylinositol-4,5-bisphosphate. J. Biol. Chem. 273:13022–13029. [DOI] [PubMed] [Google Scholar]
- Legg, J.W., C.A. Lewis, M. Parsons, T. Ng, and C.M. Isacke. 2002. A novel PKC-regulated mechanism controls CD44 ezrin association and directional cell motility. Nat. Cell Biol. 4:399–407. [DOI] [PubMed] [Google Scholar]
- Liu, S., and M.H. Ginsberg. 2000. Paxillin binding to a conserved sequence motif in the α4 integrin cytoplasmic domain. J. Biol. Chem. 275:22736–22742. [DOI] [PubMed] [Google Scholar]
- Liu, S., S.M. Thomas, D.G. Woodside, D.M. Rose, W.B. Kiosses, M. Pfaff, and M.H. Ginsberg. 1999. Binding of paxillin to α4 integrins modifies integrin-dependent biological responses. Nature. 402:676–681. [DOI] [PubMed] [Google Scholar]
- Longley, R.L., A. Woods, A. Fleetwood, G.J. Cowling, J.T. Gallagher, and J.R. Couchman. 1999. Control of morphology, cytoskeleton and migration by syndecan-4. J. Cell Sci. 112:3421–3431. [DOI] [PubMed] [Google Scholar]
- Makarem, R., P. Newham, J.A. Askari, L.J. Green, J. Clements, M. Edwards, M.J. Humphries, and A.P. Mould. 1994. Competitive binding of vascular cell adhesion molecule-1 and the HepII/IIICS domain of fibronectin to the integrin α4β1. J. Biol. Chem. 269:4005–4011. [PubMed] [Google Scholar]
- Manser, E., T.H. Loo, C.G. Koh, Z.S. Zhao, X.Q. Chen, L. Tan, I. Tan, T. Leung, and L. Lim. 1998. PAK kinases are directly coupled to the PIX family of nucleotide exchange factors. Mol. Cell. 1:183–192. [DOI] [PubMed] [Google Scholar]
- Martel, V., C. Racaud-Sultan, S. Dupe, C. Marie, F. Paulhe, A. Galmiche, M.R. Block, and C. Albiges-Rizo. 2001. Conformation, localization, and integrin binding of talin depend on its interaction with phosphoinositides. J. Biol. Chem. 276:21217–21227. [DOI] [PubMed] [Google Scholar]
- McCarthy, J.B., M.K. Chelberg, D.J. Mickelson, and L.T. Furcht. 1988. Localization and chemical synthesis of fibronectin peptides with melanoma adhesion and heparin binding activities. Biochemistry. 27:1380–1388. [DOI] [PubMed] [Google Scholar]
- McCarthy, J.B., A.P.N. Skubitz, Z. Qi, X.-Y. Yi, D.J. Mickelson, D.J. Klein, and L.T. Furcht. 1990. RGD-independent cell adhesion to the carboxy-terminal heparin-binding fragment of fibronectin involves heparin-dependent and -independent activities. J. Cell Biol. 110:777–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNamee, H.P., D.E. Ingber, and M.A. Schwartz. 1993. Adhesion to fibronectin stimulates inositol lipid synthesis and enhances PDGF-induced inositol lipid breakdown. J. Cell Biol. 121:673–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostafavi-Pour, Z., J.A. Askari, J.D. Whittard, and M.J. Humphries. 2001. Identification of a novel heparin binding site in the alternatively spliced IIICS region of fibronectin: roles of integrins and proteoglycans in cell adhesion to fibronectin splice variants. Matrix Biol. 20:63–73. [DOI] [PubMed] [Google Scholar]
- Mould, A.P., and M.J. Humphries. 1991. Identification of a novel recognition sequence for the integrin α4β1 in the COOH-terminal heparin-binding domain of fibronectin. EMBO J. 10:4089–4095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mould, A.P., L.A. Wheldon, A. Komoriya, E.A. Wayner, K.M. Yamada, and M.J. Humphries. 1990. Affinity chromatographic isolation of the melanoma adhesion receptor for the IIICS region of fibronectin and its identification as the integrin α4β1. J. Biol. Chem. 265:4020–4024. [PubMed] [Google Scholar]
- Mould, A.P., A. Komoriya, K.M. Yamada, and M.J. Humphries. 1991. The CS5 peptide is a second site in the IIICS region of fibronectin recognized by the integrin α4β1. Inhibition of α4β1 function by RGD peptide homologues. J. Biol. Chem. 266:3579–3585. [PubMed] [Google Scholar]
- Mould, A.P., J.A. Askari, S.E. Craig, A.N. Garratt, J. Clements, and M.J. Humphries. 1994. Integrin α4β1-mediated melanoma cell adhesion and migration on vascular cell adhesion molecule-1 (VCAM-1) and the alternatively spliced IIICS region of fibronectin. J. Biol. Chem. 269:27224–27230. [PubMed] [Google Scholar]
- Mould, A.P., J.A. Askari, S. Aota, S. Yamada, K.M. Yamada, A. Irie, Y. Takada, H.J. Mardon, and M.J. Humphries. 1997. Defining the topology of integrin α5β1-fibronectin interactions using inhibitory anti-α5 and anti-β1 monoclonal antibodies: Evidence that the synergy sequence of fibronectin is recognized by the amino-terminal repeats of the α5 subunit. J. Biol. Chem. 272:17283–17292. [DOI] [PubMed] [Google Scholar]
- Murakami, M., A. Horowitz, S. Tang, J.A. Ware, and M. Simons. 2002. Protein kinase C (PKC) δ regulates PKCα activity in a syndecan-4-dependent manner. J. Biol. Chem. 277:20367–20371. [DOI] [PubMed] [Google Scholar]
- Ng, T., D. Shima, A. Squire, P.I.H. Bastiaens, S. Gschmeissner, M.J. Humphries, and P.J. Parker. 1999. a. PKCα regulates β1 integrin-dependent cell motility through association and control of integrin traffic. EMBO J. 18:3909–3923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng, T., A. Squire, G. Hansra, F. Bornancin, C. Prevostel, A. Hanby, W. Harris, D. Barnes, S. Schmidt, H. Mellor, et al. 1999. b. Imaging protein kinase Cα activation in cells. Science. 283:2085–2089. [DOI] [PubMed] [Google Scholar]
- Ng, T., M. Parsons, W.E. Hughes, J. Monypenny, D. Zicha, A. Gautreau, M. Arpin, S. Gschmeissner, P.J. Verveer, P.I. Bastiaens, and P.J. Parker. 2001. Ezrin is a downstream effector of trafficking PKC-integrin complexes involved in the control of cell motility. EMBO J. 20:2723–2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh, E.-S., A. Woods, and J.R. Couchman. 1997. a. Syndecan-4 proteoglycan regulates the distribution and activity of protein kinase C. J. Biol. Chem. 272:8133–8136. [DOI] [PubMed] [Google Scholar]
- Oh, E.-S., A. Woods, and J.R. Couchman. 1997. b. Multimerization of the cytoplasmic domain of syndecan-4 is required for its ability to activate protein kinase C. J. Biol. Chem. 272:11805–11811. [DOI] [PubMed] [Google Scholar]
- Oh, E.-S., A. Woods, S.-T. Lim, A.W. Theibert, and J.R. Couchman. 1998. Syndecan-4 proteoglycan cytoplasmic domain and phosphatidylinositol-4,5-bisphosphate coordinately regulate protein kinase C activity. J. Biol. Chem. 273:10624–10629. [DOI] [PubMed] [Google Scholar]
- Parekh, D.B., W. Ziegler, and P.J. Parker. 2000. Multiple pathways control protein kinase C phosphorylation. EMBO J. 19:496–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons, M., M.D. Keppler, A. Kline, A. Messent, M.J. Humphries, R. Gilchrist, I.R. Hart, C. Quittau-Prevostel, W.E. Hughes, P.J. Parker, and T. Ng. 2002. Site-directed perturbation of PKC-integrin interaction blocks carcinoma cell chemotaxis. Mol. Cell. Biol. 22:5897–5911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podar, K., Y.T. Tai, B.K. Lin, R.P. Narsimhan, M. Sattler, T. Kijima, R. Salgia, D. Gupta, D. Chauhan, and K.C. Anderson. 2002. Vascular endothelial growth factor-induced migration of multiple myeloma cells is associated with β1 integrin- and PI3-kinase-dependent PKCα activation. J. Biol. Chem. 277:7875–7881. [DOI] [PubMed] [Google Scholar]
- Saoncella, S., F. Echtermeyer, F. Denhez, J.K. Nowlen, D.F. Mosher, S.D. Robinson, R.O. Hynes, and P.F. Goetinck. 1999. Syndecan-4 signals cooperatively with integrins in a Rho-dependent manner in the assembly of focal adhesions and actin stress fibers. Proc. Natl. Acad. Sci. USA. 96:2805–2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenwaelder, S.M., and K. Burridge. 1999. Bidirectional signaling between the cytoskeleton and integrins. Curr. Opin. Cell Biol. 11:274–286. [DOI] [PubMed] [Google Scholar]
- Sharma, A., J.A. Askari, M.J. Humphries, E.Y. Jones, and D.I. Stuart. 1999. Crystal structure of a heparin and integrin-binding segment of human fibronectin. EMBO J. 18:1468–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin, J., W. Lee, D. Lee, B.-K. Koo, I. Han, Y. Lim, A. Woods, J.R. Couchman, and E.-S. Oh. 2001. Solution structure of the dimeric cytoplasmic domain of syndecan-4. Biochemistry. 40:8471–8478. [DOI] [PubMed] [Google Scholar]
- Srivastava, J., K.J. Procyk, X. Iturrioz, and P.J. Parker. 2002. Phosphorylation is required for PMA- and cell-cycle-induced degradation of protein kinase Cδ. Biochem. J. 368:349–355. First published on September 4, 2002; 10.1042/BJ20020737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner, C.E. 2000. Paxillin interactions. J. Cell Sci. 113:4139–4140. [DOI] [PubMed] [Google Scholar]
- Wang, F., V.M. Weaver, O.W. Petersen, C.A. Larabell, S. Dedhar, P. Briand, R. Lupu, and M.J. Bissell. 1998. Reciprocal interactions between β1-integrin and epidermal growth factor receptor in three-dimensional basement membrane breast cultures: a different perspective in epithelial biology. Proc. Natl. Acad. Sci. USA. 95:14821–14826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayner, E.A., A. Garcia-Pardo, M.J. Humphries, J.A. McDonald, and W.G. Carter. 1989. Identification and characterization of the T lymphocyte adhesion receptor for an alternative cell attachment domain (CS-1) in plasma fibronectin. J. Cell Biol. 109:1321–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods, A., and J.R. Couchman. 1992. Protein kinase C involvement in focal adhesion formation. J. Cell Sci. 101:277–290. [DOI] [PubMed] [Google Scholar]
- Woods, A., and J.R. Couchman. 1994. Syndecan 4 heparan sulfate proteoglycan is a selectively enriched and widespread focal adhesion component. Mol. Biol. Cell. 5:183–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods, A., and J.R. Couchman. 2001. Syndecan-4 and focal adhesion function. Curr. Opin. Cell Biol. 13:578–583. [DOI] [PubMed] [Google Scholar]
- Woods, A., J.R. Couchman, S. Johansson, and M. Höök. 1986. Adhesion and cytoskeletal organization of fibroblasts in response to fibronectin fragments. EMBO J. 5:665–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada, K.M., and B. Geiger. 1997. Molecular interactions in cell adhesion complexes. Curr. Opin. Cell Biol. 9:76–85. [DOI] [PubMed] [Google Scholar]