

Abstract
Skeletal muscle growth requires multiple steps to form large multinucleated muscle cells. Molecules that stimulate muscle growth may be therapeutic for muscle loss associated with aging, injury, or disease. However, few factors are known to increase muscle cell size. We demonstrate that prostaglandin F2α (PGF2α) as well as two analogues augment muscle cell size in vitro. This increased myotube size is not due to PGF2α-enhancing cell fusion that initially forms myotubes, but rather to PGF2α recruiting the fusion of cells with preexisting multinucleated cells. This growth is mediated through the PGF2α receptor (FP receptor). As the FP receptor can increase levels of intracellular calcium, the involvement of the calcium-regulated transcription factor nuclear factor of activated T cells (NFAT) in mediating PGF2α-enhanced cell growth was examined. We show that NFAT is activated by PGF2α, and the isoform NFATC2 is required for PGF2α-induced muscle cell growth and nuclear accretion, demonstrating the first intersection between prostaglandin receptor activation and NFAT signaling. Given this novel role for PGF2α in skeletal muscle cell growth, these studies raise caution that extended use of drugs that inhibit PG production, such as nonsteroidal antiinflammatory drugs, may be deleterious for muscle growth.
Keywords: FP receptor; calcium; myonuclei; cell fusion; NSAIDS
Introduction
Skeletal myogenesis follows an ordered set of cellular events involving cell cycle exit of myoblasts, their subsequent differentiation, and fusion to form multinucleated myofibers in vivo or myotubes in vitro. In most cases, mammalian muscle growth requires the fusion of differentiated muscle cells with the growing multinucleated muscle cell (Darr and Schultz, 1989; Rosenblatt and Parry, 1992; Phelan and Gonyea, 1997; Barton-Davis et al., 1999; Horsley et al., 2001; Mitchell and Pavlath, 2001). By adding additional nuclei to muscle cells during growth, an increased number of nuclei are contained within one cytoplasm, allowing each nucleus to regulate more cytoplasm (Allen et al., 1999). These fusion events allow increased protein synthesis and increases in cell size. Understanding the molecular pathways that regulate muscle growth are important for treating muscle disorders and loss of muscle mass during aging. However, few molecules are known to stimulate increased muscle cell fusion and skeletal muscle growth.
Prostaglandins (PGs)* are paracrine signaling molecules that are synthesized from arachidonic acid in response to cytokines, cell injury, or growth factors (Funk, 2001). The synthesis of PGs involves the metabolism of arachidonic acid by cyclooxygenase enzymes into an intermediate PG. Specific PG synthases convert this intermediate PG into the primary PG molecules (PGE2, PGF2α, PGI2, and PGD2). Once produced, PGs are secreted and mediate signaling through G protein–coupled receptors that are distinct for each PG. Activation of PG receptors leads to an array of effects in a range of cell and tissue types, including skeletal muscle.
PGs have been implicated in skeletal muscle growth. For skeletal muscle to grow, a population of myoblasts must be available to differentiate and fuse with the myofiber. Different PGs can control proliferation (Zalin, 1987), differentiation (Schutzle et al., 1984), as well as fusion of myoblasts (Zalin, 1977; David and Higginbotham, 1981; Entwistle et al., 1986; Rossi et al., 1989). Once myotubes are formed, muscle cell size continues to increase through enhanced protein synthesis. PGs regulate this stage of growth by altering both protein degradation and protein synthesis within myotubes (Rodemann and Goldberg, 1982; Palmer, 1990; Vandenburgh et al., 1990). Consistent with a general role for PGs in skeletal muscle growth, inhibition of PG production blocks growth of myofibers in vivo (Templeton et al., 1986; McLennan, 1987). These data suggest that PGs regulate muscle cell growth by influencing multiple steps of myogenesis.
Signaling pathways that are activated by calcium are important for skeletal muscle growth (Abbott et al., 1998; Dunn et al., 1999; Musaro et al., 1999; Semsarian et al., 1999; Delling et al., 2000; Friday et al., 2000; Horsley et al., 2001; Mitchell et al., 2002). PGs have been shown to activate increases in intracellular calcium within a variety of muscle cell types (Asboth et al., 1996; Chen et al., 1997; Yew et al., 1998; Yousufzai and Abdel-Latif, 1998). Specifically, PGF2α and PGE2 can activate increases in intracellular calcium through their receptors, PGF2α receptor (FP) and EP1/EP3, respectively (Breyer et al., 2001). One calcium-regulated pathway involved in skeletal muscle growth is the family of transcription factors, nuclear factor of activated T cells (NFAT; Horsley and Pavlath, 2002). Several NFAT isoforms are expressed in skeletal muscle, and the regulation of individual NFAT isoforms appears to occur at the level of nuclear translocation (Abbott et al., 1998). For instance, the NFATC2 isoform is activated only in newly formed or nascent myotubes but not at other stages of myogenesis (Abbott et al., 1998). Previously, we have shown that the NFATC2 isoform is important for skeletal muscle growth (Horsley et al., 2001), but upstream activators of this pathway have not been elucidated.
Because PGF2α can increase intracellular calcium and calcium signaling pathways are important for numerous stages of myogenesis that contribute to muscle growth, we hypothesized that PGF2α may regulate skeletal muscle growth. Although PGF2α can regulate the final stages of muscle growth by inducing protein synthesis (Vandenburgh et al., 1990), we sought to investigate the role of PGF2α in other steps of myogenesis that require calcium, such as differentiation (Shainberg et al., 1969; Morris and Cole, 1979) and fusion (Shainberg et al., 1969; Knudsen and Horwitz, 1977). We show that PGF2α enhances myonuclear accretion after the initial formation of myotubes, leading to increases in myotube size. Furthermore, the growth induced by PGF2α occurs through an NFAT-dependent pathway. These data implicate not only a novel function for PGF2α in skeletal muscle growth but also a novel intersection between prostaglandin and NFAT signaling pathways.
Results
PGF2α increases muscle cell size and nuclear number
To test the hypothesis that PGF2α has a role in the growth of skeletal muscle cells, differentiating primary muscle cultures were treated with different doses of PGF2α or with a stable synthetic analogue of PGF2α, 17-phenyl trinor PGF2α (17-phPGF2α). After 24 h, the majority of cells are differentiated and have formed a few multinucleated cells. Although no difference is apparent between the vehicle- and drug-treated groups at 24 h, after 48 h, drug-treated myotubes are larger in size as compared with vehicle (Fig. 1 A), suggesting that PGF2α can regulate muscle growth.
Figure 1.

PGF 2α increases myotube size by facilitating muscle cell fusion. (A) Primary myoblasts were induced to differentiate in the presence of vehicle (10−6 M PGF2α or 10−7 M 17-phPGF2α) and immunostained for EMyHC at the indicated times. Bar, 60 μm. (B) The DNA content of myotube cultures treated with vehicle or 10−6 M PGF2α was quantified after 48 h in DM. (C) The percentage of nuclei within EMyHC-positive cells was analyzed after treatment with vehicle or 10−6 M PGF2α at the indicated times. (D) The percentage of nuclei within myotubes was calculated after treatment with vehicle or 10−6 M PGF2α at the indicated times. (E) Cells were treated with the indicated doses of PGF2α for 48 h, and the number of nuclei within individual myotubes was counted. Myotubes were grouped into two categories, and the percentage of myotubes in each category was determined. Data are mean ± SEM of three independent cell isolates. *Significantly different from vehicle, P < 0.05. (F) Cells were treated with the indicated doses of 17-phPGF2α for 48 h, and the number of nuclei within individual myotubes was counted as in E. Data are mean ± SEM of three independent experiments. *Significantly different from vehicle, P < 0.05. (G) Cells were treated with vehicle or 10−6 M PGF2α at 0, 24, or both 0 and 24 h. Nuclear number assays were performed as in E, and the percentage of myotubes with five or more nuclei is shown. Data are mean ± SEM of three independent experiments. *Significantly different from vehicle, P < 0.05.
The formation of a multinucleated cell requires multiple cellular processes including the formation of an adequate number of myoblasts through cell proliferation, their differentiation, and subsequent membrane fusion. To determine if PGF2α increases cell proliferation and/or cell survival in our assay, the DNA content was quantified. No difference exists in the DNA content between PGF2α and vehicle-treated cells (Fig. 1 B). Differentiation was assessed in vehicle and PGF2α-treated cultures at 24 and 48 h by immunostaining the cultures with embryonic myosin heavy chain (EMyHC), a marker of differentiation, and counting the number of nuclei contained within EMyHC-positive cells. The percentage of differentiated cells does not differ between vehicle- or PGF2α-treated cultures at 24 or 48 h (Fig. 1 C). In addition, the fusion index was determined. The percentage of nuclei in myotubes is not different between vehicle- or PGF2α-treated cultures (Fig. 1 D). These data suggest that PGF2α does not affect myoblast proliferation or survival, differentiation, or fusion to lead to muscle growth.
After the initial fusion of myoblasts that forms a multinucleated cell, cell growth occurs through the fusion of differentiated muscle cells with the nascent myotube to increase myonuclear number and cell size (Bate, 1990; Horsley et al., 2001). Although the fusion index determines the percentage of the total cell population that has fused within muscle cultures, it is not a measure of the number of myonuclei within individual myotubes. By analyzing the myonuclear number within individual myotubes, cell fusion that contributes to muscle growth can be determined. To determine if PGF2α increases muscle cell size by enhancing addition of myonuclei to existing myotubes, the number of nuclei in individual myotubes was determined in cultures treated with vehicle and PGF2α for 48 h. With vehicle treatment, an equal percentage of myotubes are present with two to four nuclei as those with five or more nuclei (Fig. 1 E). However, with 10−6 M PGF2α treatment, a significant increase occurs in the percentage of myotubes with five or more nuclei with a parallel decrease in the percentage of myotubes with two to four nuclei. This dose of PGF2α has been shown to give maximal effects in assays using cardiac and smooth muscle cells (Adams et al., 1996; Griffin et al., 1998; Kunapuli et al., 1998; Yew et al., 1998; Katsuyama et al., 2002). Other doses of PGF2α tested do not significantly differ from vehicle. Similarly, treatment of cells with 17-phPGF2α also increases myonuclear number to the same extent as PGF2α but at lower doses (Fig. 1 F), which is consistent with its higher affinity and greater metabolic stability (Lake et al., 1994; Pierce et al., 1997). These data suggest that PGF2α increases cell fusion with myotubes to facilitate increases in muscle cell size.
To further study the effect of PGF2α on increases in myonuclear number, differentiating muscle cells were treated with PGF2α at different stages of fusion. To determine if PGF2α can act at the initial stages of cell fusion, cells were only treated with PGF2α at the onset of differentiation at 0 h in differentiation media (DM). To determine if PGF2α acts during later fusion events, cells were only treated at 24 h, a time when cells are beginning to fuse and a few multinucleated muscle cells are present (Fig. 1 A). In both cases, the nuclear number of individual myotubes was analyzed at 48 h, as in Fig. 1 E. When PGF2α is administered at the onset of differentiation (0 h), no significant difference exists in the percentage of myotubes with five or more nuclei as compared with vehicle-treated cultures (Fig. 1 G). However, when PGF2α is administered at 24 h, the percentage of myotubes with five or more nuclei is significantly higher than vehicle-treated cells. This difference is comparable to the increase in nuclear number when PGF2α is added at both 0 and 24 h. These data further confirm that PGF2α acts at later stages of muscle cell fusion to allow an increase in muscle cell size.
Activation of the FP receptor induces cell growth
PGF2α primarily mediates its cellular effects by binding with high affinity (Ki= 3.4 nM) to the FP prostanoid receptor (Breyer et al., 2001). However, PGF2α can also bind with lower affinity to EP1 (Ki= 1,300 nM) and EP3 (Ki= 75 nM) receptors. To determine if muscle growth induced by the addition of PGF2α occurs through the FP receptor, cells were treated with a specific FP agonist (fluprostenol) that has a similar affinity for the FP receptor as PGF2α (Ki= 3.8 nM) but does not bind to other prostanoid receptors (Breyer et al., 2001). Fluprostenol induces an increase in muscle cell size (Fig. 2 A) as well as an increase in myonuclear number to the same extent as PGF2α at similar doses (Fig. 2 B).
Figure 2.

PGF 2α-mediated muscle growth occurs through the FP receptor. (A) Primary myoblasts were induced to differentiate in the presence of vehicle, 10−6 M fluprostenol, or 10−6 M AL-8810 and immunostained for EMyHC after 48 h. Bar, 60 μm. (B) Cells were treated with the indicated doses of fluprostenol for 48 h and analyzed as in Fig. 1 E. (C) After 24 h in DM, cells were treated with indicated doses of AL-8810 for 24 h and analyzed as in Fig. 1 E. Data are mean ± SEM of three independent experiments. *Significantly different from vehicle, P < 0.05.
To determine if endogenous PGF2α regulates myonuclear accretion and acts through the FP receptor, cells were treated with a selective, competitive FP antagonist, AL-8810 (Ki = 426 nM; Griffin et al., 1998, 1999), after 24 h in DM corresponding to the later stages of fusion. In the presence of ≥10−7 M AL-8810, myotube cultures contain few cells with five or more nuclei (Fig. 2 C). Together, these data suggest that PGF2α-induced muscle growth is mediated through the FP receptor and that endogenous PGF2α is required for muscle growth.
NFAT activity is required for muscle growth by PGF2α
PGF2α signaling is known to increase levels of intracellular calcium in a variety of cell types including smooth muscle and cardiac muscle (Yew et al., 1998; Yousufzai and Abdel-Latif, 1998). NFAT is a family of calcium-regulated transcription factors that has been implicated in skeletal muscle growth (Musaro et al., 1999; Horsley et al., 2001; Kegley et al., 2001). To investigate whether NFAT is involved in PGF2α-induced skeletal muscle growth, cells were infected with a retrovirus encoding VIVIT, a specific peptide inhibitor of NFAT activation. VIVIT acts by preventing the interaction between NFAT and calcineurin but not between calcineurin and other substrates (Aramburu et al., 1999; Friday et al., 2000; Friday and Pavlath, 2001). Primary muscle cells infected with control retrovirus exhibit an increase in cell size when treated with 10−6 M PGF2α (Fig. 3 A), similar to uninfected cells (Fig. 1 A). In contrast, cells infected with VIVIT retrovirus do not increase cell size when treated with PGF2α. To quantify these observations, we analyzed the cultures with the nuclear number assay used in Fig. 1. Cells infected with control retrovirus and treated with PGF2α show a significant increase in the percentage of myotubes with five or more nuclei. However, cells infected with the VIVIT retrovirus and treated with PGF2α are similar in nuclear number to nontreated cells. These data implicate that a calcineurin- and NFAT-dependent signaling pathway is involved in skeletal muscle growth induced by PGF2α.
Figure 3.

PGF 2α increases myotube size through an NFAT-dependent pathway. (A) Primary myoblasts were infected either with control retrovirus (Cntl RV) or with a retrovirus expressing VIVIT, a peptide inhibitor of NFAT (VIVIT RV). Cells were induced to differentiate in the presence of vehicle or 10−6 M PGF2α for 48 h, and then immunostained for EMyHC. Bar, 60 μm. (B) Nuclear number assays were performed on the retrovirally infected cultures as in Fig. 1 E. Data are mean ± SEM of three independent experiments. *Significantly different from vehicle, P < 0.05.
PGF2α activates nuclear translocation and transcriptional activity of NFAT
Given the requirement of NFAT in PGF2α-induced muscle growth, direct activation of NFAT signaling by PGF2α was analyzed in muscle cells. First, cells were transiently transfected with an NFATC2-GFP fusion construct and treated with 10−6 M PGF2α after 24 h in DM. Without treatment, cells expressing NFATC2-GFP exhibit GFP throughout the cell (Fig. 4 A). Stimulation with PGF2α results in translocation and accumulation of NFAT in nuclei of myotubes. This nuclear translocation of NFAT is blocked by treatment with cyclosporine A (CsA), suggesting that calcineurin is activated by PGF2α. In addition, PGF2α-induced nuclear translocation of NFATC2 is inhibited by cotreatment with the FP antagonist, AL-8810. Thus, stimulation of the FP receptor is responsible for NFAT activation and not a nonspecific increase in intracellular calcium at this dose of PGF2α. Transcriptional activity of NFAT was analyzed also in cells containing a NFAT responsive luciferase reporter construct (Fig. 5 B). Luciferase activity is increased in cells treated with 10−6 M PGF by approximately ninefold. Together, these data indicate that PGF2α activates NFAT signaling in skeletal muscle.
Figure 4.

PGF 2α activates nuclear translocation and transcriptional activity of NFAT in muscle cells. (A) After 24 h in DM, transiently transfected cells expressing an NFATC2-GFP fusion protein were treated with 10−6 M PGF2α alone or in combination with 10−6 M CsA or 10−6 M AL-8810. Arrowheads indicate the location of nuclei in both GFP and Hoescht images. Data are representative of three independent experiments. Bar, 60 μm. (B) Myoblasts were transiently transfected with an NFAT- responsive reporter and after 24 h in DM, stimulated with 10−6 M PGF2α alone or in combination with 10−6 M CsA. Data are reported as fold increase in luciferase activity over basal. Each bar represents the mean ± SEM of four independent experiments each performed in triplicate. *Significantly different from basal and PGF2α and CsA, P < 0.05.
Figure 5.
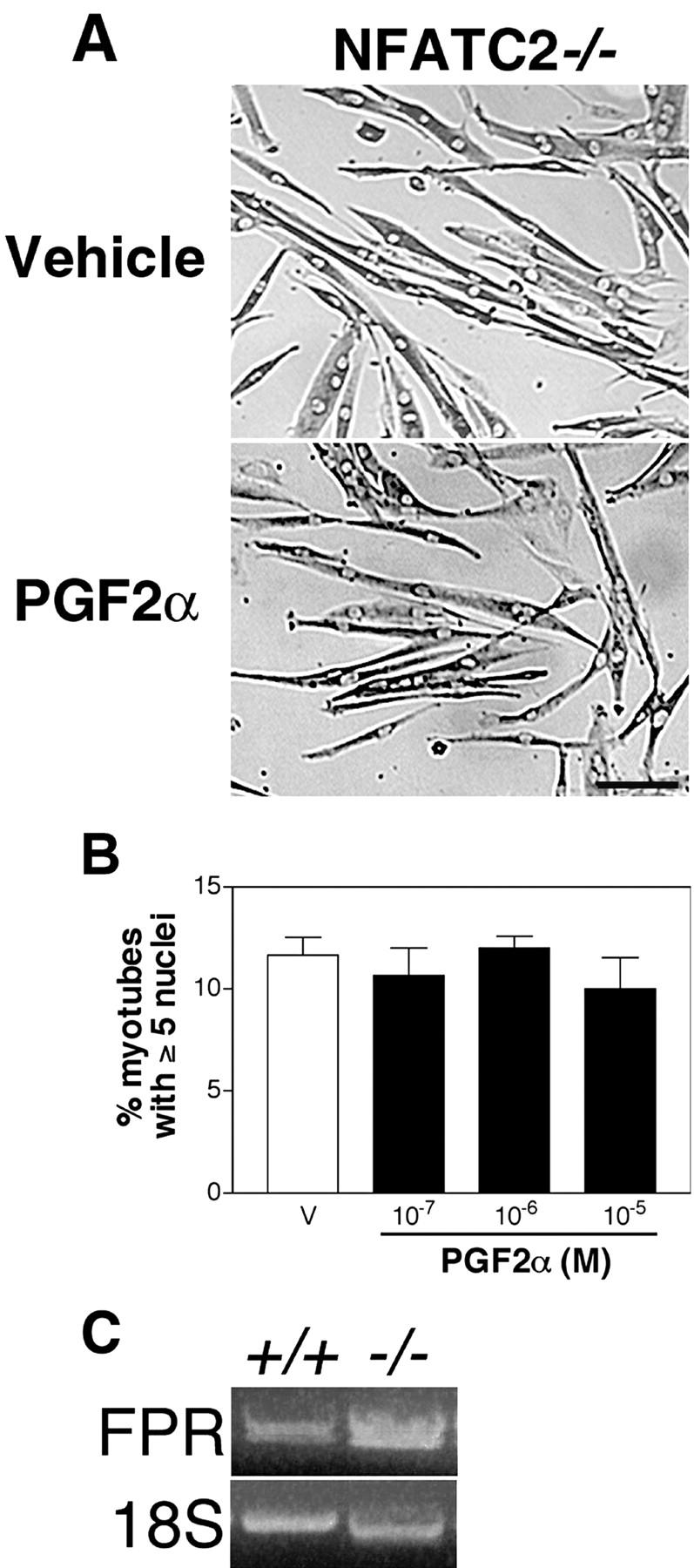
PGF 2α does not induce myotube growth or fusion of NFATC2 − / − muscle cells. (A) NFATC2−/− myoblasts were induced to differentiate and treated with vehicle or 10−6 M PGF2α for 48 h, followed by immunostaining for EMyHC. Bar, 60 μm. (B) Nuclear number assays were performed as in Fig. 1 E on NFATC2−/− cultures treated with vehicle or PGF2α at a range of doses. Data are the mean ± SEM of three independent cell isolates. (C) FP receptor mRNA expression was examined by RT-PCR after 24 h in DM in wild-type and NFATC2−/− muscle cells. Representative ethidium bromide staining of acrylamide gel is shown with 18S rRNA as an internal control.
PGF2α affects myotube size through NFATC2
Three isoforms of NFAT are expressed in skeletal muscle cells (Abbott et al., 1998). Because the NFATC2 isoform regulates skeletal muscle growth (Horsley et al., 2001) and is activated by PGF2α (Fig. 4), we investigated whether NFATC2 is required for cell growth induced by PGF2α by examining NFATC2−/− muscle cells treated with PGF2α. NFATC2−/− cells differentiate and initially fuse normally but have a defect in further myonuclear accretion that prevents muscle cell growth (Horsley et al., 2001). Addition of PGF2α to NFATC2−/− cultures does not increase myotube size (Fig. 5 A) or nuclear number (Fig. 5 B) at a range of doses, suggesting that NFATC2 is required for PGF2α-induced growth.
The inability of NFATC2−/− muscle cells to grow in response to PGF2α may result from reduced expression of the FP receptor in these mutant cells. To determine if NFATC2 regulates the expression of the FP receptor, mRNA levels of the FP receptor were examined in wild-type and NFATC2−/− cells using RT-PCR. As shown in Fig. 5 C, the FP receptor is expressed in both wild-type and NFATC2−/− cells. Thus, NFATC2 is not necessary for the expression of the FP receptor but rather functions downstream of FP receptor activation in skeletal muscle cells.
To further test whether PGF2α requires NFATC2 to induce muscle growth, we determined if expression of a recombinant NFATC2 in NFATC2−/− muscle cells could rescue the inability of these cells to increase in cell size and nuclear number in response to PGF2α. When treated with 10−6 M PGF2α, NFATC2−/− muscle cells infected with control retrovirus do not increase in size (Fig. 6 A) or in the percentage of myotubes with five or more nuclei (Fig. 6 B). Consistent with previous results (Horsley et al., 2001), expression of a recombinant NFATC2 in NFATC2−/ − muscle cells restores myotube growth. However, NFATC2−/− cells expressing a recombinant NFATC2 and treated with PGF2α exhibit a greatly enhanced cell size relative to similarly treated wild-type cells that is associated with an increase in nuclear number. These data further demonstrate that PGF2α-induced muscle growth is mediated through NFATC2-dependent pathways.
Figure 6.

PGF2 α induces hypertrophy of NFATC2 −/− myotubes expressing recombinant NFATC2. (A) Primary NFATC2−/− myoblasts were infected either with control (Cntl RV) or recombinant NFATC2 retrovirus (NFATC2 RV). Cells were induced to differentiate and treated with vehicle or 10−6 M PGF2α. After 48 h, the cells were immunostained for EMyHC. Bar, 60 μm. (B) Nuclear number assays were performed as in Fig. 1 E. Data are the mean ± SEM of three independent experiments. *Significantly different from vehicle-treated cells infected with Cntl RV, P < 0.05; **Significantly different from vehicle treated cells infected with NFATC2 RV, P < 0.05.
Discussion
The majority of molecules known to regulate muscle cell fusion in mammals mediate the initial formation of a multinucleated muscle cell (Knudsen, 1992; Yagami-Hiromasa et al., 1995; Barnoy et al., 1996; Gorza and Vitadello, 2000). Recent work in Drosophila (Rau et al., 2001) and in mammals (Horsley et al., 2001) has identified genes that regulate initial fusion events that form a myotube distinctly from the cell fusion that occurs with an existing myotube. These data suggest that the formation of a large multinucleated muscle cell involves two stages of fusion. Thus, initially, a subset of mononucleated muscle cells fuse with each other to form small nascent myotubes containing several nuclei. Subsequently, additional differentiated muscle cells fuse with the nascent myotube and muscle growth occurs. In this paper, we show that PGF2α does not act on the initial fusion of muscle cells that forms myotubes because PGF2α does not affect the percentage of nuclei in myotubes (Fig. 1 D); rather, PGF2α is a novel regulator of the second stage of muscle cell fusion as it increases the number of nuclei within myotubes (Fig. 1, E and F). Treatment of muscle cells after the formation of myotubes has begun is sufficient to induce this increase in myonuclear number (Fig. 1 G). Thus, in contrast to other molecules, PGF2α mediates muscle cell growth by enhancing myonuclear accretion in the nascent myotube. Because myoblast fusion requires multiple steps (Wakelam, 1985), PGF2α could enhance myonuclear accretion by effecting cell motility, alignment, recognition, adhesion, or membrane union.
PGs are a large family of molecules and only a few specific PGs have been studied in myogenesis. Previously, the effect of PGF2α on muscle cell fusion was examined with doses of PGF2α higher than 10−6 M (Rossi et al., 1989). No effect was seen with these doses, which is consistent with our results in Fig. 1 E. Other PGs have been shown to regulate muscle cell fusion. PGE2 and PGE1 regulate fusion by inhibiting and increasing the initial fusion of myoblasts, respectively (Zalin, 1977; Entwistle et al., 1986; Rossi et al., 1989). Consistent with a general role for PGs in muscle cell fusion and growth, inhibition of PG synthesis with inhibitors of the cyclooxygenase enzymes inhibits myoblast fusion (Zalin, 1977; David and Higginbotham, 1981; Entwistle et al., 1986) and the growth of myofibers in vivo (Templeton et al., 1986; McLennan, 1987). Given the differential effect of individual PG molecules on skeletal muscle cells, the synthesis of PG molecules is likely coordinated to control multiple steps of muscle growth.
What is the signal transduction pathway by which PGF2α induces muscle growth? Our data are consistent with a role for the PGF2α receptor, FP. Treatment of cells with fluprostenol, a specific FP receptor agonist, can activate increases in myotube size and nuclear number, similar to PGF2α (Fig. 2). Muscle growth is inhibited by AL-8810, a specific FP receptor antagonist. In addition, the FP receptor is expressed in skeletal muscle cells at the time of growth and fusion (Fig. 5 C). Several lines of evidence indicate that downstream of FP receptor activation a calcineurin/NFAT-dependent pathway is the key requirement for PGF2-induced muscle growth. Expression of a specific inhibitor of NFAT activation by calcineurin completely abrogates the effects of PGF2α on cell growth. In addition, PGF2α induces nuclear translocation of NFATC2 in a calcineurin and FP receptor-dependent manner. Furthermore, increased cell size or myonuclear number does not occur in response to PGF2α in NFATC2−/− cells, but is restored when recombinant NFATC2 is introduced into the cells. Indirectly, the time in myogenesis in which PGF2α is effective also supports a role for the involvement of NFAT. The timing of PGF2α action on myotube growth (Fig. 1 G) concurs with the timing of NFATC2 activation in skeletal muscle cells (Abbott et al., 1998), and is consistent with a role for NFATC2 during the second stage of cell fusion during myotube growth (Horsley et al., 2001). Together, these data strongly support a requirement for NFATC2 in PGF2α-induced skeletal muscle growth, and implicate a calcium signaling pathway downstream of PGF2α in skeletal muscle growth. The mechanism by which NFATC2 regulates muscle growth is unknown. Few NFAT-regulated genes are known in cell types outside of the immune system (Horsley and Pavlath, 2002). The identification of the genes regulated by the PGF2α-NFATC2 signaling pathway in skeletal muscle may reveal novel mechanisms of muscle growth.
Activity is a potent stimulus for muscle growth. Such muscle stimulation leads to release of PGF2α (Vandenburgh et al., 1995; Trappe et al., 2001) as well as activation of transcription factors such as NFAT (Liu et al., 2001; Kubis et al., 2002). The PGF2α-NFATC2 pathway described here may contribute to regulating muscle growth in vivo. PGF2α stimulation of cells overexpressing recombinant NFATC2 leads to enhanced cell fusion, and dramatic increases in cell size or hypertrophy (Fig. 6). PGF2α can induce hypertrophy of other muscle cell types. Hypertrophic growth of cardiac myocytes in vitro is stimulated by PGF2α (Adams et al., 1996; Lai et al., 1996; Kunapuli et al., 1998). In addition, exogenous PGF2α can stimulate vascular smooth muscle hypertrophy (Dorn et al., 1992). Because cardiac and smooth muscle are mononucleated cells, hypertrophy of these cell types does not require cell fusion but involves protein accumulation and thus differs from hypertrophy of skeletal muscle cells, which involves both cell fusion and increased protein accumulation. Other studies have demonstrated that PGF2α does increase protein synthesis in skeletal muscle cells (Vandenburgh et al., 1990). By activating both cell fusion and protein synthesis, PGF2α may regulate hypertrophy of skeletal muscle in vivo.
The function of the PGF2α-NFATC2 pathway in skeletal muscle may be clinically relevant. Nonsteroidal antiinflammatory drugs such as celebrex and ibuprofen are a widespread treatment for inflammation and pain relief. Often these drugs are prescribed after surgery or muscle injury. As these drugs are inhibitors of cyclooxygenase enzyme activity, PG synthesis may be decreased and lead to deleterious effects in skeletal muscle (Mishra et al., 1995), as recently shown for bone (Simon et al., 2002; Zhang et al., 2002). Our data suggest that by blocking the production of PGF2α in skeletal muscle, muscle growth and repair may be impaired in patients taking nonsteroidal antiinflammatory drugs after muscle atrophy, disease, or injury.
Materials and methods
Cell culture
Primary myoblast cultures were prepared from tibalis anterior muscles of three adult wild-type and three NFATC2−/− Balb/c mice (Horsley et al., 2001) and purified to >99% as described previously (Rando and Blau, 1994; Pavlath et al., 1998). Growth media consisted of Ham's F10, 20% FBS, 5 ng/ml bFGF, 100 U/ml penicillin G, and 100 μg/ml streptomycin. Differentiation was induced by plating cells on E-C-L (Upstate Biotechnology)–coated dishes and switching the media to a low serum DM in DME with either 2% horse serum or insulin-transferrin-selenium-A supplement (GIBCO BRL).
Plasmid production, retroviral infection, and transient transfections
The GFP-VIVIT and NFATC2 retroviral constructs have been described previously (Friday et al., 2000; Horsley et al., 2001). An NFATC2-GFP construct was created by PCR amplification of NFATC2 cDNA from the vector pREP4-NFATC2 to generate a 2.8-kb product (Ranger et al., 2000). The forward primer consisted of an EcoRI site followed by bases 224–241 of the human NFATC2 mRNA (GenBank/EMBL/DDBJ accession no. U43342), whereas the reverse primer contained a XmaI site and bases 2978–2995, which deleted the native stop codon of NFATC2. The amplified NFATC2 cDNA was then cloned into pEGFP-N1 (CLONTECH Laboratories, Inc.), preceding the EGFP coding sequence. An NFAT-αMHC enhancer-luciferase construct containing the minimal α-MHC promoter linked to nine copies of a consensus NFAT binding site without an associated AP-1 site was a gift from J. Molkentin (Children's Hospital Medical Center, Cincinnati, OH).
Production of infectious retrovirus and infection of primary myoblasts were performed as described previously (Abbott et al., 1998). Cells were subject to two rounds of infection with an efficiency of gene transfer of >90% based on visualization of green fluorescent protein. Experiments using the NFATC2 retrovirus were begun 48 h after the final infection when maximum levels of gene expression were achieved. In contrast, experiments using VIVIT retrovirus were begun 24 h after the final infection, before maximum levels of gene expression in order for normal myotube formation to occur.
For transient transfections, myoblasts were plated in 6-well dishes at 2.5 × 105 cells/well. For each well, 4 μg DNA was complexed with 6 μl Lipofectamine 2000 (Promega) for 20 min at room temperature in a total volume of 500 μl in Ham's F10. The DNA–Lipofectamine mixture was added to wells containing 2 ml Ham's F10 and incubated for 4 h at 37°C after which the cells were re-fed with fresh growth media.
Drug treatment
Primary myoblasts were plated at 2 × 105 cells per well of 6-well dishes. After 2 h, cells were placed in DM with either vehicle (0.095% ethanol), PGF2α (Sigma-Aldrich), 17-phPGF2α (Cayman Chemical), or fluprostenol (BIOMOL Research Laboratories, Inc.), and drugs were replenished at 24 h unless otherwise noted. Cells were treated with AL-8810 (Sigma-Aldrich) after 24 h in DM. Doses were chosen and used in the range where maximal effects were shown in a variety of assays using cardiac (Adams et al., 1996; Kunapuli et al., 1998; Yew et al., 1998) and smooth muscle cells (Griffin et al., 1998; Katsuyama et al., 2002).
Differentiation and fusion assays
After 24 or 48 h in DM, cells were fixed in either ice-cold methanol or 3.7% formaldehyde for 10 min and nonspecific binding was blocked with TNB buffer (NEN Life Science Products) for 1 h at room temperature. The cells were incubated with an antibody against EMyHC (F1.652, neat hybridoma supernatant; Developmental Studies Hybridoma Bank) for 1 h at room temperature. Cells were washed in PBS with 0.1% Tween, and then incubated in biotinylated goat anti–mouse IgG (1:200; Jackson ImmunoResearch Laboratories). Antibody binding was detected using Vectastain Elite ABC reagent (Vector Laboratories) and diaminobenzidine.
To analyze differentiation, the number of nuclei in EMyHC-positive cells was counted and expressed as a percentage of the total number of nuclei analyzed (250). The fusion index was determined by dividing the number of nuclei within myotubes (two or more nuclei) by the total number of nuclei analyzed (100–250). Fusion was also analyzed by performing nuclear number assays. The number of nuclei within individual myotubes was counted for 50–100 myotubes. Myotubes were grouped into two categories: those with two to four nuclei and those with five or more nuclei. The percentage of myotubes in each category was calculated.
DNA quantification
The DNA content of whole cell lysates was quantified after 48 h in DM for vehicle and PGF2α-treated cells. Cells were washed twice in ice-cold PBS, scraped off of the dish, and collected by centrifugation at 10,000 g at 4°C. Cell lysates were resuspended in 250 μl of saline phosphate buffer (0.05 M NaPO4, and 2 M NaCl, pH 7.4) and frozen. Samples were thawed and sonicated for 15 s, and a 50-μl aliquot of each sample was added to the buffer containing 0.5 μg/ml Hoechst 33258 (Molecular Probes). DNA concentration was determined by measuring the emission at 465 nm after excitation at 365 nm (Labarca and Paigen, 1980) using an Amicon-Bowman luminescence spectrophotometer (Spectronic Instruments). Calf thymus DNA was used to construct the standard curve.
FP receptor RT-PCR
RNA was isolated from wild-type and NFATC2−/− muscle cells after 24 h in DM using TRIzol reagent (Life Technologies). RT-PCR was performed for each sample using specific primers for the murine FP receptor (GenBank/EMBL/DDBJ accession no. D17433): sense, 5′-CACAACCTGCCAGACGAGAACC-3′; antisense, 5′-ATGGGCAGCACAGCCACGAAC-3′; 452 base pair. 18S rRNA was used as an internal control in each sample using QuantumRNA 18S primers (Ambion). The amplification cycle for the FP receptor consisted of 94°C for 5 min, followed by 35 cycles of 94°C for 30 s, 61.5°C for 30 s, 72°C for 45 s, and termination at 72°C for 5 min. The amplification cycle for the 18S rRNA consisted of 94°C for 5 min, followed by 30 cycles of 94°C for 30 s, 57°C for 30 s, 72°C for 30 s, and termination at 72°C for 5 min. The products were resolved by electrophoresis on a 1.5% agarose gel and visualized by ethidium bromide staining.
NFATC2 nuclear translocation
24 h after transient transfection, cells were plated at 2 × 105 cells per well of ECL-coated 6-well plates, and switched to DM after 2 h. After 24 h in DM, cells were placed in DME for 3 h, treated with 10−6 M PGF2α alone or in combination with 10−6 M CsA or 10−6 M AL-8810 for 30 min, and fixed with 3.7% formaldehyde.
Reporter assays
24 h after transient transfection with an NFAT reporter construct, myoblasts were plated at 7.5 × 104 cells per well of ECL-coated 24-well plate. The medium was replaced with DME and the cells were allowed to differentiate for 24 h. Cells were stimulated with 10−6 M PGF2α alone or in combination with 10−6 M CsA for 5 h and assayed for luciferase as described previously (Abbott et al., 1998).
Statistics
To determine significance between two groups, comparisons were made using t tests. Analyses of multiple groups were performed using one-way ANOVA with Bonferroni's post test using GraphPad Prism version 3.0a for Macintosh (GraphPad Software). For all statistical tests, the 0.05 level of confidence was accepted for statistical significance.
Acknowledgments
We wish to thank Drs. J. Molkentin for the NFAT-αMHC enhancer luciferase construct and L. Glimcher for the pREP4-NFATC2 construct.
This work is supported by National Institutes of Health, grants AR-47314, DE-13040, and AR-48884 (to G.K. Pavlath). V. Horsley is supported by NIH Training grant T32-GM08367.
Footnotes
Abbreviations used in this paper: CsA, cyclosporine A; DM, differentiation media; EMyHC, embryonic myosin heavy chain; FP receptor, PGF2α receptor; NFAT, nuclear factor of activated T cells; PG, prostaglandin; 17-phPGF2α, 17-phenyl trinor PGF2α.
References
- Abbott, K.L., B.B. Friday, D. Thaloor, T.J. Murphy, and G.K. Pavlath. 1998. Activation and cellular localization of the cyclosporine A-sensitive transcription factor NF-AT in skeletal muscle cells. Mol. Biol. Cell. 9:2905–2916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams, J.W., D.S. Migita, M.K. Yu, R. Young, M.S. Hellickson, F.E. Castro-Vargas, J.D. Domingo, P.H. Lee, J.S. Bui, and S.A. Henderson. 1996. Prostaglandin F2 alpha stimulates hypertrophic growth of cultured neonatal rat ventricular myocytes. J. Biol Chem. 271:1179–1186. [DOI] [PubMed] [Google Scholar]
- Allen, D.L., R.R. Roy, and V.R. Edgerton. 1999. Myonuclear domains in muscle adaptation and disease. Muscle Nerve. 22:1350–1360. [DOI] [PubMed] [Google Scholar]
- Aramburu, J., M.B. Yaffe, C. Lopez-Rodriguez, L.C. Cantley, P.G. Hogan, and A. Rao. 1999. Affinity-driven peptide selection of an NFAT inhibitor more selective than cyclosporin A. Science. 285:2129–2133. [DOI] [PubMed] [Google Scholar]
- Asboth, G., S. Phaneuf, G.N. Europe-Finner, M. Toth, and A.L. Bernal. 1996. Prostaglandin E2 activates phospholipase C and elevates intracellular calcium in cultured myometrial cells: involvement of EP1 and EP3 receptor subtypes. Endocrinology. 137:2572–2579. [DOI] [PubMed] [Google Scholar]
- Barnoy, S., T. Glasner, and N.S. Kosower. 1996. The role of calpastatin (the specific calpain inhibitor) in myoblast differentiation and fusion. Biochem. Biophys. Res. Commun. 220:933–938. [DOI] [PubMed] [Google Scholar]
- Barton-Davis, E.R., D.I. Shoturma, and H.L. Sweeney. 1999. Contribution of satellite cells to IGF-I induced hypertrophy of skeletal muscle. Acta Physiol. Scand. 167:301–305. [DOI] [PubMed] [Google Scholar]
- Bate, M. 1990. The embryonic development of larval muscles in Drosophila. Development. 110:791–804. [DOI] [PubMed] [Google Scholar]
- Breyer, R.M., C.K. Bagdassarian, S.A. Myers, and M.D. Breyer. 2001. Prostanoid receptors: subtypes and signaling. Annu. Rev. Pharmacol. Toxicol. 41:661–690. [DOI] [PubMed] [Google Scholar]
- Chen, W., T. Andom, P. Bhattacherjee, and C. Paterson. 1997. Intracellular calcium mobilization following prostaglandin receptor activation in human ciliary muscle cells. Curr. Eye Res. 16:847–853. [DOI] [PubMed] [Google Scholar]
- Darr, K.C., and E. Schultz. 1989. Hindlimb suspension suppresses muscle growth and satellite cell proliferation. J. Appl. Physiol. 67:1827–1834. [DOI] [PubMed] [Google Scholar]
- David, J.D., and C.A. Higginbotham. 1981. Fusion of chick embryo skeletal myoblasts: interactions of prostaglandin E1, adenosine 3′:5′ monophosphate, and calcium influx. Dev. Biol. 82:308–316. [DOI] [PubMed] [Google Scholar]
- Delling, U., J. Tureckova, H.W. Lim, L.J. De Windt, P. Rotwein, and J.D. Molkentin. 2000. A calcineurin-NFATc3-dependent pathway regulates skeletal muscle differentiation and slow myosin heavy-chain expression. Mol. Cell. Biol. 20:6600–6611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorn, G.W. II, M.W. Becker, and M.G. Davis. 1992. Dissociation of the contractile and hypertrophic effects of vasoconstrictor prostanoids in vascular smooth muscle. J. Biol. Chem. 267:24897–24905. [PubMed] [Google Scholar]
- Dunn, S.E., J.L. Burns, and R.N. Michel. 1999. Calcineurin is required for skeletal muscle hypertrophy. J. Biol. Chem. 274:21908–21912. [DOI] [PubMed] [Google Scholar]
- Entwistle, A., D.H. Curtis, and R.J. Zalin. 1986. Myoblast fusion is regulated by a prostanoid of the one series independently of a rise in cyclic AMP. J. Cell Biol. 103:857–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friday, B.B., and G.K. Pavlath. 2001. A calcineurin- and NFAT-dependent pathway regulates Myf5 gene expression in skeletal muscle reserve cells. J. Cell Sci. 114:303–310. [DOI] [PubMed] [Google Scholar]
- Friday, B.B., V. Horsley, and G.K. Pavlath. 2000. Calcineurin activity is required for the initiation of skeletal muscle differentiation. J. Cell Biol. 149:657–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funk, C.D. 2001. Prostaglandins and leukotrienes: advances in eicosanoid biology. Science. 294:1871–1875. [DOI] [PubMed] [Google Scholar]
- Gorza, L., and M. Vitadello. 2000. Reduced amount of the glucose-regulated protein GRP94 in skeletal myoblasts results in loss of fusion competence. FASEB J. 14:461–475. [DOI] [PubMed] [Google Scholar]
- Griffin, B.W., P.E. Magnino, I.H. Pang, and N.A. Sharif. 1998. Pharmacological characterization of an FP prostaglandin receptor on rat vascular smooth muscle cells (A7r5) coupled to phosphoinositide turnover and intracellular calcium mobilization. J. Pharmacol. Exp. Ther. 286:411–418. [PubMed] [Google Scholar]
- Griffin, B.W., P. Klimko, J.Y. Crider, and N.A. Sharif. 1999. AL-8810: a novel prostaglandin F2 alpha analog with selective antagonist effects at the prostaglandin F2 alpha (FP) receptor. J. Pharmacol. Exp. Ther. 290:1278–1284. [PubMed] [Google Scholar]
- Horsley, V., and G.K. Pavlath. 2002. NFAT: ubiquitous regulator of cell differentiation and adaptation. J. Cell Biol. 156:771–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horsley, V., B.B. Friday, S. Matteson, K.M. Kegley, J. Gephart, and G.K. Pavlath. 2001. Regulation of the growth of multinucleated muscle cells by an NFATC2-dependent pathway. J. Cell Biol. 153:329–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsuyama, M., C. Fan, and C. Yabe-Nishimura. 2002. NADPH oxidase is involved in prostaglandin F2alpha-induced hypertrophy of vascular smooth muscle cells: induction of NOX1 by PGF2alpha. J. Biol. Chem. 277:13438–13442. [DOI] [PubMed] [Google Scholar]
- Kegley, K.M., J. Gephart, G.L. Warren, and G.K. Pavlath. 2001. Altered primary myogenesis in NFATC3(−/−) mice leads to decreased muscle size in the adult. Dev. Biol. 232:115–126. [DOI] [PubMed] [Google Scholar]
- Knudsen, K.A. 1992. Membrane fusion. Membrane Fusion. J. Wilschut and D. Hoekstra, editors. Marcel Dekker, Inc., New York. 601–626.
- Knudsen, K.A., and A.F. Horwitz. 1977. Tandem events in myoblast fusion. Dev. Biol. 58:328–338. [DOI] [PubMed] [Google Scholar]
- Kubis, H.P., R.J. Scheibe, J.D. Meissner, G. Hornung, and G. Gros. 2002. Fast-to-slow transformation and nuclear import/export kinetics of the transcription factor NFATc1 during electrostimulation of rabbit muscle cells in culture. J. Physiol. 541:835–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunapuli, P., J.A. Lawson, J.A. Rokach, J.L. Meinkoth, and G.A. FitzGerald. 1998. Prostaglandin F2α (PGF2α) and the isoprostane, 8, 12-iso-isoprostane F2α-III, induce cardiomyocyte hypertrophy. Differential activation of downstream signaling pathways. J. Biol. Chem. 273:22442–22452. [DOI] [PubMed] [Google Scholar]
- Labarca, C., and K. Paigen. 1980. A simple, rapid, and sensitive DNA assay procedure. Anal. Biochem. 102:344–352. [DOI] [PubMed] [Google Scholar]
- Lai, J., H. Jin, R. Yang, J. Winer, W. Li, R. Yen, K.L. King, F. Zeigler, A. Ko, J. Cheng, et al. 1996. Prostaglandin F2 alpha induces cardiac myocyte hypertrophy in vitro and cardiac growth in vivo. Am. J. Physiol. 271:H2197–H2208. [DOI] [PubMed] [Google Scholar]
- Lake, S., H. Gullberg, J. Wahlqvist, A.M. Sjogren, A. Kinhult, P. Lind, E. Hellstrom-Lindahl, and J. Stjernschantz. 1994. Cloning of the rat and human prostaglandin F2 alpha receptors and the expression of the rat prostaglandin F2 alpha receptor. FEBS Lett. 355:317–325. [DOI] [PubMed] [Google Scholar]
- Liu, Y., Z. Cseresnyes, W.R. Randall, and M.F. Schneider. 2001. Activity-dependent nuclear translocation and intranuclear distribution of NFATc in adult skeletal muscle fibers. J. Cell Biol. 155:27–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLennan, I.S. 1987. Hormonal regulation of myoblast proliferation and myotube production in vivo: influence of prostaglandins. J. Exp. Zool. 241:237–245. [DOI] [PubMed] [Google Scholar]
- Mishra, D.K., J. Friden, M.C. Schmitz, and R.L. Lieber. 1995. Anti-inflammatory medication after muscle injury. A treatment resulting in short-term improvement but subsequent loss of muscle function. J. Bone Joint Surg. Am. 77:1510–1519. [DOI] [PubMed] [Google Scholar]
- Mitchell, P.O., and G.K. Pavlath. 2001. A muscle precursor cell-dependent pathway contributes to muscle growth after atrophy. Am. J. Physiol. Cell Physiol. 281:C1706–C1715. [DOI] [PubMed] [Google Scholar]
- Mitchell, P.O., S.T. Mills, and G.K. Pavlath. 2002. Calcineurin differentially regulates maintenance and growth of phenotypically distinct muscles. Am. J. Cell Physiol. 282:C984-C992. [DOI] [PubMed] [Google Scholar]
- Morris, G.E., and R.J. Cole. 1979. Calcium and the control of muscle-specific creatine kinase accumulation during skeletal muscle differentiation in vitro. Dev. Biol. 69:146–158. [DOI] [PubMed] [Google Scholar]
- Musaro, A., K.J. McCullagh, F.J. Naya, E.N. Olson, and N. Rosenthal. 1999. IGF-1 induces skeletal myocyte hypertrophy through calcineurin in association with GATA-2 and NF-ATc1. Nature. 400:581–585. [DOI] [PubMed] [Google Scholar]
- Palmer, R.M. 1990. Prostaglandins and the control of muscle protein synthesis and degradation. Prostaglandins Leukot. Essent. Fatty Acids. 39:95–104. [DOI] [PubMed] [Google Scholar]
- Pavlath, G.K., D. Thaloor, T.A. Rando, M. Cheong, A.W. English, and B. Zheng. 1998. Heterogeneity among muscle precursor cells in adult skeletal muscles with differing regenerative capacities. Dev. Dyn. 212:495–508. [DOI] [PubMed] [Google Scholar]
- Phelan, J.N., and W.J. Gonyea. 1997. Effect of radiation on satellite cell activity and protein expression in overloaded mammalian skeletal muscle. Anat. Rec. 247:179–188. [DOI] [PubMed] [Google Scholar]
- Pierce, K.L., T.J. Bailey, P.B. Hoyer, D.W. Gil, D.F. Woodward, and J.W. Regan. 1997. Cloning of a carboxyl-terminal isoform of the prostanoid FP receptor. J. Biol. Chem. 272:883–887. [DOI] [PubMed] [Google Scholar]
- Rando, T.A., and H.M. Blau. 1994. Primary mouse myoblast purification, characterization, and transplantation for cell-mediated gene therapy. J. Cell Biol. 125:1275–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranger, A.M., L.C. Gerstenfeld, J. Wang, T. Kon, H. Bae, E.M. Gravallese, M.J. Glimcher, and L.H. Glimcher. 2000. The nuclear factor of activated T cells (NFAT) transcription factor NFATp (NFATc2) is a repressor of chondrogenesis. J. Exp. Med. 191:9–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rau, A., D. Buttgereit, A. Holz, R. Fetter, S.K. Doberstein, A. Paululat, N. Staudt, J. Skeath, A.M. Michelson, and R. Renkawitz-Pohl. 2001. Rolling pebbles (rols) is required in Drosophila muscle precursors for recruitment of myoblasts for fusion. Development. 128:5061–5073. [DOI] [PubMed] [Google Scholar]
- Rodemann, H.P., and A.L. Goldberg. 1982. Arachidonic acid, prostaglandin E2 and F2 alpha influence rates of protein turnover in skeletal and cardiac muscle. J. Biol. Chem. 257:1632–1638. [PubMed] [Google Scholar]
- Rosenblatt, J.D., and D.J. Parry. 1992. Gamma irradiation prevents compensatory hypertrophy of overloaded mouse extensor digitorum longus muscle. J. Appl. Physiol. 73:2538–2543. [DOI] [PubMed] [Google Scholar]
- Rossi, M.J., M.A. Clark, and S.M. Steiner. 1989. Possible role of prostaglandins in the regulation of mouse myoblasts. J. Cell. Physiol. 141:142–147. [DOI] [PubMed] [Google Scholar]
- Schutzle, U.B., M.J. Wakelam, and D. Pette. 1984. Prostaglandins and cyclic AMP stimulate creatine kinase synthesis but not fusion in cultured embryonic chick muscle cells. Biochim. Biophys. Acta. 805:204–210. [DOI] [PubMed] [Google Scholar]
- Semsarian, C., M.J. Wu, Y.K. Ju, T. Marciniec, T. Yeoh, D.G. Allen, R.P. Harvey, and R.M. Graham. 1999. Skeletal muscle hypertrophy is mediated by a Ca2+-dependent calcineurin signalling pathway. Nature. 400:576–581. [DOI] [PubMed] [Google Scholar]
- Shainberg, A., G. Yagil, and D. Yaffe. 1969. Control of myogenesis in vitro by Ca2+ concentration in nutritional medium. Exp. Cell Res. 58:163–167. [DOI] [PubMed] [Google Scholar]
- Simon, A., M.B. Manigrasso, and J.P. O'Connor. 2002. Cyclo-oxygenase 2 function is essential for bone fracture healing. J. Bone Miner. Res. 17:963–976. [DOI] [PubMed] [Google Scholar]
- Templeton, G.H., M. Padalino, and R. Moss. 1986. Influences of inactivity and indomethacin on soleus phosphatidylethanolamine and size. Prostaglandins. 31:545–559. [DOI] [PubMed] [Google Scholar]
- Trappe, T.A., J.D. Fluckey, F. White, C.P. Lambert, and W.J. Evans. 2001. Skeletal muscle PGF(2α) and PGE(2) in response to eccentric resistance exercise: influence of ibuprofen acetaminophen. J. Clin. Endocrinol. Metab. 86:5067–5070. [DOI] [PubMed] [Google Scholar]
- Vandenburgh, H.H., S. Hatfaludy, I. Sohar, and J. Shansky. 1990. Stretch-induced prostaglandins and protein turnover in cultured skeletal muscle. Am. J. Physiol. 259:C232–C240. [DOI] [PubMed] [Google Scholar]
- Vandenburgh, H.H., J. Shansky, R. Solerssi, and J. Chromiak. 1995. Mechanical stimulation of skeletal muscle increases prostaglandin F2 alpha production, cyclooxygenase activity, and cell growth by a pertussis toxin sensitive mechanism. J. Cell. Physiol. 163:285–294. [DOI] [PubMed] [Google Scholar]
- Wakelam, M.J. 1985. The fusion of myoblasts. Biochem. J. 228:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagami-Hiromasa, T., T. Sato, T. Kurisaki, K. Kamijo, Y. Nabeshima, and A. Fujisawa-Sehara. 1995. A metalloprotease-disintegrin participating in myoblast fusion. Nature. 377:652–656 (see comments). [DOI] [PubMed] [Google Scholar]
- Yew, S.F., K.A. Reeves, and B. Woodward. 1998. Effects of prostaglandin F2 α on intracellular pH, intracellular calcium, cell shortening and L-type calcium currents in rat myocytes. Cardiovasc. Res. 40:538–545. [DOI] [PubMed] [Google Scholar]
- Yousufzai, S.Y., and A.A. Abdel-Latif. 1998. Tyrosine kinase inhibitors suppress prostaglandin F2α-induced phosphoinositide hydrolysis, Ca2+ elevation and contraction in iris sphincter smooth muscle. Eur. J. Pharmacol. 360:185–193. [DOI] [PubMed] [Google Scholar]
- Zalin, R.J. 1977. Prostaglandins and myoblast fusion. Dev. Biol. 59:241–248. [DOI] [PubMed] [Google Scholar]
- Zalin, R.J. 1987. The role of hormones and prostanoids in the in vitro proliferation and differentiation of human myoblasts. Exp. Cell Res. 172:265–281. [DOI] [PubMed] [Google Scholar]
- Zhang, X., E.M. Schwarz, D.A. Young, J.E. Puzas, R.N. Rosier, and R.J. O'Keefe. 2002. Cyclooxygenase-2 regulates mesenchymal cell differentiation into the osteoblast lineage and is critically involved in bone repair. J. Clin. Invest. 109:1405–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]