

Abstract
Extracellular matrix (ECM) fragments or cryptic sites unmasked by proteinases have been postulated to affect tissue remodeling and cancer progression. Therefore, the elucidation of their identities and functions is of great interest. Here, we show that matrix metalloproteinases (MMPs) generate a domain (DIII) from the ECM macromolecule laminin-5. Binding of a recombinant DIII fragment to epidermal growth factor receptor stimulates downstream signaling (mitogen-activated protein kinase), MMP-2 gene expression, and cell migration. Appearance of this cryptic ECM ligand in remodeling mammary gland coincides with MMP-mediated involution in wild-type mice, but not in tissue inhibitor of metalloproteinase 3 (TIMP-3)–deficient mice, supporting physiological regulation of DIII liberation. These findings indicate that ECM cues may operate via direct stimulation of receptor tyrosine kinases in tissue remodeling, and possibly cancer invasion.
Keywords: ECM; MMP-2 gene expression; microarray; receptor tyrosine kinase; TIMP-3 knockout mouse
Introduction
Disruption of ECM architecture via degradation of its component macromolecules by proteolytic enzymes, such as metalloproteinases, plays a crucial role in tissue remodeling. Proteolysis creates migratory paths, frees signaling molecules including growth factors latently immobilized within the ECM, and generates biologically active neo-epitopes and ECM fragments (Foda and Zucker, 2001; Kleinman et al., 2001; Simian et al., 2001; Stetler-Stevenson and Yu, 2001; Egeblad and Werb, 2002). These cues may direct the invasion of surrounding stroma by normal epithelial cells during tissue regeneration and remodeling, or trigger mobility of cancerous epithelial cells in the initial phases of metastasis.
Basement membranes (BMs)* are thin sheets of specialized ECM supporting epithelial cell layers. A major component of epithelial BM, laminin-5 (Ln-5), has been implicated in cell migration and tumor invasion. Ln-5 has a cruciform structure with one long and three short arms. The coiled-coil structure of the long arm is formed by three chains (α3, β3, and γ2) covalently linked via interchain disulfide bonds (Yurchenco and Cheng, 1993). The rodlike regions in the short arms are composed of EGF-like repeats intercalated with globular domains (Engel et al., 1994). Several laboratories have reported enhanced expression of Ln-5, especially its γ2 subunit, at sites of tumor cell penetration in histological specimens of breast, colon, gastric, and other types of cancers (Pyke et al., 1994; Kagesato et al., 2001; Yamamoto et al., 2001; Niki et al., 2002). Ln-5 could facilitate tumor invasion by promoting at least two relevant cellular functions; adhesion and migration.
Recently, we have shown that binding of the globular LG3 domain (α3 chain) of Ln-5 to integrin α3β1 mediates cell adhesion and cell migration (Shang et al., 2001). Adhesion may also be promoted by the Ln-5 receptor α6β4 integrin, which is involved in formation of hemidesmosomes (Borradori and Sonnenberg, 1999). Robust cell migration on Ln-5 was shown to be triggered by cleavage of its γ2-chain by MMP-2 (Giannelli et al., 1997) and MT1-MMP (Koshikawa et al., 2000; Gilles et al., 2001). The suggested cleavage sites imply the liberation of the entire Ln-5 γ2 short arm (domains III, IV, and V) as well as of domains IV and V, or domain III alone. A fragment comprising domains IV and V has recently been found to mediate integration of Ln-5 into the ECM and to promote cell adhesion (Gagnoux-Palacios et al., 2001). Domain III is comprised of EGF-like repeats, and for this reason seems especially interesting. Not only is its structure reminiscent of epidermal growth factor receptor (EGFR) ligands, but several studies with synthetic peptides or laminin-1 (Ln-1) fragments containing EGF-like repeats have demonstrated effects on cell migration (Salo et al., 1999; Kleinman et al., 2001), possibly involving EGFR (Panayotou et al., 1989). Given the multi-domain architecture of Ln-5, it seems conceivable that other receptors in addition to integrins interact with one of its many potential ligand sites to mediate its diverse cellular functions.
Here, we address the hypothesis that domain III of the Ln-5 γ2-chain is a cryptic migratory signal liberated by MMP action. We have produced domain III recombinantly (rDIII), attempted to assign the migratory activity of cleaved Ln-5 to this specific fragment, and investigated a possible engagement of EGFR. Also, we have studied the in vivo proteolytic generation of this ECM fragment.
Results
Expression and characterization of recombinant γ2 subunit DIII
Previously, we showed that MMP-2 (Giannelli et al., 1997) and MT1-MMP cleave the γ2 subunit of Ln-5 (Koshikawa et al., 2000). The positions of the MT1-MMP cleavage sites have now been identified (Fig. 1 A; unpublished data).
Figure 1.
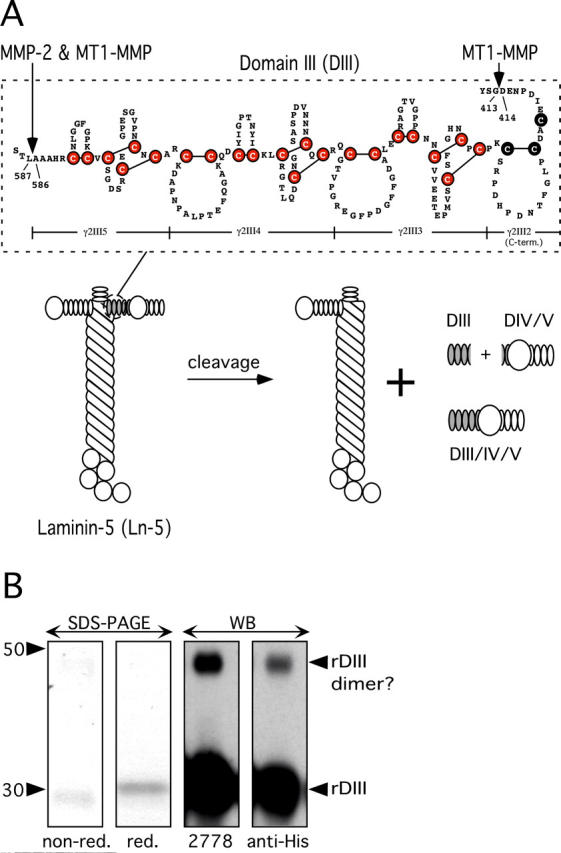
Characterization of DIII of Ln-5 γ2-chain. (A) Schematic depiction of DIII. rDIII encompasses the most COOH-terminal 3.5 EGF-like repeats (γ2III5, 4, 3, and COOH-terminal part of 2) of rat Ln-5 γ2-chain DIII. Six cysteines (filled circles) highlight the EGF-like domain signature, and the LE repeat signature is characterized by eight cysteines and an additional loop. The position of rDIII within the Ln-5 cruciform structure and the products of MMP cleavage are shown. (B) Characterization of rDIII using SDS-PAGE and WB. In SDS-PAGE (two left panels), rDIII resolved as a single band under both nonreducing and reducing conditions. Purified rDIII was judged to be better than 95% homogenous by Coomassie blue staining. In WB of reducing PAGE (two right panels), the rDIII band was recognized by both 2778 and anti-His-tag, as expected. A much fainter, higher mol wt band was also visible, which is likely dimerized rDIII. The apparent mol wt was calculated based on pre-stained mol wt standard SeeBlue® (Invitrogen).
Because γ2-chain DIII is bracketed by MMP cleavage sites and its structure is reminiscent of EGFR ligands, we tested the hypothesis that MMP proteolytic fragments such as DIII may have biological activity. We produced rDIII in baculovirus, using rat Ln-5 γ2 cDNA as a template. The NH2-terminal boundary of rDIII (D414 E415 N416) coincides with the MT1-MMP cleavage site (YSG⇓DEN), whereas the COOH terminus is 17 residues downstream from the MMP-2 cleavage site (AAA⇓LTS), which is also recognized by MT1-MMP (Fig. 1 A). rDIII includes an NH2-terminal stretch of five amino acids (ADLGS) derived from the baculovirus vector, and a COOH-terminal His6 tag.
Mass spectrometry (matrix-associated laser desorption ionization) revealed a mol wt of 21,531 for purified rDIII, in excellent agreement with its calculated mol wt of 21,525. However, rDIII resolved as a major 30-kD band under reducing conditions and as an ∼20-kD band under nonreducing conditions in SDS-PAGE (Fig. 1 B). This anomalous migration agrees with previous findings for Ln-1 fragments containing EGF-like repeats (Mayer et al., 1993). In Western blots (WBs) of reducing PAGE gels, both antibodies 2778 and anti-His recognized a 30-kD band. Both antibodies reacted with a minor 50-kD band, likely a dimerized form of rDIII. To further characterize the 30-kD band, its NH2-terminal sequence was determined by automated Edman degradation, and its first seven amino acids perfectly matched the expected sequence of the cloned rDIII (unpublished data). Together, these results confirm the identity of the recombinant protein rDIII.
Cell surface binding of rDIII
To determine whether rDIII binds to the cell surface, MDA-MB-231 breast cancer cells were incubated with rDIII, followed by excess 2778 and fluorescently labeled anti-IgG secondary antibody. Flow cytometry showed dose-dependent staining of rDIII-treated cells. Saturation of rDIII–cell surface interactions occurred at ∼4.5 μM rDIII, resulting in fluorescence ∼12-fold higher than the control (Fig. 2 A). Detection of rDIII with anti-His antibody gave similar results (unpublished data). Another recombinant His-tagged γ2 fragment, encompassing DIII to DV (rDIII-V), did not bind to MDA-MB-231 cell surfaces (unpublished data).
Figure 2.
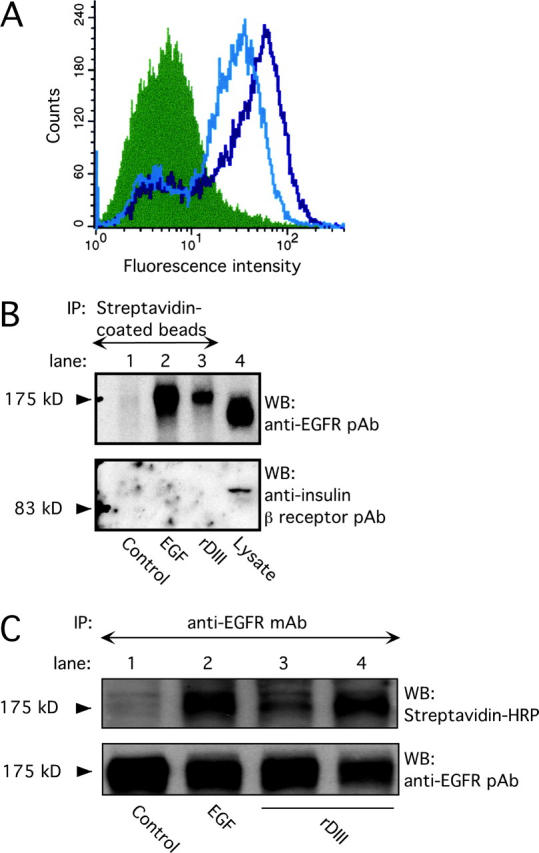
Binding of rDIII to EGFR. (A) rDIII binding to cell surfaces detected by flow cytometry. MDA-MB-231 cells were incubated with 4.5 (open black histogram) or 2 μM (open gray) rDIII or control rabbit IgG (filled), followed by 2778, and the appropriate Alexa®-conjugated secondary antibody. (B) Recovery of biotin–rDIII–EGFR complexes with streptavidin-coated beads. 1.5 μM biotinylated rDIII or 0.75 μM EGF was incubated with MDA-MB-231 cells, followed by cross-linking with BS3. After detergent solubilization, cell lysates were precipitated with streptavidin-coated beads. WB of adsorbed material with EGFR pAb (top) detected a distinct band of 175 kD in samples containing rDIII (lane 3) or EGF (lane 2), but not in control samples (lane 1, no ligand). To control for EGFR expression and specificity of cross-linking to EGFR, total MDA-MB-231 cell lysates were loaded in lane 4 and stripped blots were treated with anti-insulin receptor β antibody (bottom), respectively. (C) Immunoprecipitation of biotin–rDIII–EGFR complexes with antibodies to EGFR. Cells were treated with biotinylated rDIII or EGF and BS3, and cell lysates were immunoprecipitated with EGFR mAb. Samples were analyzed by WB using streptavidin-HRP and ECL. A distinct band at 175 kD was visible for samples containing EGF (lane 2, 0.75 μM) or rDIII (lane 3, 1.0 μM, and lane 4, 1.5 μM; top). There is no corresponding band in the control lane (lane 1; no ligand). Note, the resolution of the gradient gels used is not sufficient to distinguish between EGF or rDIII bound to EGFR, where the former would be expected to run at ∼180 kD and the latter at ∼195 kD. To ensure equal loading in each lane, the filter was stripped and reprobed with EGFR pAb (bottom).
rDIII binding to EGFR
To investigate whether DIII binds to a specific receptor, rDIII was biotinylated and incubated with MDA-MB-231 cells in the presence of bis(sulfosuccinimidyl) suberate (BS3). Cells were then washed, detergent solubilized, and the lysates were allowed to react with streptavidin-coated beads. Because rDIII is EGF-like, its binding to EGFR (ErbB1) was tested. The material adsorbed to the streptavidin beads was separated by PAGE and analyzed by WB using an EGFR-specific antibody (Fig. 2 B). A band with a mol wt matching that of EGFR (175,000) was detected in the presence of rDIII, whereas in the absence of ligand, no band was evident. As further controls, parallel incubations were performed with biotinylated mouse EGF, resulting again in a band of 175 kD, and blotting with an anti-insulin β-receptor antibody detected insulin receptor in total lysate but not in streptavidin-precipitated material.
In complementary experiments, biotin-tagged EGF and rDIII were cross-linked, and EGFR was immunoprecipitated with anti-EGFR mAb. WB with HRP streptavidin (Fig. 2 C) revealed a band at 175 kD in the ligand-containing samples, but not in the control sample. A stronger signal from biotin-tagged rDIII was seen when the concentration of rDIII was increased. Stripping the blot and reprobing with anti-EGFR antibody demonstrated the presence of EGFR in all samples. These results indicate that rDIII binds to EGFR.
EGFR autophosphorylation upon treatment with rDIII
MDA-MB-231 cells treated with rDIII for 5 min in the presence of BS3 showed distinct EGFR phosphorylation (Fig. 3 A). BS3 alone did not induce EGFR phosphorylation. In control experiments without cross-linker, significant EGFR phosphorylation was detected in samples containing rDIII (Fig. 3 B) or EGF, whereas control samples devoid of ligand failed to show EGFR phosphorylation. These data show that rDIII stimulates EGFR phosphorylation.
Figure 3.

Induction of EGFR tyrosine phosphorylation by rDIII. (A) Incubation of MDA-MB-231 cells with 185 nM rDIII for 5 min (lane 4, top) stimulated phosphorylation of EGFR. There is no EGFR stimulation for the 2-min rDIII sample (lane 3), or the 5-min no ligand control (lane 1). To exclude nonspecific effects due to cross-linking, cells were exposed to BS3 in the absence of ligand (lane 2). To ensure that equal amounts of EGFR protein were loaded, blots were stripped and reprobed with EGFR pAb (bottom). (B) EGFR phosphorylation by 185 nM rDIII (lane 1, top) and 1.7 nM EGF (lane 2) for 5 min in the absence of BS3. For control, ligand was omitted (lane 3), and the loading controls are shown in the bottom panel.
Competition receptor binding between rDIII and EGF
To test the specificity of rDIII binding to EGFR, MDA-MB-231 cells were incubated with rDIII in the presence of increasing concentrations of mouse EGF, and analyzed by flow cytometry. As shown in Fig. 4 A, EGF decreased the fluorescence signal of bound rDIII in a dose-dependent fashion. This competitive displacement of rDIII by EGF strongly supports the specificity of rDIII binding to EGFR.
Figure 4.
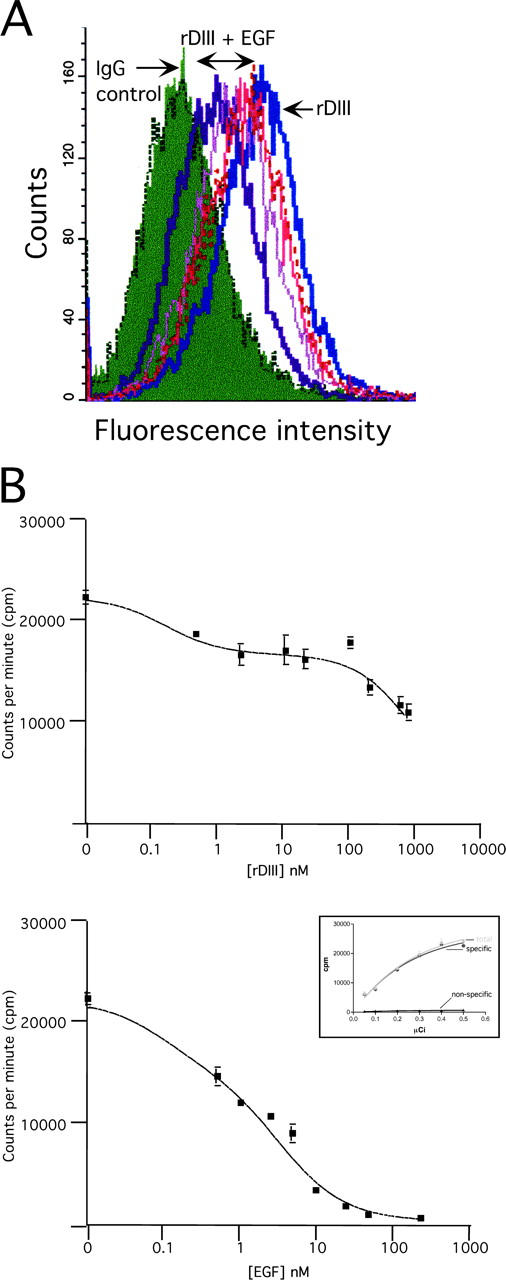
Competitive binding of rDIII and EGF. (A) Flow cytometry. MDA-MB-231 cells were incubated with 2.00 μM rDIII, in the presence of increasing concentrations of EGF (0.45, 0.85, 1.25, and 2.00 μM). rDIII binding to the cell surface was detected with anti-His tag and Alexa® 488 antibodies. The fluorescence signal for rDIII gradually decreases with increasing EGF concentrations. (B) Displacement of cell surface-bound I125-EGF by rDIII. MDA-MB-231 cells were incubated with I0.5 nM 125-EGF and increasing concentrations of cold rDIII (top) or EGF (bottom). The 0.5 nM (≈0.15 μCi) working concentration of I125-EGF was determined by calculating the specific binding of I125EGF (“specific”) based on total and nonspecific binding of I125-EGF to MDA-MB-231 cells (inset in bottom panel). Cells were incubated with increasing concentrations of I125-EGF in the absence (total binding; “total”) or presence of an excess amount (330 nM) of unlabeled EGF (nonspecific binding; “nonspecific”).
In a reverse experiment, MDA-MB-231 cells were incubated with 125I-EGF and increasing concentrations of either cold rDIII (Fig. 4 B, top), or EGF (bottom). Data were analyzed and depicted as competition curves (Fig. 4 B). In accordance with our own judgement the software “Fit option” chose the two-site competition equations as best fits for EGF and rDIII with P-values of P < 0.0001. The calculated IC50 for the competition assays shown in Fig. 4 B were as follows: for EGF, IC50_1 = 0.1 nM and IC50_2 = 3.7 nM; for rDIII, IC50_1 = 0.18 nM and IC50_2 = 1185 nM. In all displacement assays performed, the IC50_1 for rDIII was consistently between 0.17 and 0.2 nM, and the IC50_2 between 349 and 1487 nM. Variability of receptor status of MDA-MB-231 cells in various assays may be responsible for the unsteady IC50_2 of rDIII. In our hands, rDIII is not able to cause a >50% inhibition of 125I-EGF binding. It is possible that the strong tendency of rDIII to dimerize at higher concentrations and/or the recombinant nature of rDIII, with a COOH-terminal His6 tag and an NH2-terminal stretch of vector-derived amino acids, may account for reduced binding ability of rDIII. In summary, these results suggest that rDIII competes for 125I-EGF binding to more than one EGFR or EGFR/ErbB dimer, one of which has low affinity for rDIII.
Induction of extracellular signal–regulated kinase phosphorylation by rDIII
MAPK activation is a well characterized downstream signaling event that follows stimulation of the growth factor receptor EGFR (Chen et al., 2001). In MCF-7 breast cancer cells (Fig. 5 A), extracellular signal–regulated kinase 1 (ERK1; p44) and ERK2 (p42) were rapidly phosphorylated upon rDIII treatment. Phosphorylation levels peaked 5 min after stimulation, were maintained for 10–20 min, and returned to background levels after 30 min. Stimulation of ERK1/2 by EGF demonstrated similar kinetics, with a pronounced peak at 5 min after stimulation (Fig. 5 C). In contrast to rDIII and EGF, rDIII-V did not stimulate ERK1/2 (Fig. 5 D). In MDA-MB-231 cells (Fig. 5 B), phosphorylated forms of ERK1/2 were also detected on rDIII or EGF stimulation, but with the following distinctive features: there was a more prominent phospho-ERK2 band consistent with a higher expression of total ERK2 compared with ERK1. Constitutive levels of phosphorylated ERK1/2 in nonstimulated MDA-MB-231 cells were higher than in MCF-7 cells, as reported previously (Seddighzadeh et al., 1999). rDIII-induced ERK1/2 phosphorylation was completely blocked by AG1478, a selective inhibitor of EGFR tyrosine kinase, and by 528, an EGFR-blocking antibody (Fig. 5 E). These data strongly support our hypothesis that rDIII induces ERK1/2 activation via binding to EGFR.
Figure 5.

Stimulation of ERK1/2 phosphorylation by rDIII. Time course of ERK1/2 activation after exposure to rDIII. Before lysate preparation, MCF-7 (A) or MDA-MB-231 (B) cells were treated with rDIII for the indicated time periods at 37°C. The ratio of phosphorylated ERK1/2 bands (P-ERK1/2, top panels) to total ERK1/2 protein bands (ERK1/2, bottom panels) was quantified. The control signal (no ligand) was set to 1 and the relative ERK phosphorylation intensity calculated and depicted as bar graphs (bottom panels). (A) ERK phosphorylation was performed with two distinct, purified preparations of rDIII protein (Prep. A and Prep. B). (B) One representative experiment and the mean ± SD (n = 3) of relative ERK1/2 phosphorylation intensity is depicted. ERK1/2 activation induced by EGF (C) but not by control protein rDIII-V (D). MCF-7 cells were stimulated with EGF (C) for up to 30 min or with rDIII, rDIII-V or EGF for 5 min and phosphorylated ERK1/2 were detected as described in the legend to A. As compared with rDIII (lane 3, D) and EGF (lane 4, D,) no phosphorylation signal above control level (lane 1, D) was seen with rDIII-V (lane 2, D). (E) Dependency of ERK1/2 activation on EGFR. Before stimulation for 5 min with either rDIII (lane 5) or EGF (lane 2), MCF-7 cells were preincubated with either AG1478 or 528. Both EGFR inhibitors diminish phosphorylation of ERK1/2 (top panel) by rDIII (lanes 6 and 7) or EGF (lanes 3 and 4). The top bands (∼25 kD) in lane 4 (EGF + 528) and 7 (rDIII + 528) originates from the IgG light chain of 528. The total amount of loaded ERK1/2 protein is shown in the bottom panel.
Stimulation of EGFR, but not ERK1/2 phosphorylation, by intact Ln-5
To investigate accessibility of DIII for receptor binding within intact Ln-5, MDA-MB-231 cells were incubated with rat Ln-5 under conditions similar to those used for rDIII and EGF. After 10 min, EGFR phosphorylation was stimulated by EGF and to a lesser extent by Ln-5 (Fig. 6) , whereas at 90 min after stimulation, more pronounced receptor phosphorylation was observed with Ln-5 than with EGF. However, Ln-5 did not stimulate ERK1/2 phosphorylation at any time point investigated (5, 20, 30, and 90 min) in either MDA-MB-231 or MCF-7 cells, and this result was independent of whether the cells were in suspension or adherent (unpublished data). Thus, Ln-5–stimulated EGFR phosphorylation differs from that seen with rDIII in at least two respects; slower kinetics and absence of downstream ERK1/2 activation, suggesting that MMP-liberated DIII may have signaling properties distinct from those of intact Ln-5.
Figure 6.

Induction of EGFR tyrosine phosphorylation by intact Ln-5. Treatment of MDA-MB-231 cells with 1.7 nM EGF for 10 min at 37°C results in significant phosphorylation of 175 kD EGFR (lane 2, left panels) over control (lane 1, no ligand), whereas 2.5 nM purified Ln-5 causes only weak EGFR phosphorylation (lane 3). Stimulation of cells for 90 min (right panels) with Ln-5 (lane 3) results in an EGFR phosphorylation signal well above control (lane 1). In contrast, incubation of cells for 90 min in the presence of EGF (lane 2) diminished the signal toward background level (lane 1).
Induction of MMP-2 gene expression by rDIII
Activated ERKs phosphorylate downstream transcription factors that regulate gene expression (Hazzalin and Mahadevan, 2002). We investigated a possible effect of rDIII on gene expression using cDNA microarrays, which carried probes for members of the integrin, protein tyrosine kinase and MMP families, MMP inhibitors, and ECM molecules (Seftor et al., 2001). Spiking experiments with exogenous control RNAs allowed a 2.0-fold change in signal intensity to be considered statistically significant (Bilban et al., 2002). In two independent experiments (two chips), we found that MMP-2, MMP-9, and phosphoinositide 3-kinase gene expression were up-regulated on rDIII treatment, whereas urokinase plasminogen activator expression was down-regulated (Fig. 7 A, only MMP-2 data are shown). Enhanced expression of MMP-2 by rDIII treatment was confirmed by semi-quantitative RT-PCR (Fig. 7 B). Induction of MMP-2 gene expression was also observed in cells stimulated with EGF, but not with intact Ln-5. To investigate whether rDIII-induced MMP-2 gene expression is EGFR dependent, cells were incubated with AG1478 and LA1 before treatment with rDIII. Blockage of EGFR function by these compounds inhibited induction of MMP-2 gene expression by rDIII. These results provide independent evidence that rDIII interacts with EGFR and triggers downstream signaling, resulting in altered gene expression.
Figure 7.
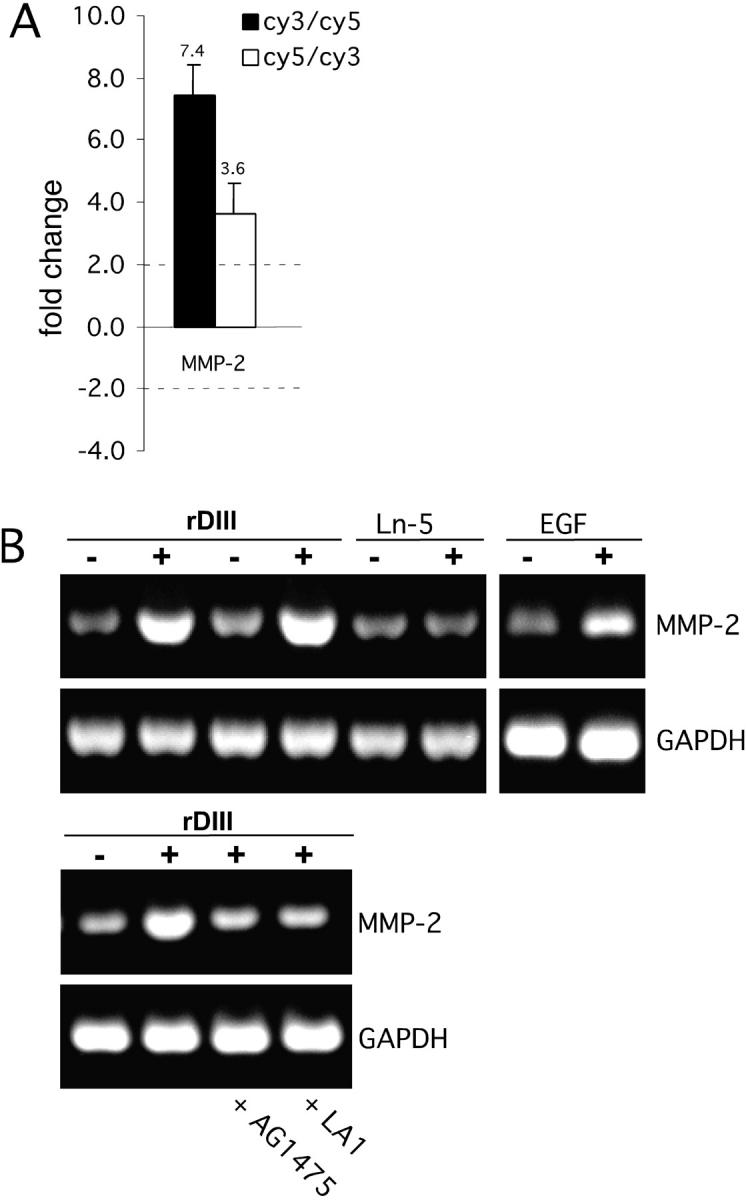
Effects of rDIII on gene expression. (A) Microarray hybridization. Amplified RNA prepared from MCF-7 cells was cultured in the presence (cy3) or absence (cy5) of rDIII and cohybridized to the microarray containing 83 human cDNAs related to cancer cell metastasis (filled histogram). The open histogram represents RNA isolates where the cy3 and cy5 labels were switched. Ratios below 1.0 were inverted and multiplied by −1 to aid in their interpretation. The MMP-2 gene expression signal exceeded a twofold signal intensity change threshold, independent of the dye label orientation. Results are expressed as the mean ± SD from six fluorescence signals. (B) Semi-quantitative RT-PCR. Changes in MMP-2 expression were assessed on the influence of 185 nM rDIII, 2.5 nM Ln-5, or 0.17 nM EGF, with or without AG1478 or LA1. DMSO was added to the controls. As a control for normalization, GAPDH was amplified similarly to MMP-2. Amplified cDNAs of MMP-2 (504 bp) and GAPDH (516 bp) were resolved by agarose gel electrophoresis and visualized by ethidium bromide staining.
Stimulation of cell migration by rDIII via EGFR
Because EGFR and ERK1/2 have motogenic properties (Klemke et al., 1997; Xie et al., 1998), we tested rDIII for its ability to affect cell motility. In Transwell migration assays with MDA-MB-231 cells on Ln-5, addition of rDIII resulted in a 1.5–2-fold increase in migratory activity. Though the increase in migration was modest, it was highly reproducible in multiple independent experiments. More pronounced stimulation of migration on Ln-5 was observed in rDIII-treated MCF-7 and MCF10A cells (Fig. 8 A).
Figure 8.
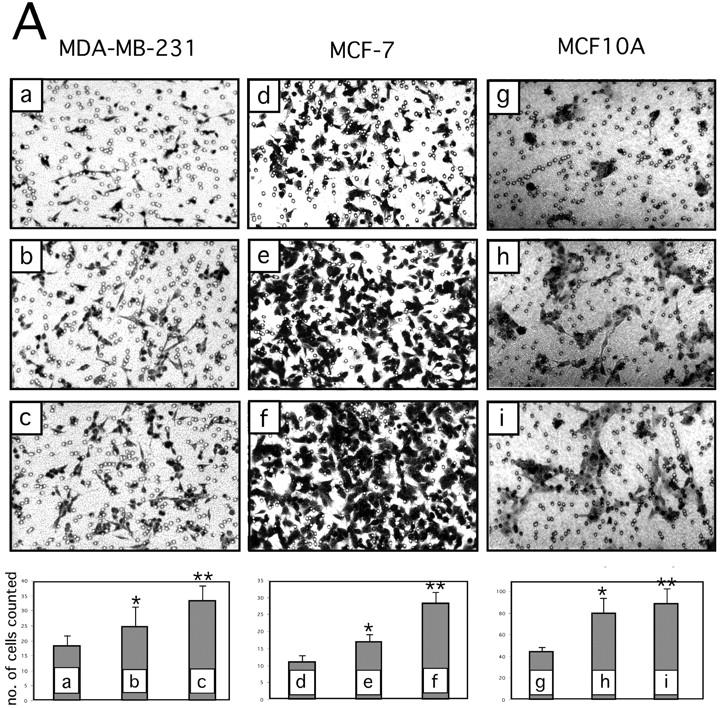
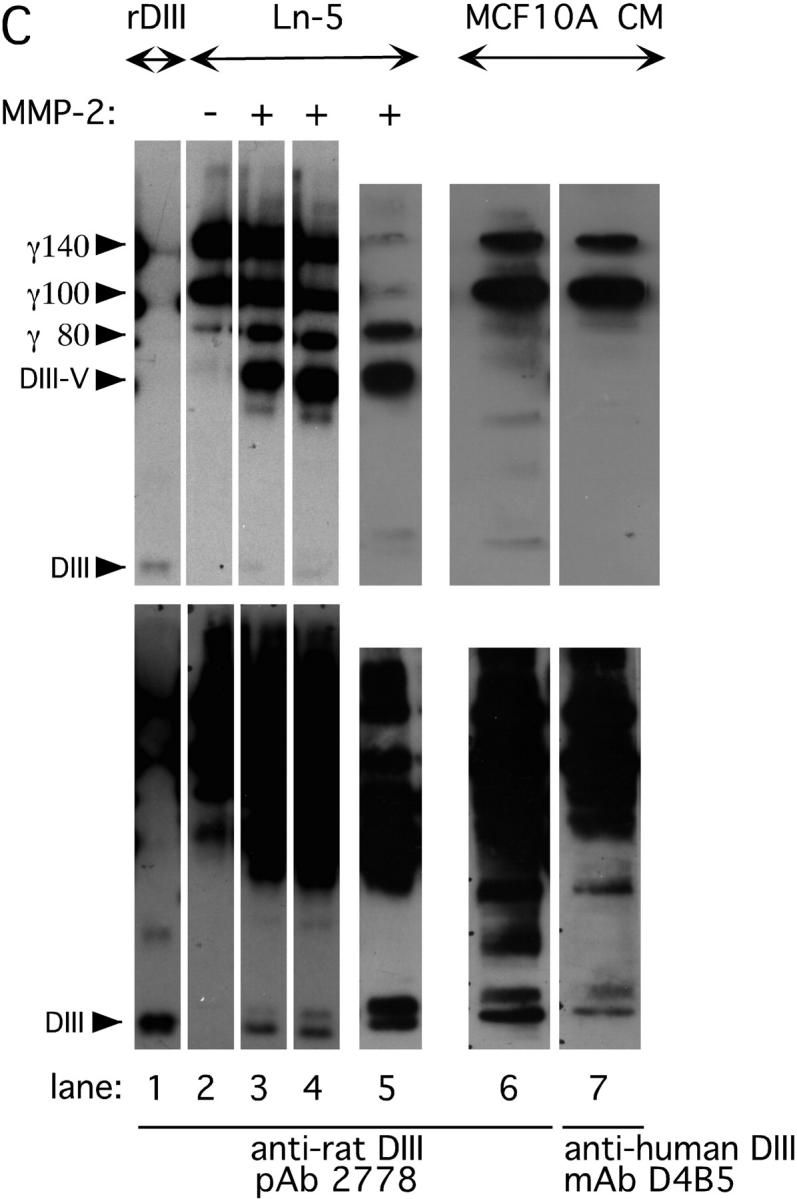

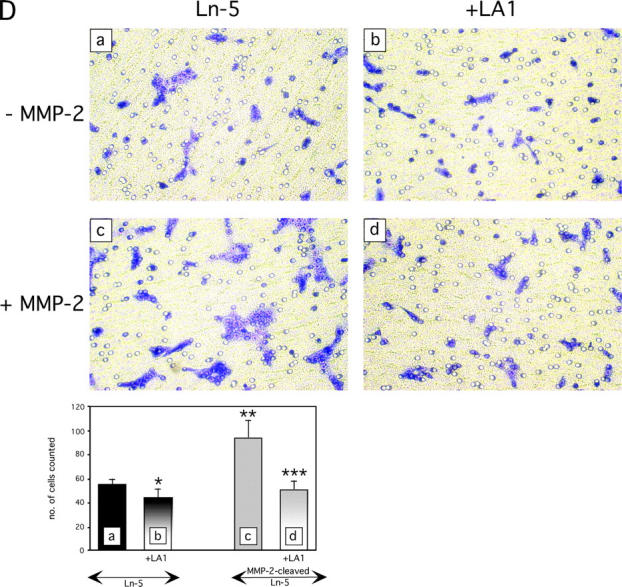
Breast cell migration stimulated by rDIII. (A) Micrographs of the lower surfaces of Transwell filters after migration of MDA-MB-231 (a–c), MCF-7 (d–f), or MCF10A (g–i) cells on coated Ln-5. The micrographs a, d, and g show migration on coated Ln-5 only. rDIII was added at increasing concentrations to the bottom (MDA-MB-231, 6 nM, b and 12 nM, c) or top chambers (MCF-7, 70 nM, e 185 nM, f and MCF10A, 70 nM, h and 460 nM, i). The magnification used to count and photograph migrated cells may differ between cell lines and assays depending on how heavily cells migrated. The results are summarized in the corresponding bar graphs. MDA-MB-231: *p (b and a) = 0.0044, **p (c and a) = 2.18E-08, ANOVA p = 6.38E-08; MCF-7: *p (e and d) = 2.96E-07, **p (f and d) = 3.07E-11, ANOVA p = 2.36E-16; MCF10A: *p (h and g) = 6.94E-07, **p (i and g) = 1.41E-07, ANOVA p = 3.43E-11. Micrographs shown are representative for several independent experiments. (B) Migration of MDA-MB-231 cells on coated Ln-5 challenged with rDIII. The effect of rDIII or EGF on MDA-MB-231 cell migration in the absence and presence of LA1, as well as constitutive migration on Ln-5 (in the absence of any stimuli) and its dependency on EGFR is depicted. Note that if membranes were not coated with Ln-5, MDA-MB-231 cells did not migrate at all (not depicted). To normalize values, each data point (cell number migrated per well) was divided by the average cell number migrated per well that was determined for Ln-5 (relative cell migration). Average cell number of Ln-5 was set to 1. Values in bar graphs represent the mean ± SD of at least three independent experiments. No change detected in 5 out of 5 assays with *p (b and a) = 0.97 - 0.16 (null- hypothesis confirmed). Statistically significant changes were found in ** 7 out of 8, *** 3 out of 3, **** 3 out of 3 and ***** 3 out of 4 assays with **p (c and a) = 1.9E-06 – 0.0147; ***p (d and c) = 8.8E-07 – 3.6E-06; ****p (e and a) = 1.37E-06 – 1.6E-05; *****p (f and e) = 8.4E-06 – 2.2E-05. (C) DIII is a cleavage product of Ln-5 and is detectable in conditioned medium of human MCF10A cells. WB of MMP-2 cleaved Ln-5 and concentrated conditioned medium were detected with 2778 (lanes 1–6), or D4B5 (lane 7). The bottom and top WBs are identical, except that the bottom panels are overexposed, depicting DIII more clearly. Cleavage of Ln-5 with MMP-2 for 2 h (lane 3), 17 h (lane 4), and 24 h (lane 5) results in the appearance of the γ80 chain, DIII-V, and DIII. In conditioned medium from MCF10A cells DIII is detectable using both 2778 (lane 6) and D4B5 (lane 7). For comparison, purified, non-MMP treated Ln-5, which is mainly composed of the γ140 and γ100 chains, was loaded in lane 2 and rDIII in lane 1. (D) Stimulated migration of MMP-2 cleaved Ln-5 depends on EGFR. MCF10A cells were allowed to migrate on Transwell membranes, which were coated with either uncleaved (top panels; −MMP-2) or MMP-2-cleaved Ln-5 (bottom panels; +MMP-2). Cells remained untreated (left panels, Ln-5), or were treated with LA1 for 30 min before seeding. LA1 was added where indicated (right panels, +LA1). The corresponding bar graph is shown with *p (b and a) = 0.0030; **p (c and a) = 7.27E-05; ***p (d and a) = 0.1333 and ***p (d and c) = 1.39E-05.
To determine whether enhancement of migration by rDIII was dependent on rDIII binding to EGFR, Transwell assays were performed in the presence of antibody LA1 (Fig. 8 B). LA1 did not affect constitutive MDA-MB-231 cell migration on coated Ln-5; however, it blocked the stimulatory effect of rDIII or EGF. These results support the conclusion that rDIII promotes cell migration in breast cells by engaging EGFR.
Stimulation of cell migration by cleaved Ln-5 via EGFR
MMP-2–cleaved Ln-5 acts as a more potent migratory substrate than intact Ln-5 (Giannelli et al., 1997). Thus, addition of rDIII to cells that are migrating on intact Ln-5 is expected to mimic the situation where MMPs generate Ln-5 cleavage products, such as DIII, which in turn may affect cell motility. Knowing that constitutive migration on Ln-5 does not engage EGFR, whereas rDIII or EGF induced migration on Ln-5 depends on EGFR (Fig. 8 B), we investigated whether migration on MMP-2–cleaved Ln-5 depends on EGFR. To address this issue, we first demonstrated that cleavage of Ln-5 by MMP-2 indeed liberates DIII as detected with antibody 2778 (Fig. 8 C). Furthermore, we detected DIII with both antibodies 2778 and D4B5 in conditioned medium from MCF10A cells.
Having shown that DIII is fully released on cleavage of Ln-5 by MMP-2, MCF10A cells were allowed to migrate on Transwell chamber membranes, where they clearly displayed enhanced migration on cleaved Ln-5 (Giannelli et al., 1997). The increase in motility on cleaved Ln-5 (Fig. 8 D) was inhibited in cells pretreated with LA1. In contrast, cells seeded on uncleaved Ln-5 showed no enhanced migration and were not significantly affected by the presence of LA1. These results strongly suggest that the EGFR dependency of cells migrating on cleaved Ln-5 is due to the presence of EGFR-engaging fragments such as DIII.
Detection of liberated DIII at physiologically significant stages of mammary gland development
Our in vitro data support the hypothesis that DIII is a cryptic migratory ECM cue released upon MMP-2 cleavage of Ln-5. To investigate the physiological relevance of these data, we tested for liberated DIII in the involuting mammary gland. The stages of mammary gland involution are well characterized with respect to MMP expression (Talhouk et al., 1992; Lund et al., 1996). We investigated the presence of DIII in wild-type (WT) as well as tissue inhibitor of metalloproteinase 3 (TIMP-3)–null (−/−) mice because deficiency of MMP inhibitors should favor DIII liberation. TIMP-3 binds tightly to the ECM (Fata et al., 2001; Leco et al., 2001) and interferes strongly with localized proteolysis of ECM molecules such as Ln-5. To detect DIII, we used mAb D4B5, which recognizes an epitope present on mouse DIII only when it is liberated from Ln-5 (i.e., it is not reactive with intact mouse Ln-5 γ2-chain). Mammary gland tissue lysates from mice killed on day 10 of lactation as well as day 1, 2, 3, 4, and 7 of involution were subjected to SDS-PAGE and WB. In WT mice, DIII is detectable in involuting tissue starting at day 2 of involution, but it is not visible at day 1 of involution or day 10 of lactation (Fig. 9) . In contrast, in the −/− mice, the DIII fragment is clearly detectable at day 10 of lactation, as well as day 1 of involution. At day 2, 3, 4, and 7 of involution, approximately equal levels of DIII were apparent in both WT and −/− tissue.
Figure 9.

Detection of DIII in lysates of mammary gland tissue. Mammary lysates were resolved on a 4–12% Bis-Tris gradient gel in MES buffer under reducing conditions. D4B5 detected a distinct band of ∼20 kD that very likely is DIII (top panel). In contrast to WT, DIII could be found at day 10 of lactation as well as at day 1 of involution in TIMP-3–null (−/−) tissue. After day 1 of involution, DIII was detectable at all time points of involution in both the involuting and lactating mice (day 2–7 of involution). Stripping and reprobing the blot with 2778 revealed unscheduled fragmentation of Ln-5 γ2 chain (bottom panel). Fragmentation occurred in −/− 1 d earlier than in WT mammary glands (day 1 of involution, WT, −/−). The exact identity of the various fragments in the range of 30–100 kD is not known. The Ln-5 γ2 chain profile at day 10 of lactation as well as at day 7 of involution does not differ between WT and −/− tissue, when detected with 2778 (not depicted). To ensure equal loading in each lane, blots were stripped and reprobed with Actin mAb (MAB 1501, CHEMICON International; middle panel).
To confirm these results, we used a pAb that detects the intact Ln-5 γ2 subunit and some proteolytic fragments. With this reagent, DIII was not readily detected. Nonetheless, degraded γ2 was visible at day 1 of involution in TIMP-3–deficient mice, but not in WT mice. Interestingly, at day 3 of involution, γ2 fragments were more abundant in WT than −/− tissues.
The fact that we were able to detect DIII in tissues under conditions of high MMP activity indicates the physiological relevance of this fragment. Furthermore, the patterns of appearance of DIII in mammary gland are consistent with previous reports in which TIMP-3–deficient involuting mammary tissue was found to show low levels of activated MMP-2 as early as day 1 of involution, as opposed to WT involuting tissue, where MMP activity could not be detected before day 3 of involution (Fata et al., 2001).
Discussion
We describe a molecular mechanism responsible for stimulating cell migration on MMP-cleaved Ln-5. The involvement of the EGF-like domain DIII of the Ln-5 γ2 subunit is demonstrated by the following data: (1) MMP-2 and MT1-MMP cleave Ln-5 (Giannelli et al., 1997; Koshikawa et al., 2000; unpublished data) excising DIII; (2) rDIII binds specifically EGFR; (3) rDIII activates downstream signaling, including EGFR and ERK1/2 phosphorylation; and (4) rDIII stimulates cell motility and changes in gene expression.
These findings point to several novel elements. First, they show that MMPs can initiate precise sequences of molecular events, including ligand-receptor binding, signal transduction, and changes in cell behavior. Second, they indicate that EGF-like ligands for receptor tyrosine kinases (RTKs) may be solid phase, i.e., contained within the structure of the ECM itself (Swindle et al., 2001), or may be released as small diffusible factors. Third, they imply that laminins exert their diverse functions by interaction with different receptor families (Kleinman et al., 2001), such as RTKs, integrins (Belkin and Stepp, 2000), and dystroglycan (Henry and Campbell, 1999).
Several independent lines of investigation demonstrated that rDIII unequivocally binds the RTK EGFR. By flow cytometry, rDIII (but not rDIII-V) bound to the cell surface in a saturable and dose-dependent manner and its binding was inhibited by EGF. Cross-linking of rDIII to the cell surface resulted in formation of a complex between rDIII and EGFR. rDIII specifically displaced cell surface bound EGF, the best known ligand for EGFR and the prototypical member of the EGF-like family of growth factors (Yarden, 2001). This family (Thompson et al., 1996) also includes TGFα, amphiregulin (AR), heparin-binding EGF-like growth factor, betacellulin, and epiregulin. EGF-like ligands bind distinct heterodimers formed by pairing of various members of the EGFR family (i.e., ErbB-2, -3, and -4; Schlessinger, 2000). Whether or not rDIII binds to such heterodimers is currently under investigation. rDIII displacement data fit well to a two-site model, and it is tempting to speculate that EGF and DIII may bind to partially overlapping sets of ErbB receptors, distinguished by subunit composition and/or affinity. This would also explain the fact that DIII cannot displace EGF binding >50%. In addition, phosphorylation of EGFR and ERK activation of rDIII binding occurs with kinetics that parallel those triggered by EGF (Kholodenko et al., 1999).
Our IC50 measurements suggest an affinity of rDIII for EGFR in the nanomolar range (IC50_1, 0.17–0.2 nM; IC50_2, 349–1487 nM), which is consistent with the rDIII concentrations used for our other studies (92.5–185 nM MAPK, 185 nM EGFR, 185 nM MMP-2, 6–460 nM migration). Affinity values in this range are common for ECM molecules or fragments. For instance, the Kd of Ln-5 for its main receptor, the α3β1 integrin, is >600 nM (Eble et al., 1998), and the affinity of a tenascin EGF-like fragment for EGFR is in the micromolar range (1–6 μM; Swindle et al., 2001). The IC50 of the physiological EGFR ligand AR varies depending on its COOH-terminal amino acid residue extension (rAR84, 840 nM; rAR87, 20 nM; rAR92, 43 nM; Thompson et al., 1996). Thus, our results provide an encouraging platform for studying DIII–EGFR interactions in physiological systems.
The structure of rDIII corroborates its EGFR binding property. Both EGF and laminin-type EGF (LE) repeats are highly conserved folds commonly found in growth factors and ECM proteins (Engel et al., 1994). Several LE domains have been reported to have biological activities. For instance, LE repeats of laminin γ2- and γ3-chains stimulate neurite outgrowth (Koch et al., 2000). Antibodies to DIII of Ln-5 γ2-chain inhibited migration of squamous cell carcinoma cells by ∼50% (Salo et al., 1999). A proteolytic fragment of Ln-1, encompassing multiple LE domains, stimulated MMP synthesis (Turpeenniemi-Hujanen et al., 1986), cell growth (Terranova et al., 1986), and induced EGFR phosphorylation and S6 kinase activity (Novak-Hofer and Thomas, 1984). We have extended these observations by connecting the biological activities of rDIII to receptor binding and downstream signaling.
The observations that rDIII-V failed to bind to cell surfaces and induce ERK phosphorylation and that intact Ln-5 stimulated EGFR phosphorylation with slow kinetics, but had no effect on ERK phosphorylation, suggest that fully released DIII may be a better ligand for EGFR. This would agree with findings on EGF and TGFα, which harbor crucial amino acids for EGFR binding in their COOH- and possibly NH2-terminal regions (Ogiso et al., 2002) and also with the growing list of EGF-like molecules, proteolytically liberated out of large multi-domain proteins as small diffusible ligands (Kennedy et al., 1994; Qi et al., 1999; Koch et al., 2000).
In experiments testing whether rDIII binding to EGFR affects cell growth, our results were inconclusive and need additional investigation. However, we demonstrated that rDIII affects cell motility. In cultured cells, rDIII produced a dose-dependent enhancement of cell migration, which may profoundly affect direction and positioning of cells within tissues. Its physiological relevance was suggested by the finding that cleavage of Ln-5 by MMP-2 releases several fragments, including DIII. Furthermore, MMP-enhanced cell migration on Ln-5 was EGFR dependent. Interestingly, branching morphogenesis during lung development requires MMP-2 and MT1-MMP, key proteases downstream of EGFR signaling, which may act via generation of bioactive ECM fragments (Kheradmand et al., 2002).
Cell migration and enhanced invasive potential have been reported to coincide with induction and up-regulation of MMPs on engagement of EGFR by EGF and other EGFR ligands (Kondapaka et al., 1997; Ellerbroek et al., 1998; McCawley et al., 1998; Rosenthal et al., 1998). The EGFR dependent up-regulation of the MMP-2 gene by rDIII is particularly interesting because it suggests a positive feedback loop regulating tightly coordinated ECM degradation. Collagen-induced activation of DDR2 resulted in enhanced MMP-1 expression, leading to a negative feedback loop that may control collagen degradation (Vogel et al., 1997). Similarly, decorin was found to be proteolytically degraded by MMP-2, -3, and -7 (Imai et al., 1997), to engage EGFR (Csordas et al., 2000), and to regulate MMP-1 gene expression in cells adhering to ECM vitronectin (Huttenlocher et al., 1996).
To investigate EGFR stimulation by DIII under physiological conditions, we studied post-lactational involution of the mammary gland, a tissue remodeling system whose dependence on MMP action and cell migration is well understood. We detected DIII in the mammary gland at stages when MMP remodeling of the ECM is known to occur (Talhouk et al., 1992; Lund et al., 1996; Fata et al., 2001). The appearance of DIII paralleled both increased MMP activity and degradation of BM during involution. DIII was not detectable during lactation, when little remodeling or MMP activity is expected. These findings fit well with a previous study demonstrating that γ2x is absent during lactation but appears during involution (Giannelli et al., 1999). In TIMP-3–null mice that lack a critical MMP inhibitor, DIII was present both during lactation and at early stages of involution. Future studies will focus on extrapolation of our data to the murine mammary gland system to link the appearance of DIII to in vivo EGFR stimulation.
In summary, we demonstrate that a biologically active ligand for EGFR is excisable from the ECM macromolecule Ln-5 by MMPs. Our findings advance the understanding of the complex molecular interactions between ECM, MMPs, and RTKs in tissue remodeling and cancer invasion.
Materials and methods
Antibodies
Anti-His tag (anti-His) mAb was purchased from QIAGEN. Anti–rat DIII antibody 2778 was generated by injecting rabbits with rDIII, and anti–human DIII mAb D4B5 was obtained from CHEMICON International. antibodies 528 (Santa Cruz Biotechnology, Inc.) and LA-1 (Upstate Biotechnology) block EGFR ligand binding and were used interchangeably.
Construction and expression of rDIII
DIII cDNA was amplified by PCR using a template that spans the 3′ 2900 bp of the rat Ln-5 γ2-chain gene. The forward primer (FPRIIIBHI; 5′-CGCGGATCCGACGAGAATCCTGACATTGAG-3′) contained the BamHI site, and the reverse primer encoded both an EcoRI site and a COOH-terminal His6 tag (RPRIIIHIS + ERI; 5′-CGGGAATTCTCAGTGATGATGATGATGATGCTGGTCCATCTGAGTCTTCAC-3′). The PCR product had the expected size of ∼720 bp and was cloned into the BamHI/EcoRI sites, five amino acids downstream of the gp67 (acidic glycoprotein gp67) secretion signal cleavage site, provided by the baculovirus transfer vector pACGP67-B (BD Biosciences). rDIII/pACGP67-B was sequenced and cotransfected into Sf9 insect cells with BaculoGold® DNA (BD Biosciences). Amplified high titer recombinant baculovirus was used to infect High Five™ insect cells. After 3 d, supernatants were harvested and secreted rDIII protein was purified using Ni-NTA agarose (QIAGEN). Purity of rDIII was judged by Coomassie blue staining.
Flow cytometry
Cells (0.5–1 × 106) were washed with HBSS, 0.1% BSA, and incubated with 1–45 μM rDIII or rDIII-V in 50 μl. After 1 h on ice and washing, bound rDIII was stained with anti-His or 2778 antibody, followed either by fluorescently labeled anti–mouse or –rabbit antibody Alexa® 488 (IgG (H+L) F(ab')2, Molecular Probes, Inc.). Washed cells were resuspended in PBS, 1% HCHO and analyzed with FACScan™ (Becton Dickinson).
Immunoprecipitation and cross-linking
Mouse EGF (Sigma-Aldrich) and rDIII were labeled with biotin (EZ-Link™ Sulfo-NHS-LC-Biotin; Pierce Chemical Co.). MDA-MB-231 cells (2 × 107) were washed with binding buffer (DME, 0.2% BSA, and 1 mM glutamine) and incubated on ice for 2 h with biotinylated ligands. The cross-linker BS3 (Pierce Chemical Co.) was added at 1 mM, incubated 2 h on ice, and quenched with 20 mM Tris, pH 7.5, for 15 min at RT. Cells were washed with PBS and extracted with lysis buffer (20 mM Tris, pH 7.5, 150 mM NaCl, 1% Triton X-100, 1 mM EDTA, 1 mM PMSF, and 1 μg/ml each of aprotinin, pepstatin, and leupeptin). Lysates were cleared by centrifugation and immunoprecipitated with anti-EGFR mAb (clone EGFR.1; BD Biosciences) at 4°C. After incubation with 50 μl of protein G Sepharose beads (Boehringer), immune complexes were washed, centrifuged, and proteins were separated by SDS-PAGE (4–12% gradient) under reducing conditions and analyzed by WB.
Precipitation with magnetic beads was done as described in the paragraph above with the following modifications: MDA-MB-231 lysates were incubated with Dynabeads® M-280, streptavidin-coated beads (Dynal). The beads were collected using a magnetic device and washed. Proteins were eluted by heating the beads with SDS-sample buffer (5 min at 95°C), and the supernatant was analyzed by SDS-PAGE and WB. Detection was performed with an anti-EGFR pAb (sc-03; Santa Cruz Biotechnology, Inc.) and HRP anti–rabbit secondary antibody at 1:300 and 1:10,000 dilutions, respectively.
EGFR phosphorylation
Serum-starved MDA-MB-231 cells were detached with 10 mM EDTA/PBS, washed, and resuspended in binding buffer. 107 cells were kept in suspension at 37°C for 30 min, then EGF or rDIII was added and cells were incubated at 37°C for 2 or 5 min. Ligands were omitted from negative controls. Cross-linking was performed as described in Immunoprecipitation and cross-linking. Cells were lysed for 1 h on ice in lysis buffer with 100 mM NaF and 1 mM Na3VO4. Lysates were immunoprecipitated with anti-EGFR mAb, and detection was performed with anti-phosphotyrosine mAb (PY-20; Transduction Laboratories) at a 1:2,000 dilution, followed by HRP goat anti–mouse IgG and ECL (PerkinElmer). To confirm equal loading in each lane, blots were stripped (65 mM Tris, pH 6.7, 2% SDS, and 100 mM β-mercaptoethanol at 55°C for 45 min) and reprobed with anti-EGFR pAb (sc-03; Santa Cruz Biotechnology, Inc.) or mAb (Ab-12, Cocktail R19/48; Neomarker). In some cases, BS3 was omitted. With Ln-5, cells were treated for up to 90 min.
I125-EGF displacement
MDA-MB-231 cells were trypsinized, washed 3× with a buffer containing DME, 25 mM Hepes, 0.2% BSA, 0.3 mM 1,10 phenanthroline, and 0.16 mM PMSF, and were then diluted to 2 × 106 ml. 500 μl cells were transferred into coated reaction tubes. Unlabeled competitor molecule (EGF or rDIII) and I125-mouse EGF (100 μCi/μg; Amersham Biosciences) were added and cells were incubated on ice for 3 h. Triplicate 150 μl aliquots of the cell label mixture were layered onto 300 μl of a 1:1 mixture of AR-20 and Addid-200 silicon oils (1,013 kg/cm3 density; Wacker Chemie). Unbound radioactivity was removed by centrifugation for 1 min at 500 g, followed by removal of the tube tip at the height of the oil layer. The cell pellet was subjected to γ-counting, and values are presented in cpm. The data were processed using PRISM software (GraphPad Software, Inc.). The IC50 was calculated using the linear regression model and either one-site or two-site competition equations to obtain best fit values. To evaluate each fit, e.g., best fit values, 95% confidence interval and standard error values were considered, and both equations were compared using the “Fit to two equations and compare with an F-test” option. The F-test was used to determine a P-value.
ERK1/2 phosphorylation
Cells were grown to 60% confluency and serum-starved for 15 h. Fresh serum-free medium containing 0.5–2 μM AG1478 (Calbiochem) or 5–10 μg/ml antibody 528 was added. DMSO was added to the control. After 30 min at 37°C, rDIII, rDIII-V, Ln-5, or EGF was added and cells were incubated at 37°C. Cells were washed in PBS and lysed for 1 h on ice in 0.5 ml lysis buffer containing one tablet of complete protease inhibitor cocktail (Roche) per 50 ml. Lysates were cleared by centrifugation for 10 min. Each sample was divided in half and analyzed for total ERK1/2 content (rabbit anti-ERK1/2 IgG; Santa Cruz Biotechnology, Inc.) or for phosphorylation (mAb to phosphorylated ERK1/2; New England Biolabs, Inc.) by WB.
Cell migration
Migration was performed in Transwell chambers (8-μm pores; Corning Costar) coated on the underside with 0.25 μg/ml rat Ln-5 for MDA-MB-231 cells, and with 1 μg/ml for MCF-7 and MCF10A cells at 4°C overnight. Rat Ln-5 was purified from spent 804G cell media (Hormia et al., 1995). Filters were washed, blocked with 5% skim milk, PBS containing 0.2% Tween 20, and 0.02% azide for 2 h at RT, and washed again. Membranes were placed into the lower chamber containing 600 μl serum-free DME. Trypsinized cells were washed and plated at 0.6–1 × 105 cells/well. rDIII, EGF, or antibody LA1 (5–10 μg/ml) were added to the lower and/or upper chambers at various concentrations to obtain maximum stimulation (see legend to Fig. 8). Chambers were incubated 6 or 18 h at 37°C. Nonmigratory cells were removed from the upper membrane with a cotton swab. Filters were fixed, stained with HEMA 3® Stain Set (Biochemical Sciences), and cells were counted in eight random fields on each of two filters for each condition. The significance of each migration assay was evaluated using the t test assuming unequal variances. P-values lower than 0.05 were considered statistically significant. To determine statistical significance between more than two groups, ANOVA was used.
cDNA microarray
MCF-7 cells at 60% confluency were serum-starved for 8 h and incubated for 24 h serum-free. 1 μg total RNA was isolated with RNeasy mini kit (QIAGEN) and amplified by in vitro transcription (Luo et al., 1999). 5 μg amplified RNA was labeled with Cy3 or Cy5-dCTP (Amersham Biosciences). Fluorescent cDNAs were hybridized to a custom cDNA microarray chip. Fluorescent images were captured using a laser scanner (ScanArray™ 5000; GSI Lumonics).
Semi-quantitative RT-PCR
The One-step RT-PCR™ kit (QIAGEN) was used with 2 μg total MCF-7 RNA, and the MMP-2 primers 5′-TCAGATCCGTGGTGAGATCTT-3′ and 5′-GCTCTTCAGACTTTGGTTCTC-3′. As a control, GAPDH was amplified using the primers 5′-TGAAGGTCGGAGTCAACGGAT-3′ and 5′-GTCATGAGTCCTTCCACGATA-3′. For each cDNA, the optimal cycle number was determined to ensure that DNA amplification was in the linear range.
Cleavage of Ln-5 by MMP-2
0.5–1.0 μg of rat Ln-5 (Hormia et al., 1995) was incubated with 0.1 μM of p-aminophenyl-mercuric acetate (APMA)–activated MMP-2 for 17 h at 37°C in 50 mM Tris, pH 7.5, 0.005% Brij-35, and 5 mM CaCl2 in a volume of 30 μl. Each reaction mixture was analyzed by SDS-PAGE and WB.
Collection of mammary tissue
Induction of synchronized involution and collection of mammary tissue was performed as described previously (Fata et al., 2001). The second and third thoracic mammary glands were homogenized in lysis buffer (10 mM Tris, pH 7.6, 5 mM EDTA, 50 mM NaCl, 1% Triton X-100, 30 mM Na4P2O7, 200 μM Na3VO4, 1 mM PMSF, 5 μg/ml aprotinin, 1 μg/ml pepstatin, and 2 μg/ml leupeptin) for WB. The homogenates were centrifuged for 20 min at 4°C. Protein concentration was determined using Dc Bio-Rad assay reagent. Supernatants and insoluble pellets were stored at −70°C until analyzed by SDS-PAGE and WB.
Acknowledgments
We thank Vasso Apostopopulus for advice with FACS analyses, Roger Beerli for reagents, Achim Brinker for Biacore studies, Lukas Buehler for statistical analysis, Francine Frasier for technical support, Deirdre O'Sullivan for editorial assistance and Gary Schoenhals for critical comments on the manuscript and help with figures.
This work was supported by National Institutes of Health grants to V. Quaranta (GM46902 and CA47858), and by a Deutsche Forschungsgemeinschaft fellowship to S. Schenk (DFG SCHE20/30).
Footnotes
Abbreviations used in this paper: AR, amphiregulin; BM, basement membrane; EGFR, epidermal growth factor receptor; ERK, extracellular signal–regulated kinase; LE, laminin-type EGF; Ln-1, laminin-1; Ln-5, laminin-5; MMP, matrix metalloproteinase; rDIII, recombinant DIII; RTK, receptor tyrosine kinase; TIMP-3, tissue inhibitor of metalloproteinase 3; WT, wild-type.
References
- Belkin, A.M., and M.A. Stepp. 2000. Integrins as receptors for laminins. Microsc. Res. Tech. 51:280–301. [DOI] [PubMed] [Google Scholar]
- Bilban, M., L.K. Buehler, S. Head, G. Desoye, and V. Quaranta. 2002. Normalizing DNA microarray data. Curr. Issues. Mol. Biol. 4:57–64. [PubMed] [Google Scholar]
- Borradori, L., and A. Sonnenberg. 1999. Structure and function of hemidesmosomes: more than simple adhesion complexes. J. Invest. Dermatol. 112:411–418. [DOI] [PubMed] [Google Scholar]
- Chen, Z., T.B. Gibson, F. Robinson, L. Silvestro, G. Pearson, B. Xu, A. Wright, C. Vanderbilt, and M.H. Cobb. 2001. MAP Kinases. Chem. Rev. 101:2449–2476. [DOI] [PubMed] [Google Scholar]
- Csordas, G., M. Santra, C.C. Reed, I. Eichstetter, D.J. McQuillan, D. Gross, M.A. Nugent, G. Hajnoczky, and R.V. Iozzo. 2000. Sustained down-regulation of the epidermal growth factor receptor by decorin. A mechanism for controlling tumor growth in vivo. J. Biol. Chem. 275:32879–32887. [DOI] [PubMed] [Google Scholar]
- Eble, J.A., K.W. Wucherpfennig, L. Gauthier, P. Dersch, E. Krukonis, R.R. Isberg, and M.E. Hemler. 1998. Recombinant soluble human alpha 3 beta 1 integrin: purification, processing, regulation, and specific binding to laminin-5 and invasin in a mutually exclusive manner. Biochemistry. 37:10945–10955. [DOI] [PubMed] [Google Scholar]
- Egeblad, M., and Z. Werb. 2002. New functions for the matrix metalloproteinases in cancer progression. Nat. Rev. Cancer. 2:161–174. [DOI] [PubMed] [Google Scholar]
- Ellerbroek, S.M., L.G. Hudson, and M.S. Stack. 1998. Proteinase requirements of epidermal growth factor-induced ovarian cancer cell invasion. Int. J. Cancer. 78:331–337. [DOI] [PubMed] [Google Scholar]
- Engel, J., V.P. Efimov, and P. Maurer. 1994. Domain organizations of extracellular matrix proteins and their evolution. Development. 120:35–42. [Google Scholar]
- Fata, J.E., K.J. Leco, E.B. Voura, H.Y. Yu, P. Waterhouse, G. Murphy, R.A. Moorehead, and R. Khokha. 2001. Accelerated apoptosis in the Timp-3-deficient mammary gland. J. Clin. Invest. 108:831–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foda, H.D., and S. Zucker. 2001. Matrix metalloproteinases in cancer invasion, metastasis and angiogenesis. Drug Discov. Today. 6:478–482. [DOI] [PubMed] [Google Scholar]
- Gagnoux-Palacios, L., M. Allegra, F. Spirito, O. Pommeret, C. Romero, J.P. Ortonne, and G. Meneguzzi. 2001. The short arm of the laminin gamma2 chain plays a pivotal role in the incorporation of laminin 5 into the extracellular matrix and in cell adhesion. J. Cell Biol. 153:835–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannelli, G., J. Falk-Marzillier, O. Schiraldi, W.G. Stetler-Stevenson, and V. Quaranta. 1997. Induction of cell migration by matrix metalloprotease-2 cleavage of laminin-5. Science. 277:225–228. [DOI] [PubMed] [Google Scholar]
- Giannelli, G., A. Pozzi, W.G. Stetler-Stevenson, H.A. Gardner, and V. Quaranta. 1999. Expression of matrix metalloprotease-2-cleaved laminin-5 in breast remodeling stimulated by sex steroids. Am. J. Pathol. 154:1193–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilles, C., M. Polette, C. Coraux, J.M. Tournier, G. Meneguzzi, C. Munaut, L. Volders, P. Rousselle, P. Birembaut, and J.M. Foidart. 2001. Contribution of MT1-MMP and of human laminin-5 gamma2 chain degradation to mammary epithelial cell migration. J. Cell Sci. 114:2967–2976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazzalin, C.A., and L.C. Mahadevan. 2002. MAPK-regulated transcription: a continuously variable gene switch? Nat. Rev. Mol. Cell Biol. 3:30–40. [DOI] [PubMed] [Google Scholar]
- Henry, M.D., and K.P. Campbell. 1999. Dystroglycan inside and out. Curr. Opin. Cell Biol. 11:602–607. [DOI] [PubMed] [Google Scholar]
- Hormia, M., J. Falk-Marzillier, G. Plopper, R.N. Tamura, J.C. Jones, and V. Quaranta. 1995. Rapid spreading and mature hemidesmosome formation in HaCaT keratinocytes induced by incubation with soluble laminin-5r. J. Invest. Dermatol. 105:557–561. [DOI] [PubMed] [Google Scholar]
- Huttenlocher, A., Z. Werb, P. Tremble, P. Huhtala, L. Rosenberg, and C.H. Damsky. 1996. Decorin regulates collagenase gene expression in fibroblasts adhering to vitronectin. Matrix Biol. 15:239–250. [DOI] [PubMed] [Google Scholar]
- Imai, K., A. Hiramatsu, D. Fukushima, M.D. Pierschbacher, and Y. Okada. 1997. Degradation of decorin by matrix metalloproteinases: identification of the cleavage sites, kinetic analyses and transforming growth factor-beta1 release. Biochem. J. 322:809–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagesato, Y., H. Mizushima, N. Koshikawa, H. Kitamura, H. Hayashi, N. Ogawa, M. Tsukuda, and K. Miyazaki. 2001. Sole expression of laminin gamma 2 chain in invading tumor cells and its association with stromal fibrosis in lung adenocarcinomas. Jpn. J. Cancer Res. 92:184–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy, T.E., T. Serafini, J.R. de la Torre, and M. Tessier-Lavigne. 1994. Netrins are diffusible chemotropic factors for commissural axons in the embryonic spinal cord. Cell. 78:425–435. [DOI] [PubMed] [Google Scholar]
- Kheradmand, F., K. Rishi, and Z. Werb. 2002. Signaling through the EGF receptor controls lung morphogenesis in part by regulating MT1-MMP-mediated activation of gelatinase A/MMP2. J. Cell Sci. 115:839–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kholodenko, B.N., O.V. Demin, G. Moehren, and J.B. Hoek. 1999. Quantification of short term signaling by the epidermal growth factor receptor. J. Biol. Chem. 274:30169–30181. [DOI] [PubMed] [Google Scholar]
- Kleinman, H.K., J. Koblinski, S. Lee, and J. Engbring. 2001. Role of basement membrane in tumor growth and metastasis. Surg. Oncol. Clin. N. Am. 10:329–338. [PubMed] [Google Scholar]
- Klemke, R.L., S. Cai, A.L. Giannini, P.J. Gallagher, P. de Lanerolle, and D.A. Cheresh. 1997. Regulation of cell motility by mitogen-activated protein kinase. J. Cell Biol. 137:481–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch, M., J.R. Murrell, D.D. Hunter, P.F. Olson, W. Jin, D.R. Keene, W.J. Brunken, and R.E. Burgeson. 2000. A novel member of the netrin family, beta-netrin, shares homology with the beta chain of laminin: identification, expression, and functional characterization. J. Cell Biol. 151:221–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondapaka, S.B., R. Fridman, and K.B. Reddy. 1997. Epidermal growth factor and amphiregulin up-regulate matrix metalloproteinase-9 (MMP-9) in human breast cancer cells. Int. J. Cancer. 70:722–726. [DOI] [PubMed] [Google Scholar]
- Koshikawa, N., G. Giannelli, V. Cirulli, K. Miyazaki, and V. Quaranta. 2000. Role of cell surface metalloprotease MT1-MMP in epithelial cell migration over laminin-5. J. Cell Biol. 148:615–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leco, K.J., P. Waterhouse, O.H. Sanchez, K.L. Gowing, A.R. Poole, A. Wakeham, T.W. Mak, and R. Khokha. 2001. Spontaneous air space enlargement in the lungs of mice lacking tissue inhibitor of metalloproteinases-3 (TIMP-3). J. Clin. Invest. 108:817–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund, L.R., J. Romer, N. Thomasset, H. Solberg, C. Pyke, M.J. Bissell, K. Dano, and Z. Werb. 1996. Two distinct phases of apoptosis in mammary gland involution: proteinase-independent and -dependent pathways. Development. 122:181–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo, L., R.C. Salunga, H. Guo, A. Bittner, K.C. Joy, J.E. Galindo, H. Xiao, K.E. Rogers, J.S. Wan, M.R. Jackson, and M.G. Erlander. 1999. Gene expression profiles of laser-captured adjacent neuronal subtypes. Nat. Med. 5:117–122. [DOI] [PubMed] [Google Scholar]
- Mayer, U., R. Nischt, E. Poschl, K. Mann, K. Fukuda, M. Gerl, Y. Yamada, and R. Timpl. 1993. A single EGF-like motif of laminin is responsible for high affinity nidogen binding. EMBO J. 12:1879–1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCawley, L.J., P. O'Brien, and L.G. Hudson. 1998. Epidermal growth factor (EGF)- and scatter factor/hepatocyte growth factor (SF/HGF)- mediated keratinocyte migration is coincident with induction of matrix metalloproteinase (MMP)-9. J. Cell. Physiol. 176:255–265. [DOI] [PubMed] [Google Scholar]
- Niki, T., T. Kohno, S. Iba, Y. Moriya, Y. Takahashi, M. Saito, A. Maeshima, T. Yamada, Y. Matsuno, M. Fukayama, et al. 2002. Frequent co-localization of cox-2 and laminin-5 gamma2 chain at the invasive front of early-stage lung adenocarcinomas. Am. J. Pathol. 160:1129–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novak-Hofer, I., and G. Thomas. 1984. An activated S6 kinase in extracts from serum- and epidermal growth factor-stimulated Swiss 3T3 cells. J. Biol. Chem. 259:5995–6000. [PubMed] [Google Scholar]
- Ogiso, H., R. Ishitani, O. Nureki, S. Fukai, M. Yamanaka, J.H. Kim, K. Saito, A. Sakamoto, M. Inoue, M. Shirouzu, and S. Yokoyama. 2002. Crystal structure of the complex of human epidermal growth factor and receptor extracellular domains. Cell. 110:775–787. [DOI] [PubMed] [Google Scholar]
- Panayotou, G., P. End, M. Aumailley, R. Timpl, and J. Engel. 1989. Domains of laminin with growth-factor activity. Cell. 56:93–101. [DOI] [PubMed] [Google Scholar]
- Pyke, C., J. Romer, P. Kallunki, L.R. Lund, E. Ralfkiaer, K. Dano, and K. Tryggvason. 1994. The gamma 2 chain of kalinin/laminin 5 is preferentially expressed in invading malignant cells in human cancers. Am. J. Pathol. 145:782–791. [PMC free article] [PubMed] [Google Scholar]
- Qi, H., M.D. Rand, X. Wu, N. Sestan, W. Wang, P. Rakic, T. Xu, and S. Artavanis-Tsakonas. 1999. Processing of the notch ligand delta by the metalloprotease Kuzbanian. Science. 283:91–94. [DOI] [PubMed] [Google Scholar]
- Rosenthal, E.L., T.M. Johnson, E.D. Allen, I.J. Apel, A. Punturieri, and S.J. Weiss. 1998. Role of the plasminogen activator and matrix metalloproteinase systems in epidermal growth factor- and scatter factor-stimulated invasion of carcinoma cells. Cancer Res. 58:5221–5230. [PubMed] [Google Scholar]
- Salo, S., H. Haakana, S. Kontusaari, E. Hujanen, T. Kallunki, and K. Tryggvason. 1999. Laminin-5 promotes adhesion and migration of epithelial cells: identification of a migration-related element in the gamma2 chain gene (LAMC2) with activity in transgenic mice. Matrix Biol. 18:197–210. [DOI] [PubMed] [Google Scholar]
- Schlessinger, J. 2000. Cell signaling by receptor tyrosine kinases. Cell. 103:211–225. [DOI] [PubMed] [Google Scholar]
- Seddighzadeh, M., J.N. Zhou, U. Kronenwett, M.C. Shoshan, G. Auer, M. Sten-Linder, B. Wiman, and S. Linder. 1999. ERK signalling in metastatic human MDA-MB-231 breast carcinoma cells is adapted to obtain high urokinase expression and rapid cell proliferation. Clin. Exp. Metastasis. 17:649–654. [DOI] [PubMed] [Google Scholar]
- Seftor, R.E., E.A. Seftor, N. Koshikawa, P.S. Meltzer, L.M. Gardner, M. Bilban, W.G. Stetler-Stevenson, V. Quaranta, and M.J. Hendrix. 2001. Cooperative interactions of laminin 5 gamma2 chain, matrix metalloproteinase-2, and membrane type-1-matrix/metalloproteinase are required for mimicry of embryonic vasculogenesis by aggressive melanoma. Cancer Res. 61:6322–6327. [PubMed] [Google Scholar]
- Shang, M., N. Koshikawa, S. Schenk, and V. Quaranta. 2001. The LG3 module of laminin-5 harbors a binding site for integrin alpha3beta1 that promotes cell adhesion, spreading, and migration. J. Biol. Chem. 276:33045–33053. [DOI] [PubMed] [Google Scholar]
- Simian, M., Y. Hirai, M. Navre, Z. Werb, A. Lochter, and M.J. Bissell. 2001. The interplay of matrix metalloproteinases, morphogens and growth factors is necessary for branching of mammary epithelial cells. Development. 128:3117–3131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stetler-Stevenson, W.G., and A.E. Yu. 2001. Proteases in invasion: matrix metalloproteinases. Semin. Cancer Biol. 11:143–152. [DOI] [PubMed] [Google Scholar]
- Swindle, C.S., K.T. Tran, T.D. Johnson, P. Banerjee, A.M. Mayes, L. Griffith, and A. Wells. 2001. Epidermal growth factor (EGF)-like repeats of human tenascin-C as ligands for EGF receptor. J. Cell Biol. 154:459–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talhouk, R.S., M.J. Bissell, and Z. Werb. 1992. Coordinated expression of extracellular matrix-degrading proteinases and their inhibitors regulates mammary epithelial function during involution. J. Cell Biol. 118:1271–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terranova, V.P., M. Aumailley, L.H. Sultan, G.R. Martin, and H.K. Kleinman. 1986. Regulation of cell attachment and cell number by fibronectin and laminin. J. Cell. Physiol. 127:473–479. [DOI] [PubMed] [Google Scholar]
- Thompson, S.A., A. Harris, D. Hoang, M. Ferrer, and G.R. Johnson. 1996. COOH-terminal extended recombinant amphiregulin with bioactivity comparable with naturally derived growth factor. J. Biol. Chem. 271:17927–17931. [DOI] [PubMed] [Google Scholar]
- Turpeenniemi-Hujanen, T., U.P. Thorgeirsson, C.N. Rao, and L.A. Liotta. 1986. Laminin increases the release of type IV collagenase from malignant cells. J. Biol. Chem. 261:1883–1889. [PubMed] [Google Scholar]
- Vogel, W., G.D. Gish, F. Alves, and T. Pawson. 1997. The discoidin domain receptor tyrosine kinases are activated by collagen. Mol. Cell. 1:13–23. [DOI] [PubMed] [Google Scholar]
- Xie, H., M.A. Pallero, K. Gupta, P. Chang, M.F. Ware, W. Witke, D.J. Kwiatkowski, D.A. Lauffenburger, J.E. Murphy-Ullrich, and A. Wells. 1998. EGF receptor regulation of cell motility: EGF induces disassembly of focal adhesions independently of the motility-associated PLCgamma signaling pathway. J. Cell Sci. 111:615–624. [DOI] [PubMed] [Google Scholar]
- Yamamoto, H., F. Itoh, S. Iku, M. Hosokawa, and K. Imai. 2001. Expression of the gamma(2) chain of laminin-5 at the invasive front is associated with recurrence and poor prognosis in human esophageal squamous cell carcinoma. Clin. Cancer Res. 7:896–900. [PubMed] [Google Scholar]
- Yarden, Y. 2001. The EGFR family and its ligands in human cancer. Signalling mechanisms and therapeutic opportunities. Eur. J. Cancer. 37:S3–S8. [DOI] [PubMed] [Google Scholar]
- Yurchenco, P.D., and Y.S. Cheng. 1993. Self-assembly and calcium-binding sites in laminin. A three-arm interaction model. J. Biol. Chem. 268:17286–17299. [PubMed] [Google Scholar]