

Abstract
Cell motility on ECM critically depends on the cellular response to force from the matrix. We find that force-dependent reinforcement of αv/β3-integrin–mediated cell–matrix connections requires the receptor-like tyrosine phosphatase α (RPTPα). RPTPα colocalizes with αv-integrins at the leading edge during early spreading, and coimmunoprecipitates with αv-integrins during spreading on fibronectin and vitronectin. RPTPα-dependent activation of Src family kinases, in particular activation of Fyn, is required for the force-dependent formation of focal complexes and strengthening of αv/β3-integrin–cytoskeleton connections during the initial phase of ECM contact. These observations indicate that Src family kinases have distinct functions during adhesion site assembly, and that RPTPα is an early component in force-dependent signal transduction pathways leading to the assembly of focal complexes on both fibronectin and vitronectin.
Keywords: phosphatase; force; src family kinases; focal contact; mechanotransduction
Introduction
As cells encounter new regions of the substrate, they develop bonds with new matrix molecules for migration, matrix remodeling, or traction force generation in the area of these adhesion sites. Physical force is critical in the development of the cell–matrix contacts at all stages (Geiger et al., 2001). ECM molecules are bound to integrins, which initiate intracellular signaling events causing integrins to connect with the cytoskeleton (Felsenfeld et al., 1996). As the rearward transport of the cytoskeleton develops force on the connection, the connection is reinforced in concert with the binding of focal contact proteins (Riveline et al., 2001; Galbraith et al., 2002). This strengthening of integrin–cytoskeleton linkages is blocked by the tyrosine phosphatase inhibitor phenylarsine oxide (Choquet et al., 1997), suggesting the involvement of tyrosine phosphatases in force transduction pathways.
Integrins form the transmembrane link between the cytoskeleton and ECM molecules (Giancotti and Ruoslahti, 1999). They are composed of two subunits, α and β, and each αβ combination has its own binding and signaling properties. Interestingly, some integrins can bind to several ECM ligands (Giancotti and Ruoslahti, 1999). The αv/β3-integrin, originally described as the vitronectin (VN)* receptor, binds to a variety of plasma and ECM proteins, including vitronectin, fibronectin (FN), fibrinogen, von Willebrand's factor, and bone sialic protein 1 (Krutzsch et al., 1999). Most of these ligands encode an RGD sequence that is presumed to represent the binding site for αv/β3-integrins. αv/β3-integrins are major components of focal contacts on FN and VN and localize to the tip of focal contacts where centripetal extension occurs (Felsenfeld et al., 1999). αv/β3-integrins associate with c-Src (Hruska et al., 1995) and the β3-cytoplasmic tail of αv/β3-integrins has been shown to mediate activation of c-Src in osteoclasts (Feng et al., 2001).
The family of transmembrane receptor-like protein tyrosine phosphatases (RPTPs) includes a number of proteins sharing the characteristics of an extracellular domain and intracellular PTP-homology domains. RPTPs have been implicated in the regulation of integrin-mediated events (Petrone and Sap, 2000). The RPTP CD45 associates with β2-integrins and the absence of CD45 enhances the adhesion to FN (Shenoi et al., 1999). In addition, the RPTP LAR localizes to adhesion sites in regions undergoing disassembly (Serra-Pages et al., 1995). Another member of this family that regulates adhesion events is RPTPα. Overexpression of RPTPα increases substrate adhesion and prevents cell rounding induced by insulin or EGF (Moller et al., 1995; Harder et al., 1998). More intriguingly, gene inactivation of RPTPα not only delays spreading on FN but also impairs activation of Src family kinases (SFK; Ponniah et al., 1999; Su et al., 1999). No soluble or cell-surface–anchored ligands are known to bind the extracellular domain of RPTPα (Petrone and Sap, 2000). It has been suggested that RPTPα forms a symmetrical-inhibited dimer in which a helix-turn-helix wedge element from one monomer is inserted into the catalytic cleft of the other monomer (Jiang et al., 2000). Upon activation, RPTPα associates with SFK such as c-Src, Fyn, and c-Yes (Zheng et al., 1992; Harder et al., 1998; Su et al., 1999). RPTPα dephosphorylates the negative regulatory tyrosine phosphate (Tyr529 in murine c-Src), causing c-Src activation (den Hertog et al., 1993; Zheng et al., 2000).
Here, we show that RPTPα associates with αv/β3-integrins during early spreading. In addition, we show that this association is required for the activation of SFK, the assembly of focal complexes, and the strengthening of integrin–cytoskeleton bonds on both FN and VN. Moreover, we provide evidence that RPTPα is a critical part of an early force-dependent signal transduction cascade.
Results
αv/β3-integrin–mediated cell spreading depends on RPTPα
To further characterize the role of RPTPα in integrin function, we compared the spreading of fibroblasts derived from RPTPα-expressing (RPTPα+/+) or RPTPα-deficient (RPTPα−/−) mice on FN and VN. Cell spreading on FN depended on the expression of RPTPα (Fig. 1 A; Su et al., 1999). On VN substrates, RPTPα+/+ cells or RPTPα−/− cells expressing wild-type RPTPα (RPTPα−/−wt) spread within 30 min after plating (Fig. 1 A). Although RPTPα−/−wt cells reached on average only a level of RPTPα protein expression of 55%, compared with RPTPα+/+ cells (as judged by scanning densitometry [unpublished data]; Fig. 1 C), this level restored the phenotype. In contrast, RPTPα−/− cells or empty vector (RPTPα−/−vec)-transfected cells plated on VN substrates or RPTPα+/+ cells plated on control substrates (BSA) showed significantly less spreading (Fig. 1, A and D). Interestingly, there was no difference in the spreading ability on laminin (LA), suggesting a substrate dependency (Fig. 1 B).
Figure 1.

Cell spreading depends on RPTPα and αv/β3-integrins. (A) Percentage of spread cells on either FN or VN was quantified 30 min after plating. (left) Spreading of RPTPα+/+ cells (+/+) compared with RPTPα−/− cells (−/−). (right) RPTPα−/−wt cells (−/−wt) compared with RPTPα−/−vec cells (−/−vec). Results shown are the mean ± SD of three to five independent experiments. (B) RPTPα+/+ and RPTPα−/− cells were plated, and the percentage of spread cells on LA was quantified 30 min after plating. Results shown are the mean ± SD of three independent experiments. (C) Equal amounts of protein from each cell line were analyzed by Western blotting using RPTPα, αv-, and β3-integrin antibodies. Micrographs shown are representative of at least three independent experiments. (D) RPTPα+/+ cells were plated with or without GPen (0.5 mM) and anti–αv- and anti–β3- integrin antibodies (25 μg/ml); the percentage of spread cells on VN was quantified after 30 min and compared with control substrates coated with BSA. Results shown are the mean ± SD of three independent experiments. (E) Percentage of spread cells in the presence of GPen or anti–αv- or anti–β3-integrin antibodies was compared after 30 min on FN. (left) Spreading of RPTPα+/+ cells compared with RPTPα−/− cells. (right) RPTPα−/−wt cells compared with RPTPα−/−vec cells. Results shown are the mean ± SD of three independent experiments.
Because FN can interact with αv/β3-integrins, spreading on FN substrates may involve the αv/β3-integrin. This integrin was expressed on all cells studied, as indicated by protein expression (Fig. 1 C) and FACS® analysis (unpublished data). To determine the contribution of αv/β3-integrins, we plated cells on FN substrates with or without adhesion blocking, monoclonal anti-αv or anti-β3 antibodies (both 25 μg/ml), or the cyclic peptide GPenGRGDSPCA (GPen; 0.5 mM). At this concentration, GPen is a selective, competitive inhibitor of the αv/β3-integrin that inhibits activation of intracellular signaling (Etienne-Manneville and Hall, 2001) and does not block α5/β1-integrins (Pierschbacher and Ruoslahti, 1987). Indeed, anti-αv or anti-β3 antibodies, as well as GPen, drastically reduced cell spreading on VN substrates (Fig. 1 D). In contrast, FN pretreatment with either GPen or with anti-αv or anti-β3 antibodies led to a twofold reduction of spread RPTPα+/+ and RPTPα−/−wt cells 30 min after plating (Fig. 1 E). Surprisingly, there was no further reduction in the number of spread RPTPα−/− or RPTPα−/−vec cells after pretreatment with GPen or with anti-αv or anti-β3 antibodies (Fig. 1 E). Together, these results indicate that the RPTPα-dependent differences in cell spreading on FN and VN substrates are mediated by the αv/β3-integrin.
RPTPα transiently colocalizes with αv-integrins
Next, we wanted to determine whether RPTPα colocalizes with αv/β3-integrins. RPTPα−/− cells were transiently transfected with an RPTPα construct in which the D2 phosphatase domain was replaced by YFP (Buist et al., 2000). To confirm functionality, we performed spreading assays with RPTPα-YFP– transfected RPTPα−/− cells and found a spreading ability similar to the RPTPα+/+ cells (Fig. 2 B). Indirect immunofluorescence was used to visualize the αv-integrins and paxillin. Confocal microscopy revealed that RPTPα colocalized with αv-integrins and paxillin at the leading edge on both FN and VN 30 min after plating in spreading cells (Fig. 2 A, a–e and k–o). After 240 min, all cells were spread and RPTPα was diffusely localized in the membrane (Fig. 2 A, f–j and p–t), whereas colocalization of αv-integrins with paxillin occurred in the area of focal contacts (Fig. 2 A, j and t). On LA, there was no localization of RPTPα to the leading edge in spreading cells (Fig. 2 C). Thus, we find evidence of a transient association between RPTPα and αv/β3-integrins during the early phases of focal complex formation on both FN and VN.
Figure 2.
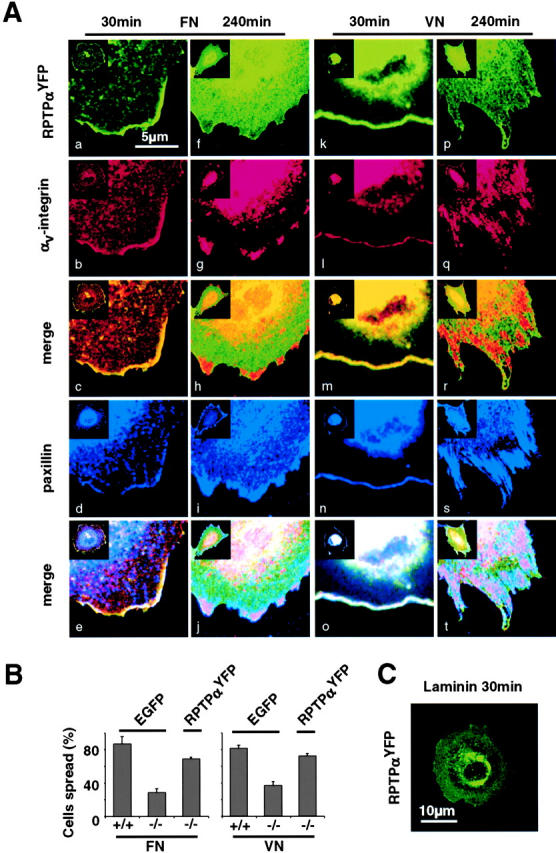
RPTPα colocalizes with αv/β3-integrins at early times. (A) RPTPα−/− cells were transiently transfected with RPTPα-YFP and spread for 30 and 240 min, fixed and stained with anti–αv-integrin and paxillin antibodies. Distribution was analyzed on either FN (a–j) or VN (k–t) after 30 min (left column) and 240 min (right column). Merges show the overlay of RPTPα and αv-integrins (c, h, m, and r) and the overlay of RPTPα, αv-integrins, and paxillin (e, j, o, and t). Micrographs shown are representative of at least five independent experiments. (B) RPTPα+/+ cells (+/+) were transiently transfected with EGFP, and RPTPα−/− cells (−/−) were transiently transfected with either EGFP or RPTPα-YFP and spread for 30 min, and the percentage of spread fluorescent cells on either FN (left) or VN (right) was quantified. Results shown are the mean ± SD of three independent experiments. (C) RPTPα−/− cells were transiently transfected with RPTPα-YFP and spread for 30 min on LA. Picture shown is representative of two independent experiments.
RPTPα transiently associates with αv-integrins and cooperates with αv/β3-integrins in the activation of downstream signaling on both FN and VN
To further investigate the relationship between RPTPα and αv/β3-integrins, we performed coimmunoprecipitation studies. We were able to coimmunoprecipitate RPTPα in anti–αv-integrin immunoprecipitates of spreading RPTPα+/+ cells after pretreatment with a water-soluble cross-linker on both FN and VN. This association was restricted to the early phases of spreading because there was no coimmunoprecipitation after 240 min. Furthermore, activation of αv/β3-integrins was necessary for the formation of the complex because cells kept in suspension for 60 min or cells pretreated with GPen did not show association of RPTPα with αv/β3-integrins. In addition, this interaction was specific for αv-integrins because anti–α5-integrin immunoprecipitates did not contain RPTPα (Fig. 3 A).
Figure 3.

RPTPα and αv/β3-integrins form a complex and cooperate in the activation of SFK. (A) RPTPα+/+ (+/+) and RPTPα−/− cells (−/−) were plated for 15 min on FN or VN with or without GPen. Alternatively, cells were plated for 240 min on FN or were kept in suspension for 60 min. Cells were subsequently cross-linked, lysed, and subjected to immunoprecipitation with (+) or without (−) addition of anti–αv- or anti–α5-integrin antibodies. Western blotting was performed with either anti-RPTPα (top) or anti–αv- and α5- integrin (bottom) antibodies. Micrographs shown are representatives of at least two independent experiments. (B, top) RPTPα+/+ (+/+), RPTPα−/− (−/−), and RPTPα−/−wt cells (−/−wt) were spread for 15 min on FN or VN with or without GPen, lysed, and equal amounts of protein were analyzed by Western blotting using a phosphospecific anti-SFK antibody (Tyr416), a de-phosphospecific anti–c-Src antibody (Tyr 527) or an anti–c-Src antibody. (bottom) Relative Tyr416 autophosphorylation of SFK was quantified using scanning densitometry and normalized to control cells (+/+). Results shown are the mean ± SD of three independent experiments.
To elucidate the functional relevance of an association of αv/β3-integrins with RPTPα, we performed Western blotting with a phosphospecific anti-SFK antibody (autophosphorylation on Tyr416). On both, FN and VN, SFK activity was three times lower in RPTPα−/− cells than it was in the RPTPα+/+ cells. Interestingly, the pretreatment of RPTPα+/+ cells with GPen significantly reduced SFK activation 15 min after plating. However, there was no further reduction in the amount of SFK activity in the RPTPα−/− cells on either substrate after pretreatment with GPen. Consistent results were obtained using an antibody specific for the dephosphorylated tyrosine 527 in c-Src. In addition, reexpression of wild-type RPTPα restored the ability to activate SFKs (Fig. 3 B). Together, these results suggest functional cooperation of αv/β3-integrins with RPTPα in the activation of SFKs. Because the antibody we used recognizes several SFKs, we cannot determine if SFKs other than c-Src are activated, but the RPTPα substrates Fyn and c-Yes are likely possibilities.
RPTPα modulates the formation of focal complexes in association with αv/β3-integrins via activation of Fyn
To study more directly the interplay between RPTPα and αv/β3-integrins, we observed the distribution of GFP-tagged paxillin on different substrates. Paxillin is a focal adhesion–associated adaptor protein that is a known marker for focal adhesions and focal complexes (Beningo et al., 2001). GFP-paxillin distribution in distinct adhesion sites versus cytoplasmic localization was analyzed in spread cells 30 min after plating. In RPTPα+/+ cells on both substrates, GFP-paxillin distributed to peripheral stripes in the majority of cells (Fig. 4, A [a and g] and B). In contrast, in RPTPα−/− cells, there was more than twofold lower focal complex or contact formation at early times. GFP-paxillin remained mainly cytoplasmic and localized to the leading edge (Fig. 4, A [d and j] and B). To determine the function of the αv/β3-integrin in this process, we plated GFP-paxillin–transfected cells on FN and VN in the presence of GPen. In RPTPα+/+ cells on both substrates, there was significantly less formation of distinct adhesion sites with GPen, as judged by paxillin assembly (Fig. 4, A [b and h] and B). In contrast, there was no further reduction in the number of cells exhibiting formation of adhesion sites in the RPTPα−/− cells with GPen (Fig. 4, A [e and k] and B). As expected, GPen drastically reduced the ability of both RPTPα+/+ and RPTPα−/− cells to spread on VN (Fig. 4, A [h and k] and B).
Figure 4.
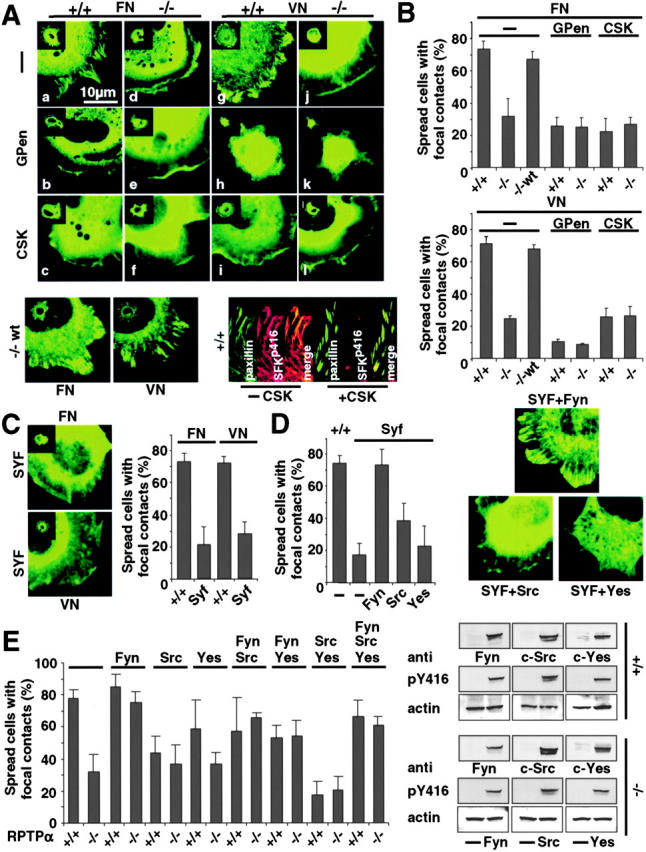
Formation of focal complexes through RPTPα and αv/β3-integrins via SFK. (A, top) RPTPα+/+ (+/+) and RPTPα−/− cells (−/−) were transfected with GFP-paxillin and spread for 30 min on FN or VN. The distribution of GFP-paxillin was compared 30 min after plating (a, d, g, and j). Cells were pretreated with GPen (b, e, h, and k) or cotransfected with CSK (c, f, i, and l), and the number of cells with either focal complexes or contacts was quantified. (bottom, left) RPTPα−/−wt cells (−/−wt) were allowed to spread on FN or VN and paxillin assembly in distinct adhesion sites was quantified after 30 min. (bottom, right) RPTPα+/+ cells were cotransfected with (+CSK) or without CSK and GFP-paxillin, fixed, and stained with anti–phospho-SFK. Micrographs shown are representative of at least three independent experiments. (B) Percentage of spread cells showing GFP-paxillin assembly in distinct adhesion sites was quantified versus cells exhibiting cytoplasmic distribution of GFP-paxillin on either FN (top) or VN (bottom). Results shown are the mean ± SD of five independent experiments. (C) Wild-type cells (c-Src+/+) (+/+) or c-Src-Fyn-Yes–deficient cells (SYF) were plated on FN or VN for 15 min, and paxillin assembly in distinct adhesion sites was quantified. Results shown are the mean ± SD of three independent experiments. (D) SYF cells were plated on FN for 15 min with or without cotransfection of GFP-paxillin with wild-type Fyn, c-Src, or c-Yes. GFP-paxillin assembly in distinct adhesion sites was quantified as described above. Results shown are the mean ± SD of three independent experiments. (E, left) RPTPα+/+ (+/+) and RPTPα−/− cells (−/−) were plated on FN for 30 min with or without cotransfection of wild-type Fyn, c-Src, or c-Yes and GFP-paxillin. The percentage of cells showing GFP-paxillin assembly in distinct adhesion sites was quantified as described above. Results shown are the mean ± SD of at least five independent experiments. (right) RPTPα+/+ (+/+) and RPTPα−/− cells (−/−) were transiently transfected with wild-type Fyn, c-Src, or c-Yes, subsequently lysed, and equal amounts of protein were analyzed by Western blotting using anti-Fyn, c-Src, or c-Yes antibodies or a phosphospecific anti-SFK antibody (Tyr416). Membranes were stripped and probed for actin to confirm protein content. Micrographs shown are representative of two independent experiments.
To observe the RPTPα-dependent role of SFK activation in the assembly of adhesion sites, GFP-paxillin and c-terminal Src kinase (CSK) were cotransfected into RPTPα+/+ and RPTPα−/− cells. CSK counteracts the effect of RPTPα on the activation of SFK by phosphorylating the inhibitory tyrosines, which are dephosphorylated by RPTPα. Inhibition of SFK activation in RPTPα+/+ cells led to a threefold reduction in the number of cells showing focal complexes or contacts (Fig. 4, A [c and i] and B). Strikingly, there was no further decrease in the RPTPα−/− cells (Fig. 4, A [f and l] and B). To confirm that overexpression of CSK actually led to reduction in the amount of SFK activity, RPTPα+/+ cells were cotransfected with GFP-paxillin and CSK and stained with an anti–phospho-SFK antibody. As expected, CSK almost completely prevented the activation of SFK (Fig. 4 A, bottom right). To verify the effect of CSK on the formation of focal complexes, cells deficient for the ubiquitously expressed SFKs, c-Src, Fyn, and c-Yes (SYF cells) were plated for 15 min on FN and VN. Quantification of spread cells showing formation of either focal complexes or contacts revealed a twofold reduction for the SYF cells compared with wild-type fibroblasts (Fig. 4 C). To differentiate which SFK is responsible for the assembly signal, we cotransfected c-Src, Fyn, c-Yes, and GFP-paxillin into SYF cells. Interestingly, cotransfection with Fyn led to full rescue in the ability to form distinct adhesion sites. In sharp contrast, the transfection of either c-Src or c-Yes did not restore the formation of focal complexes. To transfer these findings, we cotransfected c-Src, Fyn, and c-Yes alone and combinations thereof with GFP-paxillin into RPTPα+/+ and RPTPα−/− cells. Interestingly, the coexpression of Fyn led to a dramatic increase in the number of cells exhibiting focal complexes or contacts. In striking contrast, coexpression of c-Src led to reduction in focal contact formation at early times in RPTPα+/+ cells and only to a small increase in RPTPα−/− cells. Coexpression of c-Yes did not affect the formation of early adhesion sites in RPTPα+/+ cells significantly. c-Yes had an effect similar to c-Src because the combination of Fyn with either c-Src or c-Yes reduced the number of cells having distinct adhesion sites in RPTPα−/− cells and coexpression of c-Src and c-Yes dramatically reduced the formation of focal complexes or contacts in RPTPα+/+ cells at early times (Fig. 4 E). Expression and activation of the transfected SFK was confirmed by Western blotting with antibodies against the individual SFK and a phosphospecific anti-SFK antibody (Fig. 4 E). Together, these data strongly indicate that the assembly of early focal complexes, as well as contacts, depends on the αv/β3-integrin and RPTPα-dependent activation of Fyn.
Loss of RPTPα does not affect integrin–ligand interactions
The sensitivity of cell spreading on ECM substrates to RPTPα expression raises the possibility that RPTPα also modulates integrin–ligand interactions (Hughes and Pfaff, 1998). To study more directly the role of RPTPα in regulating integrin–ligand interactions, we placed ligand-coated beads on the upper surface of spreading fibroblasts with a laser trap (<0.5 μm from the leading edge of the cell). This allows the quantification of changes in ligand binding and the dynamics of traction force generation by integrins (Choquet et al., 1997), which are parameters that are essential to cell spreading and migration (Lauffenburger and Horwitz, 1996). Beads were coated with a recombinant fragment of FN (FNIII7-10), purified human VN, or BSA. Beads coated with either FNIII7-10 or VN bound to the surface with a similar frequency and significantly better than BSA-coated control beads (Fig. 5 A). The binding of VN-coated beads was reduced to the level of control beads by pretreatment with GPen (Fig. 5 A). When FNIII7-10–coated beads were placed and held on the upper surface of cells in the presence of GPen, there was a significant reduction of bead binding on both RPTPα+/+ and RPTPα−/− cells (Fig. 5 A). Together, these results suggest that integrin–ligand interactions are not modulated by RPTPα activity. The contrast between the impaired cell spreading and the unaffected bead binding indicates that RPTPα may regulate the strength or dynamics of integrin–cytoskeleton interactions.
Figure 5.

RPTPα deficiency does not affect integrin–ligand interactions, but leads to weak integrin–cytoskeleton linkages. (A) FN (left)- or VN (right)-coated beads (1-μm diam) bound specifically to the upper surface of RPTPα+/+ (+/+) and RPTPα−/− cells (−/−). Percentages of bound beads after 2 s were equalized with or without addition of GPen. Binding was compared with control beads coated with BSA. Results shown are the mean ± SD of three independent experiments. (B) Beads coated with FN (left) or VN (right) were placed on the upper surface of RPTPα+/+, RPTPα−/−, and RPTPα−/−wt cells, and the MSD was quantified after beads left the trap field (500 nm). Results shown are the mean ± SD of three independent experiments.
Loss of RPTPα inhibits the strengthening of integrin–cytoskeleton bonds
To evaluate the strength of integrin–cytoskeleton bonds, we placed and held beads on the cell surface using a laser trap. We have shown previously that FN-coated beads bind to cell-surface integrins and are drawn out of the trap in association with the retrograde moving actin cytoskeleton (Choquet et al., 1997). If the bead is not restrained by the trap after placement on the lamellipodium, it can be dislocated by the laser trap, whereas it is not possible to dislocate a bead that was restrained by the tweezers and pulled out of the trap by the cell. Thus, the cell regulates the strength of integrin–cytoskeleton linkages in response to the rigidity of the laser trap. If there is a defect in this reinforcement process, the bead is only loosely attached to the underlying cytoskeleton, resulting in a greater diffusion rate perpendicular to the direction of rearward movement. This can be quantified using the mean square displacement (MSD) of the bead diffusion (Qian et al., 1991). Interestingly, after placement of FNIII7-10– and human VN-coated beads onto RPTPα−/− cells, the MSD of the bead during retrograde transport is two to three times larger than in the RPTPα+/+ cells (Fig. 5 B). To confirm the dependency on RPTPα expression, we performed the same experiments with RPTPα−/−wt cells. Indeed, reexpression of RPTPα drastically reduced the MSD (Fig. 5 B). Interestingly, the MSD of VN-coated beads is up to six times larger than that for FN-coated beads, suggesting that VN-coated beads are only weakly linked to the cytoskeleton. On the basis of these results, we suggest that RPTPα is a selective activator of linkage assembly during the establishment of new adhesion sites.
Reinforcement of integrin–cytoskeleton linkages depends on RPTPα, αv/β3-integrins, and activation of Fyn
To further analyze the reinforcement process, we centrifuged onto cells (5 min/50 g) large beads (5.9-μm diam) coated with either FNIII7-10 or human VN. In contrast to the small beads used for the laser trap experiments, the large beads can cause the establishment of adhesion sites and, therefore, reinforcement by cellular contractions independent of laser trap restraint (Galbraith et al., 2002). Interestingly, 74% of the FNIII7-10–coated beads showed accumulation of GFP-paxillin to the bead periphery after 30 min on RPTPα+/+ cells (46% of VN-coated beads; unpublished data). In contrast, only 14% of the FNIII7-10–coated beads (7% of VN-coated beads; unpublished data) on RPTPα−/− cells showed redistribution of paxillin to the binding site (Fig. 6, A and B) . Next, we wanted to examine the function of SFK and αv/β3-integrins in the reinforcement process. Interestingly, pretreatment with GPen reduced the fraction of FNIII7-10–coated beads accumulating paxillin on RPTPα+/+ cells to 18%, but caused no significant reduction in the number of accumulating beads on the RPTPα−/− cells (11%; Fig. 6, A and B). Because the pretreatment with GPen prevented specific binding of VN-coated beads to the upper surface of RPTPα+/+ and RPTPα−/− cells (Fig. 5 A), there was no accumulation of paxillin around VN-coated beads in any case (unpublished data). Coexpression of CSK with paxillin decreased the number of FNIII7-10–coated beads, causing accumulation of paxillin to 16% in RPTPα+/+ cells, whereas there was no further reduction in RPTPα−/− cells (8%; Fig. 6, A and B). Consistent results could be obtained with VN-coated beads (8% for RPTPα+/+ cells and 5% for RPTPα−/− cells; unpublished data). We have shown earlier that RPTPα-dependent activation of Fyn is needed for the formation of focal complexes and contacts, suggesting that it might affect reinforcement. Indeed, coexpression of Fyn with GFP-paxillin led to a significant increase in the number of RPTPα−/− cells accumulating paxillin around beads (68%). In clear contrast, the coexpression of c-Src or c-Yes did not increase the number of accumulating beads in RPTPα−/− cells, but coexpression of c-Src reduced the number of cells responding to the beads in RPTPα+/+ cells to 34% (Fig. 6 C). Reintroduction of wild-type RPTPα into RPTPα−/− cells restored the ability to assemble paxillin at the binding site (Fig. 6 B). To confirm ligand specificity, we used Con A–coated beads on RPTPα+/+ cells, which showed accumulation of GFP-paxillin at the site of adhesion in only 7% of the beads, possibly due to unspecific activation of integrin receptors (Fig. 6 B). To exclude the possibility of a volume effect around the beads, we transfected RPTPα+/+ cells with EGFP alone, which did not increase the signal intensity around the beads in any case (Fig. 6 B, bottom right). Together, these results are consistent with the idea that RPTPα- and αv/β3-integrin–dependent reinforcement of integrin–cytoskeleton linkages is mediated by activation of SFK, in particular, activation of Fyn.
Figure 6.
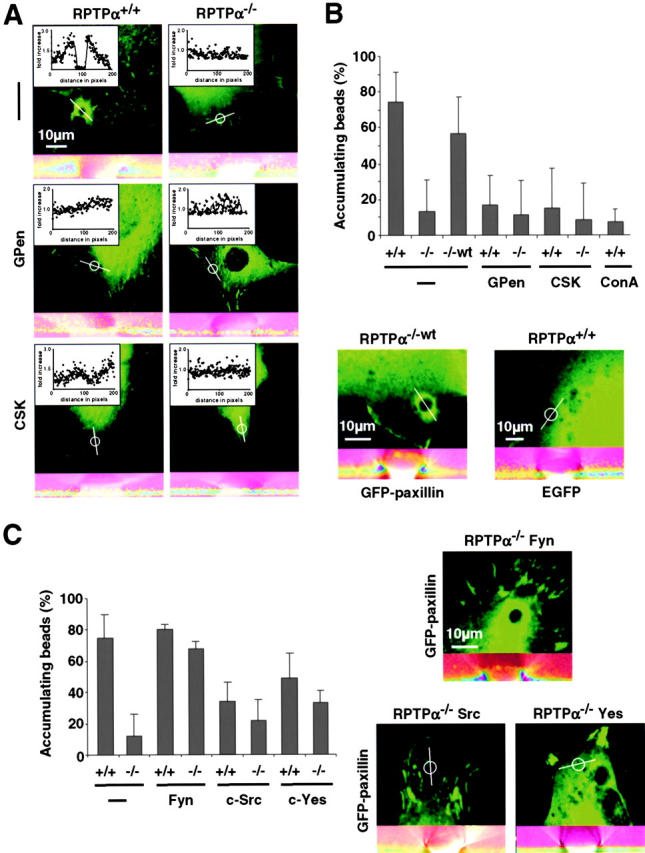
Reinforcement requires RPTPα- and αv/β3-integrin–dependent activation of SFK. (A, top) RPTPα+/+ and RPTPα−/− cells were transfected with GFP-paxillin and were spread for 30 min. Large beads (5.9-μm diam) coated with FN were spun onto the cells and incubated for 30 min. The fraction of bound beads causing accumulation (fluorescence intensity >2× surrounding; inset) of paxillin was determined. Confocal stacks were resliced along the indicated line over the beads position (bottom of each panel), shown are overlays of GFP-paxillin fluorescence intensity in pseudo-colors and the differential interference contrast image. (middle) RPTPα+/+ and RPTPα−/− cells were pretreated with GPen. (bottom) RPTPα+/+ and RPTPα−/− cells were cotransfected with CSK and analyzed as described above. (B, top) Percentage of FN- or Con A–coated beads causing accumulation of paxillin. Results shown are the mean ± SD of three independent experiments. (bottom) RPTPα−/−wt cells (−/−wt) 30 min after application of beads. (bottom, right) RPTPα+/+ cells transfected with EGFP alone 30 min after application of beads. (C) RPTPα+/+ (+/+) and RPTPα−/− cells (−/−) were spread for 30 min with or without cotransfection of wild-type Fyn, c-Src, or c-Yes and GFP-paxillin. Accumulation was analyzed as described above. (left) Percentage of FN-coated beads causing accumulation of paxillin. (right) Representative micrographs of RPTPα−/− cells coexpressing Fyn (top), c-Src (bottom, left), c-Yes (bottom, right), and GFP-paxillin 30 min after application of FN-coated beads.
Response to force requires expression of RPTPα
It has been suggested that phosphatases are involved in the process of mechanotransduction and our data are consistent with the idea that the phosphatase RPTPα is required for force-dependent reinforcement of integrin–cytoskeleton linkages. To further address this hypothesis, we designed a laser trap experiment that allowed the visualization of GFP-tagged protein dynamics in living cells and tested whether the application of force to small ligand-coated beads (1-μm diam) induces the accumulation of focal contact proteins to the adhesion site. As predicted, there was no accumulation of paxillin to the binding site in RPTPα+/+ cells when the trap was turned off after placement of beads (0%, n = 25; Fig. 7 A, bottom left). Strikingly, the restraint of FNIII7-10-coated beads with the trap caused accumulation of GFP-paxillin to the binding site within seconds in RPTPα+/+ cells (82%, n = 22; Fig. 7 A, top). The paxillin accumulation began as a distinct spot underneath the bead after application of force. However, as the bead was pulled out of the trap by the retrograde motion of the cytoskeleton, force was exerted on the bead, and the pattern of paxillin assembly changed to a ring around the bead (as seen with the large beads). When RPTPα−/− cells were tested, significantly fewer cells accumulated paxillin to the site of interaction, with (15%, n = 20) or without (0%, n = 24; unpublished data) sustained application of force by the trap (Fig. 7 B). To confirm a dependency on the expression of RPTPα, we performed these experiments with RPTPα−/−wt cells. As expected, reexpression of RPTPα restored the ability to respond to applied forces with the accumulation of paxillin (65%, n = 23; Fig. 7 C). These data strongly indicate that RPTPα is part of force-dependent signal transduction events, and that it is a crucial component in this process.
Figure 7.
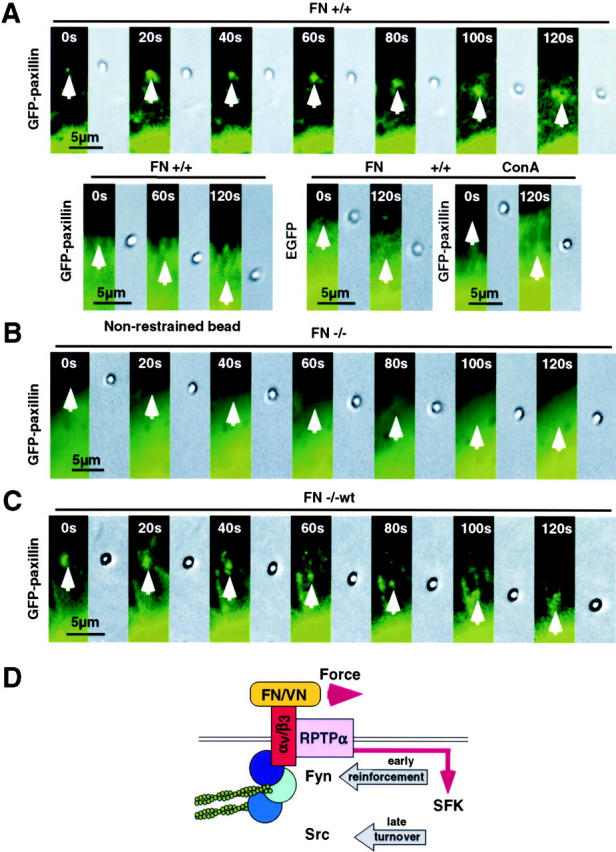
Response to force requires expression of RPTPα. (A, top) Accumulation of GFP-paxillin in RPTPα+/+ cells (+/+) in serial micrographs of rearward moving beads coated with FN (1-μm diam) after placement on the upper surface and escape out of the trap. Beads position is indicated by an arrow. (bottom, left) Beads were placed on the upper surface and GFP-paxillin assembly was quantified without application of force in RPTPα+/+ cells. (bottom, right) Serial micrographs of RPTPα+/+ cells transfected with EGFP alone (left) and of GFP-paxillin transfected RPTPα+/+ cells after placement of Con A–coated beads (right). (B) Time-lapse micrographs of GFP-paxillin–expressing RPTPα−/− cells (−/−) after placement and escape out of the trap of FN-coated beads. (C) Time-lapse micrographs of rearward moving FN beads after placement and escape out of the trap on RPTPα−/−wt cells (−/−wt). (D) Model for the force-dependent assembly of focal complexes. First, upon formation of active lamellipodia, a complex of αv/β3-integrins and RPTPα is formed, localizing to the edge of the lamellipodium. Second, force application to αv/β3-integrins leads to RPTPα-dependent activation of SFK. Third, SFK-activation promotes the assembly and the reinforcement of focal complexes at early times. Finally, as focal complexes mature, SFK activity is also required for turnover of adhesion sites.
Discussion
Our results indicate that RPTPα interacts with αv/β3-integrins, either directly or via adaptor molecules. Although we were able to coimmunoprecipitate a complex of RPTPα and αv/β3-integrins only after cross-linking, the combination of these results with colocalization and cooperation in the activation of SFK suggests the formation of a functional complex. It has been shown for a number of integrins that lateral association with other membrane proteins, such as CD47, is part of the regulatory machinery-controlling integrin function (Brown and Frazier, 2001). Furthermore, RPTPα can interact in cis with other transmembrane receptors such as contactin (Zeng et al., 1999). In addition, it has been reported that αv/β3-integrins localize in a rac-dependent process to lamellipodia, where new matrix adhesions are formed (Kiosses et al., 2001). Furthermore, αv/β3-integrin–dependent signaling is involved in the reinforcement of integrin–cytoskeleton linkages (Felsenfeld et al., 1999), and it has been reported that αv/β3-integrins influence FN receptor (α5/β1-integrin)–dependent migration toward FN (Blystone et al., 1999).
RPTPα is a well-characterized activator of SFK (Ponniah et al., 1999; Su et al., 1999). Although previous studies have suggested that SFKs are not involved in the assembly of focal contacts (Bockholt and Burridge, 1995; Klinghoffer et al., 1999), those results were obtained after several hours of incubation on a substratum, where we also find normal focal contact formation in RPTPα−/− and SYF cells (unpublished data). Our data obtained with coexpression of CSK or with the SYF cells suggest that SFK could transmit a signal for focal complex assembly as well as disassembly. It has been shown recently that SFKs are needed for the maintenance of focal contacts (Li et al., 2002) as well as for the tyrosine phosphorylation of proteins in early focal contacts (Volberg et al., 2001). c-Src has been shown to regulate focal contact turnover (Fincham and Frame, 1998; Felsenfeld et al., 1999). On the other hand, Fyn has been reported to be involved in the organization of the cytoskeleton (Stein et al., 1994; Thomas et al., 1995), and is thought to be responsible for cell volume–dependent changes in the phosphorylation of cytoskeletal and focal contact proteins (Kapus et al., 2000). In addition, Fyn has been implicated in Rho-mediated cell–cell adhesion events (Calautti et al., 2002). Our data indicate that SFK, although activated at the same time by the same enzyme, can have opposing effects on the formation of focal contacts. Fyn expression reconstituted almost wild-type levels, whereas expression of c-Src reduced the response to force and the formation of focal complexes. These results are of particular interest because they show that the decrease in force responsiveness with RPTPα deficiency can be partially compensated by high levels of Fyn. These data suggest parallel paths of force sensing in structural components (Sawada and Sheetz, 2002), which might be the target of force-dependent, RPTPα-mediated activation of Fyn. Therefore, the role of SFK remains controversial, especially during the very beginning of contact formation, and a dual role of SFK in assembly, as well as disassembly/turnover, of focal contacts has to be taken into account.
Recently, it has been shown that overexpression of RPTPα leads to an increased cell–substrate adhesion that is associated with increased levels of tyrosine phosphorylation of paxillin, c-Src, and/or focal adhesion kinase (Harder et al., 1998). In addition, it has been demonstrated that expression of RPTPα corresponds with a low tumor grade in breast cancer (Ardini et al., 2000). Our results indicate that RPTPα accelerates the assembly of adaptor and structural proteins at adhesion sites, thereby stabilizing the link between integrins and the cytoskeleton. It is probably not RPTPα itself that is interacting with these proteins; rather, RPTPα induces the appropriate signals. Regulated force generation is essential for cell spreading and migration. For spreading to proceed, adhesion sites must be continually remodeled (Sheetz et al., 1999). Our data raise the possibility that the lack of appropriate signals leads to weak linkages and results in an impaired plasticity in the integrin–cytoskeleton linkages needed for rapid cell spreading.
How cells respond to matrix rigidity through force sensing and convert physical cues to biochemical signals is still largely unknown. Besides the mechanical stimuli leading to enlargement of cell adhesions sites (Riveline et al., 2001), other stimuli affect cell behavior including stretch (MacKenna et al., 1998; Sawada et al., 2001) and flow-induced shear stress. Both transmembrane and cytoskeletal mechanisms of force sensing and force transduction have been postulated (Meyer et al., 2000; Sawada and Sheetz, 2002). In addition, force-dependent unfolding of cryptic protein–protein interaction sites in matrix molecules (Zhong et al., 1998) and recruitment of integrins, leading to activation of MAPKs (MacKenna et al., 1998), has been suggested. Specifically, αv/β3-integrins have been implicated in the transmission of mechanical signals (Wilson et al., 1995; Goldschmidt et al., 2001). Nonetheless, the mechanisms leading to the transformation of force into a biochemical signal are only poorly understood. The data presented here suggest that RPTPα localizes with αv/β3-integrins in a complex and that is a critical component in the force-dependent activation of subsequent signaling steps. Although RPTPα has an extracellular domain, which has been shown to interact in cis with other transmembrane proteins (Zeng et al., 1999), there are no known ligands either soluble or cell-surface bound (Petrone and Sap, 2000). RPTPα may homodimerize on the cell surface (Jiang et al., 2000) and the inactive, dimeric form could be activated by the application of force via αv/β3-integrins, leading to mechanical separation of the dimer. However, the precise mechanism leading to the activation of RPTPα remains speculative.
In mice missing RPTPα, there are severe hippocampal abnormalities and learning defects consistent with a developmental deficit in radial neuronal migration (unpublished observation). The similarity with Fyn deficiency (Grant et al., 1992) and the documented role of α3- and αv-integrins in this process (Anton et al., 1999) underlines a possible role for neuronal RPTPα in mechanotransduction at points of glial/neuronal contact during migration of neurons.
Materials and methods
Cell culture
RPTPα fibroblasts were described elsewhere (Su et al., 1999). c-Src-Fyn-Yes–deficient cells and c-Src+/+ wild-type fibroblasts were obtained from the American Type Culture Collection. All cells were maintained in DME supplemented with 10% (vol/vol) FBS in a humidified atmosphere of 5% CO2/95% air at 37°C and passaged every 4 d.
Spreading assays
40 μg/ml LA, 5 μg/ml VN, and the 120-kD chymotryptic fragment of FN (6.6 μg/ml) were adsorbed onto tissue culture plastic and blocked with DME/0.5% BSA. Control substrates were blocked without the application of ECM. Cells were briefly trypsinzed and trypsin was inactivated with 1 mg/ml soy trypsin inhibitor in DME. Cells were washed twice (DME/0.5% BSA) and incubated for 30 min with or without GPen (0.5 mM) or anti-αv or anti-β3 antibodies (25 μg/ml) before plating. Cells were fixed with 2% paraformaldehyde (PFA) and stained with 0.5% toluidine blue in PFA. 20 fields were counted in each well (20×; Olympus). For paxillin distribution assays, cells were transiently transfected using Fugene 6 (3:1) with GFP-paxillin (0.5 μg/ml), CSK (0.5 μg/ml), wild-type Src, Fyn, or Yes (each 0.5 μg/ml) and plated in the presence or absence of GPen. The number of spread cells exhibiting accumulation of GFP-paxillin in distinct adhesion sites (focal complexes or contacts) versus cells having cytoplasmic localization of GFP-paxillin was counted in two independent sets of experiments by independent observers. Pictures were taken using confocal microscopy (100×; Fluoview 300; Olympus).
Immunoprecipitations and Western blotting
For immunoprecipitations, extracellular proteins of 16 × 106 cells were cross-linked in the presence of 2 mM dithiobis succinimidyl propionate for 15 min and lysed on ice in 50 mM Tris-HCl, 150 mM NaCl, 50 mM NaF, 1 mM Na3VO4, 1% NP-40, 1 mM PMSF, 10 μg/ml aprotinin, and 10 μg/ml leupeptin, pH 7.6 (lysis buffer). The lysates were incubated with polyclonal anti- αv or anti-α5 antibodies for 2 h at 4°C on a rotating wheel with protein A–Sepharose beads added for the second hour. Beads were washed twice in lysis buffer and twice in wash buffer (50 mM Tris-HCl, pH 8.0) and resuspended in 2× SDS-PAGE sample buffer. For whole cell lysates, cells were lysed and lysates were diluted in 3× SDS-PAGE sample buffer. Equal amounts of proteins were further analyzed by SDS-PAGE followed by Western blotting using polyclonal anti-RPTPα, anti-αv, anti-β3, phosphospecific anti-SFK, or monoclonal anti–c-Src and dephosphospecific anti-SRC antibodies (clone 28) with immunoreactive bands being visualized by ECL detection.
Immunofluorescence staining
Fibroblasts were transfected with EYFP-RPTPα or GFP-paxillin (0.5 μg/ml) and plated as described in Spreading assays. Cells were fixed 30 min after plating in 4% PFA for 20 min and permeabilized with 0.2% Triton X-100. Cells were incubated with monoclonal antipaxillin (mouse) and anti-αv antibodies (hamster) and a polyclonal phosphospecific anti-SFK antibody for 1 h followed by detection with Alexa-labeled (488, 568, and 647 nm) secondary antibodies. Samples were further analyzed by confocal microscopy (Fluoview 300; Olympus).
Bead assays
0.91- and 5.9-μm beads were coated as described previously (Felsenfeld et al., 1996). In brief, carbodiimide-derivitized latex beads were coupled with ovalbumin (800 μg per 0.5-ml beads). Beads were derivitized with biotin and coated with avidin neutralite (150 μg per 50-μl beads). Finally, beads were coupled with biotinylated FN (FNIII 7-10; 0.61 μg per μg/μl beads) or VN (0.83 μg per μg/μl beads) and blocked with BSA-biotin (67 μg per 50-μl beads).
For bead binding assays, cells were briefly lifted and plated on LA-coated coverslips. Beads were held for 3 s 0.2–0.5 μm from the leading edge, using a 100-mW (40 pN/μm) optical gradient laser trap setup (Axiovert 100TV; Carl Zeiss Microlmaging, Inc.) that was calibrated as described elsewhere (Choquet et al., 1997). For MSD assays, ligand-coated beads were held in the laser trap on the cell surface for 30 s or until the bead had moved >500 nm from the trap center. The x and y coordinates were determined from video micrographs using single particle tracking routines performed using Isee Software (Invision Corporation/Silicon Graphics O2). Tracking accuracy was 5–10 nm for 1-μm beads. The MSD was calculated using an algorithm modified from Qian et al. (1991).
For reinforcement assays using large beads (5.9 μm), cells were transiently transfected with GFP-paxillin, and ligand-coated beads were spun (5 min/50 g) onto the cells. Binding was assessed by confocal microscopy and image analysis using ImageJ (1.24 d; http://rsb.info.nih.gov/ij/). Beads were scored if the signal intensity was more than twice surrounding area within 10 μm from the leading edge. For real-time paxillin recruitment assays, cells were transiently transfected and treated as described above. Beads were held on the cell surface until the bead had moved out of the trap. Pictures were taken using a cooled CCD camera (Coolsnapfx) mounted to the trap set up (model IX70; Olympus) and analyzed using ImageJ.
Materials
Monoclonal anti-αv and -β3 antibodies were from BD Biosciences. Polyclonal anti-αv, anti-α5, and anti-β3 antibodies were from CHEMICON International, Inc. The monoclonal antipaxillin antibody was from Transduction Laboratories. The anti-Src antibody (Ab327) was from Oncogene Research Products. The phosphospecific (Y416) anti-SFK antibody was from Cell Signaling. The de-phosphospecific (Y527) anti-Src antibody (clone 28) was a gift of H. Kawakatsu (University of California, San Francisco, San Francisco, CA). The polyclonal anti-RPTPα was produced as described elsewhere (Su et al., 1999). The anti-Fyn antibody was from Upstate Biotechnology, the anti-Yes antibody was from Transduction Laboratories. ECL reagent, peroxidase-coupled anti–rabbit and anti–mouse IgG antibodies, and protein A–Sepharose beads were from Amersham Biosciences. Dithiobis succinimidyl propionate was from Pierce Chemical Co. PVDF membranes for Western Blotting were obtained from Millipore. GPen was from Sigma-Aldrich. Alexa red (568 and 647)– and Alexa green (488)–labeled anti–hamster, anti–mouse, and anti–rabbit IgG antibodies were from Molecular Probes. All beads were from Polyscience. Fugene 6 was from Roche Diagnostics. The plasmid containing the pRK5GFP-paxillin was a gift of K.M. Yamada (National Institute of Dental and Craniofacial Research, Bethesda, MD). pSGRPTPα-516-YFP was provided by J. den Hertog (Netherlands Institute for Developmental Biology, Utrecht, Netherlands). pECMV-CSK was a gift of E.E. Marcantonio (Columbia University, New York, NY). pUSEsrcwt was from Upstate Biotechnology, pCMV5fyn was a gift of M. Resh (Memorial Sloan-Kettering Cancer Center, New York, NY), pMIKwt-yes was a gift of M. Sudol (Mount Sinai Medical Center, New York, NY). All other reagents were of the purest grade available.
Acknowledgments
We are grateful to J. Sable for expert technical assistance and support. We thank also T. Baer, G. Giannone, A. Meshel, K. Miller and Y. Sawada for comments on the manuscript.
This work was supported by the National Institutes of Health (to M.P. Sheetz and J. Sap) and the Deutsche Forschungsgemeinschaft (G. von Wichert).
Footnotes
Abbreviations used in this paper: CSK, c-terminal Src kinase; FN, fibronectin; GPen, GPenGRGDSPCA; LA, laminin; MSD, mean square displacement; RPTPα, receptor-like protein tyrosine phosphatase α; SFK, Src family kinase; VN, vitronectin.
References
- Anton, E.S., J.A. Kreidberg, and P. Rakic. 1999. Distinct functions of alpha3 and alpha(v) integrin receptors in neuronal migration and laminar organization of the cerebral cortex. Neuron. 22:277–289. [DOI] [PubMed] [Google Scholar]
- Ardini, E., R. Agresti, E. Tagliabue, M. Greco, P. Aiello, L.T. Yang, S. Menard, and J. Sap. 2000. Expression of protein tyrosine phosphatase alpha (RPTPalpha) in human breast cancer correlates with low tumor grade, and inhibits tumor cell growth in vitro and in vivo. Oncogene. 19:4979–4987. [DOI] [PubMed] [Google Scholar]
- Beningo, K.A., M. Dembo, I. Kaverina, J.V. Small, and Y.L. Wang. 2001. Nascent focal adhesions are responsible for the generation of strong propulsive forces in migrating fibroblasts. J. Cell Biol. 153:881–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blystone, S.D., S.E. Slater, M.P. Williams, M.T. Crow, and E.J. Brown. 1999. A molecular mechanism of integrin crosstalk: αvβ3 suppression of calcium/calmodulin-dependent protein kinase II regulates α5β1 function. J. Cell Biol. 145:889–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bockholt, S.M., and K. Burridge. 1995. An examination of focal adhesion formation and tyrosine phosphorylation in fibroblasts isolated from src-, fyn-, and yes- mice. Cell Adhes. Commun. 3:91–100. [DOI] [PubMed] [Google Scholar]
- Brown, E.J., and W.A. Frazier. 2001. Integrin-associated protein (CD47) and its ligands. Trends Cell Biol. 11:130–135. [DOI] [PubMed] [Google Scholar]
- Buist, A., C. Blanchetot, L.G. Tertoolen, and J. den Hertog. 2000. Identification of p130cas as an in vivo substrate of receptor protein-tyrosine phosphatase alpha. J. Biol. Chem. 275:20754–20761. [DOI] [PubMed] [Google Scholar]
- Calautti, E., M. Grossi, C. Mammucari, Y. Aoyama, M. Pirro, Y. Ono, J. Li, and G.P. Dotto. 2002. Fyn tyrosine kinase is a downstream mediator of Rho/PRK2 function in keratinocyte cell–cell adhesion. J. Cell Biol. 156:137–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choquet, D., D.P. Felsenfeld, and M.P. Sheetz. 1997. Extracellular matrix rigidity causes strengthening of integrin-cytoskeleton linkages. Cell. 88:39–48. [DOI] [PubMed] [Google Scholar]
- den Hertog, J., C.E. Pals, M.P. Peppelenbosch, L.G. Tertoolen, S.W. de Laat, and W. Kruijer. 1993. Receptor protein tyrosine phosphatase alpha activates pp60c-src and is involved in neuronal differentiation. EMBO J. 12:3789–3798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etienne-Manneville, S., and A. Hall. 2001. Integrin-mediated activation of Cdc42 controls cell polarity in migrating astrocytes through PKCzeta. Cell. 106:489–498. [DOI] [PubMed] [Google Scholar]
- Felsenfeld, D.P., D. Choquet, and M.P. Sheetz. 1996. Ligand binding regulates the directed movement of beta1 integrins on fibroblasts. Nature. 383:438–440. [DOI] [PubMed] [Google Scholar]
- Felsenfeld, D.P., P.L. Schwartzberg, A. Venegas, R. Tse, and M.P. Sheetz. 1999. Selective regulation of integrin–cytoskeleton interactions by the tyrosine kinase Src. Nat. Cell Biol. 1:200–206. [DOI] [PubMed] [Google Scholar]
- Feng, X., D.V. Novack, R. Faccio, D.S. Ory, K. Aya, M.I. Boyer, K.P. McHugh, F.P. Ross, and S.L. Teitelbaum. 2001. A Glanzmann's mutation in beta 3 integrin specifically impairs osteoclast function. J. Clin. Invest. 107:1137–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fincham, V.J., and M.C. Frame. 1998. The catalytic activity of Src is dispensable for translocation to focal adhesions but controls the turnover of these structures during cell motility. EMBO J. 17:81–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galbraith, C.G., K.M. Yamada, and M.P. Sheetz. 2002. The relationship between force and focal complex development. J. Cell Biol. 159:695–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger, B., A. Bershadsky, R. Pankov, and K.M. Yamada. 2001. Transmembrane crosstalk between the extracellular matrix–cytoskeleton crosstalk. Nat. Rev. Mol. Cell Biol. 2:793–805. [DOI] [PubMed] [Google Scholar]
- Giancotti, F.G., and E. Ruoslahti. 1999. Integrin signaling. Science. 285:1028–1032. [DOI] [PubMed] [Google Scholar]
- Goldschmidt, M.E., K.J. McLeod, and W.R. Taylor. 2001. Integrin-mediated mechanotransduction in vascular smooth muscle cells: frequency and force response characteristics. Circ. Res. 88:674–680. [DOI] [PubMed] [Google Scholar]
- Grant, S.G., T.J. O'Dell, K.A. Karl, P.L. Stein, P. Soriano, and E.R. Kandel. 1992. Impaired long-term potentiation, spatial learning, and hippocampal development in fyn mutant mice. Science. 258:1903–1910. [DOI] [PubMed] [Google Scholar]
- Harder, K.W., N.P. Moller, J.W. Peacock, and F.R. Jirik. 1998. Protein-tyrosine phosphatase alpha regulates Src family kinases and alters cell-substratum adhesion. J. Biol. Chem. 273:31890–31900. [DOI] [PubMed] [Google Scholar]
- Hruska, K.A., F. Rolnick, M. Huskey, U. Alvarez, and D. Cheresh. 1995. Engagement of the osteoclast integrin alpha v beta 3 by osteopontin stimulates phosphatidylinositol 3-hydroxyl kinase activity. Endocrinology. 136:2984–2992. [DOI] [PubMed] [Google Scholar]
- Hughes, P.E., and M. Pfaff. 1998. Integrin affinity modulation. Trends Cell Biol. 8:359–364. [DOI] [PubMed] [Google Scholar]
- Jiang, G., J. den Hertog, and T. Hunter. 2000. Receptor-like protein tyrosine phosphatase alpha homodimerizes on the cell surface. Mol. Cell. Biol. 20:5917–5929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapus, A., C. Di Ciano, J. Sun, X. Zhan, L. Kim, T.W. Wong, and O.D. Rotstein. 2000. Cell volume-dependent phosphorylation of proteins of the cortical cytoskeleton and cell-cell contact sites. The role of Fyn and FER kinases. J. Biol. Chem. 275:32289–32298. [DOI] [PubMed] [Google Scholar]
- Kiosses, W.B., S.J. Shattil, N. Pampori, and M.A. Schwartz. 2001. Rac recruits high-affinity integrin alphavbeta3 to lamellipodia in endothelial cell migration. Nat. Cell Biol. 3:316–320. [DOI] [PubMed] [Google Scholar]
- Klinghoffer, R.A., C. Sachsenmaier, J.A. Cooper, and P. Soriano. 1999. Src family kinases are required for integrin but not PDGFR signal transduction. EMBO J. 18:2459–2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krutzsch, H.C., B.J. Choe, J.M. Sipes, N. Guo, and D.D. Roberts. 1999. Identification of an alpha(3)beta(1) integrin recognition sequence in thrombospondin-1. J. Biol. Chem. 274:24080–24086. [DOI] [PubMed] [Google Scholar]
- Lauffenburger, D.A., and A.F. Horwitz. 1996. Cell migration: a physically integrated molecular process. Cell. 84:359–369. [DOI] [PubMed] [Google Scholar]
- Li, L., M. Okura, and A. Imamoto. 2002. Focal adhesions require catalytic activity of SRC family kinases to mediate integrin-matrix adhesion. Mol. Cell. Biol. 22:1203–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKenna, D.A., F. Dolfi, K. Vuori, and E. Ruoslahti. 1998. Extracellular signal-regulated kinase and c-Jun NH2-terminal kinase activation by mechanical stretch is integrin-dependent and matrix-specific in rat cardiac fibroblasts. J. Clin. Invest. 101:301–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer, C.J., F.J. Alenghat, P. Rim, J.H. Fong, B. Fabry, and D.E. Ingber. 2000. Mechanical control of cyclic AMP signalling and gene transcription through integrins. Nat. Cell Biol. 2:666–668. [DOI] [PubMed] [Google Scholar]
- Moller, N.P., K.B. Moller, R. Lammers, A. Kharitonenkov, E. Hoppe, F.C. Wiberg, I. Sures, and A. Ullrich. 1995. Selective down-regulation of the insulin receptor signal by protein-tyrosine phosphatases alpha and epsilon. J. Biol. Chem. 270:23126–23131. [DOI] [PubMed] [Google Scholar]
- Petrone, A., and J. Sap. 2000. Emerging issues in receptor protein tyrosine phosphatase function: lifting fog or simply shifting? J. Cell Sci. 113:2345–2354. [DOI] [PubMed] [Google Scholar]
- Pierschbacher, M.D., and E. Ruoslahti. 1987. Influence of stereochemistry of the sequence Arg-Gly-Asp-Xaa on binding specificity in cell adhesion. J. Biol. Chem. 262:17294–17298. [PubMed] [Google Scholar]
- Ponniah, S., D.Z. Wang, K.L. Lim, and C.J. Pallen. 1999. Targeted disruption of the tyrosine phosphatase PTPalpha leads to constitutive downregulation of the kinases Src and Fyn. Curr. Biol. 9:535–538. [DOI] [PubMed] [Google Scholar]
- Qian, H., M.P. Sheetz, and E.L. Elson. 1991. Single particle tracking. Analysis of diffusion and flow in two-dimensional systems. Biophys. J. 60:910–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riveline, D., E. Zamir, N.Q. Balaban, U.S. Schwarz, T. Ishizaki, S. Narumiya, Z. Kam, B. Geiger, and A.D. Bershadsky. 2001. Focal contacts as mechanosensors: externally applied local mechanical force induces growth of focal contacts by an mDial-dependent and ROCK-independent mechanism. J. Cell Biol. 153:1175–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawada, Y., and M.P. Sheetz. 2002. Force transduction by Triton cytoskeletons. J. Cell Biol. 156:609–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawada, Y., K. Nakamura, K. Doi, K. Takeda, K. Tobiume, M. Saitoh, K. Morita, I. Komuro, K. De Vos, M. Sheetz, and H. Ichijo. 2001. Rap1 is involved in cell stretching modulation of p38 but not ERK or JNK MAP kinase. J. Cell Sci. 114:1221–1227. [DOI] [PubMed] [Google Scholar]
- Serra-Pages, C., N.L. Kedersha, L. Fazikas, Q. Medley, A. Debant, and M. Streuli. 1995. The LAR transmembrane protein tyrosine phosphatase and a coiled-coil LAR-interacting protein co-localize at focal adhesions. EMBO J. 14:2827–2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheetz, M.P., D. Felsenfeld, C.G. Galbraith, and D. Choquet. 1999. Cell migration as a five-step cycle. Biochem. Soc. Symp. 65:233–243. [PubMed] [Google Scholar]
- Shenoi, H., J. Seavitt, A. Zheleznyak, M.L. Thomas, and E.J. Brown. 1999. Regulation of integrin-mediated T cell adhesion by the transmembrane protein tyrosine phosphatase CD45. J. Immunol. 162:7120–7127. [PubMed] [Google Scholar]
- Stein, P.L., H. Vogel, and P. Soriano. 1994. Combined deficiencies of Src, Fyn, and Yes tyrosine kinases in mutant mice. Genes Dev. 8:1999–2007. [DOI] [PubMed] [Google Scholar]
- Su, J., M. Muranjan, and J. Sap. 1999. Receptor protein tyrosine phosphatase alpha activates Src-family kinases and controls integrin-mediated responses in fibroblasts. Curr. Biol. 9:505–511. [DOI] [PubMed] [Google Scholar]
- Thomas, S.M., P. Soriano, and A. Imamoto. 1995. Specific and redundant roles of Src and Fyn in organizing the cytoskeleton. Nature. 376:267–271. [DOI] [PubMed] [Google Scholar]
- Volberg, T., L. Romer, E. Zamir, and B. Geiger. 2001. pp60(c-src) and related tyrosine kinases: a role in the assembly and reorganization of matrix adhesions. J. Cell Sci. 114:2279–2289. [DOI] [PubMed] [Google Scholar]
- Wilson, E., K. Sudhir, and H.E. Ives. 1995. Mechanical strain of rat vascular smooth muscle cells is sensed by specific extracellular matrix/integrin interactions. J. Clin. Invest. 96:2364–2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng, L., L. D'Alessandri, M.B. Kalousek, L. Vaughan, and C.J. Pallen. 1999. Protein tyrosine phosphatase α (PTPα) and contactin form a novel neuronal receptor complex linked to the intracellular tyrosine kinase fyn. J. Cell Biol. 147:707–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng, X.M., Y. Wang, and C.J. Pallen. 1992. Cell transformation and activation of pp60c-src by overexpression of a protein tyrosine phosphatase. Nature. 359:336–339. [DOI] [PubMed] [Google Scholar]
- Zheng, X.M., R.J. Resnick, and D. Shalloway. 2000. A phosphotyrosine displacement mechanism for activation of Src by PTPalpha. EMBO J. 19:964–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong, C., M. Chrzanowska-Wodnicka, J. Brown, A. Shaub, A.M. Belkin, and K. Burridge. 1998. Rho-mediated contractility exposes a cryptic site in fibronectin and induces fibronectin matrix assembly. J. Cell Biol. 141:539–551. [DOI] [PMC free article] [PubMed] [Google Scholar]