

Abstract
The RNA-editing enzyme adenosine deaminase that acts on RNA (ADAR1) deaminates adenosines to inosines in double-stranded RNA substrates. Currently, it is not clear how the enzyme targets and discriminates different substrates in vivo. However, it has been shown that the deaminase domain plays an important role in distinguishing various adenosines within a given substrate RNA in vitro. Previously, we could show that Xenopus ADAR1 is associated with nascent transcripts on transcriptionally active lampbrush chromosomes, indicating that initial substrate binding and possibly editing itself occurs cotranscriptionally. Here, we demonstrate that chromosomal association depends solely on the three double-stranded RNA-binding domains (dsRBDs) found in the central part of ADAR1, but not on the Z-DNA–binding domain in the NH2 terminus nor the catalytic deaminase domain in the COOH terminus of the protein. Most importantly, we show that individual dsRBDs are capable of recognizing different chromosomal sites in an apparently specific manner. Thus, our results not only prove the requirement of dsRBDs for chromosomal targeting, but also show that individual dsRBDs have distinct in vivo localization capabilities that may be important for initial substrate recognition and subsequent editing specificity.
Keywords: RNA-editing; RNA processing; lampbrush chromosomes; Xenopus laevis
Introduction
Adenosine deaminases that act on RNA (ADARs)* are a family of RNA-editing enzymes that catalyze the hydrolytic deamination of adenosines to inosines in a diverse group of mostly double-stranded RNA substrates. As inosine basepairs like guanosine, the editing event frequently leads to a codon exchange when the substrate is an mRNA (Bass, 1997). Having first been described in Xenopus oocytes and embryos, ADAR-like activity has subsequently been found in every metazoan tissue tested, and to date, three distinct ADAR enzymes are cloned and characterized from various organisms, termed ADAR1, ADAR2, and ADAR3 (Keegan et al., 2001). In addition to RNA editing, ADAR1 was recently suggested to be involved in the regulation of nuclear translation (Herbert et al., 2002). Structurally, all ADARs possess a conserved deaminase domain in their COOH terminus required for enzymatic activity as well as one or several copies of the double-stranded RNA-binding domain (dsRBD) in their central region. In addition, ADAR1 proteins have a long NH2 terminus that contains two tandemly arranged Z-DNA–binding domains (ZBDs), termed Z-α and Z-β (Keegan et al., 2001; see Fig. 1).
Figure 1.

Schematic representation of Xenopus ADAR1 and mutant constructs used in this paper. The 1,271–amino acid Xenopus ADAR1 protein is depicted to scale at the top with mutant constructs shown underneath. Subregions of the protein are indicated as follows: REP, 11-aa peptide repeats; ZBD, Z-DNA binding domain; NLS, nuclear localization signal; dsRBDs, double-stranded RNA-binding domains; and Deaminase, catalytic deaminase domain. The ability of constructs to label chromosomes (Chr) and enrich at the special loop (Sp. loop) is shown on the right and indicated either as positive (+), negative (−), or patchy (+/−). Deletion of the deaminase domain (Δ deaminase) had no effect on chromosomal labeling and special loop enrichment. Expression of the central part of the protein from the end of the ZBD up to the end of dsRBD3 (dsRBD1-2-3) resulted in the same localization pattern as the wild-type protein, which is indicated by chromosomal association and special loop enrichment. Conversely, removal of the three dsRBDS from the full-length protein (Δ dsRBDs) resulted in a loss of all nuclear staining. Replacement of the endogenous dsRBDs with single dsRBDs (dsRBD2), duplicated dsRBDs (dsRBD2-2) or triplicated dsRBDs (dsRBD2-2-2) failed to restore normal chromosomal association. All constructs gave patchy labeling (+/−) and none enriched at the special loop (for simplicity, only dsRBD2 containing constructs are shown).
Substrates of ADARs include viral RNAs and endogenous transcripts. Viral RNAs are frequently edited promiscuously, but can also be edited specifically affecting only a few residues. However, endogenous substrates are mostly edited specifically (Bass, 1997). During nonspecific editing, up to 50% of adenosines can be converted into inosines with the extent of editing being affected both by neighboring bases and the length of contiguous duplex regions present (Lehmann and Bass, 1999). Such editing is thought to be part of a cellular antiviral defense program; a view that is supported by the observed transcriptional interferon induction of mammalian ADAR1 (Patterson et al., 1995).
In contrast, site-specific editing is less well understood and has only been described for a few RNAs including those encoding several subunits of the mammalian glutamate-gated ion channel family and the serotonin 2C receptor (Burns et al., 1997; Higuchi et al., 2000). Interestingly, both ADAR1 and ADAR2 edit these transcripts, but preferentially deaminate different adenosines within them. Consequently, a key question regarding the function of these enzymes is not only how they specifically recognize substrate RNAs in vivo but also how they target a particular adenosine within a given transcript (Hurst et al., 1995; Lehmann and Bass, 2000).
To date, all described substrates have, or are predicted to have, an extensive duplex structure that defines the site of editing. This, in turn, suggests a central role of the dsRBDs for substrate binding that has been confirmed by the observed reduction or loss of enzymatic activity when individual dsRBDs are deleted or mutated. Mutational analysis also shows that dsRBDs are functionally nonequivalent with some domains being dispensable for enzyme function, whereas others are essential (Lai et al., 1995; Liu and Samuel, 1996). Therefore, this raises the possibility that both the number and type of dsRBDs found in different ADAR proteins does contribute to overall substrate specificity. In support of this, chimeric proteins, where the first two dsRBDs from ADAR1, are replaced by the two dsRBDs from the RNA-dependent protein kinase PKR loose editing selectivity on endogenous substrates (Liu et al., 2000). Moreover, recent in vitro work on ADAR2 suggests that the dsRBDs help to define the editing site by increasing conformational flexibility of residues adjacent to the edited site. This is thought to allow base flipping of the adenosine, which has been shown to occur during the deamination reaction (Yi-Brunozzi et al., 2001).
However, structural analyses reveal that dsRBDs interact with the RNA sugar–phosphate backbone without making base-specific contacts (Ryter and Schultz, 1998; Ramos et al., 2000). Therefore, dsRBDs are assumed to recognize specific structures rather than a consensus sequence. Nonetheless, because the structure of an RNA is also governed by the underlying sequence, binding of dsRBDs to specific structures might also select for certain sequences. Furthermore, a lack of sequence specificity does not preclude individual dsRBDs from having distinct in vivo functions. For instance, the five dsRBDs of the Drosophila Staufen protein have been shown to exhibit individual functions with regard to translational control and localization of substrate RNAs (Micklem et al., 2000). Moreover, it was recently shown that human ADAR1 has an NLS that overlaps almost entirely with the third dsRBD (Eckmann et al., 2001). Also, nuclear export or regulation of nuclear import has been attributed to dsRBDs (Brownawell and Macara, 2002). Similarly, some dsRBDs can act as homodimerization domains (unpublished data). Therefore, it is becoming increasingly apparent that proteins containing multiple dsRBDs may use each domain individually in vivo, thus, maximizing functional diversity, possibly by interacting with other protein components (for review see Doyle and Jantsch, 2002).
In addition to dsRBDs, the deaminase domain has been shown to be important for substrate recognition by ADARs. This domain can also be found conserved in a related family of tRNA-editing enzymes known as adenosine deaminases that act on tRNA (ADATs). ADATs specifically edit adenosines in the anticodon regions of several tRNAs (for review see Gerber and Keller, 2001). Unlike ADARs, they lack dsRBDs, ZBDs, or any other described nucleic acid binding motifs, but seemingly act as heteromers. Therefore, it is believed that deaminase domains themselves can confer substrate specificity, thus raising the possibility that the same may be the case for ADARs. Furthermore, it was recently shown that deaminase domains of ADAR1 and ADAR2 have a dominant role in defining substrate specificity compared with the dsRBDs. When the deaminase domains were exchanged between ADAR1 and ADAR2, the resulting chimeric proteins had a preference to edit reporter constructs according to the origin of the deaminase domain that they contained. However, editing was not restored to endogenous levels, underscoring the importance of additional elements such as dsRBDs for proper enzyme function. Moreover, editing was only monitored for a particular substrate site, but not on a genome-wide level (Wong et al., 2001).
In light of this, and the previously described findings, we reasoned that although ADAR's dsRBDs may not be required for final identification of target adenosines, they may bind specifically to favorable substrate structures in vivo, thus giving an initial level of discrimination at the chromosomal level. Such discrimination may further be enhanced through the differences exhibited between individual dsRBDs. Therefore, we set out to determine which elements within Xenopus ADAR1 are required for chromosomal localization and which role individual dsRBDs might play in transcript association. We had shown previously that ADAR1 can be found associated with the nascent RNP matrix on Xenopus lampbrush chromosomes, but also with a particular chromosomal structure termed the “special loop” (Eckmann and Jantsch, 1999). The special loop is transcriptionally silent and seemingly represents a storage site for ADAR1 and other RNA-binding proteins possibly to regulate intranuclear enzyme concentration (unpublished data). In contrast, localization of ADAR1 on all other chromosomal sites, reflects the enzyme's ability to associate with the nascent RNP matrix found on transcriptionally active chromosome loops.
Lampbrush chromosomes are giant meiotic chromosomes where individual transcription units can be visualized under the normal light microscope. The nascent RNP matrix and attached proteins can also be discriminated on transcriptionally active genes. Thus, differences in RNP composition of individual genes can be monitored microscopically.
Results
The catalytic deaminase domain is dispensable for chromosomal localization
ADATs lack any dsRBDs, yet the different members of this protein family can edit individual adenosines in tRNAs specifically (Gerber and Keller, 2001). Furthermore, it was recently shown that the deaminase domains of ADAR1 and ADAR2 play an essential role in determining substrate specificity (Wong et al., 2001). To assess the role of the deaminase domain in chromosomal localization, we removed the 3′ end of the ADAR1.1 cDNA immediately downstream of the third dsRBD, thus deleting this domain (Fig. 1) . This Δ-deaminase construct was myc-tagged at both ends, in vitro transcribed and the resulting RNA microinjected into Xenopus oocytes. After allowing in vivo translation to occur, lampbrush chromosome preparations were made and the intranuclear expression of the construct was followed by immunofluorescence using the monoclonal anti–myc 9E10 antibody (mAb 9E10). To allow a comparison with full-length ADAR1, all slides were costained for the endogenous protein using the SAT3 antiserum described previously (Eckmann and Jantsch, 1999). In addition, full-length myc-tagged ADAR1.1 was injected as a positive control. This construct was described previously and had been shown to mimic endogenous staining (Eckmann and Jantsch, 1999).
Removal of the deaminase domain had no effect on chromosomal localization with all chromosomes being efficiently labeled. When compared with the endogenous protein, the Δ-deaminase construct was distributed on individual chromosomes similar to endogenous ADAR1 with all loops labeled by the endogenous protein also being labeled by the injected construct. Such colocalization was also observed for the full-length construct (Fig. 2 A). Expression of injected constructs was verified by Western blotting of nuclei and cytoplasmic extracts using mAb 9E10 (Fig. 2 B). However, it should be noted that the SAT3 antiserum was generated against the NH2 terminus of Xenopus ADAR1. Therefore, the SAT3 antiserum can also detect the injected Δ-deaminase and full-length constructs. Nonetheless, SAT3 staining looked virtually identical in both injected and uninjected control oocytes (Eckmann and Jantsch, 1999; unpublished data).
Figure 2.
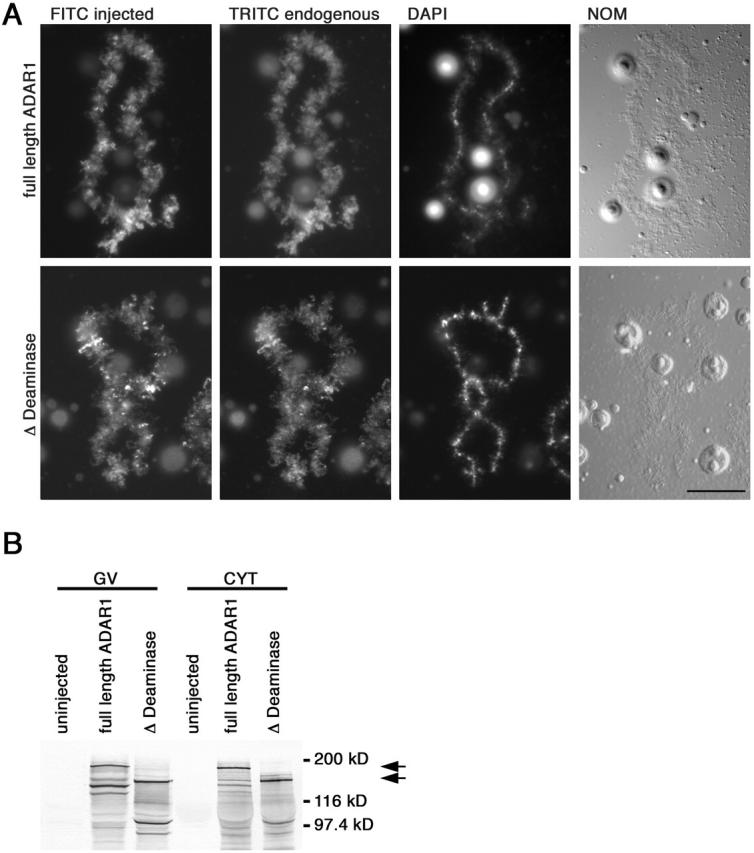
Deletion of the deaminase domain does not affect chromosomal localization. (A) RNA transcribed from full-length myc-tagged ADAR1 and a myc-tagged construct where the deaminase domain had been removed was injected individually into Xenopus oocytes. Lampbrush chromosome preparations were made and in vivo translation of each RNA was followed using the anti–myc mAb 9E10 and a secondary FITC-labeled antibody (FITC). Simultaneously, all preparations were stained for endogenous ADAR1 using the SAT3 antisera that was detected with a secondary TRITC-labeled antibody (TRITC). Injection of full-length ADAR1 (top) resulted in normal chromosomal localization compared with the endogenous protein. This was also the case when the deaminase domain was removed (bottom, Δ Deaminase), indicating at least at the chromosomal level that this domain does not play a role in targeting ADAR1 to nascent transcripts. Preparations were also stained with DAPI, and images of chromosomes were taken by differential interference contrast (NOM). Bar, 20 μm. (B) Western blot analysis of oocyte germinal vesicles (GV) and cytoplasms (CYT) from uninjected oocytes and oocytes expressing myc-tagged ADAR1 constructs to verify expression. Both myc-tagged versions express well and accumulate in the nucleus and are easily detected with the mAb 9E10. Cytoplasmic signals are seen 24 h after injection, but diminish over time as the protein is transported into the nucleus. In comparison, no signal is detected in uninjected oocytes indicating the specificity of mAb 9E10 for the injected constructs. Translated products correspond nicely to the predicted molecular masses of 175 kD (full-length ADAR1) and 130 kD (Δ Deaminase) indicated by arrows. Breakdown products can be observed at 120 kD (full-length) and 100 kD (Δ Deaminase). However, molecular mass calculations indicate that proteolytic cleavage has to occur upstream of the dsRBDs and, therefore, does not affect the RNA-binding capacity of the resulting fragments.
The double-stranded RNA-binding domains are essential and sufficient for chromosomal targeting
Having found that the deaminase domain was dispensable for chromosomal localization, we next wanted to define what other regions could be responsible for targeting the protein to chromosomes and transcriptionally active chromosomal loops. Previously, we deleted the majority of the NH2 terminus up to and including most of the ZBD and found that this had no effect on chromosomal localization (Eckmann and Jantsch, 1999). Therefore, it was apparent that the central region of the protein containing the NLS and three dsRBDs was most likely responsible for chromosomal and transcript association.
To test this we took two approaches. First, we deleted the three dsRBDs from the full-length myc-tagged ADAR1, fusing the ZBD, NLS, and the deaminase domain in frame. As expected, this construct failed to stain any intranuclear structures (Fig. 3) , whereas Western blots of nuclei and cytoplasms clearly showed that the construct had properly expressed and accumulated in the nucleus (unpublished data). Second, the converse experiment was performed by injecting a construct that contained only the NLS and the three dsRBDs. A fragment that spanned from the end of the ZBD, up to and including all of the third dsRBD, was myc-tagged at either end. Injection of this construct resulted in chromosomal labeling, and colocalization with the endogenous protein including enrichment at the special loop on chromosome 3 (Eckmann and Jantsch, 1999). Although this construct contained all three dsRBDs and the NLS, we were also aware that it contained the COOH terminus of the ZBD. To ensure that these extra amino acids had no effect on chromosomal targeting, the remainder of the Z-β domain was removed. Again, normal chromosomal labeling and colocalization with endogenous ADAR1 including enrichment at the special loop was observed (Fig. 3).
Figure 3.

The dsRBDs are necessary and sufficient for chromosomal association. Lampbrush chromosome preparations were made from oocytes injected with RNA transcribed from either of two myc-tagged constructs. In vivo translation of both constructs was followed using mAb 9E10 and a secondary FITC-labeled antibody (FITC). Expression of the central part of the protein from the end of the Z-β domain up to the end of dsRBD3 (top, dsRBD1-2-3) results in normal chromosomal association and special loop enrichment. In contrast, removal of the three dsRBDs from the full-length protein (bottom, Δ dsRBDs) causes loss of chromosomal and intranuclear staining. All preparations were costained for endogenous ADAR1 using the SAT3 antiserum and detected with a secondary TRITC-labeled antibody (TRITC). Enrichment at the special loop on the third chromosome is marked by arrows. Preparations were also stained with DAPI, and images of chromosomes were taken by differential interference contrast (NOM). Bar, 20 μm.
Individual dsRBDs can target ADAR1 to transcription units
Knowing that both the deaminase domain and the ZBDs were not required for chromosomal localization and that a minimum construct containing the NLS and dsRBDs would label chromosomes normally, we turned our attention to defining the role of the dsRBDs in more detail. We were especially keen to determine the role individual dsRBDs play in chromosomal localization given the fact that it had been demonstrated that dsRBDs, from this and other proteins, can differ functionally in vivo (Micklem et al., 2000; Eckmann et al., 2001; Strehblow et al., 2002). Therefore, we tested constructs that either contained single dsRBDs or tandemly arranged duplications or triplications of a specific domain. Proteins containing either duplications or triplications of individual dsRBDs were assumed to bind RNA with a greater affinity and might, therefore, accentuate potential differences exhibited by individual dsRBDs. Constructs were generated using a PCR strategy that maintained the precise spacing between each dsRBD present in the wild-type protein. Maintaining the proper spacing between individual domains seemed important, as it had been shown that splice variants of human ADAR1 with altered spacings between dsRBDs have different editing capabilities (Liu et al., 1997, 1999; Liu and Samuel, 1999). To simplify all clonings, unique restriction sites were introduced in the full-length myc-tagged construct flanking the dsRBDs, thus allowing their easy removal and replacement by individual domains. Furthermore, GFP was introduced as a second tagging system to allow the simultaneous injection and subsequent detection of two different constructs within the same oocyte.
To evaluate the RNA-binding ability of these constructs in vitro and to compare them with each other, all dsRBD combinations (single, duplications, and triplications) were expressed as GST fusion proteins and tested in Northwestern assays using rI/rC as a probe. At the same time, the second dsRBD of Xenopus laevis RNA-binding protein A (XlrbpA) was used as a positive control for RNA-binding as this domain binds RNA with very high affinity (Krovat and Jantsch, 1996) and had been used previously as an internal standard to quantify the RNA-binding capacity of the individual dsRBDs of Xenopus ADAR1 (Brooks et al., 1998). To determine the amount of recombinant protein in the extracts, a second gel was run in parallel and stained with Coomassie brilliant blue. The autoradiogram and Coomassie-stained gels were quantified using laser densitometry, and the amount of RNA bound was normalized to the amount of recombinant protein present in the extracts (Fig. 4) .
Figure 4.
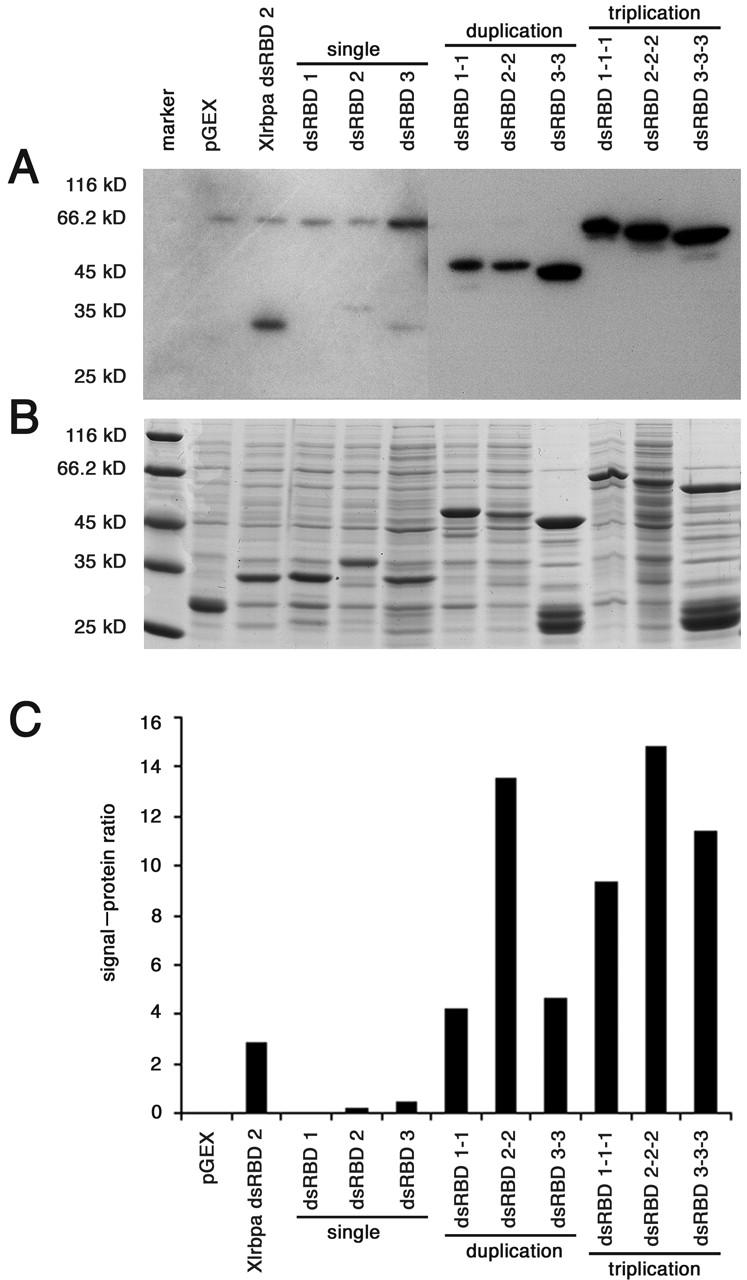
Northwestern analysis of dsRBDs constructs. The RNA-binding assay of bacterially expressed dsRBD-GST fusion proteins was performed with rI/rC (A). In parallel, the same extracts were run on a gel and stained with Coomassie brilliant blue to allow quantification of the recombinant protein (B). The ratio of signal to protein was quantified by laser densitometry and is depicted graphically (C). The number and type of dsRBDs are indicated. Also shown is the empty pGEX vector used as a negative control, whereas the second dsRBD of XlrbpA is included as a positive control. Of the single domains, dsRBD2 and dsRBD3 were found to be the best RNA binders. Constructs containing duplications of individual dsRBDs showed a higher affinity for rI/rC, whereas triplicating individual dsRBDs further enhanced this affinity.
Among the single domains, dsRBD2 and dsRBD3 were found to be the best RNA binders reflecting our previously published data (Brooks et al., 1998). Duplications of individual dsRBDs did bind rI/rC better than a single dsRBD, whereas triplicated domains had an even higher affinity for double-stranded RNA (Fig. 4).
To assess the role of individual domains in chromosomal targeting, we initially compared the distribution of single domain constructs with that of the endogenous protein. In these experiments, no single domain was able to mimic endogenous staining including enrichment at the special loop. Interestingly, however, any individual domain was capable of restoring at least some level of chromosomal localization. Unlike the distribution of full-length ADAR1, constructs bearing dsRBD1 or dsRBD2 predominantly highlighted a few loops on all chromosomes (Fig. 5) , whereas the dsRBD3-containing construct showed a rather weak and relatively homogeneous staining (unpublished data). Moreover, chromosomal loops that were labeled by individual domains were always stained by the endogenous antibody, but not vice versa (Fig. 5). To ensure that the observed labeling was not due to insufficient incubation time, and thus, reduced translation product levels, oocytes were cultured for up to 10 d. Assessment at different time points showed that labeling increased slightly over the first 48-h period, but remained constant for the remainder of the experiment. Additionally, Western blots of injected oocytes were made to control for equal and sufficient protein production (unpublished data).
Figure 5.
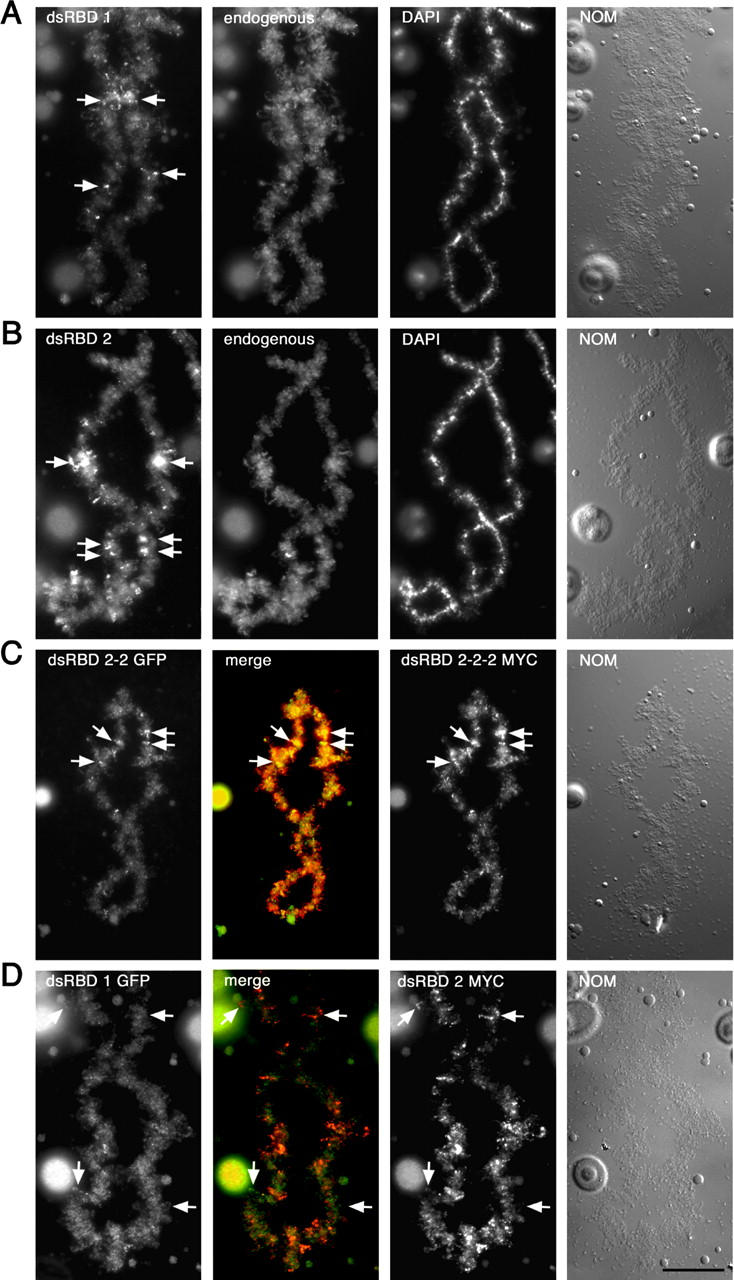
Individual dsRBDs can lead to ADAR1 enrichment on different transcription units. RNA transcribed from full-length myc-tagged ADAR1 constructs with either a single copy of dsRBD1 (A) or dsRBD2 (B) in place of the three endogenous dsRBDs was injected into oocytes. Translation of both constructs was followed using mAb 9E10 and a secondary FITC-labeled antibody. Simultaneously, all preparations were stained for endogenous ADAR1 using the SAT3 antisera (endogenous). Preparations were also stained with DAPI, and images of chromosomes were taken by differential interference contrast (NOM). Both dsRBD constructs were able to restore at least some level of chromosomal labeling, but at reduced levels compared with the endogenous ADAR1 staining. Interestingly, labeling often appeared at the same position along the arms of each homologue (A and B, arrows) indicating specific targeting to the same chromosomal loops. (C) To determine the influence of overall RNA–binding strength, constructs containing a duplication (dsRBD 2-2 GFP) or a triplication (dsRBD 2-2-2 MYC) of dsRBD were tagged with GFP and myc, respectively, injected, and detected within the same oocyte. Both constructs did label the same chromosomal sites (C, arrows), thus leading to a homogeneous yellow labeling in the overlay (merge). (D) To directly compare two individual dsRBDs on the same chromosome, RNA, transcribed from GFP-tagged ADAR1 containing a single dsRBD1, was coinjected with RNA made from a myc-tagged ADAR1 construct containing a single dsRBD2. Translation of the GFP-tagged construct was followed using appropriate antibodies. The dsRBD2-containing construct shows specific enrichment at a few chromosomal sites (D, top arrows), whereas the dsRBD1-containing construct shows a more homogeneous chromosomal staining. However, a few sites are specifically highlighted by the dsRBD1-containing constructs and, thus, appear green in the merged image (D, bottom arrows). Merged images of dsRBD1 and dsRBD2 labeling are shown (merge) as well as images of the chromosomes taken by differential interference contrast (NOM). Bar, 20 μm
Interestingly, chromosomal loops labeled by the injected constructs frequently appeared at the same position along the arms of each meiotically paired homologue (Fig. 5, arrows). Therefore, it is obvious that these constructs specifically target the same loops on each homologue. On the one hand, this suggests that individual dsRBDs might be capable of specifically binding to certain RNAs. On the other hand, labeling of a few selected loops could also be the result of different RNA concentrations and turnover rates at these chromosomal sites. Therefore, a construct containing only a single dsRBD and thus being a weak RNA binder might only accumulate at sites of high RNA concentrations, giving the impression of specific chromosomal targeting. To discriminate between these possibilities, we took two approaches. First, we reasoned that if the observed labeling of homologous sites was simply due to weak RNA binding, increasing the numbers of individual dsRBDs in a construct, and hence the total RNA-binding capability, should restore normal chromosomal labeling. Conversely, if chromosomal targeting was truly specific, labeling of homologous sites by such a construct would continue. Second, if individual dsRBDs are capable of targeting different chromosomal loci, it should be possible to visualize this directly in vivo by comparing the localization of different dsRBD constructs on the same chromosome.
Increasing RNA binding does not restore normal chromosomal labeling
To assess the role RNA-binding plays in chromosomal targeting, we wanted to compare the localization of constructs with different RNA-binding abilities. To maintain a potential binding specificity, we simultaneously injected constructs containing either duplications or triplications of the same dsRBD. Northwestern assays had shown that constructs carrying duplications or triplications of the same dsRBD showed a gradually increasing RNA-binding ability (Fig. 4). To allow a simultaneous detection of the resulting proteins, constructs carrying a duplication of a particular dsRBD were tagged with GFP, whereas constructs carrying a triplication of the same dsRBD were attached to the myc tag. Interestingly, efficient translation was only observed when the RNAs encoding these constructs were injected consecutively. In contrast, simultaneous injection of both constructs resulted in reduced translation and, thus, inefficient detection of either construct on chromosomes. However, it should be noted that the outcome of the experiments was not influenced by the order in which the two constructs were injected (unpublished data). Surprisingly, neither duplication nor triplication of any dsRBD did restore normal chromosomal labeling. Even though additional dsRBDs did lead to a slight increase in overall chromosomal localization, targeting appeared specific with chromosomes being labeled at identical positions along the length of each homologue by both duplication and triplication constructs (Fig. 5).
Individual dsRBDs can target different transcripts on homologous chromosomes
Thus far, our experiments could demonstrate that individual dsRBDs are capable of targeting specific chromosomal loci. Moreover, increasing the level of RNA binding slightly increased chromosomal association of the protein, but it remained restricted to a few sites and failed to restore homogeneous chromosomal localization comparable to that of wild-type ADAR1. Next, we wanted to determine whether individual dsRBDs can target to different transcripts in vivo. To accomplish this, constructs containing different dsRBDs were tagged with either GFP or myc, injected, and subsequently detected within the same oocyte. To verify that the different tags did not alter the localization of the resulting fusion proteins, we initially coinjected identical myc- and GFP-tagged constructs and compared their localization to each other. Additionally, synthesis and nuclear accumulation of the resulting constructs was monitored by Western blotting of hand-enucleated oocytes and isolated germinal vesicles. As expected, the different tagging systems showed no adverse effect on the chromosomal distribution of the resulting fusion proteins, leading to a complete overlap in the localization of both myc- and GFP-tagged protein (Fig. 6) .
Figure 6.
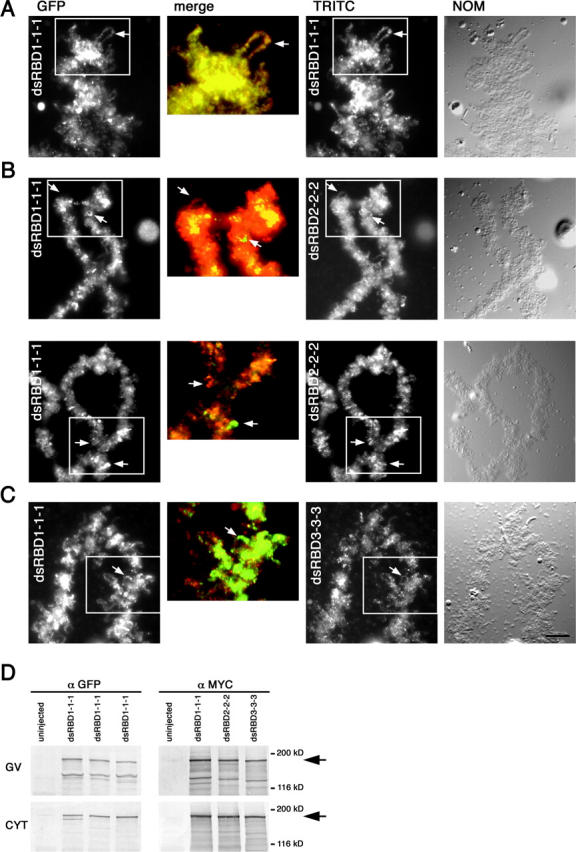
Individual dsRBDs can target ADAR1 to different subsets of loops. Double injections of RNA from different myc- and GFP triplication constructs are shown as indicated. Translation of the myc constructs was detected using mAb 9E10 and a secondary TRITC-labeled antibody (TRITC), whereas the GFP constructs were detected using an anti–rabbit GFP antibody and a secondary FITC-labeled antibody (FITC). Merged enlargements are shown between (merge). (A) Injection of identical constructs label the same chromosomal sites. Shown is a double injection of RNA made from dsRBD1 triplications that were either myc- or GFP-tagged. Both tagged versions label the chromosomes equally well and target to the same sites and loops (e.g., arrows). Such a complete overlap becomes even more apparent when similar regions of the chromosome from each channel (boxed) are enlarged, colored, and merged. (B and C) Different dsRBDs target ADAR1 to different chromosomal sites. RNA made from GFP-tagged dsRBD1 triplication was injected into oocytes followed by injection of RNA either from myc-tagged dsRBD2 triplication (B) or myc-tagged dsRBD3 triplication (C). Two different representative chromosomal sites are shown in B. Each construct labels chromosomes differently with some sites being labeled by one construct not being labeled by the other (arrows). Consequently, in the merged images, there is no complete overlap of staining. Together, this demonstrates the capability of individual dsRBDs specifically targeting different transcription units. Images of chromosomes taken by differential interference contrast (NOM) are also shown. Bar, 10 μm. (D) Western blot analysis of oocyte germinal vesicles (GV) and cytoplasms (CYT) from uninjected oocytes and oocytes expressing myc- and GFP-tagged ADAR1 constructs shown in A–C to verify expression. Translated products correspond nicely to the predicted molecular masses of ∼180 kD (arrows).
Initially, we compared the distribution of single dsRBD constructs to one another, whereas in subsequent experiments, the intranuclear distribution of constructs containing duplications or triplications of individual domains was compared. Differences in chromosomal distribution could already be observed with single domain constructs (Fig. 5). A construct containing a single dsRBD1 showed weak labeling on many loops along the entire length of a chromosome, but also highlighted a few loops specifically. In contrast, a construct containing a single dsRBD2 showed rather specific labeling of a few sites. Most importantly, the overlay image clearly indicated that a few sites were exclusively labeled by either one of the two constructs.
Nonetheless, the observed differences were more easily detected when the number of dsRBDs present in a construct was increased (Fig. 6). The most prominent differences were observed when comparing constructs carrying triplications of dsRBD1 with those containing triplications of dsRBD2. Moreover, as with the individual injections multiplications of dsRBDs1 or 2 led to a patchy chromosomal distribution. A direct comparison of these two constructs showed that some chromosomal loops were labeled exclusively by either of the two proteins (Fig. 6). Constructs containing multiple copies of dsRBD3, however, stained chromosomes only moderately, but relatively homogeneously (Fig. 6). This finding suggests that some dsRBDs can specifically target a subset of transcriptionally active chromosomal sites to associate with the nascent RNP matrix.
Discussion
dsRBDs are necessary and sufficient for chromosomal targeting
We set out to determine which elements within Xenopus ADAR1 are responsible for chromosomal targeting and could show that the dsRBDs alone are necessary and sufficient to accomplish this. Moreover, individual dsRBDs appear to have the capability to selectively recognize and target ADAR1 to a subset of chromosomal sites and, thus, to specific RNAs being transcribed there.
Interestingly, our data demonstrated that the two ZBDs located in the NH2 terminus of ADAR1 are neither necessary nor sufficient for chromosomal targeting. This is somewhat surprising, as it had been suggested that these domains might be involved in targeting the protein to sites of transcription where DNA might be in a Z-DNA conformation due to partial unwinding occurring during transcription (Herbert, 1996). Considering that the lateral loops on lampbrush chromosomes represent sites of active transcription, one might expect the ZBDs to enhance the accumulation of the protein at transcriptionally active loops or the underlying chromatin. However, to our estimation, removal of the ZBDs had no adverse effect on the efficiency with which the protein did localize to lampbrush chromosomes. Nonetheless, in vivo–editing assays performed on artificial substrates demonstrated recently that the ZBDs are dispensable for the editing of substrates longer than 15 bp, but are seemingly important for very short substrates (Herbert and Rich, 2001). Thus, ZBDs might be dispensable for targeting and subsequent editing of an average RNA–polymerase II transcript, but might be primarily important for the editing of endogenous, short-structured or -viral RNAs.
We could also demonstrate that the deaminase domain is dispensable for targeting ADAR1 to nascent transcripts. This is an interesting finding, especially when considering that this domain plays a dominant role in defining individual adenosines to be edited (Wong et al., 2001). Moreover, the tRNA-editing enzymes ADATs lack any dsRBDs, but can still edit their substrates specifically. Thus, it is generally assumed that deaminase domains are able to discriminate substrates by themselves. However, our results indicate that the deaminase domain in ADAR1 only allows discrimination of individual adenosines after the protein has associated with the nascent RNP matrix. The initial RNA-binding and recognition event seemingly occurs independently of the deaminase domain.
Chromosomal targeting versus editing
Throughout the course of this work, we have concentrated on determining the role different domains of ADAR1 play in mediating chromosomal localization and transcript association in vivo. In contrast, other works primarily investigated the contribution of dsRBDs on editing efficiency of a particular set of substrate sites. Maybe not surprisingly, in these works, mutation of the editing enzyme invariantly led to a decrease in editing efficiency on these substrates (Lai et al., 1995; Liu and Samuel, 1996; Liu et al., 2000).
However, here we focused on the contribution dsRBDs might play on RNA association at the genome-wide level. Therefore, our finding that different nascent transcripts can be targeted dependent on the type of dsRBD present suggests that whereas editing efficiency of a particular substrate site might decrease, other (potentially novel) sites within the genome might be targeted with increased efficiency by our mutant constructs. In this context, it is also worth mentioning that only a small number of ADAR1 substrates are known today. However, it is estimated that in rat brain, for instance, up to 10% of all mRNAs might be edited, thus leaving a large number of ADAR1 substrates to be discovered (Paul and Bass, 1998).
Nonetheless, given the large number of chromosomal sites being targeted by both wild-type and mutant-ADAR1 constructs it seems rather unlikely that all encode true editing substrates. Instead, it seems possible that ADAR1 associates with hnRNAs independent of whether they contain editing sites or not. Furthermore, association with hnRNAs might occur via interaction with other RNPs rather than by active RNA binding, a view that is supported by the observed association of ADAR1 with large RNP particles (Raitskin et al., 2001). Thus, the differences in distribution of our mutant constructs do not necessarily reflect binding to novel substrates. Nonetheless, our results clearly indicate that dsRBDs are capable of discriminating different hnRNAs in vivo.
Moreover, dsRBDs might not only contribute to substrate specificity by targeting the protein to the proper RNA. It has been suggested that dsRBD binding does help to identify the base to be edited by facilitating its accessibility for deamination (Yi-Brunozzi et al., 2001). Therefore, it is possible that binding of constructs carrying different combinations of dsRBDs could specifically allow access to distinct adenosines within an RNA so regulating editing specificity. In this case, specific exposure of a particular adenosine might not only be mediated by the different binding affinities exhibited by distinct dsRBDs, but also by the structural differences observed for the interaction of certain dsRBDs with structured RNAs (Ryter and Schultz, 1998; Ramos et al., 2000).
Recently, human ADAR1 was reported to induce protein translation within the nucleus (Herbert et al., 2002). This activity is thought to occur on the surface of the nucleolus and is independent of RNA editing. During the course of our work, we noted that Xenopus ADAR1 also associates with nucleoli and that some of our mutant constructs display differing affinities for nucleoli. However, given that many RNA-binding proteins stick to nucleoli when overexpressed, we did not pay further attention to this fact. In light of this potential novel function of ADAR,1 the phenomenon of nucleolar association may deserve future attention.
Substrate recognition by dsRBD proteins
To date, at least 20 different dsRBD-containing proteins have been identified from various species. Although some of these proteins bind dsRNA rather promiscuously, others can bind RNA very specifically, at least in vivo. Many of the proteins that are capable of specific substrate recognition contain multiple dsRBDs including the Drosophila Staufen protein and the RNA-editing enzymes of the ADAR superfamily. The role of individual dsRBDs in RNA recognition is not quite clear especially because it has been shown that dsRBDs primarily interact with the backbone of dsRNA, thus recognizing the specific structure of this RNA, but not allowing any base interaction to occur. However, it was also shown that different dsRBDs can bind dsRNAs with different affinities in vitro and that multiple dsRBDs can contribute to a cooperative binding effect (Bass et al., 1994; Krovat and Jantsch, 1996). Therefore, it had been suggested that only the combination of multiple dsRBDs might allow an RNA-specific interaction to occur by superimposing the double-stranded regions in a substrate RNA with the multiple dsRBDs in a protein (Fierro-Monti and Mathews, 2000).
Our present paper indicates that whereas the presence of multiple dsRBDs is important, dsRBD identity might contribute more to overall substrate specificity than thus far expected. Our data indicate that constructs carrying different dsRBDs localize to different nascent transcripts in vivo. In this context, it is important to notice that in all of our constructs the spacing between dsRBDs was maintained. Thus, it is rather unlikely that the different in situ localizations observed are caused by a different three-dimensional distribution of dsRBDs.
Our results also demonstrated that whereas a single copy of a particular dsRBD can target specific chromosomal sites, the extent of chromosomal labeling increases when additional copies of the same dsRBD are added. In principle, this increase in chromosomal labeling could be explained by a loss of binding specificity. However, if this was the case, one would expect the labeling to become increasingly uniform, which was not observed. Instead, it appeared that labeling increased at selected sites with a few additional sites coming up on the addition of more dsRBDs. Therefore, it seems that constructs carrying multiple copies of the same dsRBD did not loose binding specificity, but simply had an increased binding affinity while maintaining selectivity. Novel sites that were only detected by the multi-dsRBD constructs might remain below the limit of detection for the single dsRBD constructs. However, the addition of dsRBDs would increase the affinity to these sites and, therefore, allow their detection.
It is also interesting to note that each of the dsRBDs contributed differently to RNA association in vivo. Although constructs containing single, double, or triple copies of dsRBDs1 or 2 showed a speckled chromosomal distribution, constructs containing a comparable number of dsRBD3 showed a rather homogeneous staining. This, in turn, suggests that both dsRBDs1 and 2 might be able to recognize specific structural features of an RNA, thus leading to a preferential association with some RNAs, whereas dsRBD3 might bind all types of RNAs rather uniformly.
In vitro, dsRBD2 is the strongest RNA binder, whereas dsRBD3 binds RNA moderately. dsRBD1 shows very little RNA-binding capacity in vitro (Brooks et al., 1998). However, it is possible that dsRBD1 fails to associate with the generic double-stranded RNA substrate, rI/rC in vitro, but binds other, more specific substrates with much higher affinity. Similar differences in substrate binding had been observed for other dsRBDs in the past and could thus explain the observed discrepancies between in vitro and in vivo RNA association.
Finally, it must also be considered that the different in situ localizations caused by individual dsRBDs might not only be mediated by the RNA-binding specificity of those domains. Specific enrichment at some transcriptionally active loops might also be caused via a protein-mediated association of some dsRBDs with other components of the RNP matrix. Such additional functions of dsRBDs have recently been reported in several cases. Different domains in the Drosophila Staufen protein, for instance, have been shown to be required for RNA localization and translational repression (Micklem et al., 2000). Similarly, it was recently shown that some dsRBDs might act as nuclear import and others as nuclear export signals (Eckmann et al., 2001; Brownawell and Macara, 2002). Additionally, dsRBDs might be required for homo- and heterodimerization with other dsRBDs (unpublished data).
Our work shows that, whether caused exclusively via RNA binding or also through protein–protein interaction, dsRBDs are the key determinants for association of the ADAR1 protein with nascent transcripts. Furthermore, we show for the first time that individual dsRBDs seemingly mediate specific chromosomal association of the protein. Thus, it appears that dsRBDs can contribute to initial substrate discrimination at a genome wide level.
The special loop
Besides being found on the majority of nascent transcripts, ADAR1 is also enriched at a particular chromosomal site referred to as the special loop. Although out of the scope of this present work, a few general points can be made here. Like for other loops, special loop enrichment depends solely on the three endogenous dsRBDs; neither the ZBD nor the deaminase domain plays a role in mediating enrichment at this site. Interestingly, however, as soon as the endogenous dsRBDs arrangement is disturbed, enrichment at this site is lost; not even a triplication of an individual domain in the context of the full-length protein is able to enrich at the special loop. However, the special loop is transcriptionally silent and seemingly represents an intranuclear storage site for ADAR1 and other RNA-binding components. Therefore, targeting of ADAR1 to this site most likely depends on the protein's ability to associate with other RNA components rather than on its RNA binding ability.
Materials and methods
Epitope tagging of Adar1.1
A previously described myc-tagged version of ADAR1.1 containing six tandemly arranged myc tags upstream and downstream of the Adar1.1 cDNA was used for these experiments (Eckmann and Jantsch, 1999). Additionally, the 3′ untranslated region of the Xenopus NO38 cDNA (Peculis and Gall, 1992) containing a poly(A)+ tail had been cloned downstream of the 3′ mycs to stabilize injected RNAs. For this study, this construct was modified by flanking the dsRBDs with unique restriction sites to allow removal and insertion of different dsRBD combinations. A BclI site was introduced immediately upstream of the first dsRBD and a KpnI site just after the end of dsRBD3 by site-directed mutagenesis.
A GFP-tagged version of this construct was made by removing both sets of myc tags and inserting a GFP gene in place of the 3′ mycs. First, the 5′ mycs were removed by digestion with XbaI and NdeI. During removal, the original AUG annealed oligonucleotides containing a new in-frame AUG were inserted between these sites. Second, the 3′ mycs were removed by digestion with XhoI and NdeI. A GFP gene was introduced that had been amplified via PCR from the pEGFP-N2 vector (CLONTECH Laboratories, Inc.) using primers containing these restriction sites. The integrity of the final construct was checked by sequencing.
Construction of Adar1.1 deletions
Deletion of the dsRBDs was accomplished by partially digesting the myc-tagged construct with AvaI releasing a 970-bp fragment containing all three dsRBDs. The vector was filled and re-ligated, maintaining the correct reading frame. The Δ-deaminase construct was made by digesting the myc-tagged construct with EcoRV and XhoI, removing the 3′ end of the Adar1.1 ORF immediately downstream of dsRBD3 up to the start of the 3′ mycs. The EcoRV and XhoI sites were filled and re-ligated, maintaining the correct reading frame.
Minimal dsRBD constructs
Two minimal dsRBD constructs were used in this study. A construct containing the end of the ZBD up to and including all of dsRBD3 was made, partially digesting the full-length modified myc-tagged version ADAR1.1 with AvaI to release a 1528-bp fragment. The ends of this fragment were filled, and the fragment was cloned in-frame into a modified version of p-Bluescript that had been cut with BamHI, and the overhangs were filled. Downstream of the BamHI site, six tandemly arranged myc tags followed immediately by the 3′ untranslated region of the NO38 cDNA had been introduced. Clones were verified for correct insert orientation by restriction mapping and confirmed by sequencing.
The second minimal construct used removed the very end of the ZBD from the construct described above (see previous paragraph). This was achieved by digesting with XbaI (found in the polylinker) and BclI, whereby removing the 5′ end of the construct up to the start of dsRBD1. A PCR fragment was amplified using a forward primer containing an in-frame XbaI site that annealed immediately after the end of the ZBD, and a reverse primer that annealed across the BclI site at the start of the dsRBD1. The fragment was digested with XbaI and BclI and ligated into the prepared vector and verified by sequencing.
Single, double, and triplication dsRBD constructs
Individual dsRBDs were amplified from the full-length ADAR1.1 cDNA via PCR using primers immediately upstream and downstream of the domain. The 5′ primer contained an in-frame BamHI site and the 3′ primer contained an in-frame KpnI site. After digestion with these enzymes, individual domains were ligated in the modified myc-tagged version of the ADAR1.1 cDNA that had been cut with BclI and KpnI. All constructs were verified by sequencing.
For duplication of individual domains, a common two-step PCR approach was used. Individual domains were first amplified using a 5′ primer containing a BamHI site and a hybrid 3′ primer. The 3′ primer was designed in a such a way that its 5′ half was complementary to the inverse complementary sequence immediately upstream of the 5′ primer. Thus, the PCR fragment of the first step could be used as a 5′ primer in the second round of PCR in combination with a new 3′ primer containing an in-frame Kpn1 site. Primers were designed in such a way as to maintain the same spacing between dsRBDs as is present in the wild-type situation. After digestion with BamHI and KpnI, the duplicated domains were ligated into both epitope-tagged versions of Adar1.1 that had been cut with BclI and KpnI. Each construct was verified by sequencing.
Triplications of individual dsRBDs were made using restriction sites unique in the individual domain and, hence, present twice in the duplication constructs. Each duplication construct was both fully and partially digested with the relevant enzyme (dsRBD1 = ClaI; dsRBD2 = MscI; and dsRBD3 = Bsu65I). The insert released from complete digestion was ligated into the partially digested vector creating a triplication construct. Insert orientation was checked by restriction mapping and confirmed by sequencing.
Oocyte injections
Both myc- and GFP-tagged ADAR1 variants were linearized using a unique SnaBI restriction site downstream of the NO38 poly(A)+ tail. Capped run-off transcripts were synthesized in vitro from the linearized template using T3 RNA polymerase (Stratagene). Aliquots of all RNAs were checked for integrity on RNA gels by ethidium bromide staining. For single injections of myc-tagged constructs, 50 oocytes were injected with 50 ng RNA per oocyte and incubated for 24–72 h at 16°C in OR-2 (Wallace et al., 1973) to allow translation to occur. For double injections of both myc- and GFP-tagged variants, we first injected the GFP-tagged construct to achieve optimal translation. 50 ng RNA per oocyte was injected and, after a 12-h incubation (16°C in OR2), RNA transcribed from the myc-tagged ADAR1 variants was injected (50 ng per oocyte). All oocytes were incubated for a further 24–72 h at 16°C in OR2 to allow translation to occur.
LBC preparations and immunofluorescence stainings
LBC preparations and immunofluorescence stainings were performed as described previously (Wu et al., 1991). Antibodies used were as follows: anti–rabbit SAT3 (1:500) directed against Xenopus ADAR1.1 (Eckmann and Jantsch, 1999); the anti–myc mAb 9E10 (1:1) directed against the myc tag (Evan et al., 1985); and an anti–rabbit GFP antibody (1:1,000) directed against the GFP tag (a gift from P. Silver, Dana-Farber Cancer Institute, Boston, MA). For GFP detection, a secondary anti–rabbit FITC antibody was used. For all other stainings, appropriate combinations of secondary antibodies were used as indicated. Microscopic images were captured on a Zeiss fluorescence microscope equipped with an ORCA-cooled charged-coupled device camera (Hamamatsu). Images were imported into Photoshop 5 (Adobe Systems) with the help of a plug-in module (QED-Imaging).
Western blots
Oocytes were manually dissected, and nuclei (germinal vesicles, GVs) and cytoplasms were collected separately. Up to five cytoplasms were collected in NET-2 buffer (Steitz, 1989), sonicated, and centrifuged to remove insoluble material. The supernatant was mixed with an equal volume of 2× SDS sample buffer. Up to 20 GVs were directly collected into 2× SDS sample buffer. Extracts corresponding to eight GVs and one cytoplasm were loaded per lane on a 7% SDS-PAGE gel and blotted onto a polyvinylidene difluoride membrane (Osmonic Inc.). Myc-tagged proteins were detected with the mAb 9E10 and GFP-tagged proteins with a GFP mAb (Boehringer). Both primary antibodies were detected with a secondary alkaline phosphatase–labeled anti–mouse antibody (Pierce Chemical Co.) developed with the use of the chromogenic substrate NBT/BCIP.
Northwestern assays
For Northwestern assays of dsRBD constructs, the corresponding regions were cloned into a suitable pGEX vector and expressed as GST fusion proteins. Crude lysates from Escherichia coli expressing the proteins of interest were separated on protein gels, blotted, and probed with radiolabeled rI/rC as described previously (St Johnston et al., 1992). To determine the amount of RNA bound, the autoradiograms were scanned using laser densitometry. Furthermore, protein extracts were run on a second protein gel, stained with Coomassie R250 (Sigma-Aldrich), and the amount of recombinant protein was quantified by densitometry. The amount of RNA bound was normalized to the amount of recombinant protein present in the extract.
Acknowledgments
The authors would like to thank L. Pfaffstätter for excellent technical assistance and the members of the department for critical discussions. C. Eckmann is thanked for providing us with the initial constructs crucial to this work. P. Silver provided us with a polyclonal anti-GFP antiserum.
This work was supported by Austrian Science Foundation, grant SFB17-06 to M. Jantsch.
Footnotes
Abbreviations used in this paper: ADAR, adenosine deaminase that act on RNA; ADAT, adenosine deaminase that acts on tRNA; dsRBD, double-stranded RNA-binding domain; ZBD, Z-DNA–binding domain.
References
- Bass, B.L. 1997. RNA editing and hypermutation by adenosine deamination. Trends Biochem. Sci. 22:157–162. [DOI] [PubMed] [Google Scholar]
- Bass, B.L., S.R. Hurst, and J.D. Singer. 1994. Binding properties of newly identified Xenopus proteins containing dsRNA-binding motifs. Curr. Biol. 4:301–314. [DOI] [PubMed] [Google Scholar]
- Brooks, R., C.R. Eckmann, and M.F. Jantsch. 1998. The double-stranded RNA-binding domains of Xenopus laevis ADAR1 exhibit different RNA-binding behaviors. FEBS Lett. 434:121–126. [DOI] [PubMed] [Google Scholar]
- Brownawell, A.M., and I.G. Macara. 2002. Exportin-5, a novel karyopherin, mediates nuclear export of double-stranded RNA binding proteins. J. Cell Biol. 156:53–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns, C.M., H. Chu, S.M. Rueter, L.K. Hutchinson, H. Canton, E. Sanders-Bush, and R.B. Emeson. 1997. Regulation of serotonin-2C receptor G-protein coupling by RNA editing. Nature. 387:303–308. [DOI] [PubMed] [Google Scholar]
- Doyle, M., and M.F. Jantsch. 2002. New and old roles of the double-stranded RNA-binding domain. J. Struct. Biol. 140:147–153. [DOI] [PubMed] [Google Scholar]
- Eckmann, C.R., and M.F. Jantsch. 1999. The RNA-editing enzyme ADAR1 is localized to the nascent ribonucleoprotein matrix on Xenopus lampbrush chromosomes but specifically associates with an atypical loop. J. Cell Biol. 144:603–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckmann, C.R., A. Neunteufl, L. Pfaffstetter, and M.F. Jantsch. 2001. The human but not the Xenopus RNA-editing enzyme ADAR1 has an atypical nuclear localization signal and displays the characteristics of a shuttling protein. Mol. Biol. Cell. 12:1911–1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evan, G.I., G.K. Lewis, G. Ramsay, and J.M. Bishop. 1985. Isolation of monoclonal antibodies specific for human c-myc proto-oncogene product. Mol. Cell. Biol. 5:3610–3616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fierro-Monti, I., and M.B. Mathews. 2000. Proteins binding to duplexed RNA: one motif, multiple functions. Trends Biochem. Sci. 25:241–246. [DOI] [PubMed] [Google Scholar]
- Gerber, A.P., and W. Keller. 2001. RNA editing by base deamination: more enzymes, more targets, new mysteries. Trends Biochem. Sci. 26:376–384. [DOI] [PubMed] [Google Scholar]
- Herbert, A. 1996. RNA editing, introns and evolution. Trends Genet. 12:6–9. [DOI] [PubMed] [Google Scholar]
- Herbert, A., and A. Rich. 2001. The role of binding domains for dsRNA and Z-DNA in the in vivo editing of minimal substrates by ADAR1. Proc. Natl. Acad. Sci. USA. 98:12132–12137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbert, A., S. Wagner, and J.A. Nickerson. 2002. Induction of protein translation by ADAR1 within living cell nuclei is not dependent on RNA editing. Mol. Cell. 10:1235–1246. [DOI] [PubMed] [Google Scholar]
- Higuchi, M., S. Maas, F.N. Single, J. Hartner, A. Rozov, N. Burnashev, D. Feldmeyer, R. Sprengel, and P.H. Seeburg. 2000. Point mutation in an AMPA receptor gene rescues lethality in mice deficient in the RNA-editing enzyme ADAR2. Nature. 406:78–81. [DOI] [PubMed] [Google Scholar]
- Hurst, S.R., R.F. Hough, P.J. Aruscavage, and B.L. Bass. 1995. Deamination of mammalian glutamate receptor RNA by Xenopus dsRNA adenosine deaminase: similarities to in vivo RNA editing. RNA. 1:1051–1060. [PMC free article] [PubMed] [Google Scholar]
- Keegan, L.P., A. Gallo, and M.A. O'Connell. 2001. The many roles of an RNA editor. Nat. Rev. Genet. 2:869–878. [DOI] [PubMed] [Google Scholar]
- Krovat, B.C., and M.F. Jantsch. 1996. Comparative mutational analysis of the double-stranded RNA binding domains of Xenopus laevis RNA-binding protein A. J. Biol. Chem. 271:28112–28119. [DOI] [PubMed] [Google Scholar]
- Lai, F., R. Drakas, and K. Nishikura. 1995. Mutagenic analysis of double-stranded RNA adenosine deaminase, a candidate enzyme for RNA editing of glutamate-gated ion channel transcripts. J. Biol. Chem. 270:17098–17105. [DOI] [PubMed] [Google Scholar]
- Lehmann, K.A., and B.L. Bass. 1999. The importance of internal loops within RNA substrates of ADAR1. J. Mol. Biol. 291:1–13. [DOI] [PubMed] [Google Scholar]
- Lehmann, K.A., and B.L. Bass. 2000. Double-stranded RNA adenosine deaminases ADAR1 and ADAR2 have overlapping specificities. Biochemistry. 39:12875–12884. [DOI] [PubMed] [Google Scholar]
- Liu, Y., and C.E. Samuel. 1996. Mechanism of interferon action: functionally distinct RNA-binding and catalytic domains in the interferon-inducible, double-stranded RNA-specific adenosine deaminase. J. Virol. 70:1961–1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Y., and C.E. Samuel. 1999. Editing of glutamate receptor subunit B pre-mRNA by splice-site variants of interferon-inducible double-stranded RNA-specific adenosine deaminase ADAR1. J. Biol. Chem. 274:5070–5077. [DOI] [PubMed] [Google Scholar]
- Liu, Y., C.X. George, J.B. Patterson, and C.E. Samuel. 1997. Functionally distinct double-stranded RNA-binding domains associated with alternative splice site variants of the interferon-inducible double-stranded RNA-specific adenosine deaminase. J. Biol. Chem. 272:4419–4428. [DOI] [PubMed] [Google Scholar]
- Liu, Y., R.B. Emeson, and C.E. Samuel. 1999. Serotonin-2C receptor pre-mRNA editing in rat brain and in vitro by splice site variants of the interferon-inducible double-stranded RNA-specific adenosine deaminase ADAR1. J. Biol. Chem. 274:18351–18358. [DOI] [PubMed] [Google Scholar]
- Liu, Y., M. Lei, and C.E. Samuel. 2000. Chimeric double-stranded RNA-specific adenosine deaminase ADAR1 proteins reveal functional selectivity of double-stranded RNA-binding domains from ADAR1 and protein kinase PKR. Proc. Natl. Acad. Sci. USA. 97:12541–12546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micklem, D.R., J. Adams, S. Grunert, and D. St. Johnston. 2000. Distinct roles of two conserved Staufen domains in oskar mRNA localization and translation. EMBO J. 19:1366–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson, J.B., D.C. Thomis, S.L. Hans, and C.E. Samuel. 1995. Mechanism of interferon action: double-stranded RNA-specific adenosine deaminase from human cells is inducible by alpha and gamma interferons. Virology. 210:508–511. [DOI] [PubMed] [Google Scholar]
- Paul, M.S., and B.L. Bass. 1998. Inosine exists in mRNA at tissue-specific levels and is most abundant in brain mRNA. EMBO J. 17:1120–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peculis, B.A., and J.G. Gall. 1992. Localization of the nucleolar protein NO38 in amphibian oocytes. J. Cell Biol. 116:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raitskin, O., D.S. Cho, J. Sperling, K. Nishikura, and R. Sperling. 2001. RNA editing activity is associated with splicing factors in lnRNP particles: The nuclear pre-mRNA processing machinery. Proc. Natl. Acad. Sci. USA. 98:6571–6576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos, A., S. Grunert, J. Adams, D.R. Micklem, M.R. Proctor, S. Freund, M. Bycroft, D. St. Johnston, and G. Varani. 2000. RNA recognition by a Staufen double-stranded RNA-binding domain. EMBO J. 19:997–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryter, J.M., and S.C. Schultz. 1998. Molecular basis of double-stranded RNA-protein interactions: structure of a dsRNA-binding domain complexed with dsRNA. EMBO J. 17:7505–7513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St Johnston, D., N.H. Brown, J.G. Gall, and M. Jantsch. 1992. A conserved double-stranded RNA-binding domain. Proc. Natl. Acad. Sci. USA. 89:10979–10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steitz, J.A. 1989. Immunoprecipitation of ribonucleoproteins using autoantibodies. Methods Enzymol. 180:468–481. [DOI] [PubMed] [Google Scholar]
- Strehblow, A., M. Hallegger, and M.F. Jantsch. 2002. Nucleocytoplasmic distribution of human RNA-editing enzyme ADAR1 is modulated by double-stranded RNA-binding domains, a leucine-rich export signal, and a putative dimerization domain. Mol. Biol. Cell. 13:3822–3835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace, R.A., D.W. Jared, J.N. Dumont, and M.W. Sega. 1973. Protein incorporation by isolated amphibian oocytes. 3. Optimum incubation conditions. J. Exp. Zool. 184:321–333. [DOI] [PubMed] [Google Scholar]
- Wong, S.K., S. Sato, and D.W. Lazinski. 2001. Substrate recognition by ADAR1 and ADAR2. RNA. 7:846–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, Z.A., C. Murphy, H.G. Callan, and J.G. Gall. 1991. Small nuclear ribonucleoproteins and heterogeneous nuclear ribonucleoproteins in the amphibian germinal vesicle: loops, spheres, and snurposomes. J. Cell Biol. 113:465–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi-Brunozzi, H.Y., O.M. Stephens, and P.A. Beal. 2001. Conformational changes that occur during an RNA-editing adenosine deamination reaction. J. Biol. Chem. 276:37827–37833. [DOI] [PubMed] [Google Scholar]