

Abstract
Shear stress induces endothelial polarization and migration in the direction of flow accompanied by extensive remodeling of the actin cytoskeleton. The GTPases RhoA, Rac1, and Cdc42 are known to regulate cell shape changes through effects on the cytoskeleton and cell adhesion. We show here that all three GTPases become rapidly activated by shear stress, and that each is important for different aspects of the endothelial response. RhoA was activated within 5 min after stimulation with shear stress and led to cell rounding via Rho-kinase. Subsequently, the cells respread and elongated within the direction of shear stress as RhoA activity returned to baseline and Rac1 and Cdc42 reached peak activation. Cell elongation required Rac1 and Cdc42 but not phosphatidylinositide 3-kinases. Cdc42 and PI3Ks were not required to establish shear stress–induced polarity although they contributed to optimal migration speed. Instead, Rho and Rac1 regulated directionality of cell movement. Inhibition of Rho or Rho-kinase did not affect the cell speed but significantly increased cell displacement. Our results show that endothelial cells reorient in response to shear stress by a two-step process involving Rho-induced depolarization, followed by Rho/Rac-mediated polarization and migration in the direction of flow.
Keywords: Rho GTPases; actin cytoskeleton; Rho-kinase; migration; polarity
Introduction
Shear stress, the frictional force exerted by laminar blood flow, induces extensive changes in endothelial cell behavior and has been implicated in vasculogenesis, reendothelialization of vascular grafts, atherosclerosis, and angiogenesis (Drenckhahn and Ness, 1997; Urbich et al., 2002). Shear stress stimulates several signaling cascades in endothelial cells including: potassium channel activation (the earliest); elevation of inositol trisphosphate and diacylglycerol; an increase in intracellular calcium levels; G protein activation; MAPK signaling; and activation of transcription factors, such as nuclear factor κB (Davies, 1997; Azuma et al., 2001; Hoger et al., 2002). The early signaling events are followed by changes in gene expression and alignment of actin filaments and microtubules within the flow direction, resulting in changes in cell shape and directional migration (Braddock et al., 1998, Hsu et al., 2001). The nature of the shear stress–sensing mechanism is not clear as several signaling molecules including integrins (Tzima et al., 2001; Schoenwaelder et al., 2002), vascular endothelial growth factor receptor and vascular endothelial cadherin (Shay-Salit et al., 2002), stretch-activated cation channels (Brakemeier et al., 2002), K+ ion channel (Hoger et al., 2002), and platelet endothelial cell adhesion molecule-1 (Osawa et al., 2002) have been suggested to act as mechanosensors.
The Rho GTPases Rho, Rac, and Cdc42 were initially characterized as key regulators of actin cytoskeletal remodelling induced by extracellular signals, but have subsequently been shown to play many other roles in cell signaling (Ridley, 2001a,b). Rho regulates the formation of actin stress fibers, Rac is responsible for controlling membrane ruffling and the formation of lamellipodia, whereas Cdc42 controls the formation of filopodia (Ridley, 2001c; Etienne-Manneville and Hall, 2002). In migrating cells, Rac is generally required at the leading edge for lamellipodium extension and formation of new adhesions, whereas Rho controls cell contractility and tail retraction (Ridley, 2001a). Phosphatidylinositide 3-kinases (PI3Ks)* can act upstream of Rac in cell migratory responses (Ridley, 2001b), and are important for chemotaxis in some cell types (Funamoto et al., 2002; Hannigan et al., 2002). Cdc42 regulates cell orientation, for example in macrophages chemotaxing toward colony-stimulating factor-1 (Allen et al., 1998), in wound healing responses (Nobes and Hall., 1999; Etienne-Manneville and Hall, 2001), and in T cell polarization toward antigen-presenting cells (del Pozo et al., 1999). In endothelial cells, the relative contributions of Rho, Rac, and Cdc42 to migration varies depending on the stimulus. Rho is required for endothelial cell migration across transwell filters in response to sphingosine-1-phosphate, but its role in VEGF-induced migration remains controversial (Liu et al., 2001; Zeng et al., 2002). Rac is required for both sphingosine-1-phosphate– and VEGF-induced migration (Soga et al., 2001; Ryu et al., 2002; Zeng et al., 2002), whereas Cdc42 is not required for VEGF-induced migration of endothelial cells mediated by VEGFR-2 (Zeng et al., 2002). Rho GTPases have also been implicated in endothelial responses to shear stress (Li et al., 1999; Tzima et al., 2001). Rho and Cdc42 translocate to the membrane in response to shear stress and mediate the activation of transcription factor AP-1 through c-Jun NH2-terminal kinases (Li et al., 1999). Expressing dominant negative mutants of Rho and its downstream target Rho-kinase/ROCK, but not Cdc42, inhibited shear stress–induced cell alignment and stress fiber formation in confluent cultures of bovine aortic endothelial cells (Li et al., 1999). Constitutively activated mutants of RhoA and Rac1 inhibit shear stress–induced alignment of bovine aortic endothelial cells, indicating that dynamic regulation of those two proteins is important for cell orientation (Tzima et al., 2001, 2002). Recently, it was reported that shear stress rapidly stimulates conformational activation of integrin αvβ3 in confluent bovine aortic endothelial cells, followed by increased cell adhesion to extracellular matrix components and transient down-regulation of Rho activity (Tzima et al., 2001). It was postulated that this decrease in Rho activity is required for the alignment of the cells within the direction of flow.
Here, we investigate the relative contributions of RhoA, Rac1, and Cdc42 to shear stress–induced changes in cell shape and directed cell migration, and compare these with PI3Ks, Rho-kinases, and myosin light chain kinase (MLCK), which are known to be important for cell polarization and/or migration in other systems. We show for the first time that RhoA, Rac1, and Cdc42 are all activated during the early stages of endothelial actin cytoskeletal remodeling induced by shear stress. RhoA is required for initial cell contraction and depolarization, and subsequently the coordinated action of RhoA and Rac1 is required for the alignment and movement of endothelial cells in the direction of the flow. In contrast to their known roles in generating cell polarity in other cell types, neither Cdc42 nor PI3K is important for shear stress–induced cell polarization in the direction of flow, although they contribute to migration speed.
Results
Shear stress induces changes in endothelial cell shape and polarity
Subconfluent human umbilical vein endothelial cells (HUVECs) showed a two-phase response to shear stress. In the first phase, within 5 min of stimulation, there was a rapid increase in the number of actin stress fibers (Fig. 1, A and B) . During the next 15–30 min, the number of stress fibers decreased, the cells retracted (Fig. 1 C), and started to polarize within the direction of shear stress (Fig. 1 I). The degree of cell retraction correlated with the level of shear stress (unpublished data). In the second phase, between 30 min and 24 h, the cells respread and elongated within the direction of the flow (Fig. 1 D). The most dynamic changes in cell alignment were noticed between 30 min and 4 h of stimulation, when ∼40% of cells became aligned in the direction of shear stress (Fig. 1, D–F and I).
Figure 1.

Cell shape remodelling induced by 3 dyn/cm2 shear stress. A–F show F-actin staining in growing endothelial cells stimulated with shear stress. The direction of flow is indicated by an arrow (E). A shows cells in control (static) conditions, whereas B–F show cells stimulated with shear stress shear for 5 min, 15 min, 1 h, 2 h, and 4 h, respectively. Bar, 20 μm. G shows changes in the cell spread area; H shows changes in cell elongation; and I shows changes in cell alignment (orientation) over 24 h of stimulation with shear stress. Cell alignment (I) is shown as a percentage of cells that aligned within 10° of the shear direction. *, P ≤ 0.05; **, P ≤ 0.01, comparisons with static control, t test.
Although the first measurable change in cell alignment was noticed at 30 min (Fig. 1 I), we observed that HUVECs started to polarize at earlier time points; within 15 min of shear stress, 49 ± 18% (n = 150) of cells had lamellipodial protrusions in the flow direction as compared with only 18 ± 4% of cells in static controls. A similar time course of shear stress–induced lamellipodial protrusion was reported in bovine aortic cells (Li et al., 2002).
Changes in the activity of Rho, Rac, and Cdc42 accompany shear stress–induced remodeling of the actin cytoskeleton
To investigate the involvement of Rho, Rac, and Cdc42 in shear stress responses, we first measured their activity during endothelial cell adaptation to shear stress. RhoA activity increased threefold at 5 min after exposure to shear stress, and then decreased sharply below control levels within the next 10 min (Fig. 2) . RhoA activity gradually increased to 1.6-fold above basal level at 2 h after stimulation with shear stress (Fig. 2) and returned to basal levels by 4 h (not depicted). The rapid increase in RhoA activity at 5 min correlated with the formation of numerous stress fibers (Fig. 1 B) and the subsequent decrease in activity 15–30 min after stimulation correlated with the loss of stress fibers and cell rounding (Fig. 1, C, G, and H). The small increase in RhoA activity at 2 h corresponded to the stage when most of the cells were realigned within the direction of shear stress.
Figure 2.

Regulation of Rho, Rac, and Cdc42 activity by shear stress. HUVECs were subjected to shear stress for the indicated times. GTP-loading assays for RhoA, Rac1, and Cdc42 were performed as described in Materials and methods. Panels on the left show fold increase of RhoA, Rac1, and Cdc42 activity during stimulation and were calculated as the amount of GTP-bound protein relative to whole cell lysates. Corresponding representative examples of Western blots of GTP-bound proteins and total cell lysates are shown on the right. Values are means ± SD from four to five independent experiments. *, P ≤ 0.05; **, P ≤ 0.01, comparisons with static control, t test.
Rac1 and Cdc42 became activated with a similar time course. Their activity was increased between 5–30 min after stimulation with shear stress, with peak activation at 15 min (Fig. 2). Rac1 and Cdc42 activation coincided with the beginning of cell spreading and elongation, and was at its peak at the time when RhoA activity was at its lowest level.
Rac1 normally controls formation of membrane ruffles and lamellipodia, whereas Cdc42 controls formation of filopodia (Ridley, 2001a). We did not observe an increased formation of filopodia or lamellipodia at 5–30 min of exposure to shear stress as compared with static controls (Fig. 1). Static HUVECs had lamellipodia before shear stress but as described above, after cell retraction, lamellipodia were predominantly oriented at the downstream side of cells (see Fig. 1 F and Fig. 4 A). Filopodia were not observed under any conditions; in fact, introduction of constitutively active Cdc42 (V12Cdc42) into HUVECs results in the formation of very few filopodia followed by stress fibers and cell contraction (Wojciak-Stothard et al., 1998).
Figure 4.
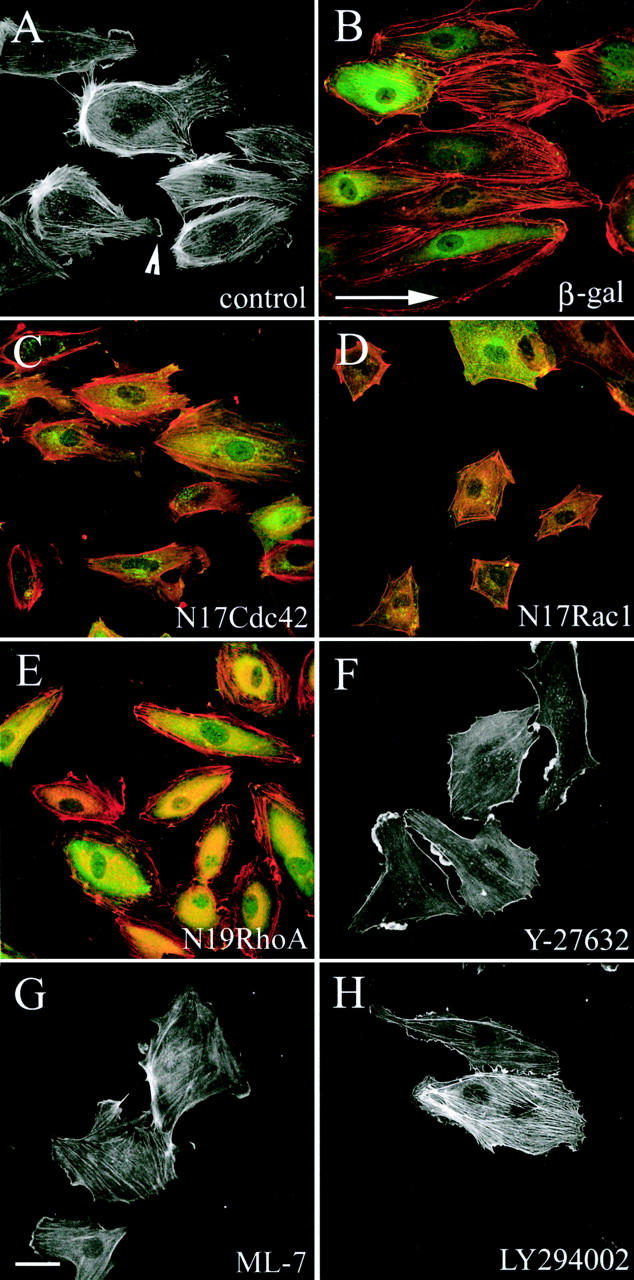
The effects of inhibitors on shear-induced remodeling of actin cytoskeleton. Cells were untreated (A) or infected with adenoviruses to express β-gal (B), N17Cdc42 (C), N17Rac1 (D), or N19RhoA (E) 16 h before stimulation with shear stress for 2 h. Alternatively, cells were treated with 5 μM Y-27632 (F), 10 μM ML-7 (G), or 10 μM LY294002 (H) 30 min before and during exposure to shear stress for 4 h. Shear direction is indicated with an arrow (B). The arrowhead (A) points to a lamellipodium formed at the downstream part. Cells were stained for F-actin (red) and anti– c-myc 9E10 antibody (green) to visualize myc epitope–tagged N19RhoA, N17Rac1, and N17Cdc42 (B–D). Bar, 20 μm.
Stress fiber formation and cell rounding depend on RhoA and Rho-kinase, whereas respreading requires Rac1 and Cdc42
To study the effects of RhoA, Rac1, and Cdc42 on shear stress–induced morphological changes, dominant negative mutants of these proteins were expressed in endothelial cells using adenoviruses. As a control for adenoviral infection, cells were infected with an adenovirus expressing β-galactosidase (β-gal; Wojciak-Stothard et al., 2001). In addition, the roles of PI3Ks, Rho-kinase, and MLCK were investigated by adding small molecule inhibitors of these enzymes.
In control cells in the absence of shear stress (static cultures), inhibition of RhoA with N19RhoA and inhibition of Rho-kinase with Y-27632 promoted cell spreading (Fig. 3 A). In contrast, N17Rac1 significantly decreased the cell spread area (P < 0.05), whereas N17Cdc42 and LY294002 did not have a significant effect on the cell spread area (Fig. 3 A). Cell elongation in static cultures was slightly reduced by Y-27632, but was not significantly changed by any of the other inhibitors (Fig. 3 B). Although N17Cdc42 expressed by adenoviral infection did not have significant effects on the shape of subconfluent endothelial cells, the protein was active as it inhibited thrombin-induced contractility in confluent cultures of endothelial cells as described previously (Wojciak-Stothard et al., 2001).
Figure 3.
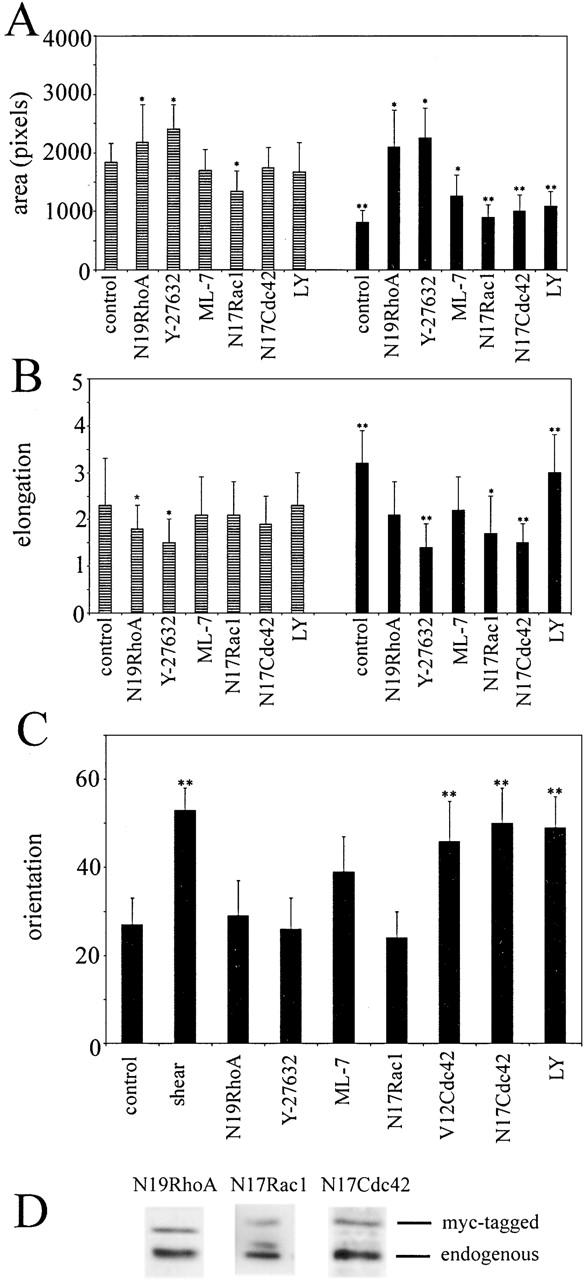
Effects of RhoA, Rac1, Cdc42 and inhibitors on shear stress–induced changes in cell spreading, elongation, and alignment. HUVECs were stimulated with shear stress for 30 min (A) or for 4 h (B and C). After stimulation, the cells were fixed and stained for F-actin, and cell spreading (A), elongation (B), or alignment (C) parameters were calculated with ImageProPlus software. The cells were untreated (controls) or were infected with adenoviruses encoding dominant negative mutants of Rho GTPases, and then subjected to shear stress 16 h after infection. The expression levels of myc-tagged N19RhoA, N17Rac1, and N17Cdc42 are shown in D. Alternatively, the inhibitors Y-27632, ML-7, and LY294002 (LY) were applied 30 min before stimulation with shear stress. Cell alignment is shown as the percentage of cells that aligned within 10° of the shear stress direction. In A–C, black bars represent cells under shear stress, and striped bars represent cells in static conditions. *, P ≤ 0.05; **, P ≤ 0.01, t test, comparison with static controls.
Stimulation with shear stress for 5–30 min induced cell rounding, resulting in a decrease in cell spread area and elongation (Fig. 1, G and H). N19RhoA and Y-27632 inhibited shear-induced cell retraction, whereas N17Rac1, N17Cdc42, and the PI3K inhibitors LY294002 and wortmannin had no effect (Fig. 3 A and not depicted). ML-7, an inhibitor of MLCK, also reduced the early shear-induced contractility of cells but to a lesser extent than N19RhoA and Y-27632 (Fig. 3 A). Elongation of untreated cells measured 4 h after stimulation with shear stress significantly increased as compared with untreated cells in static cultures. However, shear-induced cell elongation was prevented by dominant-negative mutants of RhoA (P = 0.045), Rac1 (P < 0.01), Cdc42, as well as Y-27632 (P < 0.05) but not LY294002 (P > 0.05; Fig. 3 B). In these experiments, cells were infected with adenoviruses expressing dominant-negative mutants 17–22 h before stimulation. After longer periods, as the dominant-negative proteins accumulated to higher levels, the differences between controls and the cells expressing dominant negative mutants of Rho GTPases became larger. The spread area and elongation of cells expressing N17Rac1 or N17Cdc42 was further reduced compared with untreated controls both under shear stress and in static conditions, whereas N19RhoA-expressing cells began to grow long microtubule-rich extensions (unpublished data).
Cell alignment in the direction of shear stress is dependent on RhoA and Rac1
Shear stress induced the alignment of actin stress fibers in the direction of flow within 2–4 h of shear stress in uninfected cells and in cells expressing β-gal (Fig. 4 A, control, shown at 2 h). In addition, the majority of aligned cells produced F-actin–rich lamellipodia on the downstream part of the cell (Fig. 4 A, arrowhead). As expected, N19RhoA-expressing cells showed only a few, mainly cortical, unaligned stress fibers but still had lamellipodia (Fig. 4 E). Similarly, the Rho-kinase inhibitor Y-27632 inhibited stress fiber formation (Fig. 4 F), and appeared to enhance membrane ruffling, which is consistent with a recent paper by Tsuji et al. (2002). The MLCK inhibitor ML-7 did not entirely inhibit formation of stress fibers (Fig. 4 G), suggesting that the main input into MLC phosphorylation during shear stress is via Rho-kinase rather than MLCK. Rho-kinase increases phosphorylation of MLC by inhibiting MLC phosphatase (van Nieuw Amerongen and van Hinsbergh, 2001) or by direct phosphorylation of MLC (Kaibuchi, 1999; Amano et al., 2000). In addition, Rho-kinase can regulate the actin cytoskeleton by phosphorylating LIM kinase (Maekawa et al., 1999), adducin (Kimura et al., 1998), and the Na-H exchanger (Tominaga et al., 1998), and these could also contribute to the more pronounced effect of Y-27632 compared with ML-7.
N17Cdc42 did not affect stress fiber levels or alter the shear stress–induced alignment of stress fibers (Fig. 4 C). In contrast, cells expressing N17Rac1 lacked stress fibers and lamellipodia (Fig. 4 D), consistent with previous observations showing that N17Rac1 inhibits stress fiber assembly in thrombin- and TNF-stimulated HUVECs (Wojciak-Stothard et al., 1998, 2001; Kiosses et al., 1999). The PI3K inhibitors LY294002 (Fig. 4 G) and wortmannin (not depicted) did not have a significant effect on cell alignment, stress fibers or membrane ruffling, indicating that PI3K does not act upstream of Rac in shear stress responses, unlike growth factor–induced Rac activation (Ridley, 2001b).
In addition to analyzing stress fiber alignment, we measured cell orientation in response to shear stress. At 2 h, the majority of cells were oriented with their longer axis in the direction of flow (Fig. 3 C, shear). This shear stress–induced cell alignment was inhibited by N19RhoA, Y-27632, and N17Rac1 but not N17Cdc42 or LY294002, which is consistent with the effects on stress fiber alignment (Fig. 3 C and Fig. 4). Like Y-27632, ML-7 inhibited cell alignment, although it did not entirely abolish formation of stress fibers (Fig. 4 G). These results suggest that actomyosin contractility plays an essential role in shear-induced polarization of endothelial cells. Y-27632 and ML-7 are both inhibitors of actomyosin contractility, but have been shown to affect different subsets of stress fibers in human fibroblasts (Katoh et al., 2001) and play distinct roles in the spatial regulation of MLC phosphorylation (Totsukawa et al., 2000).
The effects of constitutively activated RhoA, Rac1, and Cdc42 on actin organization and shear stress–induced alignment were consistent with Rho and Rac, but not with Cdc42, contributing to shear stress–induced cell orientation. Constitutively activated L63RhoA induced formation of stress fibers and prevented cell alignment (Fig. 5 A), in agreement with previous observations on bovine aortic endothelial cells (Tzima et al., 2001). L61Rac1 also inhibited HUVEC alignment and induced formation of lamellipodia (Fig. 5 B) similar to results on bovine aortic endothelial cells (Tzima et al., 2002). In contrast, L61Cdc42 did not have a significant effect on cell alignment (Fig. 3 C and Fig. 5 C) or elongation (not depicted), which is consistent with the lack of effect of N17Cdc42 on these responses.
Figure 5.

Effects of constitutively activated RhoA, Rac1, and Cdc42 on cell alignment under shear stress. HUVECs were transfected with expression vectors encoding GFP-L63RhoA (A), GFP-L61Rac1 (B), or GFP-L61Cdc42 (C). After 18 h, cells were exposed to shear stress for 4 h. The cells expressing GFP-tagged proteins are green and indicated with arrowheads; F-actin staining is in red. Shear direction is indicated with an arrow. Bar, 20 μm.
N17Rac1, N17Cdc42, and PI3K inhibitors reduce cell migration speed
In static cultures, HUVECs migrated randomly at an average speed of 48.36 ± 7 μm/h (n = 120). The cells expressing N17Rac1 and N17Cdc42 moved significantly slower than control cells (Fig. 6 A), whereas the cells expressing N19RhoA or treated with Y-27632 did not show a reduction in the cell speed. LY294002 and ML-7 also decreased cell speed (Fig. 6 A).
Figure 6.
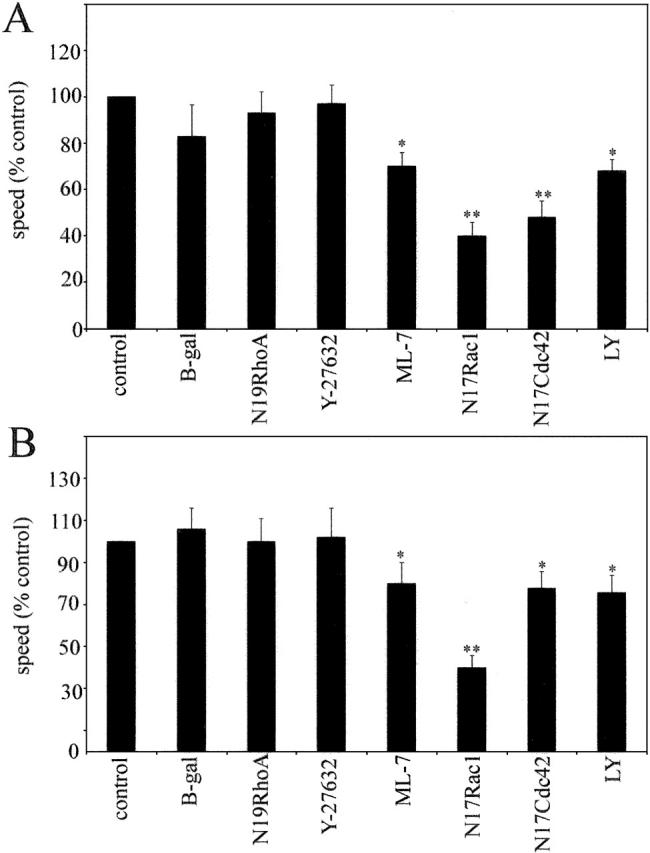
Regulation of cell migration speed by Rho signaling pathways. HUVECs cultured under static conditions (A) or under shear stress (B) were monitored by timelapse videomicroscopy for 5 h. ML-7, LY294002 (LY), and Y-27632 were added 30 min before the experiment or cells were infected with adenoviruses expressing N19RhoA, N17Rac1, and N17Cdc42 16 h before the experiment. Cells were tracked with Kinetic Imaging software and the trajectories were statistically analyzed using Mathematica software (see Materials and methods). *, P ≤ 0.05; **, P ≤ 0.01, comparison with controls, t test.
When exposed to shear stress, cells migrated preferentially within the direction of shear stress and with the flow, and migration in any other direction was strongly reduced (Fig. 6 B). The speed of cell migration was 24 ± 7 μm/h, which was significantly lower than in static cultures. Similar to cells in the absence of shear stress, N17Cdc42, N17Rac1, LY294002, and ML-7 reduced cell speed under flow, whereas N19RhoA and Y-27632 did not significantly alter migration speed (Fig. 6 B). The decrease in speed induced by ML-7, but not by Y-27632 or N19RhoA, may be a result of an inhibitory effect of ML-7 on the formation of lamellipodia (Fig. 4 G). In fibroblasts, ML-7 has also been shown to inhibit ruffling, induce cell rounding, and inhibit formation of peripheral stress fibers, whereas Y-27632 did not affect lamellipodium formation or reduce cell spreading (Katoh et al., 2001).
RhoA and Rac1 but not Cdc42 are required for shear stress–induced orientation of cell migration
As described above (Fig. 6), in the absence of shear stress endothelial cells in sparse cultures moved randomly, whereas cells under shear stress migrated predominantly within the direction of the flow (Fig. 7 , A–D). During chemotaxis, directionality has been shown to depend on Cdc42 and PI3Ks (Wang et al., 2002). In contrast, directional migration induced by shear stress was unaffected by either N17Cdc42 or LY294002/wortmannin (Fig. 7 E). Instead, orientation of cell movement was abolished in cells expressing N17Rac1 or N19RhoA, or cells treated with Y-27632 or ML-7 (Fig. 7 E), correlating with their effect on shear stress–induced cell alignment.
Figure 7.
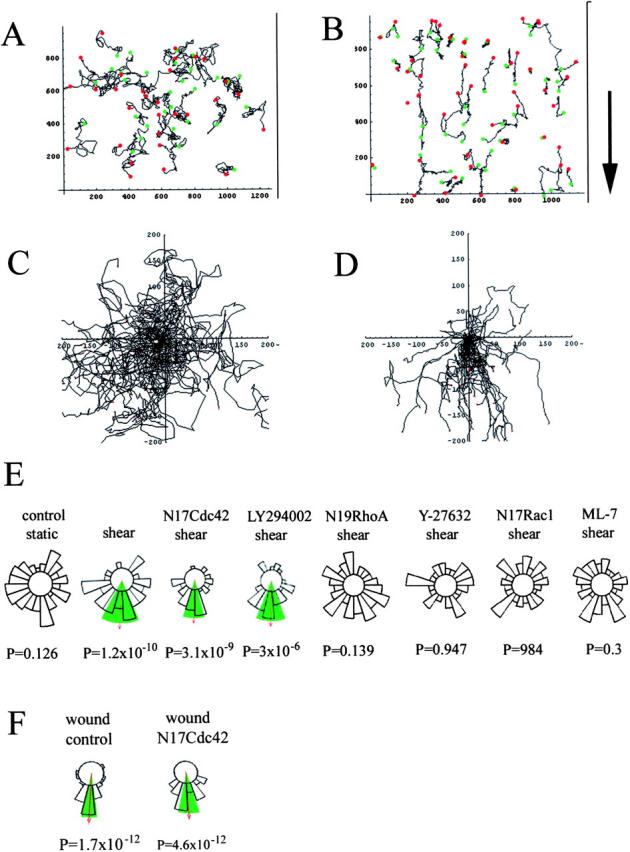
The effects of dominant negative RhoA, Rac1, and Cdc42 and inhibitors on the direction of cell migration. Cell trajectories during a 12-h experiment are shown in static conditions (A and C) or under shear stress (B and D). The starting point of each cell trajectory is plotted at the intersection of the X and Y axes (C and D). Circular histograms (E and F) show the proportion of cells migrating into each of 20 equal segments, measured when each cell had migrated 50 μm from its starting point. Cells that migrated <50 μm are excluded from this analysis. Arrow indicates the mean direction of the cell population where this is significant (P values indicate significance), and the green shaded areas mark the 95% confidence intervals of statistical significance, as calculated using a Rayleigh test. The direction of shear stress in all cases is toward the bottom of the histograms (B, arrow). For each condition, 90 cells in three independent experiments were analyzed.
Given that Cdc42 is required for cell orientation in a variety of cell types (Etienne-Manneville and Hall, 2003), we wanted to determine whether Cdc42 can contribute to directional sensing in endothelial cells, or whether it is just not important for shear stress–induced reorientation of cells. Therefore, we performed an in vitro wound healing assay, similar to that used to show a requirement for Cdc42 in fibroblast orientation and migration into wounds (Nobes and Hall, 1999). Interestingly, N17Cdc42 did not affect directionality of endothelial cell movement toward the center of the wound (Fig. 7 F), suggesting that Cdc42 is not involved in regulating the directionality of endothelial cell movement. However, as in shear stress conditions, N17Cdc42 reduced the overall speed of cell movement to 69 ± 3% of the control value in the wound healing assay.
Inhibition of RhoA and Rho-kinase increases cell displacement
To study the ability of the cells to move effectively in one direction, we determined the percentage of cells that had translocated a distance of 50 μm or more from their starting point in each experiment. Because over 20% of cells in all experimental conditions managed to migrate over this distance, it was chosen as suitable for comparisons of cell displacement. For groups of cells that migrate at similar speeds, cell displacement can be used as a measure for persistence of cell movement as only cells that migrate persistently in one direction manage to migrate over a preset horizon. In static conditions, HUVECs frequently changed direction and only 47% of cells migrated over 50 μm in 5 h (Fig. 8, A and B) . Cells expressing N19RhoA or treated with Y-27632 showed a significant increase in total cell displacement (Fig. 8 B; P < 0.01), changed direction less frequently than control cells, and had straighter trajectories, particularly in cells treated with Y-27632 (Fig. 8 A).
Figure 8.


Effects of RhoA, Rac1, Cdc42, and inhibitors on cell displacement. A shows trajectories of untreated (control) cells (left plot), cells expressing N19RhoA (middle plot) and cells pretreated with Y-27632 (right plot) under static conditions. The cells were tracked for 5 h in static conditions. B and C show total cell displacement in static conditions (B) and under shear stress (C). Total cell displacement is a percentage of the total number of cells that migrated over a preset horizon of 50 μm during 5 h of experiment (n = 90). In A, each plot shows trajectories of 30 endothelial cells in static conditions.
Under shear stress, 36 ± 6% of control cells translocated over 50 μm. This was also significantly increased by RhoA and Rho-kinase inhibitors (P < 0.01). Cell displacement in cells expressing N17Rac1 (P < 0.01), N17Cdc42, or treated with ML-7 or LY294002 (P < 0.05) was lower than in untreated cells both in static conditions and under shear stress, reflecting the reduction in cell speed (Fig. 8, B and C).
Inhibition of Rho/Rho-kinase has been shown to reduce tail detachment in a number of cell types (Alblas et al., 2001; Swetman et al., 2002). Similarly, we observed by video time-lapse microscopy that Y-27632 inhibited tail detachment in HUVECs exposed to shear stress, resulting in a transient elongation of cells when the tail remained attached and the front of the cell continued to extend (unpublished data). This behavior could contribute to the increased cell displacement as the cells expressing N19RhoA or preincubated with Y-27632 turned (changed direction) significantly less than control cells (Fig. 8 A).
Discussion
We have observed that shear stress–induced polarization and migration of endothelial cells requires the coordinated actions of RhoA, Rac1, and Cdc42. Shear stress initially activates RhoA, leading to stress fiber formation and cell contraction. Subsequently, cells respread and elongate, and this is dependent on Rac and Cdc42. Cells align and migrate in the direction of shear stress, and this polarization requires Rac and Rho. PI3Ks and Cdc42 contribute to cell migration speed but not to cell alignment, in contrast to their roles in cell polarization in other cell types.
Rho is a key regulator of shear-induced remodeling of endothelial cell shape and movement
To migrate in the direction of flow, randomly oriented cells have to change their polarity. To do this, cells could turn as they migrate. However, our results suggest that shear stress–induced cell reorientation is achieved first by early cell contraction mediated by Rho, leading to cell depolarization, and then by extension of new lamellipodia in the direction of flow. This two-stage mechanism for reorienting cells may be more efficient than turning for relatively slow-moving cells. The activation of RhoA is very transient after application of shear stress, returning to control levels by 10 min. This may explain why it was not observed in a previous paper on shear stress–induced changes in RhoA activity in confluent bovine aortic endothelial cells (Tzima et al., 2001). In addition, higher levels of shear stress were used by Tzima et al. (2001), and this could affect the timing of changes in RhoA activity.
The early requirement for Rho to induce cell contraction could explain why inhibition of Rho also prevents subsequent cell alignment in the direction of flow. However, although RhoA activity falls rapidly after the initial peak, it gradually increases over time, which is consistent with RhoA playing a long-term role in shear stress sensing. Indeed, it is known that Rho is important for sensing mechanical stress (Geiger and Bershadsky, 2001), thus, it is probable that Rho is involved in sensing and/or signaling shear stress at the back of the cell. Interestingly, although Rho is required for shear stress–induced cell polarization, it does not affect migration speed and, in fact, negatively regulates cell displacement. The contribution of RhoA/Rho-kinase to cell migration speed depends on cell type and conditions. Models for cell migration propose that RhoA and Rho-kinase act both by increasing cell body contractility and by stimulating tail detachment (Cox and Huttenlocher, 1998, Ridley, 2001a). Inhibition of RhoA/Rho-kinase in neutrophils and monocytes results in an inhibition of tail detachment and, as a result, inhibition of transendothelial migration (Alblas et al., 2001; Worthylake et al., 2001). Similarly, we observed that Y-27632 reduced tail detachment (unpublished data) but this did not affect the migration speed of endothelial cells, presumably because tail detachment is not the rate-limiting step for endothelial cell migration. On the other hand, inhibition of RhoA/Rho-kinase can promote cell motility in other cell types (Sahai et al., 2001; Chang et al., 2002; Chen et al., 2002). In reality, the balance between Rho signaling and other pathways regulating cell migration is likely to determine to what extent inhibiting Rho/Rho-kinase affects cell migration.
In contrast to Y-27632, the MLCK inhibitor ML-7 reduced lamellipodium extension, cell spreading, and the speed of cell movement. Therefore, its effects were more similar to inhibition of Rac than Rho/Rho-kinase, and probably reflect the known role of myosin IIb in lamellipodial protrusion (Diefenbach et al., 2002). Similarly, Katoh et al. (2001) have shown that in fibroblasts, ML-7 reduces cell ruffling, inhibits formation of peripheral stress fibers, and reduces cell spreading, whereas Y-27632 inhibited formation of central stress fibers but did not affect ruffling or cell spreading. In addition, MLCK and Rho-kinase play distinct roles in spatial regulation of MLC phosphorylation: Rho-kinase promotes MLC phosphorylation in the center of the cell, whereas MLCK regulates MLC phosphorylation at the cell periphery (Totsukawa et al., 2000). This difference could also contribute to the distinct effects of Y-27632 and ML-7 on endothelial responses to shear stress.
Roles of Rac and Cdc42 in endothelial cell polarization and migration
The peak shear stress–induced activation of Rac1 and Cdc42 was ∼15 min, when RhoA activity was at its lowest. At this time, cells started to polarize by producing lamellipodial protrusions in the flow direction. The overall increase in Rac1 activity was small, but it is likely that this is because Rac1 is primarily activated only at the leading edge of cells (Kraynov et al., 2000; Tzima et al., 2002).
Our observation that Cdc42 is not required for endothelial cell alignment and movement in the direction of flow was unexpected given observations on directional migration in other cell types, where Cdc42 is necessary to maintain cell directionality (Allen et al., 1998; Nobes and Hall, 1999; Etienne-Manneville and Hall, 2001). Interestingly, we found that in a wound healing assay, Cdc42 was also not required for directional migration of endothelial cells into the wound in contrast to results in fibroblasts and astrocytes (Nobes and Hall, 1999; Etienne-Manneville and Hall, 2001). It may be that in endothelial cells, Cdc42 is not molecularly linked to the PAR6–PAR3–aPKC complex involved in generating cell polarity in a variety of cell systems (Etienne-Manneville and Hall, 2003). In this context, it is interesting that in endothelial cells signaling downstream of Rac is also different to fibroblasts. For example, p21-activated kinase PAK1, a downstream target of Rac, promotes stress fiber formation and inhibits migration in endothelial cells (Kiosses et al., 1999), whereas in fibroblasts it induces loss of stress fibers and stimulates migration (Kiosses et al., 1999; Sells et al., 1999). Cdc42 could contribute to migration speed by affecting the organization of polymerized actin at the leading edge (via WASP family proteins; Cory and Ridley, 2002) and/or by affecting focal adhesion turnover through effects on PAKs (Bagrodia and Cerione, 1999).
PI3Ks in morphological responses to shear stress
PI3Ks are known to be activated by shear stress and are involved in both NO production and c-Jun NH2-terminal kinases activation (Go et al., 2001), but their roles in morphological responses to shear stress have not previously been investigated. Our results show that wortmannin/LY294002-sensitive PI3Ks are not required for shear-induced changes in cell polarity but contribute to cell migration speed. PI3Ks contribute to cell migration in a variety of cell types (Sotsios and Ward, 2000; Cantley, 2002), but only a few studies have addressed their role in cell polarization (Weiner et al., 2002). For example, in Dictyostelium and neutrophils, PI3Ks are required for both migration speed and to maintain directionality (Wang et al., 2002; Weiner et al., 2002), whereas we report here that PI3Ks contribute to migration speed but not directionality. They can contribute to cell migration at least in part by acting upstream of Rac (Weiner, 2002) but in shear stress responses of endothelial cells, inhibition of PI3Ks does not affect cell elongation, whereas inhibition of Rac does, indicating that PI3K activation is not required for all Rac-dependent responses. Similarly, PI3Ks are not required for Rac activation in fMLP-stimulated neutrophils (Geijsen et al., 1999).
Conclusions
We propose that the process of adaptation of sparse endothelial cells to shear stress can be divided into two stages regulated by RhoA, Rac1, and Cdc42. In the first stage, an increase in RhoA activity leads to the formation of stress fibers and cell contraction. This allows cells to assume a nonpolar morphology, from which they can elongate efficiently in the direction of shear stress. This elongation involves directional spreading via protrusion at the front of the cells, and is mediated by Rac and Cdc42 activation. Importantly, RhoA activity is down-regulated at this stage to allow optimal extension of protrusions. Later, RhoA activity increases, and Rho together with Rac is required to maintain polarized migration in the direction of shear stress. The fact that Cdc42 and PI3Ks are not required for cell polarization in endothelial cells but are in other cell types/conditions indicates that cells have multiple mechanisms for sensing and responding to polarity-inducing stimuli.
Materials and methods
Reagents
Human fibronectin, FITC-phalloidin, and wortmannin were obtained from Sigma-Aldrich; Y-27632 was obtained from Yoshitomi Pharmaceutical Industries Ltd.; BODIPY- and fluorescein-labeled goat anti–mouse secondary antibodies were obtained from Jackson ImmunoResearch Laboratories; anti–c-myc (9E10), anti-RhoA, and anti-Cdc42 monoclonal antibodies were obtained from Santa Cruz Biotechnology, Inc.; Moviol, ML-7, and LY294002 were obtained from Calbiochem-Novabiochem; Rac Activation Assay Kit was obtained from Upstate Biotechnology; pGEX-GST-RBD was a gift from M. Schwartz (Scripps Institute, La Jolla, CA); pGEX-GST-WASp-PBD was a gift from D. Sacks (Brigham and Women's Hospital, Boston, MA); GFP-tagged constructs of L61Rac1, L61Cdc42, and L63RhoA were a gift from Dr. N. Hotchin (School of Biosciences, University of Birmingham, Birmingham, UK).
Cell culture, adenoviral infection, transfection, and use of inhibitors
HUVECs were purchased from Biowhittaker and cultured in flasks coated with 10 μg/ml human fibronectin in EGM-2 medium (Biowhittaker) supplemented with 2% FCS. For experiments, cells were used between two and four passages.
HUVECs were plated at a density of 2 × 104 cells/ml in 9-cm2 Nunc slide flasks (Life Technologies) or on glass coverslips coated with 10 μg/ml human fibronectin. The cells were used for cell motility assays and cell shape analysis in static cultures or placed under shear stress in a parallel plate system. Adenoviral gene transfer was used to express dominant negative RhoA, Rac1, and Cdc42 proteins in HUVECs (Wojciak-Stothard et al., 2001). Experiments were performed 16–18 h after infection. GFP-tagged constitutively active forms of Rho proteins, L61Rac1, L61Cdc42, and L63RhoA, were transfected into HUVECs using a Nucleofector (Amaxa) and nucleofector transfection kit (Biowhittaker). Cells were used for experiments 16–18 h after transfection.
Rho-kinase inhibitor Y-27632 (5 μM), the MLCK inhibitor ML-7 (10 μM), or the PI3K inhibitors wortmannin (0.1 μM) and LY294002 (10 μM) were added to the cells 30 min before stimulation with shear stress. The concentration of ML-7 was established by titration and was the lowest concentration that had an effect on cell morphology during 5 h. At higher concentrations (20–40 μM), ML-7 induced rapid loss of stress fibers and cell detachment under flow conditions. The concentration selected for Y-27632 was based on prior experiments (Wojciak-Stothard et al., 2001), and the concentration for LY294002 and wortmannin in endothelial cells was based on published reports (Nakashio et al., 2002).
Shear stress
Laminar shear stress was applied to cells for various times ranging from 5 min to 24 h using parallel plate flow chambers set in series in a closed circulating system with 5% CO2 at 37°C. Parallel plate flow chambers were custom-made at Glaxo Wellcome, and circulation of the medium was produced by a peristaltic pump (Masterflex) calibrated to deliver a shear stress of 3 dyn/cm2. The chambers were assembled as described previously (Houston et al., 1999). The level of shear stress chosen for experiments corresponded to the physiological level of shear stress in venous vessels (Morawietz et al., 2000). Static controls were performed on cells not subjected to shear stress. The wall shear stress Tw, expressed (dyn/cm2), was calculated using the standard equation Tw = 3yQ/2a2b (Houston et al., 1999), where y is viscosity at 37°C (Poise), Q is the volumetric flow rate (ml/s), a is the half-channel height (cm), and b is the channel width (cm).
Rho, Rac, and Cdc42 pull-down assays
The cells were grown on fibronectin-covered 80-cm2 glass plates to subconfluency and incubated in growth factor–deprived medium with 2% FCS for 16–18 h before stimulation with shear stress. Glass plates were inserted into flow chambers and cells were subjected to a shear stress of 3 dyn/cm2 for 5–120 min after stimulation, the cells were lysed in 1× lysis buffer using 1 ml lysis buffer/plate for 30 s on ice, and GTP loading of RhoA, Rac1, and Cdc42 were measured. RhoA activity was measured with recombinant GST-RBD bound to glutathione beads (Amersham Biosciences) as described previously (Ren and Schwartz, 2000), using 100 μg GST-RBD for each sample. GTP loading of Rac1 was determined using a Rac Activation Assay Kit (Upstate Biotechnology), with 10 μg PAK1 PBD added to each sample. The activity of Cdc42 was measured with 20 μg GST-WASP-PBD/sample. GST-WASP-PBD was a gift from Dr. C. Wells (Ludwig Institute for Cancer Research, London, UK). Affinity-precipitated RhoA, Rac1, and Cdc42 proteins were resolved by SDS-PAGE and detected by Western blotting. The detailed protocol of Rac pull-down assay and preparation of lysis/wash buffer used in Rac and Cdc42 pull-down assays was provided by Upstate Biotechnology (lot 21956, Rac Activation Assay Kit).
Immunofluorescence and localization of F-actin
To visualize the distributions of F-actin and myc-tagged proteins, HUVECs were fixed with 4% formaldehyde dissolved in PBS for 10 min at room temperature and permeabilized for 6 min with 0.2% Triton X-100. Cells were incubated in 0.5% BSA in PBS for 45 min to block nonspecific antibody binding, and then incubated with mouse monoclonal 9E10 anti–c-myc antibody (1:200). Cells were incubated with fluorescein-labeled goat anti–mouse antibody and 1 μg/ml TRITC-phalloidin for 1 h. The specimens were mounted in Moviol (source). Single plane images of the cells were obtained by confocal laser scanning microscopy (model 61 LSM 510; Carl Zeiss MicroImaging, Inc.).
Analysis of cell shape and migration
To analyze cell spread area, elongation, and orientation, digitized images acquired by confocal microscopy were analyzed with Image Pro Plus (source). Cell orientation (alignment) was an angle between the long axis of the cell and the chosen direction. In the analysis of the cell shape in Image Pro Plus, a vertical line was used as a reference direction. Cell elongation was calculated as described by Dunn and Brown (1986), and was a proportion of the long axis of the cell to the shortest axis of the cell. Data are presented as means ± SD. The unpaired t test was used to compare differences between groups. Statistical significance was accepted for P < 0.05.
To analyze cell migration, cells were digitally recorded by video microscopy using a time-lapse interval of 10 min over a 5–24-h period. Cells were tracked with software (Kinetic Imaging Ltd.), and the trajectories were statistically analyzed using Mathematica software (Wolfram Research Inc.), as described previously (Allen et al., 1998). Cell displacement was evaluated by calculating the percentage of the total cells analyzed that had migrated a distance of 50 μm or more from the starting point during 5 h in each experiment. To visualize the directionality of cell movement in response to shear stress, statistical analysis of directional data was used and displayed as direction plots (Zicha et al., 1997). In brief, each trajectory was converted to a single angle representing the direction from the starting point of the trajectory to the point at which it first crossed a virtual horizon set at 50 μm. These data were summarized in a direction plot that is a circular histogram showing the number of cell directions lying within each 20° interval. The Rayleigh test for unimodial clustering of directions was applied to the data, and P < 0.01 was chosen as the criterion for rejecting the null hypothesis of random directionality (Allen et al., 1998). Where there was a significant unimodial clustering, the mean direction and its 95% confidence interval were calculated.
Wound healing assay for endothelial cell migration
HUVECs were grown to confluency on glass coverslips coated with fibronectin. Some of the cultures were infected with adenovirus to express N17Cdc42 and used for experiments after 16–18 h. A scratch wound was created in the monolayer as described previously (Nobes and Hall, 1999), and cell migration into the wound was recorded by video time-lapse microscopy.
Acknowledgments
We are particularly grateful to Dr. M. Braddock (Glaxo Welcome, Stevenage, UK) for help with the shear stress equipment; Dr. M. Schwartz for providing pGEX2T-GST-RBD; Dr. D. Sacks for providing pGEX2T-GST-Wasp-PBD; Dr. C. Wells for purifying GST-WASp-PBD; Dr. N. Hotchin for providing GFP-tagged constructs, L61Rac1, L61Cdc42, and L63RhoA; Dr. D. Cutler (Laboratory for Molecular Cell Biology, UCL, London, UK) for the use of the nucleofector; and Dr. A. Entwistle for help with confocal microscopy.
This work was supported by the British Heart Foundation Fellowship (FS/99021) and the Ludwig Institute for Cancer Research.
Footnotes
Abbreviations used in this paper: β-gal, β-galactosidase; HUVEC, human umbilical vein endothelial cell; MLCK, myosin light chain kinase; PAK, p21-activated kinase; PI3K, phosphatidylinositide 3-kinase.
References
- Alblas, J., L. Ulfman, P. Hordijk, and L. Koenderman. 2001. Activation of RhoA and ROCK are essential for detachment of migrating leukocytes. Mol. Biol. Cell. 12:2137–2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen, W.E., D. Zicha, A.J. Ridley, and G.E. Jones. 1998. A role for Cdc42 in macrophage chemotaxis. J. Cell Biol. 141:1147–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amano, M., Y. Fukata, and K. Kaibuchi. 2000. Regulation and functions of Rho-associated kinase. Exp. Cell Res. 261:44–51. [DOI] [PubMed] [Google Scholar]
- Azuma, N., N. Akasaka, H. Kito, M. Ikeda, V. Gahtan, T. Sasajima, and B.E. Sumpio. 2001. Role of p38 MAP kinase in endothelial cell alignment induced by fluid shear stress. Am. J. Physiol. Heart Circ. Physiol. 280:H189–H197. [DOI] [PubMed] [Google Scholar]
- Bagrodia, S., and R.A. Cerione. 1999. Pak to the future. Trends Cell Biol. 9:350–355. [DOI] [PubMed] [Google Scholar]
- Braddock, M., J.L. Schwachtgen, P. Houston, M.C. Dickson, M.J. Lee, and C.J. Campbell. 1998. Fluid shear stress modulation of gene expression in endothelial cells. News Physiol Sci. 13:241–246. [DOI] [PubMed] [Google Scholar]
- Brakemeier, S., I. Eichler, H. Hopp, R. Kohler, and J. Hoyer. 2002. Up-regulation of endothelial stretch-activated cation channels by fluid shear stress. Cardiovasc. Res. 53:209–218. [DOI] [PubMed] [Google Scholar]
- Cantley, L.C. 2002. The phosphoinositide 3-kinase pathway. Science. 296:1655–1657. [DOI] [PubMed] [Google Scholar]
- Chang, Y., B. Ceacareanu, M. Dixit, N. Sreejayan, and A. Hassid. 2002. Nitric oxide-induced motility in aortic smooth muscle cells: role of protein tyrosine phosphatase SHP-2 and GTP-binding protein Rho. Circ. Res. 91:390–397. [DOI] [PubMed] [Google Scholar]
- Chen, B.H., J.T. Tzen, A.R. Bresnick, and H.C. Chen. 2002. Roles of Rho-associated kinase and myosin light chain kinase in morphological and migratory defects of focal adhesion kinase-null cells. J. Biol. Chem. 277:33857–33863. [DOI] [PubMed] [Google Scholar]
- Cory, G.O., and A.J. Ridley. 2002. Cell motility: braking WAVEs. Nature. 418:732–733. [DOI] [PubMed] [Google Scholar]
- Cox, E.A., and A. Huttenlocher. 1998. Regulation of integrin-mediated adhesion during cell migration. Microsc. Res. Tech. 43:412–419. [DOI] [PubMed] [Google Scholar]
- Davies, P.F. 1997. Overview: temporal and spatial relationships in shear stress-mediated endothelial signalling. J. Vasc. Res. 34:208–211. [DOI] [PubMed] [Google Scholar]
- del Pozo, M.A., M. Vicente-Manzanares, R. Tejedor, J.M. Serrador, and F. Sanchez-Madrid. 1999. Rho GTPases control migration and polarization of adhesion molecules and cytoskeletal ERM components in T lymphocytes. Eur. J. Immunol. 29:3609–3620. [DOI] [PubMed] [Google Scholar]
- Diefenbach, T.J., V.M. Latham, D. Yimlamai, C.A. Liu, I.M. Herman, and D.G. Jay. 2002. Myosin 1c and myosin IIB serve opposing roles in lamellipodial dynamics of the neuronal growth cone. J. Cell Biol. 158:1207–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drenckhahn, D., and W. Ness. 1997. The endothelial contractile cytoskeleton. Vascular Endothelium. Physiology, Pathology and Therapeutic Opportunities. G.V.R. Born and C.J. Schwartz, editors. Schattauer, Stuttgart, Germany. 1–27.
- Dunn, G.A., and A.F. Brown. 1986. Alignment of fibroblasts on grooved surfaces described by a simple geometric transformation. J. Cell Sci. 83:313–340. [DOI] [PubMed] [Google Scholar]
- Etienne-Manneville, S., and A. Hall. 2001. Integrin-mediated activation of Cdc42 controls cell polarity in migrating astrocytes through PKCzeta. Cell. 106:489–498. [DOI] [PubMed] [Google Scholar]
- Etienne-Manneville, S., and A. Hall. 2002. Rho GTPases in cell biology. Nature. 420:629–635. [DOI] [PubMed] [Google Scholar]
- Etienne-Manneville, S., and A. Hall. 2003. Cell polarity: Par6, aPKC and cytoskeletal crosstalk. Curr. Opin. Cell Biol. 15:67–72. [DOI] [PubMed] [Google Scholar]
- Funamoto, S., R. Meili, S. Lee, L. Parry, and R.A. Firtel. 2002. Spatial and temporal regulation of 3-phosphoinositides by PI 3-kinase and PTEN mediates chemotaxis. Cell. 109:611–623. [DOI] [PubMed] [Google Scholar]
- Geiger, B., and A. Bershadsky. 2001. Assembly and mechanosensory function of focal contacts. Curr. Opin. Cell Biol. 13:584–592. [DOI] [PubMed] [Google Scholar]
- Geijsen, N., S. van Delft, J.A. Raaijmakers, J.W. Lammers, J.G. Collard, L. Koenderman, and P.J. Coffer. 1999. Regulation of p21rac activation in human neutrophils. Blood. 94:1121–1130. [PubMed] [Google Scholar]
- Go, Y.M., Y.C. Boo, H. Park, M.C. Maland, R. Patel, K.A. Pritchard, Jr., Y. Fujio, K. Walsh, V. Darley-Usmar, and H. Jo. 2001. Protein kinase B/Akt activates c-Jun NH(2)-terminal kinase by increasing NO production in response to shear stress. J. Appl. Physiol. 91:1574–1581. [DOI] [PubMed] [Google Scholar]
- Hannigan, M., L. Zhan, Z. Li, Y. Ai, D. Wu, and C.K. Huang. 2002. Neutrophils lacking phosphoinositide 3-kinase gamma show loss of directionality during N-formyl-Met-Leu-Phe-induced chemotaxis. Proc. Natl. Acad. Sci. USA. 99:3603–3608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoger, J.H., V.I. Ilyin, S. Forsyth, and A. Hoger. 2002. Shear stress regulates the endothelial Kir2.1 ion channel. Proc. Natl. Acad. Sci. USA. 99:7780–7785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houston, P., M.C. Dickson, V. Ludbrook, B. White, J.L. Schwachtgen, J.H. McVey, N. Mackman, J.M. Reese, D.G. Gorman, C. Campbell, and M. Braddock. 1999. Fluid shear stress induction of the tissue factor promoter in vitro and in vivo is mediated by Egr-1. Arterioscler. Thromb. Vasc. Biol. 19:281–289. [DOI] [PubMed] [Google Scholar]
- Hsu, P.P., S. Li, Y.S. Li, S. Usami, A. Ratcliffe, X. Wang, and S. Chien. 2001. Effects of flow patterns on endothelial cell migration into a zone of mechanical denudation. Biochem. Biophys. Res. Commun. 285:751–759. [DOI] [PubMed] [Google Scholar]
- Kaibuchi, K. 1999. Regulation of cytoskeleton and cell adhesion by Rho targets. Prog. Mol. Subcell. Biol. 22:23–38. [DOI] [PubMed] [Google Scholar]
- Katoh, K., Y. Kano, M. Amano, K. Kaibuchi, and K. Fujiwara. 2001. Stress fiber organization regulated by MLCK and Rho-kinase in cultured human fibroblasts. Am. J. Physiol. Cell Physiol. 280:C1669–C1679. [DOI] [PubMed] [Google Scholar]
- Kimura, K., Y. Fukata, Y. Matsuoka, V. Bennett, Y. Matsuura, K. Okawa, A. Iwamatsu, and K. Kaibuchi. 1998. Regulation of the association of adducin with actin filaments by Rho-associated kinase (Rho-kinase) and myosin phosphatase. J. Biol. Chem. 273:5542–5548. [DOI] [PubMed] [Google Scholar]
- Kiosses, W.B., R.H. Daniels, C. Otey, G.M. Bokoch, and M.A. Schwartz. 1999. A role for p21-activated kinase in endothelial cell migration. J. Cell Biol. 147:831–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraynov, V.S., C. Chamberlain, G.M. Bokoch, M.A. Schwartz, S. Slabaugh, and K.M. Hahn. 2000. Localized Rac activation dynamics visualized in living cells. Science. 290:333–337. [DOI] [PubMed] [Google Scholar]
- Li, S., B.P. Chen, N. Azuma, Y.L. Hu, S.Z. Wu, B.E. Sumpio, J.Y. Shyy, and S. Chien. 1999. Distinct roles for the small GTPases Cdc42 and Rho in endothelial responses to shear stress. J. Clin. Invest. 103:1141–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, S., P. Butler, Y. Wang, Y. Hu, D. Cho Han, S. Usami, J. Guan, and S. Chien. 2002. The role of the dynamics of focal adhesion kinase in the mechanotaxis of endothelial cells. Proc. Natl. Acad. Sci. USA. 99:3546–3551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, F., A.D. Verin, P. Wang, R. Day, R.P. Wersto, F.J. Chrest, D.K. English, and J.G.N. Garcia. 2001. Differential regulation of sphingosine-1-phosphate and VEGF-induced endothelial cell chemotaxis. Am. J. Respir. Cell Mol. Biol. 24:711–719. [DOI] [PubMed] [Google Scholar]
- Maekawa, M., T. Ishizaki, S. Boku, N. Watanabe, A. Fujita, A. Iwamatsu, T. Obinata, K. Ohashi, K. Mizuno, and S. Narumiya. 1999. Signaling from Rho to the actin cytoskeleton through protein kinases ROCK and LIM-kinase. Science. 285:895–898. [DOI] [PubMed] [Google Scholar]
- Morawietz, H., R. Talanow, M. Szibor, U. Rueckschloss, A. Schubert, B. Bartling, D. Darmer, and J. Holtz. 2000. Regulation of the endothelin system by shear stress in human endothelial cells. J. Physiol. 525:761–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakashio, A., N. Fujita, and T. Tsuruo. 2002. Topotecan inhibits VEGF- and bFGF-induced vascular endothelial cell migration via downregulation of the PI3K-Akt signaling pathway. Int. J. Cancer. 98:36–41. [DOI] [PubMed] [Google Scholar]
- Nobes, C.D., and A. Hall. 1999. Rho GTPases control polarity, protrusion, and adhesion during cell movement. J. Cell Biol. 144:1235–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osawa, M., M. Masuda, K. Kusano, and K. Fujiwara. 2002. Evidence for a role of platelet endothelial cell adhesion molecule-1 in endothelial cell mechanosignal transduction: is it a mechanoresponsive molecule? J. Cell Biol. 158:773–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren, X.D., and M.A. Schwartz. 2000. Determination of GTP loading on Rho. Methods Enzymol. 325:264–272. [DOI] [PubMed] [Google Scholar]
- Ridley, A.J. 2001. a. Rho GTPases and cell migration. J. Cell Sci. 114:2713–2722. [DOI] [PubMed] [Google Scholar]
- Ridley, A.J. 2001. b. Rho proteins, PI 3-kinases, and monocyte/macrophage motility. FEBS Lett. 498:168–171. [DOI] [PubMed] [Google Scholar]
- Ridley, A.J. 2001. c. Rho family proteins: coordinating cell responses. Trends Cell Biol. 11:471–477. [DOI] [PubMed] [Google Scholar]
- Ryu, Y., N. Takuwa, N. Sugimoto, S. Sakurada, S. Usui, H. Okamoto, O. Matsui, and Y. Takuwa. 2002. Sphingosine-1-phosphate, a platelet-derived lysophospholipid mediator, negatively regulates cellular Rac activity and cell migration in vascular smooth muscle cells. Circ. Res. 90:325–332. [DOI] [PubMed] [Google Scholar]
- Sahai, E., M.F. Olson, and C.J. Marshall. 2001. Cross-talk between Ras and Rho signaling pathways in transformation favours proliferation and increased motility. EMBO J. 20:755–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenwaelder, S.M., S.C. Hughan, K. Boniface, S. Fernando, M. Holdsworth, P.E. Thompson, H.H. Salem, and S.P. Jackson. 2002. RhoA sustains integrin alpha IIbbeta 3 adhesion contacts under high shear. J. Biol. Chem. 277:14738–14746. [DOI] [PubMed] [Google Scholar]
- Sells, M.A., J.T. Boyd, and J. Chernoff. 1999. p21-activated kinase 1 (Pak1) regulates cell motility in mammalian fibroblasts. J. Cell Biol. 145:837–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shay-Salit, A., M. Shushy, E. Wolfovitz, H. Yahav, F. Breviario, E. Dejana. and N. Resnick. 2002. VEGF receptor 2 and the adherens junction as a mechanical transducer in vascular endothelial cells. Proc. Natl. Acad. Sci. USA. 99:9462–9467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soga, N., J.O. Connolly, M. Chellaiah, J. Kawamura, and K.A. Hruska. 2001. Rac regulates vascular endothelial growth factor stimulated motility. Cell Commun Adhes. 8:1–13. [DOI] [PubMed] [Google Scholar]
- Sotsios, Y., and S.G. Ward. 2000. Phosphoinositide 3-kinase: a key biochemical signal for cell migration in response to chemokines. Immunol. Rev. 177:217–235. [DOI] [PubMed] [Google Scholar]
- Swetman, C.A., Y. Leverrier, R. Garg, C.H. Gan, A.J. Ridley, D.R. Katz, and B.M. Chain. 2002. Extension, retraction and contraction in the formation of a dendritic cell dendrite: distinct roles for Rho GTPases. Eur. J. Immunol. 32:2074–2083. [DOI] [PubMed] [Google Scholar]
- Tominaga, T., T. Ishizaki, S. Narumiya, and D.L. Barber. 1998. p160ROCK mediates RhoA activation of Na-H exchange. EMBO J. 17:4712–4722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Totsukawa, G., Y. Yamakita, S. Yamashiro, D.J. Hartshorne, Y. Sasaki, and F. Matsumura. 2000. Distinct roles of ROCK (Rho kinase) and MLCK in spatial regulation of MLC phosphorylation for assembly of stress fibres and focal adhesions in 3T3 fibroblasts. J. Cell Biol. 150:797–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuji, T., T. Ishizaki, M. Okamoto, C. Higashida, K. Kimura, T. Furuyashiki, Y. Arakawa, R.B. Birge, T. Nakamoto, H. Hirai, and S. Narumiya. 2002. ROCK and mDia1 antagonize in Rho-dependent Rac activation in Swiss 3T3 fibroblasts. J. Cell Biol. 157:819–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzima, E., M.A. del Pozo, S.J. Shattil, S. Chien, and M.A. Schwartz. 2001. Activation of integrins in endothelial cells by fluid shear stress mediates Rho-dependent cytoskeletal alignment. EMBO J. 20:4639–4647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzima, E., M.A. del Pozo, W.B. Kiosses, S.A Mohamed, S. Li, S. Chien, and M.A. Schwartz. 2002. Activation of Rac1 by shear stress in endothelial cells mediates both cytoskeletal reorganization and effects on gene expression. EMBO J. 21:6791-800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbich, C., E. Dernbach, A. Reissner, M. Vasa, A.M. Zeiher, and S. Dimmeler. 2002. Shear stress–induced endothelial cell migration involves integrin signaling via the fibronectin receptor subunits alpha(5) and beta(1). Arterioscler. Thromb. Vasc. Biol. 22:69–75. [DOI] [PubMed] [Google Scholar]
- van Nieuw Amerongen, G.P., and V.W. van Hinsbergh. 2001. Cytoskeletal effects of rho-like small guanine nucleotide-binding proteins in the vascular system. Arterioscler. Thromb. Vasc. Biol. 21:300–311. [DOI] [PubMed] [Google Scholar]
- Wang, F., P. Herzmark, O.D. Weiner, S. Srinivasan, G. Servant, and H.R. Bourne. 2002. Lipid products of PI(3)Ks maintain persistent cell polarity and directed motility in neutrophils. Nat. Cell Biol. 4:513–518. [DOI] [PubMed] [Google Scholar]
- Weiner, O.D. 2002. Rac activation: P-Rex1—a convergence point for PIP(3) and Gbetagamma? Curr. Biol. 12:R429–R431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner, O.D., P.O. Neilsen, G.D. Prestwich, M.W. Kirschner, L.C. Cantley, and H.R. Bourne. 2002. A PtdInsP(3)- and Rho GTPase-mediated positive feedback loop regulates neutrophil polarity. Nat. Cell Biol. 4:509–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojciak-Stothard, B., A. Entwistle, R. Garg, and A.J. Ridley. 1998. Regulation of TNF-alpha-induced reorganization of the actin cytoskeleton and cell-cell junctions by Rho, Rac, and Cdc42 in human endothelial cells. J. Cell. Physiol. 176:150–165. [DOI] [PubMed] [Google Scholar]
- Wojciak-Stothard, B., S. Potempa, T. Eichholtz, and A.J. Ridley. 2001. Rho and Rac but not Cdc42 regulate endothelial cell permeability. J. Cell Sci. 114:1343–1355. [DOI] [PubMed] [Google Scholar]
- Worthylake, R.A., S. Lemoine, J.M. Watson, and K. Burridge. 2001. RhoA is required for monocyte tail retraction during transendothelial migration. J. Cell Biol. 154:147–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng, H., D. Zhao, and D. Mukhopadhyay. 2002. KDR stimulates endothelial cell migration through heterotrimeric G protein Gq/11-mediated activation of a small GTPase RhoA. J. Biol. Chem. 277:46791–46798. [DOI] [PubMed] [Google Scholar]
- Zicha, D., G. Dunn, and G. Jones. 1997. Analyzing chemotaxis using the Dunn direct-viewing chamber. Methods Mol. Biol. 75:449–457. [DOI] [PubMed] [Google Scholar]