

Abstract
Glucokinase (GK) activity plays a key role in glucose-stimulated insulin secretion from pancreatic β cells. Insulin regulates GK activity by modulating its association with secretory granules, although little is known about the mechanisms involved in regulating this association. Using quantitative imaging of multicolor fluorescent proteins fused to GK, we found that the dynamic association of GK with secretory granules is modulated through nitric oxide (NO). Our results in cultured β cells show that insulin stimulates NO production and leads to S-nitrosylation of GK. Furthermore, inhibition of NO synthase (NOS) activity blocks insulin-stimulated changes in both GK association with secretory granules and GK conformation. Mutation of cysteine 371 to serine blocks S-nitrosylation of GK and causes GK to remain tightly bound to secretory granules. GK was also found to interact stably with neuronal NOS as detected by coimmunoprecipitation and fluorescence resonance energy transfer. Finally, attachment of a nuclear localization signal sequence to NOS drives GK to the nucleus in addition to its normal cytoplasmic and granule targeting. Together, these data suggest that the regulation of GK localization and activity in pancreatic β cells is directly related to NO production and that the association of GK with secretory granules occurs through its interaction with NOS.
Keywords: insulin; glucokinase; nitric oxide; nitric oxide synthase; fluorescence resonance energy transfer
Introduction
Glucokinase (GK)* activity is a major determinant of β cell glucose metabolism and insulin secretion (Sweet et al., 1996; Matschinsky et al., 1998; Matschinsky, 2002). Thus, a complete model of its regulation is central to our understanding of glucose homeostasis and the pathogenesis of diabetic disease states. To maintain physiological glucose responsiveness, GK activity needs to be constrained within a very narrow range (Wang and Iynedjian, 1997; Matschinsky et al., 1998). Regulation of GK expression in β cells has been widely studied and is induced mainly by glucose, although it can be modulated by other factors, including insulin (Leibiger et al., 2001; Matschinsky, 2002). Posttranslational regulation of GK has only more recently been described, and the mechanisms involved in this mode of regulation are not well known. Recent studies have shown that low levels of glucose cause an association of GK with secretory granules (Toyoda et al., 1999; Stubbs et al., 2000; Rizzo et al., 2002) and that this association correlates with a decrease in GK activity (Rizzo et al., 2002). Prolonged exposure to high glucose (>20 min) causes dissociation of GK from the granule along with conformational changes associated with activation. Glucose-stimulated GK regulation is blocked by inhibitors of insulin secretion, and insulin by itself can rapidly (<2 min) induce similar changes to GK localization and activity (Rizzo et al., 2002). This suggests that minute-to-minute regulation of GK activity and localization occurs through receptor-mediated signaling and not by interaction between glucose and GK.
The molecular mechanism of GK association with secretory granules and the processes that modulate this association are unknown. To address the mechanism of GK association with secretory granules, we examined the role of nitric oxide synthase in this regulation. Neuronal nitric oxide synthase (nNOS) is activated by a rise in intracellular calcium, which is a known response of β cells to glucose or insulin stimulation (Aspinwall et al., 2000); and nNOS is also known to be localized on insulin secretory granules (Lajoix et al., 2001). Nitric oxide (NO) has also been shown to have a stimulatory affect on glucose-stimulated insulin secretion from both cultured β cell lines and pancreatic islets (Smukler et al., 2002; Kaneko et al., 2003). However, these findings are highly controversial, and an inhibitory effect on insulin secretion has also been shown using a variety of experimental approaches. (Salehi et al., 1996; Lajoix et al., 2001; Henningsson et al., 2002). This controversy points to the need for more careful consideration of potential targets for NO in the regulation of glucose-stimulated insulin secretion in order to understand the role of NO synthase (NOS) in β cell function. GK is one potential target for regulation by NO, since GK contains several cysteines that have been shown to be critical for maintaining catalytic activity (Tiedge et al., 2000).
Results and discussion
To assess the role of NOS in the regulation of GK by insulin, we examined whether insulin could stimulate NO production in β cells. βTC3 cells were loaded with 4-amino-5-methylamino-2′,7′-difluorofluorescein diacetate (DAF-FM) (Kojima et al., 1999), an indicator dye whose fluorescence increases upon reaction with NO. A rapid increase in the fluorescence of DAF-FM was observed within minutes in insulin-treated cells compared with untreated cells (Fig. 1 A). This indicates that insulin treatment results in NO production on a time scale consistent with regulation of GK (Rizzo et al., 2002). We also examined the role of NOS activation in the regulation of GK association with secretory granules. Association of GK with secretory granules was analyzed in cells expressing a YFP-labeled GK (GK-YFP) and a CFP targeted to insulin granules by insertion into a proinsulin cDNA (Rizzo et al., 2002; Watkins et al., 2002) (Fig. 1 B). Association of GK-YFP with CFP-labeled granules can be assayed by selectively photobleaching the GK-YFP in a small region of the cell (Fig. 1 B, white box) and monitoring the FRAP to the CFP-labeled granules. Recovery of GK-YFP to CFP-labeled granules occurs at a faster rate in insulin-treated cells (Fig. 1 C), which indicates a net translocation of GK to the cytoplasm (Rizzo et al., 2002). A significant difference in the degree of fluorescence recovery between insulin-treated (5 min, 100 nM) and untreated cells could be reliably measured 2 s into recovery (Fig. 1 D). Inhibition of NOS activity using NG-nitro-l-arginine-methyl ester (L-NAME) blocked the stimulatory effects of insulin on GK-YFP FRAP, indicating a requirement for NO production. Furthermore, the effects of NOS inhibition were reversed by treatment with SNAP, an NO releasing agent. These results are consistent with a requirement for NO production in modulating GK association with secretory granules.
Figure 1.
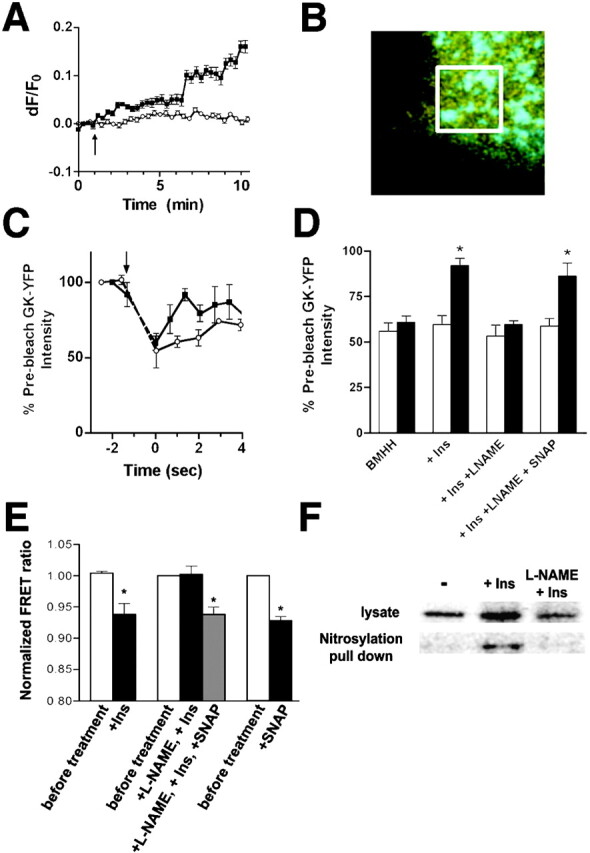
Regulation of GK by insulin requires NO. (A) βTC3 cells were starved for 4 h before loading with DAF-FM and analysis using confocal microscopy. The change in DAF intensity is represented as the change in fluorescence/initial fluorescence (dF/F0) from the average of at least 10 cells treated with 100 nM insulin (indicated by the arrow, ▪) or left untreated (○). (B) FRAP measurements were taken in cells expressing GK-YFP (yellow) and proinsulin-CFP (cyan) by selectively photobleaching YFP fluorescence in a small region (white box) of the cell containing several granules. (C) Fluorescence recovery of GK-YFP to CFP-labeled insulin granules was measured after photobleaching GK-YFP in starved cells (○) and after insulin treatment (100 nM, 5 min, ▪). The bleaching period is indicated by the arrow and broken lines. (D) FRAP measurements show fluorescence intensity of granule-associated GK-YFP immediately after photobleaching (white bars) and 2 s after photobleaching (black bars) and expressed as the percentage of prebleached fluorescence intensity. Cells were starved previously in BMHH for 3 h before insulin treatment (5 min), pretreatment with L-NAME (5 mM, 10 min) before insulin treatment (100 nM, 5 min), and after treatment with SNAP (100 μM, 1 min). Statistical significance from initial postbleach intensity (P < 0.05, t test) is denoted by an asterisk. (E) Cells expressing CFP-GK-YFP were examined for FRET by fluorescence microscopy as indicated. Statistical significance (P < 0.05 by ANOVA or t test as appropriate) is denoted by an asterisk. Cells were treated under the same conditions as in D. (F) Nitrosylated proteins were precipitated with neutravidin-agarose after biotinylation of S-nitrosylated proteins from cell lysates. Cells were treated as above where indicated. GK was detected in unreacted lysates and precipitated fractions by Western blot using an antibody to GK (Jetton and Magnuson, 1992).
The requirement for NO production in GK regulation was also tested using a fluorescence resonance energy transfer (FRET)–based assay to examine changes in GK conformation. The FRET ratio between CFP and YFP inserted on opposing ends of GK decreases in cells treated with insulin (Fig. 1 E) and correlates with increased GK activity (Rizzo et al., 2002). Treatment with L-NAME prevented the decrease in the FRET ratio observed with insulin treatment alone. A decrease in the FRET ratio was observed in cells treated with an NO releasing agent either in the presence of L-NAME and insulin or in the absence of other treatments. These results suggest that NO production is a regulator of GK and mediates the effects of insulin treatment. Glucose-stimulated activation of GK FRAP and FRET was also sensitive to treatment with L-NAME (unpublished data) and is consistent with the effect of glucose-stimulated insulin secretion on autofeedback regulation of GK (Rizzo et al., 2002).
To determine whether regulation of GK by NO is the result of direct posttranslational modification of GK, we examined whether GK was S-nitrosylated in insulin-treated cells. Nitrosylated proteins from cell lysates were chemically modified with biotin (Jaffrey et al., 2001) before isolation using neutravidin-agarose and analysis by Western blot. Nitrosylated GK was detected only in precipitates from insulin-treated cells but not from untreated cells or cells treated with L-NAME in addition to insulin (Fig. 1 F). Together with our fluorescence-based assays, these results support a model in which changes in GK localization and activity are related to S-nitrosylation of GK.
To test the role of S-nitrosylation in regulating GK, we examined whether site-directed mutagenesis of GK could block its nitrosylation and affect its regulation. Since reaction of NO with cysteines can be greatly enhanced by a consensus nitrosylation motif (Stamler et al., 1997), we examined the primary structure of GK for potential nitrosylation sites. Four such consensus sites were found in GK (C220, C364, C371, and C434), and each was subsequently mutated to serine, an amino acid that does not react with NO. The mutant GK constructs were tagged with YFP, and the S-nitrosylation of the mutated proteins was assessed (Fig. 2 A). Of the four mutations generated, only C371S eliminated GK nitrosylation, although a slight decrease in the amount of nitrosylated GK was observed for the C364S mutation. Furthermore, the C371S mutant stopped insulin-stimulated FRAP to CFP-labeled granules (Fig. 2 B) and changes in FRET (Fig. 2 C). Mutation of C364S did not have a significant effect on insulin-stimulated FRAP to CFP-labeled granules (Fig. 2 B) and changes in FRET (Fig. 2 C), suggesting that nitrosylation of C364 is not critical to GK regulation. Thus, nitrosylation of cysteine 371 plays a key role in modulating GK association with secretory granules and conformational changes that correlate with GK activation.
Figure 2.
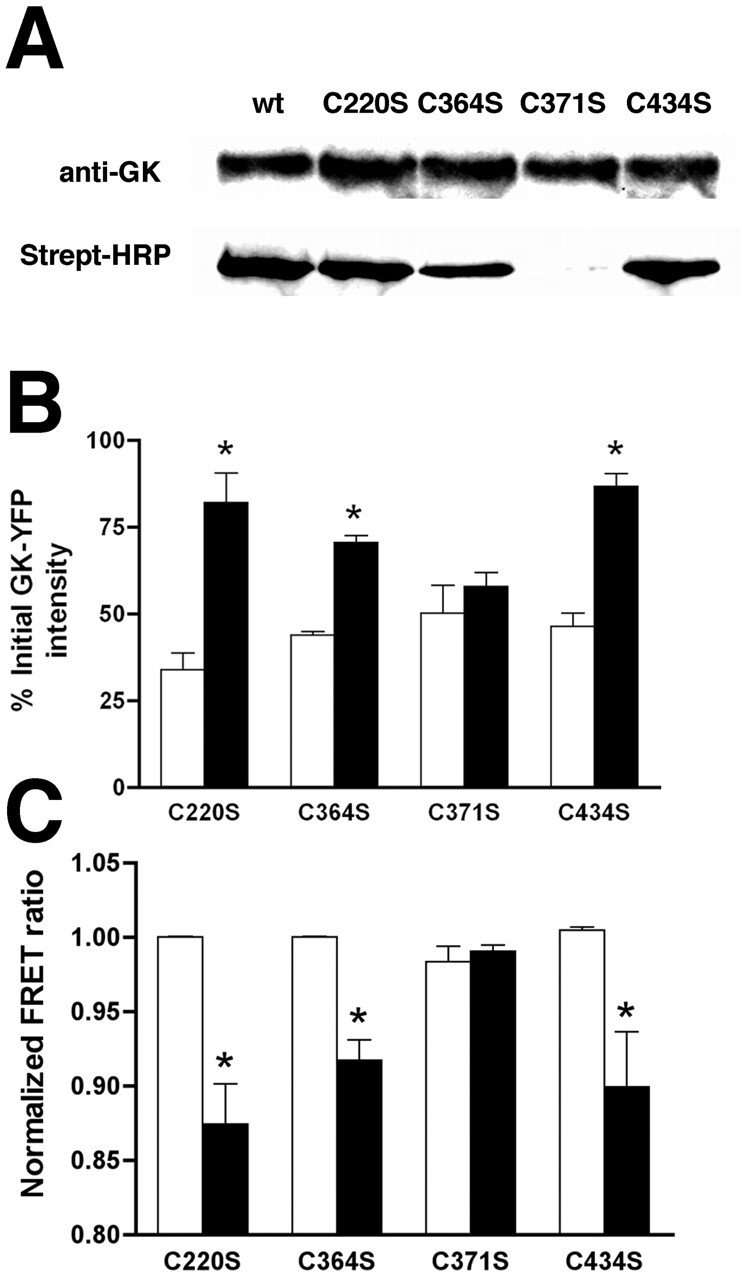
Nitrosylation of cysteine 371 is required for GK regulation. (A) Mutant GK-YFP constructs were expressed in βTC3 cells. Nitrosylated proteins were biotinylated before immunoprecipitation of GK-YFP proteins and analysis by Western blot for GK (anti-GK) or biotinylated proteins using peroxidase-conjugated streptavidin (Strept-HRP). (B) Association of the GK-YFP mutants with secretory granules was measured using FRAP in cells expressing the indicated GK-YFP construct and proinsulin-CFP. Fluorescence intensity of granule-associated GK-YFP is expressed as the percentage of prebleached intensity immediately after bleaching (white bars) and 2 s into recovery (black bars). Statistical significance from initial postbleach intensity (P < 0.05, t test) is denoted by an asterisk. (C) Mutations were made in CFP-GK-YFP and expressed in βTC3 cells. Cells were starved for 4 h in BMHH, and the normalized FRET ratio was calculated before (white bars) and after (black bars) stimulation with 100 nM insulin (5 min). Statistical significance from pretreatment FRET ratio (P < 0.05, t test) is denoted by an asterisk.
To assess the role of C371 in regulating GK activity, the kinetic parameters of recombinantly expressed and purified GK(C371S) were examined and compared with the kinetic parameters of native GK. GK-C371S was found to have an increased Km for glucose (12.5 ± 2.5 mM) and a reduced Vmax (30.6 ± 0.3 U/mg protein) compared with GK (Km = 8.7 ± 1.3 mM; Vmax = 48.1 ± 2.7 U/mg protein). Because of the reducing agents needed to stabilize recombinantly expressed GK (Tippett and Neet, 1983; Tiedge et al., 1997; Matschinsky et al., 1998), the effect of S-nitrosylation on GK activity in vitro could not be determined. However, our analysis of GK(C371S) is consistent with a previous analysis (Tiedge et al., 2000) and shows that modification of C371 alters the enzymatic activity of GK.
Since other proteins that react with NO are known to form complexes containing nNOS (Brenman et al., 1996; Fang et al., 2000; Nedvetsky et al., 2002), we examined the role of nNOS in determining GK localization. To detect interaction between GK and nNOS, we immunoprecipitated endogenous GK from cell lysates and probed for endogenous nNOS by Western blot (Fig. 3 A). We were able to detect nNOS in these precipitates and also from those of expressed GK-YFP and GK(C371S)-YFP. Incubation of GK precipitates with diethylamine nitric oxide (DEANO), a chemical that rapidly releases NO, resulted in elution of nNOS from endogenous GK and GK-YFP precipitates but not from GK(C371S)-YFP precipitates. These data show that GK association with nNOS is both consistent with GK association with secretory granules and also sensitive to the presence of NO.
Figure 3.
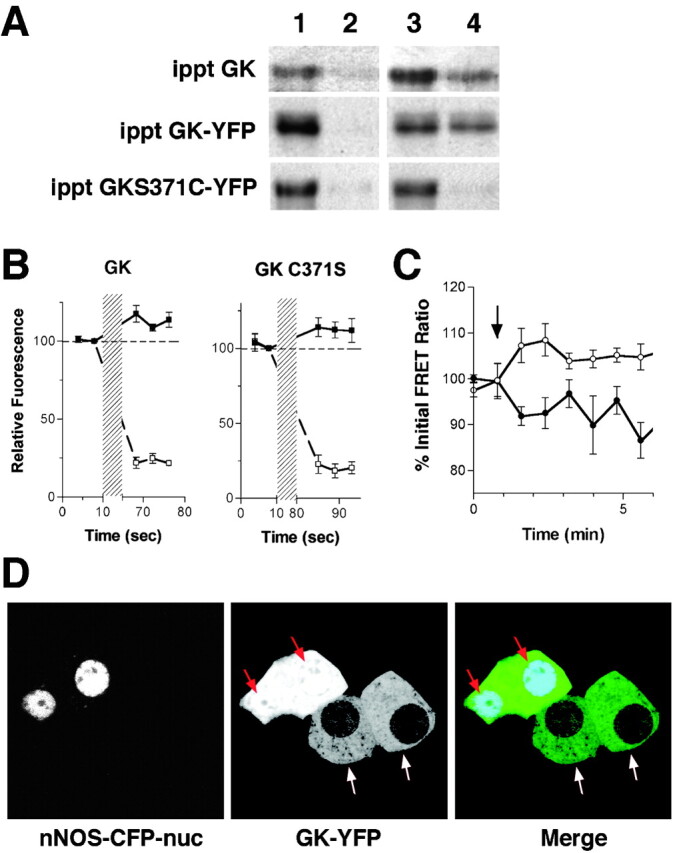
Association of GK with nNOS can direct GK localization. (A) Endogenous GK, GK-YFP, and C371S GK-YFP were immunoprecipitated from cell lysates using anti-GK antibodies or anti-GFP antibodies for YFP-tagged proteins in combination with agarose-conjugated secondary antibodies. Precipitates were then heated to 37°C for 10 min in the presence of 1 mM DEANO in PBS (lanes 3 and 4) or an equivalent volume of vehicle (DMSO) in PBS alone (lanes 1 and 2). Pellets (lanes 1 and 3) and supernatants (lanes 2 and 4) were analyzed by SDS-PAGE and Western blot using anti-nNOS antibodies. (B) Cells expressing nNOS-CFP and GK-YFP or GK(C371S)-YFP were examined by two-photon microscopy before and after photobleaching with a 514 nm Ar laser (indicated by the hatched region). Average relative fluorescence (n = 6) for cellular CFP (▪) and YFP (□) intensities were plotted versus time. A dotted line was drawn as a reference to indicate prebleach intensity. (C) FRET between nNOS-CFP and either GK-YFP (•) or GK(C371S)-YFP(○) was examined in living cells by two-photon microscopy. FRET ratios were normalized to pretreatment values (100%) before averaging (n = 6) and plotted versus time. Addition of insulin (100 nM) is indicated by the arrow. (D) Cells were cotransfected with GK-YFP and nNOS-CFP-nuc and examined by confocal microscopy. In cells expressing both constructs, GK-YFP is found colocalized with nNOS in the nucleus (red arrows). GK-YFP did not localize to the nuclei of cells that were singly transfected with GK-YFP (white arrows). In the merged panel, the CFP image was colored blue and the YFP image was colored green. Colocalization is indicated by cyan (red arrows).
Interaction between nNOS and GK was also examined in living cells by measuring changes in FRET between CFP-labeled nNOS and GK-YFP. FRET between nNOS-CFP and GK-YFP was verified by acceptor photobleaching of either GK-YFP or GK(C371S)-YFP, which caused an increase in nNOS-CFP fluorescence (Fig. 3 B). Insulin treatment resulted in a decrease of the FRET between nNOS-CFP and GK-YFP, whereas FRET between nNOS-CFP and GK(C371S)-YFP increased (Fig. 3 C). The increase in FRET between nNOS-CFP and nitrosylation-resistant GK(C371S)-YFP is most likely due to dissociation of competing endogenous GK and association of unbound GK(C371S)-YFP to the vacated NOS binding sites. These results indicate that GK and nNOS interact on secretory granules either directly or as part of a complex. To determine whether the interaction between GK and nNOS was sufficient to determine GK localization, we targeted nNOS-CFP to the nucleus. A nuclear localization sequence from simian virus 40 large T antigen (Kalderon et al., 1984; Lanford et al., 1986) was attached to the COOH terminus of nNOS-CFP and coexpressed in β cells with GK-YFP. GK-YFP localized to the nuclei of cells only when coexpressed with the nucleus-targeted nNOS-CFP (Fig. 3 D). Since the subcellular localization of GK can be determined by the localization of nNOS, it suggests that nNOS may be the primary target for GK on secretory granules.
Overall, our data indicate that the GK localization and activity in the β cell are determined by GK association with nNOS and that association is disrupted by GK nitrosylation at cysteine 371. Regulation of GK-NOS association by nitrosylation provides a sensitive means for modulating GK activity, thus affecting glucose-stimulated insulin secretion. Future work on the nature of the GK-NOS association will certainly increase our understanding of the signaling pathways that contribute to regulation of glucose-stimulated insulin secretion. The regulation of GK nitrosylation might also prove to be a useful target for pharmacological manipulation in the treatment of diabetes and other glycemic disorders. In addition, our data demonstrate that changes in protein nitrosylation can regulate protein localization, protein–protein interactions, and protein function in a highly specific and rapid way that is similar to the role of protein phosphorylation.
Materials and methods
Cell culture
βTC3 cells were cultured as described (Rizzo et al., 2002) and were transferred to media containing 1g/L glucose overnight before observation. 3 h before experimentation, cells were washed four times and incubated with BMHH buffer (125 mM NaCl, 5.7 mM KCl, 2.5 mM CaCl2, 1.2 mM MgCl2, 10 mM Hepes, pH 7.4) containing 0.1% BSA. For FRAP, CFP-GK-YFP FRET, and nitrosylation experiments, DNA plasmids containing GK-YFP constructs, proinsulin-CFP, or CFP-GK-YFP were introduced by electroporation before seeding as described previously (Rizzo et al., 2002). For studies requiring cotransfection, plasmid DNA encoding the indicated constructs were transfected using either a ratio of (2 μg nNOS-CFP:1 μg GK-YFP:6 μl FUGENE [Roche Applied Science]) or of (1 μg nNOS-CFP-nuc:2 μg GK-YFP:6 μl FUGENE). Transfection was for 4 h in serum-free media, followed by replacement with low-glucose media. Observation took place the following day.
Generation of constructs
Mutations to GK-YFP were made by 4 primer PCR (with Advantage 2 Polymerase Mix; BD Biosciences, CLONTECH Laboratories, Inc.) using the same set of end primers (sense, 5′-GGCACCAAAATCAACGGGAC-3′ and antisense, 5′-CTCGCCCTTGCTCACCAT-3′) along with primers containing the mutation (C220S sense, 5′-GACCGCCAATCTGAGGTCG-3′ and antisense, 5′-CGACCTCAGATTGGCGGTCT-3′; C364S sense, 5′-CACCGACTCCGATATCGTGC-3′ and antisense, 5′-CACGATATCGGAGTCGGTGAC-3′; C371S sense, 5′-CCGTGCCTCTGAAAGCGTG-3′ and antisense, 5′-CACGCTTTCAGAGGCACGGC-3′; C434S sense, 5′-CACCCAACTCCGAAATCACCT-3′ and antisense, 5′-GGTGATTTCGGAGTTGGGTG-3′). Mutated GK was then inserted into CFP-GK-YFP using restriction sites to replace wild-type GK. For bacterial expression, GK and GK(C371S) were excised from GK-YFP plasmids using BglII and XmaI and ligated into the BamHI and XmaI sites in pQE30 (QIAGEN). To create nNOS-CFP constructs, a silent mutation was introduced into the rat cDNA for nNOS in order to remove the AgeI restriction site using the QuickChange Site-Directed Mutagenesis Kit (Stratagene) (sense primer, 5′-GGGATGACAACCGATACCACGAGGACATC-3′ and antisense primer, 5′-GATGTCCTCGTGGTATCGGTTGTCATCCC-3′). The cDNA for the modified rat nNOS was then amplified by PCR (sense, 5′-AGCTAGCCACCATGGAAGAGAACACG-3′ and antisense, 5′-TTAACCGGTGAGCTGAAAACCTCATCTGC-3′) and inserted into the NheI and AgeI restriction sites in the pECFP-nuc vector (BD Biosciences, CLONTECH Laboratories, Inc.), and a derivative vector with the nuclear localization signal removed by BamHI and BglII digestion followed by religation. All primers were custom synthesized and purified by Integrated DNA Technologies. All plasmid constructs were purified using DNA preparation kits (QIAGEN), and the sequences of all constructs were verified by sequencing reactions performed by the Vanderbilt-Ingram Cancer Center DNA Sequencing Shared Resource.
Microscopy
βTC3 cells were labeled with 5 μM DAF-FM diacetate (Molecular Probes) for 25 min at 25°C in BSA-free BMHH. Observation was with 488 nm excitation and 505–530 nm collection by laser scanning confocal microscopy (LSM510; Carl Zeiss MicroImaging, Inc.). During observation, cells were heated to 32°C using a Bioptechs Delta T system. FRAP and intramolecular FRET measurements were performed on cultured βTC3 cells as described previously (Rizzo et al., 2002). Statistical significance (P < 0.05 by ANOVA or t test as appropriate) is denoted by an asterisk and was determined from at least three granules in the same region or at least three cells for the FRET studies as compared with pretreatment FRET ratios. Data shown is representative of at least three independent trials. Observation of nNOS-CFP-nuc and GK-YFP were performed using the same filter settings as the FRAP measurements. FRET between nNOS-CFP and GK-YFP was observed using 864 nm two-photon excitation along with a KP700/514 main beam splitter and standard CFP and YFP collection using a 40×, 1.3NA F-FLUAR oil immersion objective lens. For the FRET measurements, the temperature was maintained at 37°C using an S-M incubator (Carl Zeiss MicroImaging, Inc.) controlled by the CTI temperature regulator along with humidification and an objective heater.
Detection of nitrosylated proteins
S-nitrosylated proteins were biotinylated and isolated from cell lysates using the protocol described (Jaffrey et al., 2001). Briefly, cells were lysed in HEN buffer (250 mM Hepes, pH 7.7, 1 mM EDTA, 0.1 mM neocuproine) using 0.5% Triton X-100 and 0.5% cholate before protection of free cysteines with methyl methanethiosulfonate (Pierce Chemical Co.) and derivatization of S-nitrosylated cysteines with ascorbic acid and biotin-HPDP (Pierce Chemical Co.). Biotinylated proteins were isolated by binding to neutravidin-agarose (Pierce Chemical Co.) followed by five washes with a high salt buffer (20 mM Hepes, pH 7.7, 600 mM NaCl, 1 mM EDTA, 0.5% Triton X-100) and elution in HNE buffer (20 mM Hepes, pH 7.7, 100 mM NaCl, 1 mM EDTA) plus 100 mM 2-mercaptoethanol. Alternatively, GK-YFPs were isolated by immunoprecipitation with A.v. Peptide Antibody (BD BioSciences, Clontech Laboratories, Inc.) preconjugated to IgG agarose (Sigma-Aldrich). Western blots were then performed on eluted proteins to detect GK and nitrosylated proteins.
Kinetic analysis of GK mutations
His-tagged recombinant GK and GK(C371S) were produced in M15 (pREP4) cells and purified by Ni:NTA affinity chromatography (QIAGEN) precisely as described (Tiedge et al., 1997). Protein concentrations were determined using the Advanced Protein Assay (Cytoskeleton Inc.) at A590 according to the manufacturer's protocol. Kinetic analysis was performed on 2 mU of purified protein at 37°C using a photometric assay containing 50 mM Hepes (pH 7.2), 100 mM KCl, 10 mM MgCl2, 2.5 mM dithiothreitol, 5 mM ATP, 2 mM NAD+, 4 U/ml glucose-6-phosphate dehydrogenase from Leuconostoc mesenteroides, and varying concentrations of glucose. 1 U was defined as the amount of protein required to produce 1 μmole NADH per minute. NADH production was assayed from stopped reactions (1 ml cold 500 mM NaHCO3 per 100 μl reaction mix) as described previously (Rizzo et al., 2002), and reaction velocities were calculated from reaction time courses over 2 min as a function of glucose concentration. Km and Vmax were calculated from linear regression analysis of Hanes-Woolf plots using Prism software (Graphpad).
Immunoprecipitation
BMHH-starved cells from one 60-mm dish were collected in cold PBS, pH 7.4 (PBS, prepared from 10× solution [GIBCO BRL]), and resuspended in lysis buffer (HNE buffer with 1% cholate, 1% Triton X-100, 1× Protease Inhibitor Cocktail for use with Mammalian Cells and Tissue Extracts [Sigma-Aldrich]) for 20 min. Following removal of insoluble material by centrifugation (5 min, 3000 g), normalized amounts of protein (determined by Advanced Protein Assay; A590) were incubated for 1 h with primary antibodies for GK (Jetton and Magnuson, 1992) or GFP preconjugated to 25 μl agarose IgG in 750 μl lysis buffer. Precipitates were washed twice in lysis buffer and three times in HNE buffer before analysis by Western blot or treatment with DEANO (Molecular Probes).
Figure preparation
Figures were prepared using Adobe Photoshop® 7.0 software.
Acknowledgments
We would like to thank A. Snowman and S.H. Snyder (Johns Hopkins University, Baltimore, MD) for the nNOS cDNA. In addition we would like to thank M.A. Magnuson (Vanderbilt University, Nashville, TN) and P. Drain (University of Pittsburgh, Pittsburgh, PA) for helpful discussions about this work. We also thank M.A Magnuson and J.V. Rocheleau (Vanderbilt University) for critical reading of this manuscript.
Funding for this work was provided by National Institutes of Health grants DK60275 (M.A. Rizzo), and DK53434 and CA86283 (both to D.W. Piston), and by National Science Foundation grant BBI-9871063 (to D.W. Piston).
Footnotes
Abbreviations used in this paper: DAF-FM, 4-amino-5-methylamino-2′,7′-difluorofluorescein diacetate; DEANO, diethylamine nitric oxide; FRET, fluorescence resonance energy transfer; GK, glucokinase; L-NAME, NG-nitro-l-arginine-methyl ester; NO, nitric oxide; NOS, NO synthase; nNOS, neuronal NOS.
References
- Aspinwall, C.A., W.J. Qian, M.G. Roper, R.N. Kulkarni, C.R. Kahn, and R.T. Kennedy. 2000. Roles of insulin receptor substrate-1, phosphatidylinositol 3-kinase, and release of intracellular Ca2+ stores in insulin-stimulated insulin secretion in β-cells. J. Biol. Chem. 275:22331–22338. [DOI] [PubMed] [Google Scholar]
- Brenman, J.E., D.S. Chao, S.H. Gee, A.W. McGee, S.E. Craven, D.R. Santillano, Z. Wu, F. Huang, H. Xia, M.F. Peters, et al. 1996. Interaction of nitric oxide synthase with the postsynaptic density protein PSD-95 and alpha1-syntrophin mediated by PDZ domains. Cell. 84:757–767. [DOI] [PubMed] [Google Scholar]
- Fang, M., S.R. Jaffrey, A. Sawa, K. Ye, X. Luo, and S.H. Snyder. 2000. Dexras1: a G protein specifically coupled to neuronal nitric oxide synthase via CAPON. Neuron. 28:183–193. [DOI] [PubMed] [Google Scholar]
- Henningsson, R., A. Salehi, and I. Lundquist. 2002. Role of nitric oxide synthase isoforms in glucose-stimulated insulin release. Am. J. Physiol. Cell Physiol. 283:C296–C304. [DOI] [PubMed] [Google Scholar]
- Jaffrey, S.R., H. Erdjument-Bromage, C.D. Ferris, P. Tempst, and S.H. Snyder. 2001. Protein S-nitrosylation: a physiological signal for neuronal nitric oxide. Nat. Cell Biol. 3:193–197. [DOI] [PubMed] [Google Scholar]
- Jetton, T.L., and M.A. Magnuson. 1992. Heterogeneous expression of glucokinase among pancreatic beta cells. Proc. Natl. Acad. Sci. USA. 89:2619–2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalderon, D., B.L. Roberts, W.D. Richardson, and A.E. Smith. 1984. A short amino acid sequence able to specify nuclear location. Cell. 39:499–509. [DOI] [PubMed] [Google Scholar]
- Kaneko Y., T. Ishikawa, S. Amano, and K. Nakayama. 2003. Dual effect of nitric oxide on cytosolic Ca2+ concentration and insulin secretion in rat pancreatic β-cells. Am. J. Physiol. Cell. Physiol. 284:C1215–C1222. [DOI] [PubMed] [Google Scholar]
- Kojima, H., Y. Urano, K. Kikuchi, T. Higuchi, Y. Hirata, and T. Nagano. 1999. Fluorescent indicators for imaging nitric oxide production. Angew. Chem. Int. Ed. Engl. 38:3209–3212. [DOI] [PubMed] [Google Scholar]
- Lajoix, A.D., H. Reggio, T. Chardes, S. Peraldi-Roux, F. Tribillac, M. Roye, S. Dietz, C. Broca, M. Manteghetti, G. Ribes, et al. 2001. A neuronal isoform of nitric oxide synthase expressed in pancreatic beta-cells controls insulin secretion. Diabetes. 50:1311–1323. [DOI] [PubMed] [Google Scholar]
- Lanford, R.E., P. Kanda, and R.C. Kennedy. 1986. Induction of nuclear transport with a synthetic peptide homologous to the SV40 T antigen transport signal. Cell. 46:575–582. [DOI] [PubMed] [Google Scholar]
- Leibiger, B., I.B. Leibiger, T. Moede, S. Kemper, R.N. Kulkarni, C.R. Kahn, L.M. de Vargas, and P.P. Berggren. 2001. Selective insulin signaling through A and B insulin receptors regulates transcription of insulin and glucokinase genes in pancreatic β-cells. Mol. Cell. 7:559–570. [DOI] [PubMed] [Google Scholar]
- Matschinsky, F.M. 2002. Regulation of pancreatic beta-cell glucokinase: from basics to therapeutics. Diabetes. 51:S394–S404. [DOI] [PubMed] [Google Scholar]
- Matschinsky, F.M., B. Glaser, and M.A. Magnuson. 1998. Pancreatic beta-cell glucokinase: closing the gap between theoretical concepts and experimental realities. Diabetes. 47:307–315. [DOI] [PubMed] [Google Scholar]
- Nedvetsky, P.I., W.C. Sessa, and H.H. Schmidt. 2002. There's NO binding like NOS binding: protein-protein interactions in NO/cGMP signaling. Proc. Natl. Acad. Sci. USA. 99:16510–16512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzo, M.A., M.A. Magnuson, P.F. Drain, and D.W. Piston. 2002. A functional link between glucokinase binding to insulin granules and conformational alterations in response to glucose and insulin. J. Biol. Chem. 277:34168–34175. [DOI] [PubMed] [Google Scholar]
- Salehi, A., M. Carlberg, R. Henningson, and I. Lundquist. 1996. Islet constitutive nitric oxide synthase: biochemical determination and regulatory function. Am. J. Physiol. 270:C1634–C1641. [DOI] [PubMed] [Google Scholar]
- Smukler, S.R., L. Tang, M.B. Wheeler, and A.M. Salapatek. 2002. Exogenous nitric oxide and endogenous glucose-stimulated beta-cell nitric oxide augment insulin release. Diabetes. 51:3450–3460. [DOI] [PubMed] [Google Scholar]
- Stamler, J.S., E.J. Toone, S.A. Lipton, and N.J. Sucher. 1997. (S)NO signals: translocation, regulation, and a consensus motif. Neuron. 18:691–696. [DOI] [PubMed] [Google Scholar]
- Stubbs, M., S. Aiston, and L. Agius. 2000. Subcellular localization, mobility, and kinetic activity of glucokinase in glucose-responsive insulin-secreting cells. Diabetes. 49:2048–2055. [DOI] [PubMed] [Google Scholar]
- Sweet, I.R., G. Li, H. Najafi, D. Berner, and F.M. Matschinsky. 1996. Effect of a glucokinase inhibitor on energy production and insulin release in pancreatic islets. Am. J. Physiol. 271:E606–E625. [DOI] [PubMed] [Google Scholar]
- Tiedge, M., U. Krug, and S. Lenzen. 1997. Modulation of human glucokinase intrinsic activity by SH reagents mirrors post-translational regulation of the enzyme. Biochim. Biophys. Acta. 1337:175–190. [DOI] [PubMed] [Google Scholar]
- Tiedge, T. Richter, and S. Lenzen. 2000. Importance of cysteine residues for the stability and catalytic activity of human pancreatic beta cell glucokinase. Arch. Biochem. Biophys. 375:251–260. [DOI] [PubMed] [Google Scholar]
- Tippett, P.S., and K.E. Neet. 1983. Interconversions between different sulfhydryl-related kinetic states in glucokinase. Arch. Biochem. Biophys. 222:285–298. [DOI] [PubMed] [Google Scholar]
- Toyoda, Y., S. Yoshie, H. Shironoguchi, and I. Miwa. 1999. Glucokinase is concentrated in insulin-secretory granules of pancreatic B-cells. Histochem. Cell Biol. 112:35–40. [DOI] [PubMed] [Google Scholar]
- Wang, H., and P.B. Iynedjian. 1997. Modulation of glucose responsiveness of insulinoma beta-cells by graded overexpression of glucokinase. Proc. Natl. Acad. Sci. USA. 94:4372–4377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins, S., X. Geng, L. Li, G. Papworth, P.D. Robbins, and P. Drain. 2002. Imaging secretory vesicles by fluorescent protein insertion in propeptide rather than mature secreted peptide. Traffic. 3:461–471. [DOI] [PubMed] [Google Scholar]