

Abstract
In NIH3T3 cells, 0.001 nM of the synthetic androgen R1881 induces and stimulates association of androgen receptor (AR) with Src and phosphatidylinositol 3-kinase (Pl3-kinase), respectively, thereby triggering S-phase entry. 10 nM R1881 stimulates Rac activity and membrane ruffling in the absence of the receptor–Src–PI3-kinase complex assembly. The antiandrogen Casodex and specific inhibitors of Src and PI3-kinase prevent both hormonal effects, DNA synthesis and cytoskeletal changes. Neither low nor high R1881 concentration allows receptor nuclear translocation and receptor-dependent transcriptional activity in fibroblasts, although they harbor the classical murine AR. The very low amount of AR in NIH3T3 cells (7% of that present in LNCaP cells) activates the signaling pathways, but apparently is not sufficient to stimulate gene transcription. This view is supported by the appearance of receptor nuclear translocation as well as receptor-mediated transcriptional activity after overexpression of AR in fibroblasts. In addition, AR-negative Cos cells transiently transfected with a very low amount of hAR cDNA respond to low and high R1881 concentrations with signaling activation. Interestingly, they do not show significant transcriptional activation under the same experimental conditions. Fibroblasts are the first example of cells that respond to steroid hormones with activation of signaling pathways in the absence of endogenous receptor transcriptional activity. The data reported also show that hormone concentration can be crucial in determining the type of cell responsiveness.
Keywords: androgens; nontranscriptional effects; S-phase; membrane ruffling
Introduction
Mesenchyme plays a crucial role in neoplasia. Nontumorigenic NIH3T3 mouse embryo fibroblasts induce human prostatic PC-3 tumor growth (Camps et al., 1990). Recombination of prostatic epithelial cells and stromal components shows that tumor stroma induces tumor phenotype in epithelial cells (Olumi et al., 1999; Hayward et al., 2001). These findings support the view that the stroma is an integral part of tumors, and can exert a dominant force over the malignant phenotype under certain conditions (Matrisian et al., 2001). In addition, androgens initially act on mesenchymal cells during prostate development (Cunha et al., 1997); therefore, mesenchymal components are increasingly the focus of cancer progression studies.
The synthetic androgen R1881 immediately triggers a direct association of androgen receptor (AR)* and estrogen receptor with Src in LNCaP cells, which are derived from human prostate cancer. As a result, estradiol induces association of the same ternary complex (Migliaccio et al., 2000). This complex is required for powerful activation of the Src–Ras–extracellular signal–regulated kinase (ERK) pathway by either androgens or estradiol and subsequent DNA synthesis (Migliaccio et al., 2000). Here, we show that a very low concentration of R1881 stimulates the S-phase entry of NIH3T3 cells, whereas a high hormone concentration, which maximally stimulates DNA synthesis of LNCaP cells (Migliaccio et al., 2000), has a negligible effect.
Cell migration is an essential process during development and wound healing. Furthermore, the signaling pathway aberrations involved in the regulation of cell migration, cell–cell and cell–matrix interactions contribute to tumor invasion and metastasis. The Rho-like GTPases, including Cdc42, Rac1, and RhoA, are key regulators of signal transducing pathways that mediate the distinct cytoskeleton changes required for cell migration (Hall, 1998). We found that, in contrast to DNA synthesis, stimulation of NIH3T3 fibroblasts with high concentrations of R1881 induces rapid membrane ruffling formation. Interestingly, rapid and transient activation of Rac precedes membrane ruffling. In a previous paper, we observed that estradiol stimulation of MCF-7 cells triggers association of its receptor with Src and p85, which is the regulatory subunit of PI3-kinase. These two signaling effectors are both required for the hormone-induced G1-S progression (Castoria et al., 2001). Association of AR with Src and p85 has been analyzed at low and high androgen concentrations along with the role of they play in the NIH3T3 cells responsiveness to R1881.
In the absence of hormones AR is predominantly localized in the cytoplasm of target cells and ligand promotes its nuclear import (Zhou et al., 1994). In fibroblasts, the analysis of AR localization shows that it does not enter nuclei upon agonist stimulation. The very low levels of AR seem to be responsible for AR distribution as well as its inability to activate gene transcription in response to hormone treatment.
Results
Low androgen concentrations induce S-phase entry of quiescent NIH3T3 cells
Fibroblasts were made quiescent using 0.5% charcoal-treated serum and medium lacking phenol red, which has weak estrogen activity, for 18 h. Thereafter, 0.001 nM R1881 was added to the medium and a 48-h time course of BrdU incorporation was monitored. After in vivo labeling, cells were analyzed for S-phase entry by in situ immunofluorescence. After hormone addition to the medium, a large number of cells incorporated BrdU into DNA with a peak at 18 h (Fig. 1 A).
Figure 1.

A low androgen concentration specifically stimulates the S-phase entry of NIH3T3 cells. (A) Quiescent fibroblasts on coverslips were left untreated (○) or treated (•) for the indicated times with 0.001 nM R1881. After in vivo labeling with 100 μM BrdU, the cells were fixed, permeabilized, and stained for BrdU incorporation. DNA synthesis was calculated by the formula: percentage of BrdU-positive cells = (number of BrdU-positive cells/number of total cells) × 100. (B) The cells were left untreated or treated with the indicated compounds, labeled with BrdU, and were fixed and stained after 18 h. DNA synthesis was calculated as in A. Different experiments were performed and multiple coverslips were analyzed. The means and SEM are shown. (C) Lysates from quiescent NIH3T3, LNCaP, MCF-7, and T47D cells were immunoblotted with the antibodies against the indicated receptors. Endogenous AR in NIH3T3 cells was revealed by two different antibodies (either C-19 or N-20). The optical density/mm2 measured by densitometric analysis of blot with C-19 anti-AR antibody was 6.12 and 0.47 for AR expressed in LNCaP and NIH3T3 cells, respectively. In the lowest section of C, lysates from primary fibroblasts (mouse fibroblasts, MF, or mouse embryo fibroblasts, MEF, both at second passage) were immunoblotted with the C-19 anti-AR antibody.
Next, we examined the effect of two diverse concentrations of sex steroid hormones on BrdU incorporation of quiescent NIH3T3 cells (Fig. 1 B). BrdU-incorporating cells increased from 10 to 60% after 0.001 nM R1881 was added to the medium. In contrast, stimulation was weak after the addition of 10 nM R1881. No increase of the S-phase entry was induced by either 0.001 or 10 nM (the latter concentration is not depicted) estradiol or progestin R5020. An excess of the antiandrogen Casodex abolished the BrdU incorporation induced by the lower concentration of R1881, which implies that the androgen stimulatory effect is mediated by its receptor.
We analyzed the expression of sex steroid receptors in NIH3T3 cells using Western blot. The anti-AR antibodies (N-20 or C-19, directed at NH2- and COOH-terminal sequences, respectively) reacted with a 108-kD band in LNCaP cell lysates (Fig. 1 C). Such a migration of the AR from LNCaP cells has been previously reported (Warriar et al., 1994). Parallel analysis in NIH3T3 cell lysate revealed a major band migrating with an apparent molecular mass of 110 kD. The same migration was reported for the mouse AR from different tissues (Zhou et al., 2002). A minor band migrating at 95 kD was also recognized by the C-19 Ab. It might represent an AR-truncated isoform (Wilson and McPhaul, 1994) or a proteolytic product. Noteworthy, post-translational modifications of AR have been described previously (Kuiper et al., 1992). Androgen receptor in fibroblasts was only 7% of that in LNCaP cells (Fig. 1). Similarly, low AR levels were detected with Western blot analysis of lysates from either Src-, Ras-, or Myc-transformed NIH3T3 fibroblasts, as well as Swiss3T3 fibroblasts (unpublished data). In agreement with the finding that estradiol or progestin did not affect S-phase entry, neither estradiol receptor (ERα or ERβ) nor progesterone receptor (PgR-B or -A) was detected in NIH3T3 cell lysates (Fig. 1 C). As a positive control, we used ERα, ERβ, or PgR (B and A isoforms) present in lysates from MCF-7, LNCaP, and T47D cells, respectively (Fig. 1 C). Remarkably, a 110-kD protein immunoreacting with the C-19 anti-AR antibody was also detected by Western blot analysis of lysates from mouse female and embryo primary fibroblasts (Fig. 1 C, lower section).
Together, Fig. 1 shows that a very low concentration of androgen R1881 promotes the S-phase entry of NIH3T3 fibroblasts. This effect, which is substantially reduced by the antiandrogen Casodex, correlates with detection of a protein recognized by two different anti-AR antibodies.
Role of the extranuclear signaling effectors in androgen-stimulated S-phase entry of NIH3T3 cells
Next, we studied the regulation of androgen-induced S-phase entry in response to the expression of various extranuclear effectors. In NIH3T3 cells, we transiently transfected the following: the catalytically inactive form of Src (Lys295 changed to methionine; Src K−; Barone and Courtneidge, 1995), the kinase-dead MAPK/ERK kinase-1 (MEK-1; Ser221 changed to alanine; A221–MEK-1, Cowley et al., 1994), the dominant-negative regulatory subunit of PI3-kinase (Δp85α; Dhand et al., 1994), or the Myc-tagged kinase-dead Akt (Lys 179 changed to methionine; Akt K−; Kohn et al., 1996). 12 h after transfection, cells were made quiescent and left unstimulated or stimulated with 0.001 nM R1881 for an additional 18 h. After in vivo labeling with BrdU, the cells on coverslips were fixed and stained. Several independent transfections were performed and multiple coverslips were analyzed. The number of BrdU-positive cells expressing either Src K−, A221–MEK-1, Δp85α, or the Myc-tagged Akt K− was compared with the number of BrdU-positive nontransfected cells. Data from different experiments were pooled and statistically analyzed (Fig. 2 A). About 10% of nontransfected cells incorporated BrdU in the absence of R1881. Addition of the hormone to the medium increased the number of BrdU-positive cells to ∼60%. Overexpression of Src K−, A221–MEK-1, Δp85α, or the Myc-tagged Akt K− inhibited the androgen-stimulated S-phase entry by 75, 67, 59, and 68%, respectively. In contrast, overexpression of the wild-type form of Src (Src wt) or the wild-type form of p85α (p85α wt) did not affect the androgen-induced S-phase entry. Transfection of NIH3T3 fibroblasts with the pSG5 empty plasmid did not modify BrdU incorporation irrespective of the presence or absence of R1881 (unpublished data). Images from one representative experiment with Src K− and A221–MEK-1 are presented. The left-hand panels of Fig. 2 B show cells expressing Src K− (top) or A221–MEK-1 (bottom). The same cells did not incorporate BrdU when visualized with anti-BrdU antibody (Fig. 2 B, middle). The nuclear staining with Hoechst is also shown (Fig. 2 B, right).
Figure 2.


Requirement for the Src–ERK and PI3-kinase–Akt pathways in the androgen-stimulated S-phase entry is correlated to AR–Src–PI3-kinase association in NIH3T3 cells. Fibroblasts on coverslips were either untransfected or transfected with the plasmids expressing the indicated proteins. (A) Transfected cells were made quiescent, and then left unstimulated or stimulated with 0.001 nM R1881. BrdU was added and coverslips were analyzed for BrdU incorporation. In transfected cells, BrdU incorporation was calculated by the formula: percentage of BrdU-positive cells = (number of transfected BrdU-positive cells/number of transfected cells) × 100 and compared with BrdU incorporation of untransfected cells from the same coverslips. For each plasmid, data are derived from at least 200 scored cells. The results of more than two independent experiments have been averaged; means and SEM are shown. The statistical significance of these results was also evaluated by paired t test. P values were <0.001 for cells transfected with either Src K−, Δp85α, Akt K−, or A221–MEK-1. The difference in BrdU incorporation between the cells transfected with Src K− and those transfected with Src wt was significant (P < 0.005). Also significant (P < 0.001) was the difference in BrdU incorporation between the cells transfected with Δp85α and those transfected with p85α wt. No significance was attributed to the difference in BrdU incorporation between the cells transfected with either Src wt or p85α wt and nontransfected cells stimulated with the androgen R1881. (B) Representative images of one of the experiments in A. Fluorescence in the left panels is from reactivity with either the anti-Src mAb (top) or the anti–MEK-1 Ab (bottom). Arrows and arrowheads mark the cells transfected with either Src K− or A221–MEK-1 expressing plasmids. The central panels show staining with anti-BrdU antibody. Hoechst 33258 nuclear staining is presented in the right panels. Quiescent NIH3T3 cells were either left untreated or treated for 2 min with the indicated compounds. (C) Lysate proteins were immunoprecipitated with either control antibody (ctrl) or the 327 anti-Src monoclonal antibody (anti-Src mAb). (D) Lysate proteins were immunoprecipitated with either control antibody (ctrl) or rabbit polyclonal anti-p85 antibody (anti-p85 Ab). (C and D) Immunocomplexes were analyzed by immunoblot with antibodies against the indicated proteins. (D) By an NIH 1.61 image program, a 38% increase of AR/p85 association was detected on 0.001 nM R1881 stimulation of cells. This experiment was reproduced with similar findings. (E) Lysate proteins from NIH3T3 cells challenged for 2 min with the indicated compounds were immunoblotted with the C-19 anti-AR antibody.
To investigate further the role of Src and PI3-kinase in androgen-induced S-phase entry, we challenged NIH3T3 cells with 0.001 or 10 nM R1881 and immunoprecipitated the lysates with anti-Src (Fig. 2 C) or anti-p85 antibodies (Fig. 2 D). In Fig. 2 C, immunocomplexes were blotted with either anti-Src (top) or anti-AR antibodies (bottom). At the lower R1881 concentration, but not at a 1,000-fold excess of the antiandrogen Casodex, Src coimmunoprecipitated with the two proteins immunodetected by the C-19 anti-AR antibody in NIH3T3 cell lysates that migrated at 110 and 95 kD. Remarkably, no association of Src with AR occurred at the higher R1881 concentration. Fig. 2 D shows immunocomplexes blotted with anti-p85 (top) or anti-AR antibodies (bottom). In unchallenged cell lysates, p85 coimmunoprecipitated with the 110-kD AR. Stimulation with the lower R1881 concentration, slightly (40%) increased this coimmunoprecipitation, which was undetectable at a higher concentration of R1881 (Fig. 2). The control antibody (ctrl) did not precipitate Src (Fig. 2 C) or p85 (Fig. 2 D). The possibility that treatment of cells could modify the AR level was excluded by the finding that the same amount of AR was detected by immunoblot of lysates, irrespective of R1881 and Casodex concentrations used to stimulate NIH3T3 cells (Fig. 2 E).
These data demonstrate that, in contrast to the higher R1881 concentration, the lower concentration induces coimmunoprecipitation of Src with AR and increases AR–PI3-kinase coimmunoprecipitation. Such a coimmunoprecipitation is associated with the androgen stimulated S-phase entry.
Androgen at high concentration induces Rac activation and membrane ruffling in NIH3T3 fibroblasts
NIH3T3 cells on coverslips were serum-starved and maintained in DME lacking phenol red. In a preliminary experiment (unpublished data), the cells were challenged with 0.001 or 10 nM R1881 for various times and stained with Texas red–phalloidin to visualize F-actin. Treatment of cells with 10 nM R1881 caused membrane ruffling, which appeared as early as 10 min after stimulation and increased after 20 min. In contrast, there was no response to treatment with 0.001 nM R1881, even after 40 min of ligand stimulation. In Fig. 3 (A–C) representative images of one experiment are shown. R1881 induced pronounced membrane ruffling at 10 nM, whereas it was ineffective at 0.001 nM. In addition, the pure antiandrogen Casodex prevented the effect of 10 nM androgen (Fig. 3 D). Next, the effect of signaling inhibitors was evaluated. Both the Src inhibitor, PP2 (Fig. 3 E), and the PI3-kinase inhibitor, LY294002 (Fig. 3 F), prevented cytoskeletal response to the androgen. In addition, PP2 affected cell morphology, resulting in a more roundish phenotype.
Figure 3.

A high androgen concentration induces membrane ruffling mediated by PI3-kinase and Src. Serum-starved NIH3T3 cells on coverslips were left untreated or treated for 20 min with the indicated compounds. The chemical inhibitors PP2 and LY294002 (Qbiogene) at 5 and 10 μM, respectively, were added 10 min before the hormonal stimulation. A 1,000-fold excess of Casodex was added 5 min before the hormonal stimulation. Cells stained with Texas red–phalloidin were visualized by a fluorescent microscope. Micrographs are representative of three independent experiments.
Because Rac activation is implicated in membrane ruffling and lamellipodia formation (Ridley, 2001), we next evaluated the effect of androgen on Rac activity using a pull-down assay. Stimulation of serum-starved NIH3T3 cells with 10 nM androgen induced a rapid and transient activation of Rac, with a peak at 2 min (Fig. 4 A). Interestingly, the lower androgen concentration (0.001 nM R1881) did not affect Rac activation. Consistent with the data on the androgen-induced membrane ruffling, LY294002 and PP2 inhibited the androgen-induced Rac1 activation (Fig. 4 B).
Figure 4.

PI3-kinase and Src mediate Rac activation induced by high androgen concentration. (A) Serum-starved NIH3T3 cells were left untreated or treated for the indicated times with either 0.001 or 10 nM R1881. (B) The cells were left untreated or treated for 2 min with 10 nM R1881 in the absence or presence of PP2 or LY294002 (at 5 and 10 mM, respectively). The chemical inhibitors were added 10 min before the hormonal stimulation. Lysate proteins were submitted to a Rac pull-down assay, and the eluted proteins were immunoblotted with the anti-Rac antibody.
Activation of signaling pathway(s) is involved in the estradiol-induced cytoskeleton changes in endothelial cells (Razandi et al., 2000) and implicated in morphological changes induced by estradiol in breast cancer–derived cells (Song et al., 2002). Here, we demonstrate that there is a hormone-regulated correlation between cytoskeletal changes and Rac activation; thus, Rac represents a new signaling effector activated by steroid hormones.
Protein interacting with anti-AR antibodies does not translocate into the nuclei of NIH3T3 fibroblasts
We used in situ immunofluorescence to determine the intracellular localization of protein interacting with either the C-19 or the N-20 anti-AR antibodies. Quiescent NIH3T3 and LNCaP cells were unstimulated or stimulated with 10 nM R1881 for 30 min and stained for AR detection. Interestingly, whatever the antibody used (either C-19 or N20), fluorescence was observed in the cytoplasm of NIH3T3 cells (Fig. 5 , A–B1). Similar results (unpublished data) were obtained irrespective of signal duration (60 and 120 min) and ligand concentration (0.001 nM). Conversely, stimulation of human prostate carcinoma LNCaP cells induced nuclear import of AR in >40% of cells as observed by staining with the C-19 anti-AR antibody (Fig. 5, C and C1). Specificity of the C-19 staining in NIH3T3 cells was confirmed by the almost complete displacement of immunofluorescence observed with an excess of peptide against which the C-19 antibody is raised (Fig. 5, D and D1).
Figure 5.

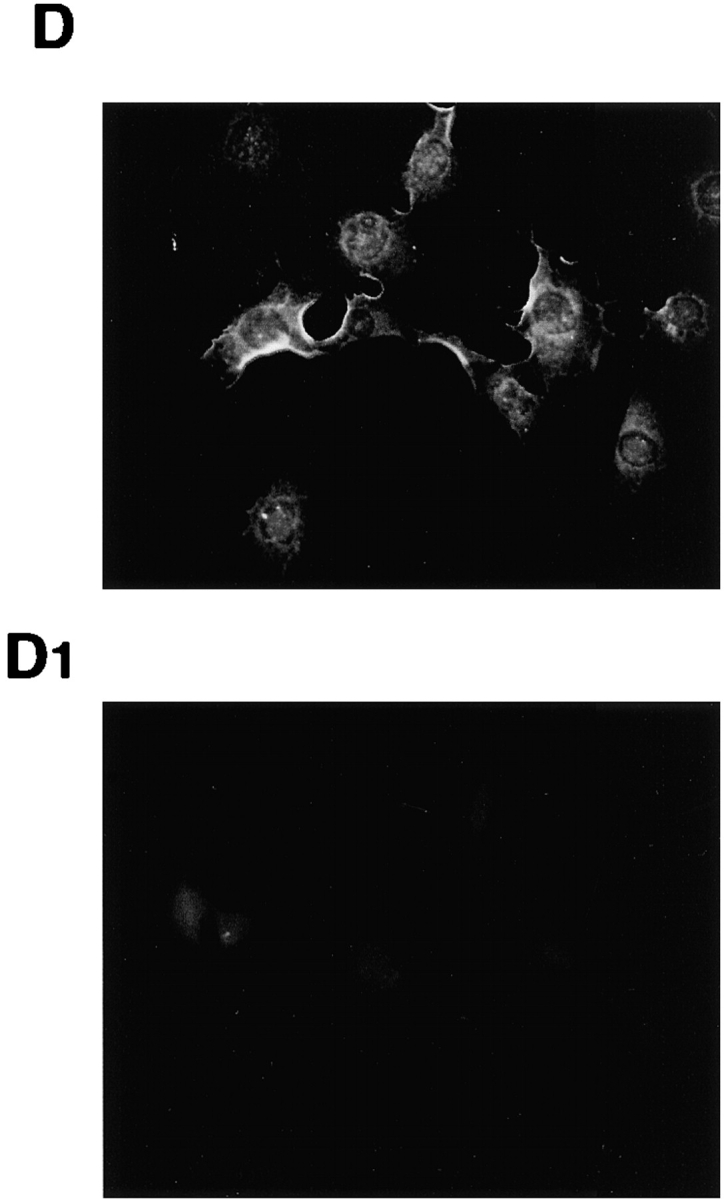
Immunostaining of AR in NIH3T3 and LNCaP cells. Quiescent NIH3T3 and LNCaP cells on coverslips were untreated or treated for 30 min with 10 nM R1881. Cells were permeabilized as described in Materials and methods, and AR was visualized by immunofluorescence using the indicated antibodies. (A–B1) Regardless of the antibody used (either C-19 or N-20), AR is localized in the extranuclear compartment of untreated (A and B) or androgen-treated NIH3T3 cells (A1 and B1). Exposure time in NIH3T3 cells was enhanced to better visualize cytoplasm AR. (C and C1) AR is localized in both the nuclear and extranuclear compartment of untreated LNCaP cells (C). 10 nM R1881 induced nuclear import of AR in LNCaP cells (C1). The cells that fall into the category of predominantly nuclear staining are marked with arrows. Immunostaining of AR from at least 200 scored LNCaP cells was also monitored, and the data were expressed as a percentage of cells that fall into the category of predominantly nuclear staining. In LNCaP cells, this value was ∼20% and shifted to ∼65% when they were stimulated for 30 min with 10 nM R1881. (D and D1) Quiescent NIH3T3 cells on coverslips were fixed and permeabilized as described in Materials and methods. AR was visualized by immunofluorescence using the C-19 anti-AR antibody alone (D) or in the presence (D1) of a 100-fold excess of a competing peptide (AR/C-19 P; Santa Cruz Biotechnology, Inc.).
Lack of androgen-stimulated transcription activity in NIH3T3 cells
The prevalently extranuclear localization of AR prompted us to analyze the effect of androgen on the reporter gene transcription. Two androgen enhancers, the 3416 and 3424 constructs (Verrijdt et al., 2000), were transiently transfected in NIH3T3 cells and the effect exerted by androgen on the reporter gene transcription was evaluated after 18 h of cell treatment with either 0.001 or 10 nM R1881. In agreement with the extranuclear localization of AR, neither high nor low androgen concentrations increased luciferase activity (Fig. 6, A and B) .
Figure 6.

Androgen-stimulated gene transcription in NIH3T3 cells before and after overexpression of hAR and hAR intracellular localization. NIH3T3 cells were transfected with either ARE 3424 (A) or 3416 (B) constructs with or without hAR-expressing plasmid. Cells were left unstimulated or stimulated for 18 h with the indicated concentrations of the androgen R1881. The luciferase activity was assayed, normalized using β-gal as an internal control, and expressed as fold induction. The same lysates were used for Western blot analysis with C-19 anti-AR Ab (C). (D and D2) NIH3T3 cells were transfected with hAR-expressing plasmid, and then made quiescent. Cells were left unstimulated (D) or stimulated for 1 h with either 0.001 nM (D1) or 10 nM R1881 (D2). Fixed cells on coverslips were permeabilized as described in Materials and methods, and hAR was visualized by immunofluorescence using the rabbit polyclonal anti-AR (N-20) antibody.
It is unlikely that these findings depend on the cell milieu because a substantial increase of luciferase activity (39- and 48-fold for the 3424 or the 3416 construct, respectively) was observed on 10 nM androgen stimulation of NIH3T3 cells overexpressing hAR (Fig. 6, A and B). Such overexpression is confirmed by Western blot analysis of lysates from NIH3T3 cells transfected with hAR cDNA in the presence of either 3416 or 3424 construct (Fig. 6 C). In addition, the cell environment does not interfere with AR nuclear translocation because the overexpressed hAR enters nuclei on a 10-nM ligand stimulation (Fig. 6 D2). Interestingly, neither gene transcription (Fig. 6, A and B) nor nuclear import (Fig. 6 D1) occurred after stimulation of transfected NIH3T3 cells with 0.001 nM R1881, implicating that both processes occur at high levels of both, receptor and ligand.
Results illustrated in Figs. 5 and 6 demonstrate that endogenous AR does not enter nuclei of NIH3T3 cells and, consequently, it is unable to activate gene transcription. Nevertheless, it does induce S-phase entry and membrane ruffling on androgen stimulation of the cells.
Identification of murine AR in NIH3T3 fibroblasts by RT-PCR
Data in the previous sections led us to further identify the AR expressed in NIH3T3 cells. We generated cDNA from poly(A)+ RNA of growing cells by reverse transcriptase. The cDNA was amplified by PCR using primers for the NH2 terminus, the DNA-binding domain, and the ligand-binding domain (LBD) of AR. The analysis by agarose gel of PCR products (Fig. 7) revealed DNA bands of the expected size of 1,300, 850, and 420 bp for the NH2 terminus, the DNA-binding domain, and the LBD (Fig. 7, A–C, left lanes) of AR, respectively. The corresponding PCR products from the AR of LNCaP cells were analyzed in parallel (Fig. 7, A–C, right lanes). DNA sequencing (unpublished data) confirmed that the PCR fragments from NIH3T3 cells contained the predicted sequence of AR NH2 terminus, DNA binding, and LBD, thereby proving that NIH3T3 cells contain the classical mouse AR.
Figure 7.

Molecular analysis of AR-RNA expressed in NIH3T3 and LNCaP cells. Total RNA and poly(A)+ RNA was obtained from growing LNCaP or NIH3T3 cells, respectively. cDNA was synthesized and amplified by PRC using specific primers for the NH2 terminus (A), DNA binding (B), and LBD (C) of mouse AR. The PCR products were submitted to agarose gel. PCR products generated without reverse transcriptase from poly(A)+ RNA of NIH3T3 cells were submitted to the same analysis, as a control (ctrl).
Low AR expression mediates signaling activation but not gene transcription in Cos cells
Our results indicate that AR expressed in NIH3T3 cells is transcriptionally inactive while it efficiently stimulates the cytosolic-coupled pathways. To verify whether the low amount of endogenous AR might be responsible for this behavior, we transiently transfected AR-negative Cos cells with two different amounts of hAR cDNA (either 500 or 1 ng) and analyzed the AR expression levels (Fig. 8 A, inset). The ability of AR to transactivate the androgen-responsive element (ARE) 3416 reporter gene was then assayed. Irrespective of the amount of expressed hAR, no transcription stimulation occurred on treatment of Cos cells with 0.001 nM R1881 (Fig. 8 A). When the same cells were stimulated with 10 nM R1881, those expressing the lowest amount of hAR showed a negligible increase of the transcriptional activity (4% of that detected in cells showing the highest amount of hAR; Fig. 8 A). In sharp contrast, Cos cells expressing the lowest amount of AR showed a strong Src activity increase after stimulation with 0.001 nM R1881. Such an increase was comparable with that observed in cells expressing high levels of AR when stimulated by 10 nM R1881 (Fig. 8 B).
Figure 8.

In Cos cells expressing very low amount of hAR, stimulation by 0.001 nM R1881 triggers the Src kinase activity but not gene transcription. Cos cells were transfected with the ARE-3416-Luc reporter gene in the absence or presence of the indicated amounts of hAR cDNA. (A) Cells were left unstimulated or stimulated for 18 h with the indicated concentrations of the androgen R1881. The luciferase activity was assayed, normalized using β-gal as an internal control, and expressed as fold induction. The same lysates were used for Western blot analysis with the C-19 anti-AR Ab (A, inset). (B) Quiescent cells were left unstimulated or stimulated for 2 min with the indicated concentrations of R1881. Lysates were either immunoblotted with the C-19 anti-AR Ab (top) or immunoprecipitated with anti-Src mAb. Immunoprecipitates were either blotted with anti-Src mAb (middle) or assayed for Src kinase activity using enolase as a substrate (bottom).
Noteworthy, like in NIH3T3 cells overexpressing hAR, in Cos cells stimulation of AR-dependent transcription requires high levels of both ligand and AR. Altogether, these data support the hypothesis that the very low AR amounts expressed in NIH3T3 cells are sufficient to activate cytoplasmic functions but inadequate to stimulate gene transcription.
Discussion
The expression and activity of steroid receptors is not restricted to epithelial cells. Mesenchyme AR is required to initiate prostatic bud formation (Cunha et al., 1987). Estrogen receptor-α is present in the breast stroma of rodents and is responsible for secretion of growth factors (epithelial cellular mitogens) in response to estradiol. Tissue recombinants with stroma and epithelium from normal as compared with ERα knockout mice showed that stromal ERα is responsible for estrogen-induced epithelial growth (Cunha et al., 1997). Moreover, the stroma of human mammary cancers contains ERβ (Jensen et al., 2001).
Androgen activates different signaling effectors in such cell types as prostate stromal cells, LNCaP cells, and osteoblasts (Peterziel et al., 1999; Migliaccio et al., 2000; Kousteni et al., 2001). We previously described signaling pathway activation and consequent DNA synthesis in various epithelial cells stimulated by the sex steroid hormones (Di Domenico et al., 1996; Migliaccio et al., 1996, 1998, 2000; Castoria et al., 1999, 2001). We now report that androgen stimulation of NIH3T3 cells induces them to synthesize DNA. This effect is androgen-specific. It correlates with the presence of a 110-kD protein interacting with two different anti-AR antibodies in NIH3T3 cells and is prevented by Casodex, a specific AR antagonist. Interestingly, a similar immunoreactive protein is recognized by the C-19 anti-AR antibody in Western blot analysis of lysates from mouse female and embryo primary fibroblasts. This indicates that AR expression is not restricted to immortalized fibroblasts.
As in LNCaP cells (Migliaccio et al., 2000), the Src–ERK pathway is required for androgen-induced DNA synthesis because dominant-negative forms of signaling effectors prevent this synthesis. We provide the first evidence that the PI3-kinase–Akt pathway also mediates androgen-induced S-phase entry. Noteworthy, estradiol activation of this pathway has the same role in mammary cancer–derived MCF-7 cells (Castoria et al., 2001). Therefore, different steroid hormones can activate the Src- and PI3-kinase–dependent pathways in different cell types and, thus, exert mitogenic effects. Interestingly, a very low (0.001 nM) R1881 concentration is required for a pronounced DNA synthesis. This, together with the low level of AR expression in fibroblasts, shows that agonist binding to a very small number of receptor molecules induces DNA synthesis. Similar conclusions have been previously reached for estrogen-induced proliferation in PR1 pituitary lactotroph tumor–derived cells (Chun et al., 1998) and low concentrations of estradiol or R1881 efficiently induce S-phase entry of MCF-7 or LNCaP cells (unpublished data). In addition, the androgen-induced S-phase entry of NIH3T3 fibroblasts is restricted to a single cell cycle (Fig. 1 A). Fluctuation of cell cycle regulators is responsible for similar findings in progestin-stimulated T47D cells (Horwitz et al., 1982).
In NIH3T3 cells stimulated with 0.001 nM R1881, Src and p85 associated with AR. Interestingly, the higher R1881 concentration weakly stimulated DNA synthesis and did not induce association of AR with Src or p85. Consequently, this interaction appears to play a role in DNA synthesis stimulation. We previously reported that the hAR–Src association is direct and requires the Src-SH3 domain and a proline stretch (372–379) of the receptor (Migliaccio et al., 2000). Therefore, it is likely that the homologous proline stretch (367–373) present in the mouse AR is responsible for the association of AR with Src in fibroblasts.
The outcome of signaling activation can depend on differences in ligand concentration (Marshall, 1995), and excessive signals inhibit the DNA synthesis entry of the cell cycle in certain systems (Olson et al., 1998). Here, we show that stimulation of NIH3T3 cells with a high (10 nM) androgen concentration leads to rapid activation of Rac, with a maximal effect within 2 min. Rac activation rapidly causes changes in the actin assembly that result in membrane ruffling. In addition to regulating cytoskeletal changes and motility of normal and neoplastic cells, Rac can act as an oncogene when overexpressed in fibroblasts. Its linkage to cell transformation occurs through the Ras and the PI3-kinase–dependent pathways, independently of Akt (Vivanco and Sawyers, 2002). The inhibition of androgen-stimulated Rac by Src and PI3-kinase inhibitors observed in this paper is in agreement with this and other papers. Class Ia PI3-kinase can act upstream of Rac (Plattner et al., 1999; Ridley, 2001) and Src has been implicated in PDGF-mediated membrane ruffling of mouse embryo fibroblasts (Plattner et al., 1999). The observation that DNA synthesis and membrane ruffling both require Src and PI3-kinase activities indicates that the signaling effectors mediate different effects that depend on different ligand concentrations. The finding that androgen concentration regulates the association between AR and signaling effectors suggests that assembly or disassembly of different modules are involved in the different androgen effects triggered by low and high hormone concentrations. The observation that high ligand concentration, whereas it dissociates the AR–Src–PI3-kinase complex, stimulates ruffling through a process that still depends on Src and PI3-kinase activities is reminiscent of the multi-adaptor Cb1 mutant action. This mutant is unable to bind signaling molecules, nevertheless, it strongly enhances the PDGF-induced membrane ruffling through a process that requires Src and PI3-kinase (Scaife et al., 2003). The observation that different androgen levels trigger different responses in NIH3T3 cells is a remarkable example of the pronounced flexibility of cell responsiveness to steroids.
An AR nontranslocable to nuclei has been detected in T cells (Benten et al., 1999) and a functional membrane testosterone receptors that modify actin cytoskeleton has been recently identified in LNCaP cells (Kampa et al., 2002). The experiments reported herein show that the AR present in NIH3T3 fibroblasts does not enter the nuclei in response to androgen, as assessed by two different anti-AR antibodies. As a consequence, it does not activate gene transcription. These findings reinforce the concept that commitment to S-phase entry (Castoria et al., 1999) and induction of membrane ruffling do not require transcriptional activity of steroid receptors.
The expression of AR in NIH3T3 cells is very low. Nuclear translocation of AR and ARE-dependent transcription are detected only after exogenous AR overexpression in the presence of high androgen concentration. Altogether, these findings suggest that the amount of AR in fibroblasts is sufficient to engage cytosol-coupled signaling effectors but not to trigger gene transcription. This hypothesis is supported by the finding that AR-negative Cos cells, after expression of a very low amount of AR, respond with Src activation on stimulation with high or low androgen concentration, whereas efficient AR-dependent gene transcription requires high ligand and AR concentration. In addition to NIH3T3 cells, other mesenchymal cells contain very low amounts of steroid receptors (Manolagas and Kousteni, 2001). Like fibroblasts, they might respond to hormones with S-phase entry, cytoskeletal changes, and other effects through activation of signaling pathways in the absence of receptor transcriptional activity. Fibroblasts emerge as a new model of hormone-responsive cells that can be exploited to better understand the early, extranuclear effects of steroid hormones.
Materials and methods
Constructs
The cDNA coding hAR was cloned into the pSG5 expression vector as reported previously (Chang et al., 1988). The wild-type and dominant-negative Δp85α were cloned into the pSG5 vector. The Myc-His–tagged dominant-negative Akt (K179M) in pUSEAmp was purchased from UBI. cDNAs encoding either the wild-type or kinase-inactive form of Src (Lys-259 changed to methionine) were cloned into pSG5 (Barone and Courtneidge, 1995). cDNA encoding the kinase-dead MEK-1 (Ser-221 changed to alanine; A221–MEK-1) was cloned into pEXV3 (Cowley et al., 1994). The 3416 construct, containing four copies of the wild-type slp-HRE2 (5′-TGGTCAgccAGTTCT-3′) and the 3424 construct (5′-TGGACAgccAGTTCT-3′), were cloned in the NheI site in pTK-TATA-Luc (Verrijdt et al., 2000).
Cell culture, transfection, and transactivation assay
Fast growing human prostate cancer LNCaP cells (FGC-LNCaP cells at seventh passage; American Type Culture Collection), low passage mouse embryo NIH3T3 fibroblasts, and Cos cells were grown as reported previously (Migliaccio et al., 1998, 2000; Castoria et al., 1999). Mouse female as well as embryo primary fibroblasts were grown at 37°C and 5% CO2 in DME supplemented with 10% FBS and antibiotics. For S-phase entry analysis, NIH3T3 cells were seeded onto gelatin-precoated coverslips at 60% confluence and made quiescent by serum starvation (Castoria et al., 1999). Cells were stimulated with the indicated concentrations of steroids (dissolved in 0.001% ethanol, final concentration). Control cells were treated with the vehicle alone. The effect of Casodex (Zeneca) on S-phase entry was monitored using a 1,000-fold excess of the antagonist. The antiandrogen was also solubilized in ethanol (0.001%, final concentration). When indicated, NIH3T3 fibroblasts were plated onto coverslips at 60% confluence in phenol red–free DME containing 10% charcoal-stripped serum, and then transfected by Superfect (QIAGEN) together with 2 μg of either Src K− or Src wt or A221–MEK-1. 12 h later, transfected cells were made quiescent (Castoria et al., 1999) and maintained in this condition for an additional 18 h.
Transfections of NIH3T3 cells with either p85α wt, Δp85α, or the Myc-tagged Akt K− have been described previously (Castoria et al., 2001). Fibroblasts were left stimulated with 0.001 nM R1881 (Zeneca) for 24 h. The androgen was dissolved in ethanol (0.001%, final concentration), and control cells were treated with the vehicle alone. For androgen-stimulated transcriptional analysis, NIH3T3 fibroblasts were plated at 70% confluence in phenol red–free DME containing 10% charcoal-stripped serum. After 48 h, the cells were transfected by Superfect with 4 μg of either 3416-pTK-TATA-Luc or 3424-pTK-TATA-Luc constructs, alone or with 1 μg pSG5-hAR–expressing plasmid. Cos cells were plated at 70% confluence in phenol red–free DME containing 5% charcoal-stripped serum. After 48 h, the cells were transfected by Superfect with 4 μg 3416-pTK-TATA-Luc, alone or with the indicated amounts of pSG5-hAR–expressing plasmid. 24 h later, transfected cells were stimulated with the indicated concentration of R1881 (dissolved in 0.001% ethanol, final concentration) for 18 h. Control cells were treated with the vehicle alone. Lysates were prepared and the luciferase activity was measured using a luciferase assay system (Promega). The results were corrected using CH110-expressed β-galactosidase activity (Amersham Biosciences). For androgen-stimulated translocation analysis, NIH3T3 fibroblasts were plated onto coverslips at 60% confluence in phenol red–free DME containing 10% charcoal-stripped serum, and transfected by Superfect with 1 μg pSG5-hAR–expressing plasmid. 24 h later, transfected cells were made quiescent, and stimulated for 1 h with the indicated concentration of R1881 (dissolved in 0.001% ethanol, final concentration). Control cells were treated with the vehicle alone.
RNA extraction, reverse transcription-PCR, and DNA sequencing
Total RNA was extracted from growing LNCaP cells using TRIzol reagent (Life Technologies) according to the manufacturer's instructions. Poly(A)+ RNA was prepared from growing NIH3T3 fibroblasts using the Oligotex mRNA kit (QIAGEN). Random primed double-strand cDNA was synthesized using the Superscript first-strand synthesis system for RT-PCR (Invitrogen). PCR was carried using Platinum Taq DNA polymerase (Invitrogen) and the oligonucleotide primer pairs TRP 731 5′-TGGCGGTCCTTCACTAATGTCAACTCCAGG-3′ together with ASP 870 5′-GTCCACGCTCACCATATGGACTTGATTAG-3′; TRP 731 5′-TGGCGGTCCTTCACTAATGTCAACTCCAGG-3′ together with 2,760 nt 5′-AGGCAGAAGACAT-CTGGAAAGGGAACAAGGTGGG-3′ for 35 cycles using the following parameters: 96°C for 30 s, 60°C for 1 min, and 72°C for 1 min. For the oligonucleotide primer pairs NH1 5′-ATGGAGGTGCAGTTAGGGCTGGGA-3′ and Tyr460 5′-ATAGGGGGCTACAGGCCCGGCATCGCTTGG-3′, and Pro 451 5′-CCAAGCGATGCCGGGCCTGTAGCCCCCTAT-3′ and Arg740 5′-CCTGGAGTTGACATTAGTGAAGGACCGCCA-3′, PCR was carried for 35 cycles using the following parameters: 96°C for 30 s, 64°C for 1 and 5 min, and 72°C for 2 min. The specificity of each pair of the oligos used was verified using the BLAST Program (Sambrook and Russell, 2001). PCR products were eluted from agarose gel with QIAEX II kit (QIAGEN) and sequenced with Thermo Sequenase radiolabeled Terminator Cycle Sequencing kit (Amersham Biosciences) using either the oligos used for PCR amplification or specific internal oligos.
Immunofluorescence, DNA synthesis analysis, and cytoskeletal changes
LNCaP cells and NIH3T3 fibroblasts on coverslips were challenged with the indicated concentration of R1881 in ethanol. Control cells were treated with the vehicle alone (0.001%, final concentration). Unless otherwise stated, cells on coverslips were washed once with PBS, fixed for 10 min with PFA (3%, wt/vol in PBS), permeabilized for 5 min with Triton X-100 (0.2%, vol/vol in PBS), and incubated for 1 h with PBS containing 1% vol/vol FCS. Coverslips were stained for in situ AR localization. Diluted (1:100 in PBS containing 0.01% BSA) anti-AR (C-19; Santa Cruz Biotechnology, Inc.) rabbit polyclonal antibody was used and the incubation was performed for 1 h at room temperature. For competition experiments, diluted (1:100 in PBS containing 20 μg C-19 peptide and 0.01% BSA) anti-AR C-19 antibody was used. After extensive washings in PBS, coverslips were incubated for 45 min with diluted (1:200 in PBS containing 0.01% BSA) Texas red–conjugated affinipure anti–rabbit IgG (Jackson ImmunoResearch Laboratories). hAR overexpressed in NIH3T3 cells was visualized using diluted (1:100 in PBS containing 0.01% BSA) anti-AR (N-20; Santa Cruz Biotechnology, Inc.) rabbit polyclonal antibody and the FITC-conjugated secondary antibody was used at 1:200 dilution in PBS containing 0.01% BSA. Endogenous AR in NIH3T3 cells was also revealed using the anti-AR (N-20; Santa Cruz Biotechnology, Inc.) rabbit polyclonal antibody. Cells on coverslips were submitted to methanol fixation (100% methanol at −20°C) as described (Spector et al., 1998). Coverslips were incubated for 1 h with PBS containing 1% vol/vol FCS and mouse AR was stained using diluted (1:50 in PBS) N-20 rabbit polyclonal antibody. The Texas red–conjugated affinipure anti–rabbit IgG (Jackson ImmunoResearch Laboratories) was used at 1:100 dilution in PBS. DNA synthesis was assayed by a 4-h pulse with 100 μM BrdU (Boehringer), and BrdU incorporation was monitored as reported in Castoria et al. (1999); Src wt or Src K− was visualized according to the same paper. A221–MEK-1, p85α wt, Δp85α, and the Myc-His–tagged Akt K− were visualized as described previously (Migliaccio et al., 2000; Castoria et al., 2001). Coverslips were washed with PBS and nuclei stained with Hoechst 33258 (Sigma-Aldrich). To determine cytoskeletal changes, quiescent NIH3T3 on coverslips were maintained for 3 h in serum-free DME and stimulated with the androgen R1881 (dissolved in 0.001% ethanol, final concentration) for the indicated times. Control cells were treated with the vehicle alone. Cells were fixed, permeabilized, washed, and stained with Texas red–labeled phalloidin (Sigma-Aldrich) as reported in Barone et al. (2001). Images were generated with an Axiophot (Carl Zeiss MicroImaging, Inc.) or a DMLB (Leica) fluorescent microscope using 40× and 100× objectives. Images were processed using either KS300 (Carl Zeiss MicroImaging, Inc.) or IM1000 (Leica) software.
Immunoprecipitation and Src and Rac assays
For Src activity assays, Cos cells transfected with hAR as indicated (see Cell culture, transfection, and transactivation assay) were made quiescent by serum starvation. The phenol red–free DME was supplemented with 0.5% charcoal-treated FCS, and the cells were left quiescent for 18 h. They were challenged with the indicated amounts of the androgen R1881 (dissolved in ethanol) and control cells were treated with the vehicle alone. Lysates were prepared as described previously (Migliaccio et al., 1998), and protein concentrations were measured using a Bio-Rad protein assay kit (Bio-Rad Laboratories). Cell lysates (2 mg/ml protein concentration) were immunoprecipitated with either the mouse monoclonal anti-Src antibody (clone 327; Calbiochem) or the rabbit polyclonal anti-p85 antibody (UBI) as reported previously (Migliaccio et al., 1998; Castoria et al., 2001). Src kinase assay was performed as described in Migliaccio et al. (1996), using acidified enolase as a substrate. For Rac assay, quiescent NIH3T3 fibroblasts at 70% confluence were maintained for 3 h in serum-free DME and stimulated with the androgen R1881 (dissolved in ethanol) for the indicated times. Control cells were treated with the vehicle alone (0.001%, final concentration). Fresh lysates (4 mg/ml of protein concentration) were prepared and incubated with the PAK-1 PBD agarose (UBI) as reported previously (Benard et al., 1999). The eluted proteins were immunoblotted for Rac detection.
Electrophoresis and immunoblotting
Electrophoresis and immunoblotting procedures were performed as described in Migliaccio et al. (1998). Rac was detected with the mouse monoclonal anti-Rac antibody (clone 23A8; UBI). The rabbit polyclonal anti-AR antibodies (either C-19 or N-20; Santa Cruz) were used to reveal AR. Src was detected with the mouse monoclonal anti-Src antibody (clone 327; Calbiochem) and p85 was immunoblotted using the rabbit polyclonal anti-p85 antibody (UBI). ERα was immunoblotted using the rat monoclonal anti-ERα (H222) antibody and ERβ using the rabbit polyclonal anti-ERβ antibody (UBI). PgR was detected using mouse monoclonal anti-PgR antibody (StressGen Biotechnologies). Immunoreactive proteins were revealed by the ECL detection system (Amersham Biosciences).
Acknowledgments
We thank F. Claessens and G. Verrijdt for 3416 and 3424 ARE-Luc constructs, J. Downward for the A221–MEK-1 and Δp85α–expressing plasmids, and B. Vanhaesebroeck for the wild-type p85α–expressing plasmid. Abbott Laboratories provided the H222 anti-ERα monoclonal antibody, and Zeneca provided the antiandrogen Casodex. We are grateful to J. Gilder (Scientific Communication) for text editing.
This work was supported by grants from Associazione Italiana per la Ricerca sul Cancro, Ministero dell'Università e della Ricerca Scientifica (Cofinanziamento MURST 2001-2002 - Fondi 40 e 60%) and Consiglio Nazionale delle Ricerche (C.N.R. - Agenzia 2000). M. Lombardi is recipient of a Fondazione Italiana per la Ricerca sul Cancro fellowship.
G. Castoria and M. Lombardi contributed equally to this work.
Footnotes
Abbreviations used in this paper: AR, androgen receptor; ARE, androgen-responsive element; ERK, extracellular signal–regulated kinase; LBD, ligand-binding domain; MEK-1, MAPK/ERK kinase-1; PgR, progesterone receptor; Pl3-kinase, phosphatidylinositol 3-kinase; p85α, p85α wt; Src, Src wt.
References
- Barone, M.V., and S.A. Courtneidge. 1995. Myc but not Fos rescue of PDGF signalling block caused by kinase-inactive Src. Nature. 378:509–512. [DOI] [PubMed] [Google Scholar]
- Barone, M.V., L. Sepe, R.M. Melillo, A. Mineo, G. Santelli, C. Monaco, M.D. Castellone, D. Tramontano, A. Fusco, and M. Santoro. 2001. RET/PTC1 oncogene signaling in PC Cl 3 thyroid cells requires the small GTP-binding protein Rho. Oncogene. 20:6973–6982. [DOI] [PubMed] [Google Scholar]
- Benard, V., B.P. Bohl, and G.M. Bokoch. 1999. Characterization of rac and cdc42 activation in chemoattractant-stimulated human neutrophils using a novel assay for active GTPases. J. Biol. Chem. 274:13198–13204. [DOI] [PubMed] [Google Scholar]
- Benten, W.P.M., M. Lieberherr, G. Giese, C. Wrehlke, O. Stamm, C.E. Sekeris, H. Mossmann, and F. Wunderlich. 1999. Functional testosterone receptors in plasma membranes of T cells. FASEB J. 13:123–133. [DOI] [PubMed] [Google Scholar]
- Camps, J.L., S.M. Chang, T.C. Hsu, S.J. Freeman, M.R. Hong, H.E. Zhau, A.C. von Eschenbach and L.W.K. Chung. 1990. Fibroblast-mediated acceleration of human epithelial tumor growth in vivo. Proc. Natl. Acad. Sci. USA. 87: 75–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castoria, G., M.V. Barone, M. Di Domenico, A. Bilancio, D. Ametrano, A. Migliaccio, and F. Auricchio. 1999. Non-transcriptional action of estrogen and progestin triggers DNA synthesis. EMBO J. 18:2500–2510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castoria, G., A. Migliaccio, A. Bilancio, M. Di Domenico, A. de Falco, M. Lombardi, R. Fiorentino, L. Varricchio, M.V. Barone, and F. Auricchio. 2001. PI3-K in concert with Src promotes the S-phase entry of estradiol-stimulated MCF-7 cells. EMBO J. 20:6050–6059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, C.S., J. Kokontis, and S.T. Liao. 1988. Molecular cloning of human and rat complementary DNA encoding androgen receptors. Science. 240:324–326. [DOI] [PubMed] [Google Scholar]
- Chun, T.Y., D. Gregg, D.K. Sarkar, and J. Gorski. 1998. Differential regulation by estrogens of growth and prolactin synthesis in pituitary cells suggests that only a small pool of estrogen receptors is required for growth. Proc. Natl. Acad. Sci. USA. 95:2325–2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowley, S., H. Paterson, P. Kemp, and C.J. Marshall. 1994. Activation of MAP kinase kinase is necessary and sufficient for PC12 differentiation and for transformation of NIH3T3 cells. Cell. 77:841–852. [DOI] [PubMed] [Google Scholar]
- Cunha, G.R., A.A. Donjacour, P.S. Cooke, S. Mee, R.M. Bigsby, S.J. Higgins, and Y. Sugimura. 1987. The endocrinology and developmental biology of the prostate. Endocr. Rev. 8:338–362. [DOI] [PubMed] [Google Scholar]
- Cunha, G.R., P. Young, Y.K. Hom, P.S. Cooke, J.A. Taylor, and D.B. Lubahn. 1997. Elucidation of a role for stromal steroid hormone receptors in mammary gland growth and development using tissue recombinants. J. Mammary Gland Biol. Neoplasia. 2:393–402. [DOI] [PubMed] [Google Scholar]
- Dhand, R., K. Hara, I. Hiles, B. Bax, I. Gout, G. Panayotou, M.J. Fry, K. Yonezawa, M. Kasuga, and M.D. Waterfield. 1994. PI3-kinase: structural and functional analysis of intersubunit interactions. EMBO J. 13:511–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Domenico, M., G. Castoria, A. Bilancio, A. Migliaccio, and F. Auricchio. 1996. Estradiol activation of human colon carcinoma-derived Caco-2 cell growth. Cancer Res. 56:4516–4521. [PubMed] [Google Scholar]
- Hall, A. 1998. Rho GTPases and the actin cytoskeleton. Science. 279:509–514. [DOI] [PubMed] [Google Scholar]
- Hayward, S.W., Y. Wang, M. Cao, Y.K. Hom, B. Zhang, G.D. Grossfeld, D. Sudilovsky, and G.R. Cunha. 2001. Malignant transformation in a nontu-morigenic human prostatic epithelial cell line. Cancer Res. 61:8135–8142. [PubMed] [Google Scholar]
- Horwitz, K.B., M.B. Mockus, and B.A. Lessey. 1982. Variant T47D human breast cancer cells with high progesterone-receptor levels despite estrogen and antiestrogen resistance. Cell. 28:633–642. [DOI] [PubMed] [Google Scholar]
- Jensen, E.V., G. Cheng, C. Palmieri, S. Saji, S. Makela, S. Van Noorden, T. Wahlstrom, M. Warner, R.C. Coombes, and J.A. Gustafsson. 2001. Estrogen receptors and proliferation markers in primary and recurrent breast cancer. Proc. Natl. Acad. Sci. USA. 98:15197–15202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampa, M., E.A. Papakonstanti, A. Hatzoglou, E.N. Stathopoulos, C. Stournaras, and E. Castanas. 2002. The human prostate cancer cell line LNCaP bears functional membrane testosterone receptors that increase PSA secretion and modify actin cytoskeleton. FASEB J. 16:1429–1431 [DOI] [PubMed] [Google Scholar]
- Kohn, A.D., S.A. Summers, M.J. Birnbaum, and R.A. Roth. 1996. Expression of a constitutively active Akt Ser/Thr kinase in 3T3-L1 adipocytes stimulates glucose uptake and glucose transporter 4 translocation. J. Biol. Chem. 271:31372–31378. [DOI] [PubMed] [Google Scholar]
- Kousteni, S., T. Bellido, L.I. Plotkin, C.A. O'Brien, D.L. Bodenner, L. Han, K. Han, G.B. DiGregorio, J.A. Katzenellenbogen, B.S. Katzenellenbogen, et al. 2001. Nongenotropic, sex-nonspecific signaling through the estrogen or androgen receptors: dissociation from transcriptional activity. Cell. 104:719–730. [PubMed] [Google Scholar]
- Kuiper, G.G., P.E. de Ruiter, and A.O. Brinkmann. 1992. Androgen receptor heterogeneity in LNCaP cells is caused by a hormone independent phosphorylation step. J. Steroid Biochem. Mol. Biol. 41:697–700. [DOI] [PubMed] [Google Scholar]
- Manolagas, S.C., and S. Kousteni. 2001. Perspective: nonreproductive sites of action of reproductive hormones. Endocrinology. 142:2200–2204. [DOI] [PubMed] [Google Scholar]
- Marshall, C.J. 1995. Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell. 80:179–185. [DOI] [PubMed] [Google Scholar]
- Matrisian, L.M., G.R. Cunha, and S. Mohla. 2001. Epithelial-stromal interactions and tumor progression: meeting summary and future directions. Cancer Res. 61:3844–3846. [PubMed] [Google Scholar]
- Migliaccio, A., M. Di Domenico, G. Castoria, A. de Falco, P. Bontempo, E. Nola, and F. Auricchio. 1996. Tyrosine kinase/p21ras/MAP-kinase pathway activation by estradiol-receptor complex in MCF-7 cells. EMBO J. 15:1292–1300. [PMC free article] [PubMed] [Google Scholar]
- Migliaccio, A., D. Piccolo, G. Castoria, M. Di Domenico, A. Bilancio, M. Lombardi, W. Gong, M. Beato, and F. Auricchio. 1998. Activation of the Src/p21ras/Erk pathway by progesterone receptor via cross-talk with estrogen receptor. EMBO J. 17:2008–2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migliaccio, A., G. Castoria, M. Di Domenico, A. de Falco, A. Bilancio, M. Lombardi, M.V. Barone, D. Ametrano, M.S. Zannini, C. Abbondanza, and F. Auricchio. 2000. Steroid-induced androgen receptor-oestradiol receptor β-Src complex triggers prostate cancer cell proliferation. EMBO J. 19:5406–5417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olumi, A.F., G.D. Grossfeld, S.W. Hayward, P.R. Carroll, T.D. Tlsty, and G.R. Cunha. 1999. Carcinoma-associated fibroblasts direct tumor progression of initiated human prostatic epithelium. Cancer Res. 59: 5002–5011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson, M.F., H.F. Paterson, and C.J. Marshall. 1998. Signals from Ras and Rho GTPases interact to regulate expression of p21Waf1/Cip1. Nature. 394:295–299. [DOI] [PubMed] [Google Scholar]
- Peterziel, H., S. Mink, A. Schonert, M. Becker, H. Klocker, and A.C. Cato. 1999. Rapid signalling by androgen receptor in prostate cancer cells. Oncogene. 18:6322–6329. [DOI] [PubMed] [Google Scholar]
- Plattner, R., L. Kadlec, K.A. DeMali, A. Kazlauskas, and A.M. Pendergast. 1999. c-Abl is activated by growth factors and Src family kinases and has a role in the cellular response to PDGF. Genes Dev. 13:2400–2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razandi, M., A. Pedram, and E.R. Levin. 2000. Estrogen signals to the preservation of endothelial cell form and function. J. Biol. Chem. 275:38540–38546. [DOI] [PubMed] [Google Scholar]
- Ridley, A. 2001. Rho proteins, PI3-kinases, and monocyte/macrophage motility. FEBS Lett. 498:168–171. [DOI] [PubMed] [Google Scholar]
- Sambrook, A., and D.W. Russell. 2001. Molecular Cloning. A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. A11.3–A11.22.
- Scaife, R.M., S.A. Courtneidge, and W.Y. Langdon. 2003. The multi-adaptor proto-oncoprotein Cbl is a key regulator of Rac and actin assembly. J. Cell Sci. 116:463–473. [DOI] [PubMed] [Google Scholar]
- Song, R.X., R.A. McPherson, L. Adam, Y. Bao, M. Shupnik, R. Kumar, and R.J. Santen. 2002. Linkage of rapid estrogen action to MAPK activation by ERalpha-Shc association and Shc pathway activation. Mol. Endocrinol. 16:116–127. [DOI] [PubMed] [Google Scholar]
- Spector, D.L., R.D. Goldman, and L.A. Leinwand. 1998. Cells: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. 103.5–103.6.
- Verrijdt, G., E. Schoenmakers, A. Haelens, B. Peeters, G. Verhoeven, W. Rombauts, and F. Claessens. 2000. Change of specificity mutations in androgen-selective enhancers. J. Biol. Chem. 275:12298–12305. [DOI] [PubMed] [Google Scholar]
- Vivanco, I., and C.L. Sawyers. 2002. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat. Rev. Cancer. 2:489–501. [DOI] [PubMed] [Google Scholar]
- Zhou, Z.X., C.I. Wong, M. Sar, and E.M. Wilson. 1994. The androgen receptor: an overview. Recent Prog. Horm. Res. 49:249–274. [DOI] [PubMed] [Google Scholar]
- Zhou, Q., R. Nie, G.S. Prins, P.T. Saunders, B.S. Katzenellenbogen, and R.A. Hess. 2002. Localization of androgen and estrogen receptors in adult male mouse reproductive tract. J. Androl. 23:870–881. [PubMed] [Google Scholar]
- Warriar, N., N. Page, M. Koutsilieris, and M.V. Govindan. 1994. Antiandrogens inhibit human androgen receptor-dependent gene transcription activation in the human prostate cancer cells LNCaP. Prostate. 24:176–186. [DOI] [PubMed] [Google Scholar]
- Wilson, C.M., and M.J. McPhaul. 1994. A and B forms of the androgen receptor are present in human genital skin fibroblasts. Proc. Natl. Acad. Sci. USA. 91:1234–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]