

Abstract
Mitochondrial genetic and metabolic stress causes activation of calcineurin (Cn), NFAT, ATF2, and NFκB/Rel factors, which collectively alter the expression of an array of nuclear genes. We demonstrate here that mitochondrial stress–induced activation of NFκB/Rel factors involves inactivation of IκBβ through Cn-mediated dephosphorylation. Phosphorylated IκBβ is a substrate for Cn phosphatase, which was inhibited by FK506 and RII peptide. Chemical cross-linking and coimmunoprecipitation show that NFκB/Rel factor–bound IκBβ forms a ternary complex with Cn under in vitro and in vivo conditions that was sensitive to FK506. Results show that phosphorylation at S313 and S315 from the COOH-terminal PEST domain of IκBβ is critical for binding to Cn. Mutations at S313/S315 of IκBβ abolished Cn binding, inhibited Cn-mediated increase of Rel proteins in the nucleus, and had a dominant-negative effect on the mitochondrial stress–induced expression of RyR1 and cathepsin L genes. Our results show the distinctive nature of mitochondrial stress–induced NFκB/Rel activation, which is independent of IKKα and IKKβ kinases and affects gene target(s) that are different from cytokine and TNFα-induced stress signaling. The results provide new insights into the role of Cn as a critical link between Ca2+ signaling and NFκB/Rel activation.
Keywords: calcineurin; IκBβ; mitochondrial stress signaling; dephosphorylation; NFkB/Rel activation
Introduction
NFκB/Rel family transcription factors are activated under various stress conditions and play critical roles in diverse cellular processes such as growth, development, immune response, inflammation, apoptosis, and oncogenesis (Beg and Baldwin, 1993; Grilli et al., 1993; Pahl, 1999). The active factors consist of homo- or heterodimers of Rel family proteins (RelA, RelB, c-Rel, p50, and p52), which bind to DNA in a sequence-specific manner through conserved Rel DNA-binding domain and mediate transcription activation or repression of target genes (Beg and Baldwin, 1993; Israel, 1995; Pahl, 1999). IκBα and IκBβ are the two major cytoplasmic inhibitory proteins, along with other less characterized factors including IκBγ, IκBɛ, and Bcl-3, which bind to Rel proteins and restrict their nuclear entry.
Major differences between IκBα and IκBβ factors have been reported in terms of tissue/cell distribution, rates of synthesis/degradation, response to different stimuli both at transcription and posttranslation levels, mechanisms by which they sequester Rel factors in the cytoplasm, and finally, their nuclear entry and exit (Chu et al., 1996; Johnson et al., 1999). Some studies show that IκBβ plays a more important role in the constitutive phase of NFκB/Rel function, whereas IκBα regulates the stress-induced activation of the factors (Tam and Sen, 2001). IKK-dependent phosphorylation at the NH2-terminal sites (Ser32 and -36 in the case of IκBα and S19 and -23 in the case of IκBβ) is an important regulatory step for ubiquitin-dependent degradation of these inhibitory factors and activation of NFκB/Rel (Ghosh and Karin, 2002). Recently, Fenwick et al. (2000) also showed that members of κB-Ras G proteins bind to IκBβ and regulate the rate of its degradation. Phosphorylation at the COOH-terminal acidic PEST domain has also been shown to be important for the function of IκBβ (Chu et al., 1996; McKinsey et al., 1997; Tran et al., 1997), though unphosphorylated inhibitory protein seems to bind to Rel proteins with equal efficiency (Thompson et al., 1995; Chu et al., 1996; Weil et al., 1997). However, the precise mechanisms of inactivation of IκBβ and the release of NFκB/Rel dimeric proteins remain unclear.
The possible regulation of the NFκB/Rel pathway by Ca2+ signaling was indirectly implied in studies showing that transcription activation of IL-2, an NFκB target gene, was adversely affected by known inhibitors of calcineurin (Frantz et al., 1994; Chu et al., 1996). However, the details of this pathway remain unclear. The Ca2+ and calmodulin-dependent phosphatase calcineurin (Cn)* plays important roles in various physiological and pathological processes including T cell activation, Ca2+-induced apoptosis, endocytosis of synaptic vesicles, muscle development, and skeletal and cardiac muscle hypertrophy (Clipstone and Crabtree, 1992; Molkentin et al., 1998; Crabtree, 1999; Lai et al., 1999; Wang et al., 1999; Rusnak and Mertz, 2000). Cn elicits these varied physiological functions by dephosphorylation of key phosphoproteins of the pathways (Crabtree, 1999; Li et al., 2000). A classic example of Cn-mediated activation is dephosphorylation of transcription factor NFATc, facilitating the nuclear localization of the active factor (O'Keefe et al., 1992; Beals et al., 1997).
Recently, we described a novel mitochondria to nucleus stress signaling in C2C12 rhabdomyocytes and human pulmonary A549 cells that involves altered Ca2+ fluxes (Biswas et al., 1999; Amuthan et al., 2002) and altered expression of several nuclear gene targets (Amuthan et al., 2001). We showed that depletion of mitochondrial DNA (mtDNA) or treatment with mitochondrial-specific inhibitors, leading to the disruption of mitochondrial membrane potential (ΔΨm), results in elevated steady-state Ca2+ ([Ca2+]c). These changes were also accompanied by a three- to fivefold increase in cytoplasmic Cn, increased nuclear NFATc level, increased cytoplasmic IκBα, and reduced RelA (p65) in the nucleus (Biswas et al., 1999). In the present study, we show that nuclear p50 and cRel are markedly increased in cells subjected to mitochondrial stress, suggesting the activation of an alternate pathway. We demonstrate that an increase in Cn activity and attendant inactivation of IκBβ are distinctive features of the mitochondrial stress–induced activation of NFκB/Rel proteins. These results for the first time provide a critical link between mitochondrial stress, Ca2+ signaling, and activation of NFκB/Rel factors. Results also demonstrate the novel features of the mitochondrial stress signaling that are distinctly different from the known cytokines and TNFα-induced signaling.
Results
Activation of NFκB/Rel factors by mitochondrial stress and by overexpression of CnA in C2C12 cells
Recently, we generated a series of C2C12 cell lines containing varying levels of mtDNA by treatment with ethidium bromide (Biswas et al., 1999). mtDNA-depleted cells and also control cells treated with carbonyl cyanide-m chlorophenylhydrazone (CCCP), a mitochondria-specific ionophore, showed disrupted ΔΨm and elevated [Ca2+]c. These changes were reversed to near control cell levels in reverted cells, whose mtDNA content was brought back to >70% of control cells by growing them in the absence of ethidium bromide. In the present study, we investigated mechanisms of mitochondrial stress–induced activation of NFκB/Rel.
Immunoblot in Fig. 1 A shows that the cytoplasmic IκBα increased two- to threefold in mtDNA-depleted C2C12 cells, whereas IκBβ reduced markedly and returned to near control cell levels in reverted cells. Additionally, the nuclear cRel and p50 levels were increased markedly in mtDNA-depleted cells and returned back to near control cell level in reverted cells. In keeping with its chaperone-like function (Tam and Sen, 2001), a significant amount of IκBα was detected in the nuclear extract from control cells that was reduced by 50–60% in the mtDNA-depleted cells (Fig. 1 A). However, there was no detectable IκBβ in the nuclear fractions of all three cell types. The levels of Na+/K+ ATPase and nuclear transcription factor YY1 used as loading controls for postnuclear and nuclear fractions, respectively, did not vary by mtDNA depletion and reversal. Finally, there was no detectable YY1 in the postnuclear fraction and Na+/K+ ATPase in the nuclear fraction, indicating the purity of the subcellular fractions used.
Figure 1.


Cytoplasmic and nuclear levels of IκBα and IκBβ in control, mtDNA-depleted, and reverted C2C12 cells. (A) Postnuclear or nuclear protein fractions (30 μg protein each) from control, mtDNA-depleted, and reverted C2C12 cells were subjected to immunoblot analysis using the antibodies indicated in the figure. YY1 and Na+/K+ ATPase were used as loading controls and also for assessing the levels of cross-contamination. Numbers in parentheses underneath stained bands represent relative band intensities. (B) Cells transfected with IκBα or IκBβ cDNAs were stained with antibodies to respective proteins and secondary antibodies Alexa 594 anti–mouse and Alexa 488 anti–rabbit for IκBα and IκBβ, respectively. One set of cells in each case was treated with LMB, 5 nM for 4 h, as indicated.
The lack of significant IκBβ in the nuclear compartment was further investigated by immunofluorescence microscopy of C2C12 cells treated with leptomycin B (LMB), an inhibitor of CRM1-mediated export of protein from nucleus to cytoplasm. As shown in Fig. 1 B, in both control and LMB-treated C2C12 cells there was no significant nuclear staining with antibody to IκBβ (Fig. 1 B). IκBα antibody yielded a distinctly different staining pattern. An intense nuclear staining was observed in cells treated with LMB, and mostly cytoplasmic staining was observed in control cells. These results support the conclusion that, unlike IκBα, IκBβ is not translocated to the nuclear compartment in C2C12 myocytes.
Immunoblot in Fig. 2 A shows a fivefold reduced cytoplasmic IκBβ and three- to sixfold increased nuclear p50 and cRel levels in CCCP-treated C2C12 cells and in mtDNA-depleted cells. In addition, it shows that all of these changes are sensitive to FK506, a Cn inhibitor (O'Keefe et al., 1992) and also Ca2+ chelator EGTA/AM. Therefore, we investigated the role of Cn in IκBβ inactivation and associated nuclear translocation of NFκB/Rel factors by overexpressing CnAα, the catalytic subunit of the enzyme in control C2C12 myocytes. FK506 treatment was used as a control to ascertain the role of Cn in IκBβ inactivation. Immunoblot in Fig. 2 B shows that overexpression of wild-type CnAα (CnA) caused a 40% reduction in cytoplasmic IκBβ levels but no reduction of IκBα. Furthermore, addition of 10 nM FK506 resulted in increased IκBβ in the cytoplasm of both control cells and cells transfected with CnA cDNA constructs. The reduced IκBβ in cells overexpressing CnA is likely due to increased degradation, since we did not detect any increase in the nuclear IκBβ (unpublished data). The results also suggest that the basal Ca2+ (∼75 nM), prevalent in control C2C12 cells (Biswas et al., 1999), is sufficient to activate ectopically expressed CnA.
Figure 2.
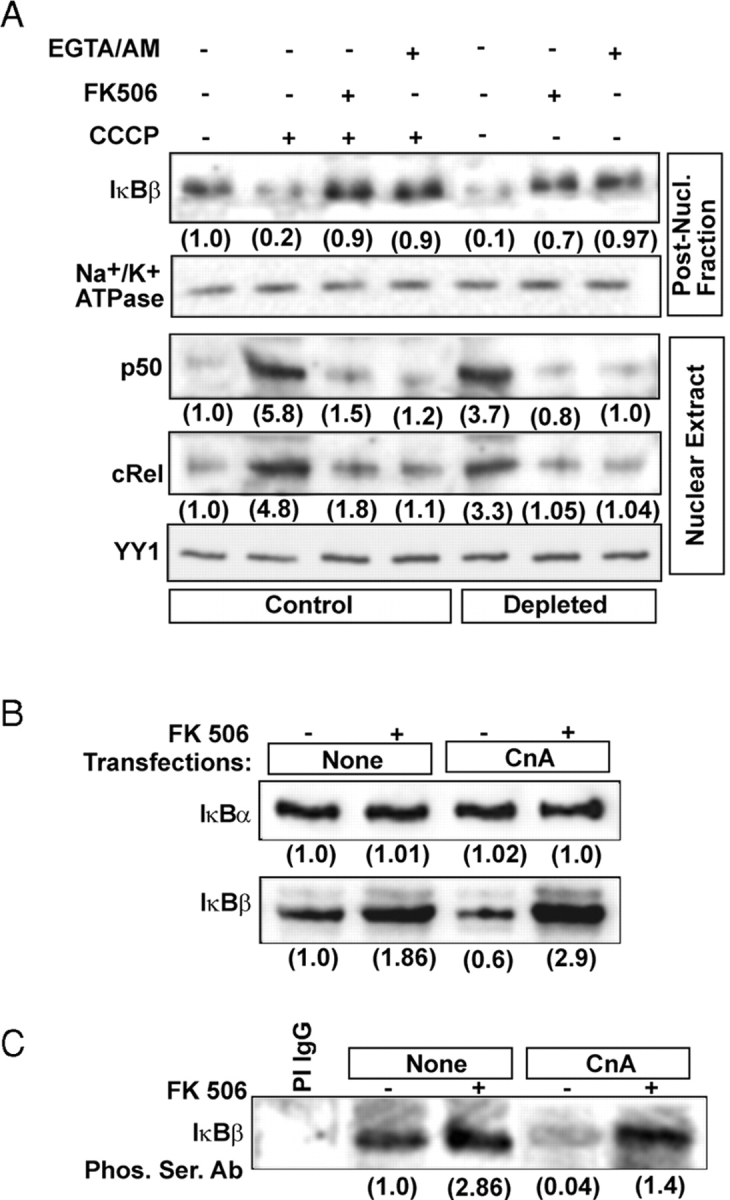

Role of Ca2 + and Cn on dephosphorylation of IκBβ and steady-state levels of NFκB/Rel proteins. (A) Control and CCCP-treated (25 μM for 2 h) C2C12 cells were incubated with or without added FK506 (10 nM) or EGTA/AM (30 μM). Alongside mtDNA-depleted C2C12 cells were also incubated for 2 h with or without inhibitors. Nuclear and postnuclear protein fractions (30 μg each) were subjected to immunoblot analysis using indicated antibodies. (B) Postnuclear fractions (30 μg each) of C2C12 cells transfected with CnA cDNA and treated with FK506 as in A were analyzed by immunoblot analysis using antibodies against IκBα or IκBβ. (C) Postnuclear fractions (0.5 mg protein) from control and transfected cells were immunoprecipitated with IκBβ antibody as described in Materials and methods. The immunoprecipitates were subjected to immunoblot analysis using Ser-phosphate antibody. (D) The in vivo effects of CnA on IκBβ phosphorylation. C2C12 cells transfected with WT and ΔPEST domain mutant IκBβ or CnA cDNAs were labeled with 32P-orthophosphate and treated with or without FK506 (10 nM) as described in Materials and methods. The top panel shows the extent of 32P labeling of IκBβ detected by autoradiography, and the middle and bottom panels show the immunoblot analyses for the levels of IκBβ and CnA, respectively. The numbers in parentheses in A–D underneath the gel bands show relative band intensities determined by imaging through Bio-Rad Laboratories Fluor-S Imager.
Fig. 2 C shows the steady-state phosphorylation status of IκBβ in cells transfected with CnA in the presence or absence of FK506. In control cells, the level of phosphorylated IκBβ increased about threefold after FK506 treatment. The level was reduced by >20-fold in cells overexpressing CnA, which was nearly completely reversed by FK506 treatment. The role of Cn in the dephosphorylation of IκBβ under in vivo conditions was investigated by metabolic labeling of endogenous or ectopically expressed IκBβ with 32P-orthophosphate. Fig. 2 D shows that phosphorylation of endogenous IκBβ was markedly inhibited by overexpressing CnA, which was effectively reversed by FK506 (lanes 1–3). Similarly, phosphorylation of ectopically expressed flag-tagged IκBβ was inhibited by cotransfection with CnA cDNA, which was effectively reversed by FK506. The PEST domain deletion mutant of IκBβ was phosphorylated at very low levels compared with WT protein. The phosphorylation of mutant protein was not affected by either overexpression of CnA or treatment with FK506 and may represent nonspecific phosphorylation at sites outside the PEST domain. These results suggest that IκBβ is subject to Cn-mediated dephosphorylation of COOH-terminal PEST domain sites.
Immunoblot in Fig. 3 A shows that transfection with CnA cDNA caused about a threefold increase in cytoplasmic CnA level and about a twofold increase in nuclear p65, p50, cRel, and NFATc levels. FK506 caused a 30–40% reduction in nuclear p65 and a more pronounced reduction (70–80%) of nuclear cRel, p50, and NFATc. Overexpression of IκBβ resulted in a 70–80% reduction in nuclear cRel and p50 and a 25–30% reduction of RelA (p65) (unpublished data). YY1 and Na+/K+ ATPase used as controls for nuclear and postnuclear proteins, respectively, were not detected significantly in reciprocal cell fractions, suggesting their purity. These results suggest that Cn plays a role in modulating the activity of NFκB/Rel family factors.
Figure 3.


Increased Cn and mitochondrial stress induce nuclear Rel protein levels. Control C2C12 cells were transfected with Cn cDNA or treated with CCCP (25 μM for 2 h) in the presence or absence of added FK506 (10 nM). (A) Nuclear and postnuclear cell fractions (30 μg protein each) were subjected to immunoblot analysis with indicated antibodies. Values in parentheses under each panel show relative band intensities. YY1 and Na+/K+ ATPase antibodies were used to monitor protein loading and cross-contamination between the two subcellular fractions. (B–E) Gel mobility shift analysis with nuclear extracts from control or transfected C2C12 cells or mtDNA-depleted cells. In B, D, and E, NFκB consensus probe was used, whereas in C ZBP-89 DNA was used as a probe. In B, supershift with indicated antibody (1–2 μl antibody) was performed. Treatment with FK506 and CCCP were as in Fig. 2.
The nuclear NFκB/Rel levels under different conditions were also estimated by gel mobility shift analysis using different DNA probes. The gel pattern in Fig. 3 B shows that nuclear extract from mtDNA-depleted cells yielded a more intense complex with NFκB DNA probe than the control nuclear extract. The DNA-bound complexes in both cases were competed by a 20-fold excess unlabeled probe. Fig. 3 C shows that under the same reaction condition the nuclear extracts from control, mtDNA-depleted, and CCCP-treated C2C12 cells yielded complexes of comparable intensity with DNA probe specific for a ubiquitously expressed transcription factor, ZBP-89 (Feo et al., 1995). These results indicate that variations in the extent of protein binding to NFκB DNA in Fig. 3 B is not due to variations in the input proteins. The gel shift pattern in Fig. 3 D shows that transfection with CnA cDNA yields an intense NFκB-specific complex comparable to that of nuclear extract from mtDNA-depleted cells (Fig. 3 B). As expected, the DNA-bound complex was supershifted with antibodies to p65, p50, and cRel. Treatment of transfected cells with FK506 caused a drastic reduction in complex formation, confirming the role of Cn in increased DNA binding in these cells. The gel pattern in Fig. 3 E shows that nuclear extract from CCCP-treated cells also yielded a more intense NFκB complex compared with extract from control C2C12 cells. These results are consistent with the results of immunoblot analysis in Fig. 2 A and Fig. 3 A.
Physical interaction and dephosphorylation of IκBβ in vitro by calcineurin
Dephosphorylation of IκBβ by Cn was tested in vitro using purified Cn and 32P-labeled IκBβ. Fig. 4 A shows that Cn alone had a negligible effect when the reaction was performed at 4°C for 10 min. Under these conditions, a combination of Cn and calmodulin had a marginal effect (Fig. 4 A, lane 4), which was enhanced by adding Ca2+ and Mn2+ (Fig. 4 A, lane 5). However, parallel reactions run at 30°C for 5 min yielded a 12–50-fold more pronounced dephosphorylation of IκBβ (Fig. 4 A, lanes 6 and 7), which was Cn dependent (Fig. 4 A, lanes 7 and 8). Immunoblot analysis of parallel reactions (Fig. 4, A and B, bottom panels) showed comparable levels of IκBβ, suggesting no protein degradation during in vitro incubation. Furthermore, dephosphorylation of IκBβ by Cn was inhibited by RII peptide in a dose-dependent manner (Fig. 4 B). These results show for the first time that Cn plays a direct role in the dephosphorylation of IκBβ.
Figure 4.
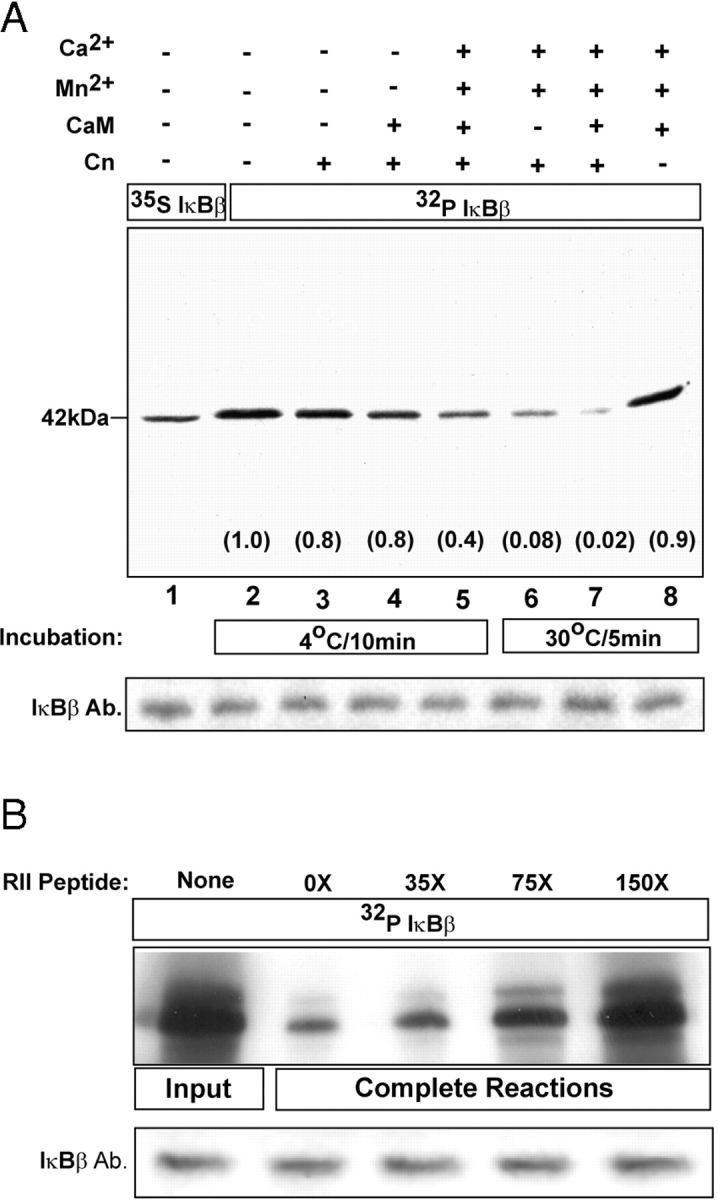
Dephosphorylation of IκBβ by calcineurin. In vitro reactions were run using purified calcineurin and 32P-labeled IκBβ (A) as described in Materials and methods. Assays were performed in duplicate at 4 or 30°C with indicated additions. (B) Dose- dependent inhibition of Cn-mediated dephosphorylation of 32P IκBβ by RII peptide (0 to 150 molar excess). For both A and B, one set of reaction was immunoprecipitated with IκBβ antibody, while a duplicate set was subjected to immunoblot analysis.
Phosphorylation of COOH-terminal PEST domain is critical for the in vivo interaction of IκBβ with calcineurin
If active phosphorylated IκBβ binds to Cn under in vivo conditions, one would expect Rel family proteins to also be part of this complex. This possibility was tested by chemical cross-linking of ectopically expressed human IκBβ using reversible cross-linker. The flag-tagged IκBβ formed two complexes, one migrating with an apparent molecular mass of 250 kD and the second with ∼150 kD (Fig. 5 , A–E). The 250-kD complex contained IκBβ, cRel, p50, RelA, and CnA (Fig. 5, A–E) as tested by immunoblot analysis. Although not shown, this complex also contained CnB and calmodulin. The faster migrating 150-kD complex, on the other hand, contained IκBβ and all three Rel proteins (Fig. 5, A–C and E) but no detectable Cn (Fig. 5 D). These complexes could be dissociated into individual protein subunits by treatment with 1 mM DTT (unpublished data). Based on their size, we infer that each complex contains one homo or heterodimer of Rel proteins. Both the 250- and 150-kD complexes also lacked significant IκBα (Fig. 4 F). However, analysis of an equivalent aliquot of cytoplasmic protein showed the presence of IκBα in the input protein. FK506 selectively inhibited the formation of the 250-kD complex but had no effect on the formation of the 150-kD complex. Transfection with PEST II domain deletion construct, hIκBβ/ΔPEST, resulted in vastly reduced 250-kD complex, whereas the level of the 150-kD complex was not affected. Similarly, mutation at S313,315A (hIκBβ/S313,315A) caused a marked reduction in the 250-kD complex to a level similar to that observed with the PEST domain–deleted protein, without altering the level of the 150-kD complex. On the other hand, mutation at S315 (hIκBβ/S315A) only marginally reduced the level of the 250-kD complex. It should be noted that the complexes with ΔPEST and S313,315A mutant hIκBβ did not contain detectable Cn (Fig. 5 D) even when the blot was overdeveloped (unpublished data). These ectopically expressed mutant IκBβ proteins, however, formed a minor 250-kD complex as detected by antibodies to p50, cRel, and IκBβ (Fig. 5, A–C and E). These latter complexes are probably due to interaction of IκBβ with other isoforms of Cn. The results show that Rel protein–bound IκBβ forms a ternary complex with Cn under in vivo conditions and that phosphorylation of PEST domain, particularly at the S313/315 positions, are important for the IκBβ–NFκB complex to bind to Cn. The PEST domain mutations did not have adverse effects on IκBβ binding to NFκB/Rel proteins as seen by no change in the level of the 150-kD complex. Results also show that FK506 inhibits the interaction of IκBβ with Cn in the formation for the 250-kD complex but does not interfere with the interaction of IκBβ with NFκB/Rel proteins. We believe that the 150-kD complex represents a static pool of the IκBβ–Rel complex that can be converted to the dynamic 250-kD complex by PEST domain phosphorylation.
Figure 5.


Ectopically expressed IκBβ forms a ternary complex with Cn and Rel family proteins. C2C12 cells were transfected with flag-tagged WT, S313,315A, or PEST domain–deleted hIκBβ cDNAs, and cytosolic protein factions (1 mg protein each) of transfected cells treated with or without FK506 (10 nM) were subjected to cross-linking with DSP as described in Materials and methods. The cross-linked products were immunoprecipitated with anti-flag antibody, and the immunoprecipitates were subjected to immunoblot analysis (A–F) with indicated antibodies. In F, an aliquot of the cross-linked product (50 μg protein) was subjected to immunoblot analysis with antibody against IκBα to ensure that the extract indeed contained the protein. (G) Physical interaction of IκBβ with Cn and Rel factors was tested by the antibody pull-down assay. Cytosolic fractions (1 mg protein each) from control C2C12 cells were immunoprecipitaed with Cn, IκBβ, or IκBα, and the immunoprecipitates were probed with a combination of indicated antibodies. (H) Interaction of IκBβ with Cn through its PEST domain was tested by antibody pull-down assay. Postnuclear fraction from C2C12 cells transfected as in G was subjected to immunoprecipitation with flag antibody, and the immunoprecipitates were probed with antibodies to IκBβ and CnAα. Numbers in parentheses in H indicate relative band intensities.
The difference in the binding affinities of IκBβ and IκBα with Cn was further ascertained by the antibody pull-down assay (Fig. 5 G). Antibody to CnA pulled down IκBβ (Fig. 5 G, lane 1), but not IκBα, at a detectable level (Fig. 5 G, lane 2). Similarly, IκBα antibody failed to coimmunoprecipitate CnA (Fig. 5 G, lane 3), whereas IκBβ antibody coimmunoprecipitated CnA (Fig. 5 G, lane 4). Lanes 5–7 show that the complex pulled down by CnA antibody also contained p65, p50, and cRel, further confirming that Cn physically interacts with the IκBβ–NFκB complex.
The role of COOH-terminal PEST domain in binding to Cn was further tested by the antibody pull-down assay. Cytoplasmic protein from C2C12 cells transfected with various hIκBβ cDNA constructs was immunoprecipitated with flag antibody, and the immunoprecipitates were analyzed for the level of Cn and IκBβ. Immunoblot in Fig. 5 H shows that flag antibody immunoprecipitated comparable levels of IκBβ from cells expressing WT and mutant (ΔPEST, S313/315A and S315A) constructs. The level of Cn coimmunoprecipitated with different IκBβ proteins varied, however. The WT IκBβ pulled down the highest level of CnA, whereas its level was significantly reduced by 60% with the S315A mutant IκBβ. However, in the case of ΔPEST and S313,315A mutant proteins, no significant CnA was pulled down. Although not shown, immunoprecipitation of in vitro translation products yielded a similar pattern of coimmunoprecipitation. These results further support the possibility that IκBβ binds to CnA through its COOH-terminal PEST domain, and S313 and 315 residues are critical for this binding.
Role of Cn and IκBβ in mitochondria to nucleus stress signaling
The physiological significance of IκBβ's interaction with Cn was investigated using the muscle tissue from CnAα knockout mouse (Zhang et al., 1996) based on the rationale that reduced Cn should result in increased cytoplasmic IκBβ and reduced nuclear NFκB/Rel. RNase protection analysis showed that the skeletal muscle from adult CnAα2/− mice contained significantly reduced CnAβ, and CnAγ mRNAs (unpublished data). Muscle extracts from CnAα2/− mouse used in this study had very low Cn activity. Immunoblot in Fig. 6 A shows detectable CnA in skeletal muscle from wild-type but not CnAα2/− mice. As expected, the nuclear extract from skeletal muscle of CnAα2/− mouse showed no detectable NFATc. The level of IκBα remains the same in muscle tissue from wild-type and CnAα2/− mice, whereas the level of antibody reactive IκBβ increased by approximately two- to threefold. Additionally, the nuclear p65 level was reduced by ∼60%, whereas the p50 and cRel levels were reduced by >70–80% in the CnAα2/− mouse tissue. These results support the view that Cn plays an important role in the regulation of the NFκB pathway in vivo.
Figure 6.
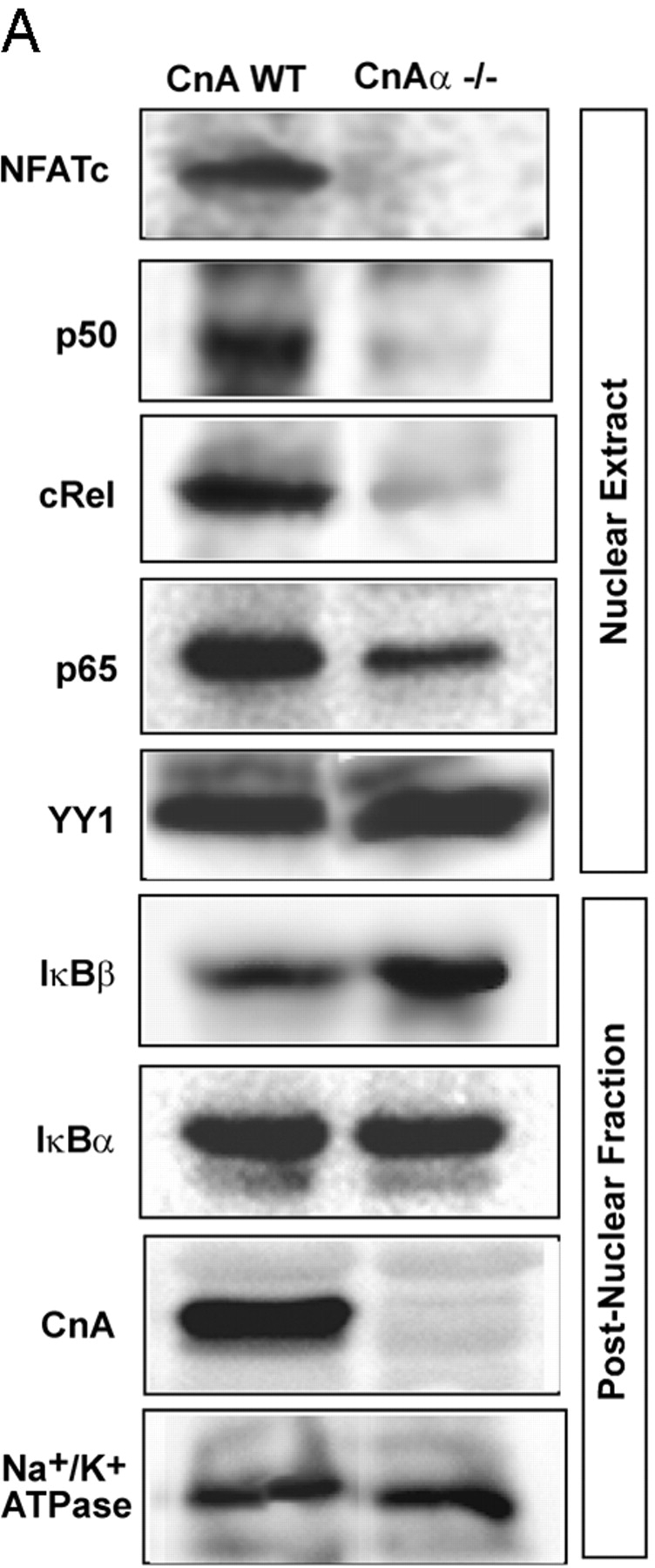

Levels of mitochondrial stress–induced NFκB pathway proteins in CnAα− / − muscle and cells treated with Ca 2+ chelators. (A) The nuclear and postnuclear fractions (25 μg protein each) of skeletal muscle tissues from wild-type and CnAα−/− mice were analyzed by immunoblot analysis with the indicated antibodies. Other details are as in the legend to Fig. 1 and as described in Materials and methods. (B) Control and mtDNA-depleted C2C12 cells treated with or without EGTA/AM and FK506 were fractionated, and postnuclear fractions were subjected to immunoblot analysis with the indicated antibodies. Values in the parentheses in B indicate relative band intensities.
Previously, we showed that ryanodine receptor (RyR1) Ca2+ channel and cathepsin L genes are up-regulated in mtDNA-depleted C2C12 cells (Biswas et al., 1999; Amuthan et al., 2001, 2002). In the present study, we assessed the role of increased [Ca2+]c in induced expression of these two potential target genes using EGTA/AM, a Ca2+ chelator, and FK506, an inhibitor of Cn. RyR1 and cathepsin L levels are increased >10-fold in both CCCP-treated and mtDNA-depleted C2C12 cells compared with untreated cells (Fig. 6 B). In support of the results in Fig. 6 A, FK506 effectively blocked the increase in both CCCP-treated and mtDNA-depleted cells. EGTA/AM also effectively blocked the increase of these proteins in both CCCP-treated and mtDNA-depleted cells. These results further support the hypothesis that mitochondrial stress operates through Ca2+ and camodulin-mediated Cn activation.
The distinctive nature of mitochondrial stress signaling and the involvement of IκBβ in the signaling cascade were investigated using fibrosarcoma cell line HT1080I, which carries a superrepressor mutant of IκBα with vastly diminished phosphorylation and proteasome-mediated degradation (Wang et al., 1996). The superrepressor protein binds to NFκB/Rel proteins with higher affinity, inhibiting their nuclear translocation and DNA binding (Wang et al., 1998). The mutant cell line HT1080I is sensitive to TNFα-mediated apoptosis because the IκBα pathway is refractory to stress response. The HT1080V cells carrying wild-type IκBα exhibit normal response to TNFα (Wang et al., 1998).
Immunoblot in Fig. 7 A shows that, as reported before for C2C12 cells, induction of mitochondrial stress by CCCP treatment caused a reduction in postnuclear IκBβ in both HT1080V and HT1080I cell lines. On the other hand, treatment with TNFα did not alter the level of cytoplasmic IκBβ. The cytoplasmic IκBα level was increased twofold by CCCP treatment only in the wild-type cells but had no effect on the mutant HT1080I cells. Similarly, TNFα treatment drastically reduced the cytoplasmic IκBα level in wild-type cells but had no effect on mutant cells. The effect on cytoplasmic IκBα became apparent in less than 1 h of TNFα treatment and remained unchanged up to 4 h of treatment (unpublished data). The cytoplasmic CnA was induced in both cell types by CCCP but not by TNFα. CCCP treatment caused a reduction in cytoplasmic cRel and p50 levels with concomitant increase of these proteins in the nuclear compartment in both cell lines. The nuclear cRel and p50 were increased in response to TNFα in wild-type cells but did not increase in mutant cell line. Finally, the nuclear fraction from both cell lines contained no detectable IκBβ. The levels of Na+/K+ ATPase used as a loading control for postnuclear fraction, and YY1 for nuclear fraction, did not vary under these conditions. These results show that both mutant and wild-type cells respond similarly to CCCP-induced mitochondrial stress, and Cn and NFκB/Rel were activated in both cases. Furthermore, results show that TNFα signaling does not involve a change in Cn level and Cn-mediated inactivation of IκBβ.
Figure 7.
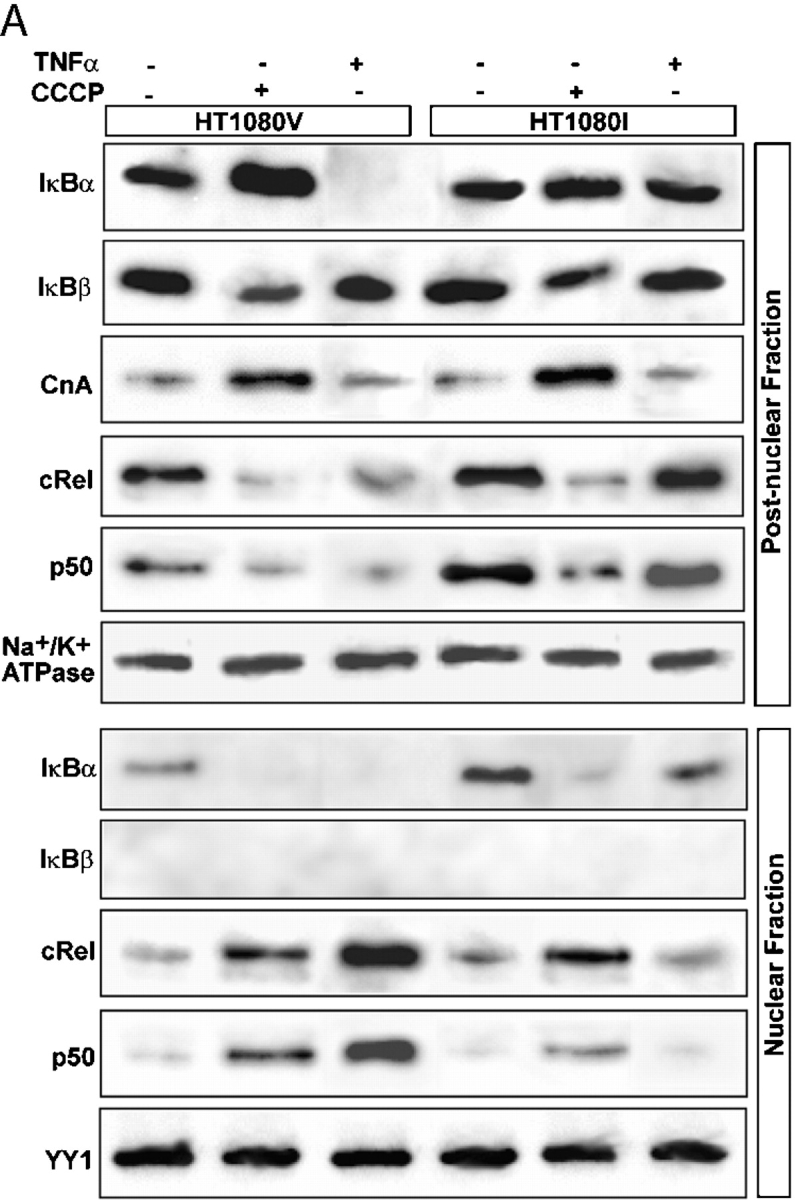

Distinctive features of mitochondrial stress–induced NFκB activation. (A) Postnuclear fractions (30 μg protein each) of HT1080V and HT1080I cells treated with or without CCCP or TNFα as described in the legend to Fig. 6 were subjected to immunoblot analysis with indicated antibodies. Results show that both RyR1 and cathepsin L are induced only by treatment with CCCP, not TNFα. (B) Control 3T3, IKKα2/−, and IKKβ2/− fibroblast cells were treated with or without CCCP (25 μM) as in A, and nuclear and cytoplasmic protein fractions (30 μg each) were subjected to immunoblot analysis with indicated antibodies. In both A and B, levels of YY1 and Na+/K+ ATPase were used as loading controls for nuclear and postnuclear fractions, respectively.
Activation of NFκB is regulated by several different kinases, although IKKα- and IKKβ-mediated phosphorylation is believed to be critical for the cytokine and TNFα-induced activation of NFκB (Zandi et al., 1998; May et al., 2000; Ghosh and Karin, 2002). We therefore tested the roles of these kinases in mitochondrial stress–induced activation of NFκB using IKKα2/− and IKKβ2/− cell lines. Fig. 7 B shows that CCCP-mediated mitochondrial stress in these cells and also control fibroblasts caused reduced cytoplasmic Rel protein levels and also IκBβ but had no effect on the cytoplasmic IκBα. Similarly, nuclear cRel, p50, and p65 in all three cell types were increased two- to fivefold in response to mitochondrial stress. These results suggest that mitochondrial stress–induced activation of NFκB may occur through a pathway other than that involving IKKα and IKKβ kinases.
Distinctive nature of TNFα and mitochondrial stress–induced NFκB targets
Previous studies from our laboratory showed that mitochondrial stress induced the expression of several marker genes (Biswas et al., 1999; Amuthan et al., 2001, 2002). In the present study, using RyR1 and cathepsin L genes as markers, we investigated whether TNFα-mediated and mitochondrial stress–induced signaling pathways affect the same or different gene targets. Immunoblot in Fig. 8 A shows that expression of both RyR1 Ca2+ channel and cathepsin L proteins were induced by CCCP treatment in both HT1080V and HT1080I cells, but the level of IP3 channel protein remained the same (Fig. 8 A). TNFα-mediated stress signaling in both HT1080V and HT1080I cells failed to induce the expression of RyR1 and cathepsin L proteins, further demonstrating the distinctive nature of the two signaling pathways. Although not shown, the induction was at the transcription level as indicated by increased mRNA levels. Furthermore, overexpression of ΔPEST domain or S313A mutants of IκBβ in cells treated with CCCP resulted in complete reversal of both RyR1 and cathepsin L gene expression (Fig. 8 B). However, overexpression of S315A IκBβ resulted in partial reversal (Fig. 8 B). These results provide direct evidence that Cn-mediated inactivation of IκBβ is critical for mitochondrial stress–induced activation of marker genes.
Figure 8.



Distinctive nuclear gene targets of mitochondrial stress signaling and involvement of IκBβ. (A) Postnuclear fractions (30 μg protein each) of HT1080V and HT1080I cells treated with or without CCCP or TNFα as described in the legend to Fig. 7 were subjected to immunoblot analysis with indicated antibodies. Results show that both RyR1 and cathepsin L are induced only by treatment with CCCP, not TNFα. (B) Control HT1080V cells or cells subjected to mitochondrial stress by treatment with CCCP (25 μM for 1 h) were transfected with wild-type or mutated IκBβ cDNA constructs as indicated. Postnuclear extracts were subjected to immunoblot analysis with indicated antibodies. (C) Immunoblot analysis of postnuclear fractions (30 μg each) of control and mtDNA-depleted C2C12 cells transfected with CnWT and hIκBβ/S313,315A constructs. The blots were developed with indicated antibodies. Numbers in parentheses indicates relative band intensities. (D) Nuclear and cytoplasmic fractions (30 μg each) of control C2C12 cells transfected with the indicated cDNAs were subjected to immunoblot analysis with antibodies to various Rel proteins or IκBβ.
The ability of PEST domain–mutated IκBβ in impeding the propagation of mitochondrial stress signaling was further tested in control C2C12 cells overexpressing CnA and also mtDNA-depleted cells that contain elevated Cn activity as follows. Initially, the effect on the marker gene expression was tested. Immunoblot in Fig. 8 C shows that transfection of control C2C12 cells with CnA yielded increased steady-state levels of RyR1 and cathepsin L proteins as shown before. The level of IP3 channel protein was not increased under these conditions. Cotransfection with S313,315A mutant IκBβ drastically reversed the levels of antibody reactive RyR1 and cathepsin L proteins in both control and mtDNA-depleted cells. In the second series of experiments, we tested the effect of overexpression of S313,315A mutant IκBβ on the nuclear and cytoplasmic distribution of Rel proteins. Immunoblot in Fig. 8 D shows that transfection of control C2C12 cells with hIκBβWT cDNA resulted in vastly reduced nuclear p65, p50, and cRel levels with simultaneous increases of these factors and also that of IκBβ in the cytoplasm. Cotransfection with CnA cDNA caused a complete reversal with increased Rel proteins in the nucleus and reduced levels in the cytoplasm. There was also a reduced cytoplasmic IκBβ, suggesting that Cn-mediated dephosphorylation may modulate its degradation. Transfection with hIκBβ/S313,315A cDNA had an effect similar to that with IκBβWT construct. However, the inhibitory effect of this mutant was not reversed by cotransfection with CnA. These results further demonstrate that Cn-mediated dephosphorylation is critical for the inactivation of IκBβ and attendant release of NFκB/Rel proteins and that the S313,315A mutant IκBβ has a dominant-negative effect on mitochondrial stress signaling.
Discussion
In recent studies, we described a new mechanism of mitochondria to nucleus stress signaling, which is initiated by mtDNA and/or membrane damage and disruption of ΔΨm. These changes caused a sustained increase in [Ca2+]c and altered expression of an array of nuclear genes (Biswas et al., 1999; Amuthan et al., 2001, 2002). The altered gene expression was also associated with changes in cell morphology and formation of invasive phenotypes in otherwise noninvasive C2C12 rhabdomyosarcoma and A549 lung carcinoma cells (Amuthan et al., 2001, 2002). The mitochondrial stress–induced biochemical changes included increased nuclear p50 and cRel and marginally reduced RelA (p65) factors. In the present study, we investigated whether the mechanism of mitochondrial stress–induced activation of NFκB/Rel factors is distinct from, or overlaps with, TNFα-mediated NFκB/Rel activation. Our results show that mitochondrial stress–induced activation of NFκB/Rel factors occurs through inactivation of IκBβ by Cn-mediated dephosphorylation and subsequent release of Rel proteins for nuclear translocation. We also demonstrate that phosphorylation at the COOH-terminal PEST region is critical for the mitochondrial stress–induced inactivation of IκBβ and attendant activation of NFκB/Rel factors. It is likely that the κB Ras binding (Fenwick et al., 2000) represents a distinct step of IκBβ regulation, possibly a downstream event regulating its degradation.
Nuclear entry and exit of IκBα and IκBβ are important parts of signal-mediated activation of NFκB/Rel. The nuclear IκBα plays an important role in the exit of Rel factors from the nucleus at the end of cytokine or other signaling events. Furthermore, LMB-sensitive CRM1 serves as an essential vehicle for the exit of nuclear IκBα to the cytoplasm (Turpin et al., 1999; Huang et al., 2000; Tam et al., 2000). IκBβ, on the other hand, lacks the CRM1 binding domain (Turpin et al., 1999; Tam et al., 2000), which raises questions about its nuclear entry and exit. In support of recent studies (Malek et al. 2001; Tam and Sen, 2001), we also failed to detect IκBβ in the nuclear compartments of C2C12 and HT1080 cells (Fig. 1, A and B, and Fig. 7 A). Therefore, IκBβ appears to be a strictly cytoplasmic protein, indicating yet another distinctive feature different from IκBα.
We present multiple lines of evidence for the physical and functional interaction of IκBβ with the catalytic subunit of Cn. Both endogenous and ectopically expressed IκBβ form a ternary complex of ∼250 kD with Cn and NFκB/Rel proteins, as tested by reversible cross-linking with dithiobis(succinimidylpropionate) (DSP). Direct proof of the functional association between IκBβ and Cn comes from in vitro experiments showing that 32P-labeled IκBβ is dephopshorylated by purified Cn in the presence of calmodulin and other necessary cofactors. In vivo and in vitro association of IκBβ with Cn and also its Cn-dependent dephosphorylation are inhibited by FK506 and RII peptide, well-known inhibitors of catalytic function of Cn (O'Keefe et al., 1992; Enz et al., 1994). Notably, the effect of FK506 is targeted to complexes containing Cn but not the 150-kD binary complexes of IκBβ and NFκB/Rel proteins. This is probably the first demonstration of a functional interaction between IκBβ and Cn.
Cn binds to diverse protein substrates including type II cAMP-dependent protein kinase, protein phosphatase inhibitor-1, myosin light chains, α-subunit of phosphorylase kinase, G substrate, DARPP-32, dynamin, tau factor, NMDA receptor, NO synthase, and NFATc (for reviews see Rusnak and Mertz, 2000; Shibasaki et al., 2002) through its phosphatase active site leading to protein dephosphorylation. A sequence domain spanning residues 81–99 of type II cAMP-dependent protein kinase, designated as RII domain, is involved in interaction with the active site of Cn. The RII peptide has been extensively used as a site-specific inhibitor of Cn phosphatase. A common feature among the protein substrates of Cn is the conservation of phosphoprotein sequence similar to the RII domain. The COOH-terminal PEST domain from the mouse and human IκBβ (sequence 306–322) shows ∼80% structural homology (β-sheet structure) of RII peptide (Blumenthal et al., 1986) and contains acidic phosphorylatin sites. Indeed, RII peptide inhibited binding of IκBβ to Cn (Fig. 4 B). Results of chemical cross-linking and antibody pull-down assay (Fig. 5, D and H) show that IκBβ, lacking the COOH-terminal PEST domain, or substituted S313 and S315 CKII target sites from this domain, failed to interact with Cn. Although not shown, even S to E substituted protein failed to bind to Cn, suggesting that phosphorylated Ser residues at 313 and 315 positions are critical for binding. Therefore, binding of IκBβ with Cn appears to vary from that of NFAT family members in two ways. First, the 13-aa segment (Cn binding peptide A) from the NH2-terminal domain of NFAT binds to Cn even in the absence of phosphorylation (Aramburu et al., 1998). Second, IκBβ binding to Cn does not appear to involve a second COOH-terminal site similar to that shown for some members of NFAT proteins (Park et al., 2000). Furthermore, mutations targeted to PEST domain phosphorylation sites of IκBβ also acted as a persistent inhibitor of Cn-mediated activation of NFκB/Rel (Fig. 8).
Previous studies show that phosphorylation at the COOH-terminal PEST domain sites (Thompson et al., 1995; Chu et al., 1996; Schwarz et al., 1996; McKinsey et al., 1997; Tran et al., 1997; Weil et al., 1997) are important for the function of IκBβ. It was shown that dephosphorylation of purified IκBβ or discrete PEST domain mutations affected its ability to bind NFκB/Rel dimmers (Chu et al., 1996; McKinsey et al., 1997). In extension of these studies, we demonstrate that mutations targeted to Ser 313 and 315 of the PEST domain abolished Cn binding but had minimal effect for binding to NFκB/Rel factors. Our results suggest that Cn-mediated dephosphorylation is critical for the inactivation of IκBβ and release of NFκB/Rel.
TNFα, interleukin 1, and other receptor-mediated signaling pathways induce the activation of NFκB/Rel factors mostly through signal-mediated phosphorylation and inactivation of IκBα, without significantly affecting the steady-state levels of IκBβ. Using the HT1080I mutant cell line, which does not respond to TNFα and interleukin-mediated signaling (Wang et al., 1996), we demonstrate that mitochondrial stress–induced activation of NFκB/Rel occurs through a mechanism distinctly different from the TNFα signaling. First, the activation of Cn and reduction in the steady-state level of IκBβ in response to CCCP treatment occur in both wild-type HT1080V cells and mutant HT1080I cell lines. However, TNFα-mediated reduction in IκBα occurs only in HT1080V cells (Fig. 8). Furthermore, TNFα treatment in both cell lines has no effect on the level of IκBβ. Second, mitochondrial stress response genes RyR1 and cathepsin L are induced in both cell types in response to CCCP, a specific inducer of mitochondrial stress but not by TNFα. Results presented in this study therefore suggest that the mitochondrial stress signaling follows a pathway distinctly different from TNFα and interleukin 1 signaling. A direct proof for the involvement IκBβ in the mitochondrial stress–induced signaling comes from experiments showing that ectopic expression of PEST domain–deleted or S313,315A–substituted IκBβ caused a dramatic reduction in the expression of RyR1 and cathepsin L genes in CCCP-treated HT1080V cells and mtDNA-depleted C2C12 cells (Fig. 8). A similar dominant-negative effect of the S313,315A mutant of IκBβ on the mitochondrial stress signaling, reversal of invasive property, and also cell morphology of mtDNA-depleted A549 and C2C12 cells was shown (Amuthan et al., 2002; unpublished data).
Several protein kinases, including CKII, GSK3, PKCδ, PKCθ, IKKα, and IKKβ regulate the activity of NFκB, though the precise mechanisms in some cases is not clear (Ghosh and Karin, 2002). For example, CKII and GSK3 are thought to modulate the DNA-binding activity of the nuclear Rel proteins by subunit phosphorylation (Ghosh and Karin, 2002), though CKII may also activate NFκB by phosphorylation of IκBβ and IκBα at their COOH-terminal PEST domain sites (Chu et al., 1996; McElhinny et al., 1996). Some studies suggest that CKII may phosphorylate free IκBα and Iκ Bβ, whereas IKKs may phosphorylate Rel protein–bound inhibitory proteins (Pando and Verma, 2000). Using a combination of gene knock out and an active site inhibitor of activator protein, NEMO, it was shown that IKKα and IKKβ play critical roles in the activation of NFκB in response to lymphotoxin β, interleukin, and TNFα-induced stress signaling (DiDonato et al., 1997; May et al., 2000; Pando and Verma, 2000; Ghosh and Karin, 2002). In further support of the distinctive nature of the mitochondrial stress–induced activation of NFκB different from TNFα-induced activation described above, CCCP, a potent inducer of mitochondrial stress, induced NFκB activation in IKKα2/− and IKKβ2/− cells. These results further suggest the possibility that the mitochondrial stress–induced activation occurs through a pathway other than that involving the IKKα- and IKKβ-mediated phosphorylation of IκBβ. Alternatively, IKKα- and IKKβ-mediated phosphorylation of COOH-terminal sites of IκBβ may be a downstream event that follows the Cn-dependent dephosphorylation of PEST domain sites.
In summary, our results provide evidence for the distinctive nature of the mitochondria to nucleus stress signaling different from various cytokines and receptor-mediated stress. Results also provide mechanistic insights into a novel-signaling pathway by which NFκB/Rel factors respond to Ca2+- and calmodulin-dependent process as summarized in Fig. 9 . Our working model proposes that activated Cn binds to the cytoplasmic IκBβ–Rel complex and catalyzes the dephosphorylation of IκBβ at its COOH-terminal PEST domain. The Cn-mediated dephosphorylation in turn causes inactivation of IκBβ, resulting in the release of active p50/cRel heterodimers for translocation into the nuclear compartment.
Figure 9.

Model depicting mitochondrial stress–induced activation of Rel/NFκB factors through Cn-mediated dephosphorylation of IκBβ.
Materials and methods
Cell culture and transfection
Control C2C12 murine myoblast cells and the mtDNA-depleted and reverted cells were generated and maintained as described previously (Biswas et al., 1999). HT1080 cells were grown in DME containing 10% FBS and 1× penicillin and streptomycin. Cells were transfected with 5 μg of expression cDNA constructs/100-mm plate using the Fugene 6 (Roche Biochemicals) transfection agent, which routinely yielded >70% transfection efficiency.
Subcellular fractionation and immunoblot analysis
Cultured cells and skeletal muscle tissue were homogenized in homogenization buffer (0.3 M sucrose, 10 mM Tris-HCl, pH 8.0, 10 mM NaCl, 3 mM MgCl2, 0.2 mM EDTA) containing phosphatase inhibitors (1 mM NaVO4, 100 μM molybdic acid, 10 mM NaF) and protease inhibitors (1 mM phenylmethylsulfonyl fluoride, 50 μg/ml each leupeptin, pepstatin, aprotinin, chymostatin, and antipain). Subcellular fractions were isolated by differential centrifugation as described previously (Biswas et al., 1999). Proteins were resolved by electrophoresis on 10 or 12% SDS–polyacrylamide gels (Laemmli, 1970) and subjected to immunoblot analysis as described by Towbin et al. (1979). Blots were developed using Super Signal West Femto maximum sensitivity substrate from Pierce Chemical Co.
Gel mobility shift assays
DNA–protein binding was assayed by gel mobility shift as described before (Amuthan et al., 2001). Binding was performed with 32P end–labeled NFκB consensus DNA (5′-AGTTGAGGGGACTTTCCAGGC-3′) of ZBP-89 DNA (5′-GGGTGGGGGGG-3′). Binding reactions (20 μl) contained ∼0.1–0.2 ng of labeled DNA (10,000–15,000 cpm), 15 μg of nuclear extract, and 2 μg of poly (dI-dC) under conditions described previously (Amuthan et al., 2001). DNA–protein complexes were resolved on 4% nondenaturing polyacrylamide gels in 0.5% × Tris-Glycine (25 mM Tris, 100 mM glycine, 1 mM EDTA, pH 8.3). Competition with 20 molar excess unlabeled DNA and antibody supershift were performed as described (Amuthan et al., 2001).
Immunoprecipitation
Immunoprecipitation was performed by the protein A–Sepharose pull-down method as described previously (Anandatheerthavarada et al., 1999) using 1 mg equivalent of cytosolic proteins from control or transfected cells. The immunoprecipitates were extracted with 2× Laemmli buffer devoid of β-mercaptoethanol at 95°C for 5 min. Immunoprecipitation of in vitro–translated proteins was performed essentially as described previously (Bhat and Avadhani, 1985).
Chemical cross-linking of NFκB with Cn
Cross-linking was performed in vitro using 1 mg of cytosolic proteins diluted to 500 μl volume with 50 mM Hepes (pH 7.5). The cross-linking reaction was performed with 500 μM reversible cross-linker, DSP (Pierce Chemical Co.) at 25°C for 30 min. Reaction was stopped by adding 100 mM Tris buffer (pH 7.2). Cross-linked products were subjected to immunoprecipitation using monoclonal flag antibody and subjected to immunoblot analysis using different antibodies to Rel factors, IκBβ, IκBα, and calcineurin.
Protein phosphatase assay
IκBβ synthesized in vitro with unlabeled amino acids was phosphorylated by adding [γ-32P]ATP and 10 U CKII (New England Biolabs Inc.). Phosphatase assays were performed in 50 μl vol using 32P-labeled proteins (80,000 cpm) as the substrate in 50 mM Hepes (pH 7.5) containing 1 mM CaCl2, 1 mM MnCl2, 3 μM calmodulin (CaM), and 1.5 μg of purified bovine brain Cn (Calbiochem). The reactions were run at 4 or 30°C for 5 min and subjected to immunoprecipitation as described above using IκBβ antibody. Immunoprecipitates were subjected to SDS–polyacrylamide gel, and the gels were imaged using GS525 Molecular Imager (Bio-Rad Laboratories). A parallel set was subjected to immunoblot analysis using IκBβ antibody for protein level.
Metabolic labeling of IκBβ in intact cells
Control C2C12 cells were transfected with CnAWT, flag-tagged IκBβWT, or S313,315A cDNAs, and at 60 h posttransfection, cells were labeled for 4 h with 32Pi (1 mCi/ml) in phosphate-depleted DME with or without added FK506 (10 nM) as described by Chu et al. (1996). Cytosolic proteins (0.5 mg each, 100,000 g supernatant fraction) were immunoprecipitated with either IκBβ or flag antibody. Immunoprecipitates were resolved by 12% SDS-PAGE, and the labeled IκBβ was visualized by autoradiography. Companion gels were subjected to immunoblot analysis using IκBβ and Cn antibodies.
Immunocytochemistry
C2C12 cells were grown on coverslips were transfected with IκBα or IκBβ cDNAs (5 μg) for 60 h. In some cases LMB (5 nM) was added during the last 4 h of culturing. Fixing and staining of cells with primary and secondary antibodies were performed as described previously (Biswas et al., 1999). Confocal microscopy was performed with a TCS laser confocal microscope (Leica).
Acknowledgments
We are thankful to Drs. Stephen J. O'Keefe (Merck & Co. Inc., Rahway, NJ), Jorge Caamano (University of Birmingham Medical School, Birmingham, UK), J.G. Seidman (Harvard Medical School, Boston, MA), Dean W. Ballard (Vanderbilt University, Nashville, TN), Robert S. Carter (Vanderbilt University), A.S. Baldwin Jr. (University of North Carolina, Chapel Hill, NC), C.-Y. Wang (University of Michigan, Ann Arbor, MI), Sankar Ghosh (Yale University, New Haven, CT), and Michael May (University of Pennsylvania) for generously providing the various expression cDNA constructs, cell lines, or transgenic mouse lines used in this study. We also thank Drs. Michael Atchison and Sergei Fuchs for their criticisms and suggestions and to Molly Higgins for editorial help.
This work was supported in part by National Institutes of Health grant CA-22762 to N.G. Avadhani.
G. Biswas and H.K. Anandatheerthavarada contributed equally to this work.
Footnotes
Abbreviations used in this paper: ΔΨm, mitochondrial membrane potential; CCCP, carbonyl cyanide-m chlorophenylhydrazone; Cn, calcineurin; DSP, dithiobis(succinimidylpropionate); LMB, leptomycin B; mtDNA, mitochondrial DNA.
References
- Amuthan, G., G. Biswas, S.Y. Zhang, A. Klein-Szanto, C. Vijayasarathy, and N.G. Avadhani. 2001. Mitochondria-to-nucleus stress signaling induces phenotypic changes, tumor progression and cell invasion. EMBO J. 20:1910–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amuthan, G., G. Biswas, H.K. Ananadatheerthavarada, C. Vijayasarathy, H.M. Shephard, and N.G. Avadhani. 2002. Mitochondrial stress-induced calcium signaling, phenotypic changes and invasive behavior in human lung carcinoma A549 cells. Oncogene. 21:7839–7849. [DOI] [PubMed] [Google Scholar]
- Anandatheerthavarada, H.K., G. Biswas, J. Mullick, N.B. Sepuri, L. Otvos, D. Pain, and N.G. Avadhani. 1999. Dual targeting of cytochrome P4502B1 to endoplasmic reticulum and mitochondria involves a novel signal activation by cyclic AMP-dependent phosphorylation at Ser128. EMBO J. 18:5494–5504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aramburu, J., F. Garcia-Cozar, A. Raghavan, H. Okamura, A. Rao, and P.G. Hogan. 1998. Selective inhibition of NFAT activation by a peptide spanning the calcineurin targeting site of NFAT. Mol. Cell. 1:627–637. [DOI] [PubMed] [Google Scholar]
- Beals, C.R., N.A. Clipstone, S.N. Ho, and G.R. Crabtree. 1997. Nuclear localization of NF-ATc by a calcineurin-dependent, cyclosporin-sensitive intramolecular interaction. Genes Dev. 11:824–834. [DOI] [PubMed] [Google Scholar]
- Beg, A.A., and A.S. Baldwin, Jr. 1993. The IκB proteins: multifunctional regulators of Rel/NF-κB transcription factors. Genes Dev. 7:2064–2207. [DOI] [PubMed] [Google Scholar]
- Bhat, N.K., and N.G. Avadhani. 1985. Transport of proteins into hepatic and nonhepatic mitochondria: specificity of uptake and processing of precursor forms of carbamoyl-phosphate synthetase I. Biochemistry. 24:8107–8113. [DOI] [PubMed] [Google Scholar]
- Biswas, G., O.A. Adebanjo, B.D. Freedman, H.K. Anandatheerthavarada, C. Vijayasarathy, M. Zaidi, M. Kotlikoff, and N.G. Avadhani. 1999. Retrograde Ca2+ signaling in C2C12 skeletal myocytes in response to mitochondrial genetic and metabolic stress: a novel mode of inter-organelle crosstalk. EMBO J. 18:522–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumenthal, D.K., K. Takio, R.S. Hansen, and E.G. Krebs. 1986. Dephosphorylation of camp-dependent protein kinase regulatory subunit (type II) by calmodulin-dependent protein phosphatase. Determinants of substrate specificity. J. Biol. Chem. 261:8140–8145. [PubMed] [Google Scholar]
- Chu, Z.L., T.A. McKinsey, L. Liu, X. Qi, and D.W. Ballard. 1996. Basal phosphorylation of the PEST domain in IκBβ regulates its functional interaction with the c-rel proto-oncogene product. Mol. Cell. Biol. 16:5974–5984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clipstone, N.A., and G.R. Crabtree. 1992. Identification of calcineurin as a key enzyme in T-lymphocyte activation. Nature. 357:695–697. [DOI] [PubMed] [Google Scholar]
- Crabtree, G.R. 1999. Generic signals and specific outcomes: signaling through Ca2+, calcineurin and NF-AT. Cell. 96:611–614. [DOI] [PubMed] [Google Scholar]
- DiDonato, J.A., M. Hayakawa, D.M. Rothwarf, E. Zandi, and M. Karin. 1997. A cytokine-responsive IkappaB kinase that activates the transcription factor NF-kappaB. Nature. 388:548–554. [DOI] [PubMed] [Google Scholar]
- Enz, A., C. Shapiro, and A. Dattler. 1994. Nonradioactive assay for protein phosphatase 2B (calcineurin) activity using a partial sequence of the subunit of cAMP-dependent protein kinase as substrate. Anal. Biochem. 216:147–153. [DOI] [PubMed] [Google Scholar]
- Frantz, B., E.C. Nordby, G. Bren, N. Steffan, C.V. Paya, R.L. Kincaid, M.J. Tocci, S.J. O'Keefe, and E.A. O'Neill. 1994. Calcineurin acts in synergy with PMA to inactivate I kappa B/MAD3, an inhibitor of NF-kappa B. EMBO J. 15:861–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenwick, C., S.Y. Na, R.E. Voll, H. Zhong, S.Y. Im, J.W. Lee, and S. Ghosh. 2000. A subclass of Ras proteins that regulate the degradation of IkappaB. Science. 287:869–873. [DOI] [PubMed] [Google Scholar]
- Feo, S., V. Antona, G. Barbieri, R. Passantino, L. Cali, and A. Giallongo. 1995. Transcription of the human beta enolase gene (ENO-3) is regulated by an intronic muscle-specific enhancer that binds myocyte-specific enhancer factor 2 proteins and ubiquitous G-rich-box binding factors. Mol. Cell. Biol. 15:5991–6002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grilli, M., J.S. Chiu, and M.J. Lenardo. 1993. NF-κB and Rel-participants in a multiform transcriptional regulatory system. Int. Rev. Cytol. 143:1–62. [DOI] [PubMed] [Google Scholar]
- Ghosh, S., and M. Karin. 2002. Missing pieces in the NF-kappaB puzzle. Cell. 109:S81–S96. [DOI] [PubMed] [Google Scholar]
- Huang, T.T., N. Kudo, M. Yoshida, and S. Miyamoto. 2000. A nuclear export signal in the N-terminal regulatory domain of IkappaBalpha controls cytoplasmic localization of inactive NF-kappaB/IkappaBalpha complexes. Proc. Natl. Acad. Sci. USA. 97:1014–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Israel, A. 1995. A role for phosphorylation and degradation in the control of NF-κB activity. Trends Genet. 11:203–205. [DOI] [PubMed] [Google Scholar]
- Johnson, C., D. Van Antwerp, and T.J. Hope. 1999. An N-terminal nuclear export signal is required for the nucleocytoplasmic shuttling of IkapppaBalpha. EMBO J. 18:6682–6693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laemmli, U.K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 227:680–685. [DOI] [PubMed] [Google Scholar]
- Lai, M.M., J.J. Hong, A.M. Ruggiero, P.E. Burnett, V.I. Slepnev, P. De Camilli, and S.H. Snyder. 1999. The calcineurin-dynamin 1 complex as a calcium sensor for synaptic vesicles endocytosis. J. Biol. Chem. 274:25963–25966. [DOI] [PubMed] [Google Scholar]
- Li, L., D. Guerini, and E. Carafoli. 2000. Calcineurin controls the transcription of Na+/Ca2+ exchanger isoforms in developing cerebellar neurons. J. Biol. Chem. 275:20903–20910. [DOI] [PubMed] [Google Scholar]
- Malek, S., Y. Chen, T. Huxford, and G. Ghosh. 2001. IκBβ but not IκBα functions as a classical cytoplasmic inhibitor of NF-κB dimers by masking both NF-κB nuclear localization sequences in resting cells J Biol. Chem. 276:45225–45235. [DOI] [PubMed] [Google Scholar]
- May, M.J., F. D'Acquisto, L.A. Madge, J. Glockner, J.S. Pober, and S. Ghosh. 2000. Selective inhibition of NF-kappaB activation by a peptide that blocks the interaction of NEMO with the IkappaB kinase complex. Science. 289:1550–1554. [DOI] [PubMed] [Google Scholar]
- McElhinny, J.A., S.A. Trushin, G.D. Bren, N. Chester, and C.V. Paya. 1996. Casein kinase II phosphorylates I kappa B alpha at S-283, S-289, S-293, and T-291 and is required for its degradation. Mol. Cell. Biol. 16:899–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinsey, T.A., Z.-L. Chu, and D.W. Ballard. 1997. Phosphorylation of the PEST domain of IκBβ regulates the function of NF-κB/IκBβ complexes. J. Biol. Chem. 272:22377–22380. [DOI] [PubMed] [Google Scholar]
- Molkentin, J.D., J.R. Lu, C.L. Antos, B. Markham, J. Richardson, J. Robbins, S.R. Grant, and E.N. Olson. 1998. A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell. 93:215–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Keefe, S.J., J. Tamura, R.L. Kincaid, M.J. Tocci, and E.A. O'Neill. 1992. FK-506- and CsA-sensitive activation of the Interleukin-2 promoter by calcineurin. Nature. 357:692–694. [DOI] [PubMed] [Google Scholar]
- Pahl, L.H. 1999. Activators and target genes of Rel/NF-κB transcription factors. Oncogene. 18:6853–6866. [DOI] [PubMed] [Google Scholar]
- Pando, M.P., and I.M. Verma. 2000. Signal-dependent and -independent degradation of free and NF-kappa B-bound IkappaBalpha. J. Biol. Chem. 275:21278–21286. [DOI] [PubMed] [Google Scholar]
- Park, S., M. Uesugi, and G.L. Verdine. 2000. A second binding site on the NFAT regulatory domain. Proc. Natl. Acad. Sci. USA. 97:7130–7135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusnak, F., and P. Mertz. 2000. Calcineurin: form and function. Physiol. Rev. 80:1483–1521. [DOI] [PubMed]
- Schwarz, E.M., D. Van Antwerp, and I.M. Verma. 1996. Constitutive phosphorylation of IkappaBalpha by casein kinase II occurs preferentially at serine 293: requirement for degradation of free IkappaBalpha. Mol. Cell. Biol. 16:3554–3559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibasaki, F., U. Hallin, and H. Uchino. 2002. Calcineurin as a multifunctional regulator. J. Biochem. 131:1–15. [DOI] [PubMed] [Google Scholar]
- Tam, W.F., and R. Sen. 2001. IκB family members function by different mechanisms. J. Biol. Chem. 276:7701–7704. [DOI] [PubMed] [Google Scholar]
- Tam, W.F., L.H. Lee, L. Davis, and R. Sen. 2000. Cytoplasmic sequestration of Rel proteins by IκBα requires CRM1-dependent nuclear export. Mol. Cell. Biol. 20:2269–2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson, J.E., R.J. Phillips, H. Erdjument-Bromage, P. Tempst, and S. Ghosh. 1995. IkB-b regulates the persistent response in a biphasic activation of NF-kB. Cell. 80:573–582. [DOI] [PubMed] [Google Scholar]
- Towbin, H., T. Staehelin, and J. Gordon. 1979. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc. Natl. Acad. Sci. USA. 76:4350–4354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran, K., M. Merika, and D. Thanos. 1997. Distinct functional properties of IκBα and IκBβ. Mol. Cell. Biol. 17:5386–5399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turpin, P., R.T. Hay, and C. Dargemont. 1999. Characterization of IκBα nuclear import pathway. J. Biol. Chem. 274:6804–6812. [DOI] [PubMed] [Google Scholar]
- Wang, C.Y., M.W. Mayo, and A.S. Baldwin, Jr. 1996. TNF- and cancer therapy-induced apoptosis: potentiation by inhibition of NF-kappaB. Science. 274:784–787. [DOI] [PubMed] [Google Scholar]
- Wang, C.-Y., M.W. Mayo, R.G. Korneluk, D.V. Goeddel, and A.S. Baldwin Jr. 1998. NF-κB antiapoptosis: Induction of TRAF1 and TRAF2 and c-IAP1 and c- IAP2 to suppress caspase-8 activation. Science. 281:1680–1683. [DOI] [PubMed] [Google Scholar]
- Wang, H.G., N. Pathan, I.M. Ethell, S. Krajewski, Y. Yamaguchi, F. Shibasaki, F. McKeon, T. Bobo, T.F. Franke, and J.C. Reed. 1999. Ca2+-induced apoptosis through calcineurin dephosphorylation of BAD. Science. 284:339–343. [DOI] [PubMed] [Google Scholar]
- Weil, R., C. Laurent-Winter, and A. Israel. 1997. Regulation of IκBβ degradation. Similarities to and differences from IκBα. J. Biol. Chem. 272:9942–9949. [DOI] [PubMed] [Google Scholar]
- Zandi, E., Y. Chen, and M. Karin. 1998. Direct phosphorylation of IkappaB by IKKalpha and IKKbeta: discrimination between free and NF-kappaB-bound substrate. Science. 281:1360–1363. [DOI] [PubMed] [Google Scholar]
- Zhang, B.W., G. Zimmer, J. Chen, D. Ladd, E. Li, F.W. Alt, G. Wiederrecht, J. Cryan, E.A. O'Neill, C.E. Seidman, et al. 1996. T cell responses in calcineurin A alpha-deficient mice. J. Exp. Med. 183:413–420. [DOI] [PMC free article] [PubMed] [Google Scholar]