

Abstract
Tight junctions are well-developed between adjacent endothelial cells of blood vessels in the central nervous system, and play a central role in establishing the blood-brain barrier (BBB). Claudin-5 is a major cell adhesion molecule of tight junctions in brain endothelial cells. To examine its possible involvement in the BBB, claudin-5–deficient mice were generated. In the brains of these mice, the development and morphology of blood vessels were not altered, showing no bleeding or edema. However, tracer experiments and magnetic resonance imaging revealed that in these mice, the BBB against small molecules (<800 D), but not larger molecules, was selectively affected. This unexpected finding (i.e., the size-selective loosening of the BBB) not only provides new insight into the basic molecular physiology of BBB but also opens a new way to deliver potential drugs across the BBB into the central nervous system.
Keywords: tight junction; central nervous system; endothelial cells; blood vessel; drug delivery
Introduction
The existence of the blood-brain barrier (BBB)* was first described by Ehrlich (1885) more than 100 yr ago. He found that dyes injected into veins stained all the organs but the brain. Since then, the BBB has been thought to protect the brain from various harmful materials circulating in the blood (for reviews see Pardridge, 1998; Rubin and Staddon, 1999). On the other hand, the BBB prevents many potential drugs from entering the central nervous system (CNS). Therefore, many researchers have tried to loosen the BBB for therapeutic purposes in various CNS disorders (for reviews see Miller, 2002; Pardridge, 2002), but limited information on the molecular basis for BBB has hampered these trials.
To establish BBB, well-developed tight junctions (TJs) between adjacent endothelial cells are indispensable, in addition to various transporters in their plasma membranes (Reese and Karnovsky, 1967; for reviews see Pardridge, 1998; Rubin and Staddon, 1999; Edwards, 2001; Wolburg and Lippoldt, 2002). TJs are one mode of cell–cell adhesion, and play a central role in sealing the intercellular space in epithelial and endothelial cellular sheets (e.g., barrier function; for reviews see Schneeberger and Lynch, 1992; Anderson and van Itallie, 1995; Tsukita et al., 2001). On ultrathin section electron microscopy, TJs appear as a zone where plasma membranes of neighboring cells focally make complete contact (Farquhar and Palade, 1965). On freeze-fracture electron microscopy, TJs are visualized as a continuous, anastomosing network of intramembranous particle strands (TJ strands or fibrils) and complementary grooves (Staehelin, 1974). These observations led to our current understanding of the three-dimensional structure of TJs: at TJs, within the lipid bilayer of each membrane, some specific integral membrane proteins aggregate linearly to constitute “TJ strands.” Individual TJ strands then laterally and tightly associate with those in the apposing membrane of adjacent cells to form paired strands, where the intercellular distance becomes almost zero (so-called “kissing points” of TJs; for review see Tsukita et al., 2001).
Several peripheral membrane proteins such as ZO-1 were reported to concentrate at the cytoplasmic surface of TJs (for review see Schneeberger and Lynch, 1992; Anderson and van Itallie, 1995; Balda and Matter, 1998; Tsukita et al., 1999, 2001). Until recently, information on TJ adhesion molecules has been lacking, but now three distinct types of integral membrane proteins are known to be localized at TJs: occludin (Furuse et al., 1993), junctional adhesion molecule (JAM) (Martin-Padura et al., 1998), and claudin (Cld; Furuse et al., 1998a). Occludin, an ∼65-kD integral membrane protein with four transmembrane domains, was identified as the first component of TJ strands (Furuse et al., 1993). However, several studies including gene knockout analyses revealed that TJ strands can be formed without occludin (Balda et al., 1996; Saitou et al., 1998). JAM with a single transmembrane domain was recently shown to associate laterally with TJ strands, but not to constitute the strands per se (Itoh et al., 2001). In contrast, Cld is now believed to be a major constituent of TJ strands (for review see Tsukita et al., 2001). Clds with molecular masses of ∼23 kD comprise a multigene family consisting of >20 members (Morita et al., 1999a; Tsukita et al., 2001). Clds also bear four transmembrane domains, but do not show any sequence similarity to occludin. Interestingly, when each Cld species was overexpressed in mouse L fibroblasts lacking endogenous Clds, exogenously expressed Cld molecules were polymerized within the plasma membrane to reconstitute “paired” TJ strands at cell–cell contact regions (Furuse et al., 1998b). Furthermore, through further detailed transfection experiments as well as immunolabeling studies, it is now widely accepted that heterogeneous Cld species constitute the backbone of TJ strands in situ; i.e., TJ strands are copolymers of heterogeneous Cld species (and also occludin; Furuse et al., 1999; Tsukita et al., 2001).
Recently, Cld-5 was found specifically in endothelial cells, in large amounts especially in the brain endothelial cells (Morita et al., 1999b), leading to the idea that Cld-5 may be directly involved in the establishment of the BBB. In this paper, to evaluate this idea, we generated Cld-5–deficient mice by homologous recombination, and found that in these mice the BBB was loosened in a size-selective manner. We believe that this work marks the first step in developing a new TJ-based method of drug delivery to the CNS.
Results
Generation of Cld-5–deficient mice
We produced mice unable to express Cld-5. Nucleotide sequencing as well as restriction mapping identified only one exon that covers the whole open reading frame of Cld-5. We constructed a targeting vector, which was designed to disrupt the Cld-5 gene by replacing its exon with the neomycin resistance gene (Fig. 1 a). Two distinct lines of mice were generated from distinct embryonic stem (ES) cell clones in which the Cld-5 gene was disrupted by homologous recombination. Southern blotting confirmed the disruption of the Cld-5 gene in heterozygous as well as homozygous mutant mice (Fig. 1 b), and RT-PCR detected no Cld-5 mRNA from the brains of homozygous mutant mice (Cld5 −/− mice; Fig. 1 c). Cld5 −/− mice were born in the expected Mendelian ratios, and looked normal macroscopically (Fig. 1 d). However, their movements gradually ceased, and they all died within 10 h of birth. Because both lines of mice showed the same phenotypes, we will mainly present data obtained from one line.
Figure 1.

Generation of Cld-5–deficient mice. (a) Restriction maps of the wild-type allele, the targeting vector, and the targeted allele of the mouse Cld-5 gene. Only one exon covers the whole open reading frame of Cld-5. The targeting vector contained the pgk neo cassette in its middle portion to delete the exon in the targeted allele. The positions of the 5′ and 3′ probes for Southern blotting are indicated as bars. E, EcoRI; Sa, SacI; K, KpnI; N, NcoI; and S, Sse8387I. (b) Genotype analyses by Southern blotting of Eco RI-digested genomic DNA from wild-type (+/+), heterozygous (+/−), and homozygous (−/−) mice for the mutant Cld-5 gene allele. Southern blotting with 5′ and 3′ probes yielded an 8.4-kb band from the wild-type allele, and a 4.7- and 3.7-kb band from the targeted allele, respectively. (c) Loss of Cld-5 mRNA in the brain of Cld-5 −/− mice examined by RT-PCR. As a control, the hypoxanthine phosphoribosyl transferase gene was equally amplified in all samples. (d) Newborn Cld-5 +/+ and Cld-5 −/− mice. Homozygous Cld-5–deficient mice were born in the expected Mendelian ratios, and looked normal macroscopically. However, their movements gradually ceased, and they all died within 10 h of birth.
Morphology of brain blood vessels in Cld-5–deficient mice
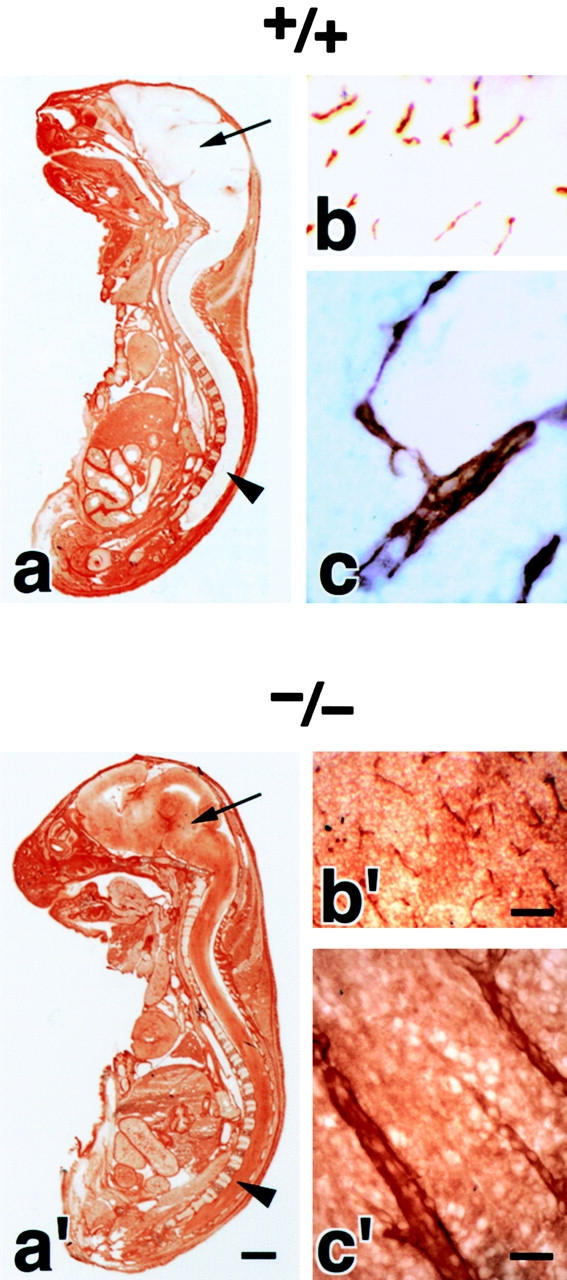
We examined the brain blood vessels morphologically in Cld5 −/− mice. As shown in Fig. 2 (a and a′), whole-mount immunostaining of 9.5-d embryos for PECAM-1, an endothelial cell marker, detected no significant abnormality in the vasculogenesis in Cld5 −/− mice. Furthermore, in both hematoxylin-eosin and anti–PECAM-1 mAb stainings of the brain sections of 18.5-d embryos, no difference was discerned between wild-type and Cld5 −/− mice (Fig. 2, b, b′, c, and c′). No signs of bleeding or edema were detected in the Cld5 −/− brain. Consistently, there was no difference in the brain tissue water content between newborn wild-type (85.6 ± 0.38%, n = 11) and Cld5 −/− mice (85.4 ± 0.30%, n = 12).
Figure 2.

Vasculogenesis and the brain histology of Cld5 −/− mice. (a and a′) Whole-mount immunostaining. Mouse 9.5-d wild-type and Cld5 −/− embryos were labeled with anti–PECAM-1 mAb. The characteristic treelike and ladderlike staining patterns of blood vessels in the head and vertebrate portions were observed both in wild-type and Cld5 −/− embryos. (b, b′, c, and c′) Hematoxylin-eosin–stained paraffin sections (b and b′) and anti–PECAM-1–stained frozen sections (c and c′) of the wild-type and Cld5 −/− brains of 18.5-d embryos. No difference was detected. Bars: (a and a′) 300 μm; (b and b′) 100 μm; (c and c′) 40 μm.
At the electron microscopic level, the blood vessels of the brain of newborn Cld5 −/− mice did not exhibit overt morphological abnormalities (Fig. 3) . Close inspection revealed that, even in Cld5 −/− mice, TJs with a normal appearance occurred at the endothelial cell–cell contact regions, showing the so-called kissing points of TJs. Considering that Cld-5 disappeared from the endothelial cells (Fig. 4) , this finding suggested that in endothelial cells of the wild-type brain, TJ strands were not only composed of Cld-5 but also other Cld species. We researched for such endothelial cell-specific Cld among Cld-1–16 using available and newly generated antibodies, and found that, in addition to Cld-5, Cld-12 was expressed in large amounts and colocalized with ZO-1 in the wild-type brain endothelial cells and also in the Cld5 −/− brain endothelial cells (Fig. 4). Thus, we concluded that in the Cld5 −/− brain, Cld-5 was simply removed from the TJs of endothelial cells, leaving TJs consisting of at least Cld-12.
Figure 3.

Ultrastructure of brain blood vessels in Cl d5 −/− mice. In the Cld5 −/− brains of 18.5-d embryo, ultrathin-section electron microscopy revealed no overt morphological abnormalities in the blood vessels. TJs were observed between adjacent endothelial cells, and at higher magnification, so-called kissing points of TJs were clearly visualized (arrowheads). Bars: (left) 4 μm; (middle) 0.2 μm; (right) 50 nm.
Figure 4.

Cld-12 in brain blood vessels. (a) Specificity of newly generated anti–Cld-12 pAb. Immunoblotting of total lysates of Escherichia coli expressing GST fusion proteins with cytoplasmic domains of Cld-1–16 confirmed its specificity. CBB, Coomassie brilliant blue staining; anti–Cld-12 pAb, immunoblotting with anti–Cld-12 pAb. (b) Frozen sections of the wild-type and Cld5 −/− brains of 18.5-d embryo were double stained with anti–ZO-1 mAb and anti–Cld-5 pAb or anti–Cld-12 pAb. Cld-5 was completely undetectable from the Cld5 −/− brain. Cld-12 was concentrated at ZO-1–positive TJs not only in the wild-type but also in the Cld5 −/− brain endothelial cells. These Cld-12 signals were abolished when the anti–Cld-12 pAb was preincubated with the GST-Cld-12 fusion protein. There appeared to be no significant difference in the intensity of Cld-12 signal between the wild-type and Cld5 −/− brain endothelial cells. The concentration of occludin at TJs of endothelial cells was not affected in the Cld5 −/− brain (unpublished data). Bar, 20 μm.
Size-selective loosening of the BBB in Cld-5–deficient mice
The important question is whether the barrier function of TJs (i.e., the BBB) is affected in the Cld5 −/− brain endothelial cells. To evaluate the BBB in Cld5 −/− mice, we performed tracer experiments: Cld5 +/− intercross littermates were obtained by Caesarian section at embryonic day 18.5, resuscitated, and perfused with tracers from the left cardiac ventricle. These littermates were later genotyped. First, we used a primary amine-reactive biotinylation reagent (443 D), which covalently cross-links to accessible proteins, as a tracer. This tracer has been successfully used so far to evaluate the permeability of TJs of epithelial cells (Chen et al., 1997; Furuse et al., 2002). 5 min after perfusion, the localization of the biotinylation reagent was examined on sagittal whole body sections using HRP-conjugated streptavidin (Fig. 5 , a and a′). In wild-type embryos, the reagent was specifically excluded from the CNS. This was indeed confirmation of Ehrlich's work, which first described the BBB (Ehrlich, 1885). In contrast, in the Cld5 −/− mice, the brain and the spinal cord showed strong and diffused HRP activity. At a higher magnification in the wild-type brain, the biotinylation reagent was retained in the blood vessels, whereas in the Cld5 −/− brain, this reagent was distributed diffusely throughout the brain parenchyma with some concentration in blood vessels (Fig. 5, b , b′, c, and c′). Fig. 6 represents the time course of the leakage of the biotinylation reagent from blood vessels in the Cld5 −/− brain. This reagent appeared to have already passed through the BBB 1 min after perfusion. These findings clearly demonstrated that the BBB was severely affected in Cld5 −/− mice.
Figure 5.
Impairment of the BBB in the Cld5 −/− brains of 18.5-d embryos. Primary amine-reactive biotinylation reagent (443 D) was perfused from the heart of resuscitated embryos. After a 5-min incubation, sagittal frozen sections from the whole body (a and a′) or the brain (b, b′, c, and c′) were incubated with HRP-conjugated streptavidin to localize the biotinylation reagent by peroxidase activity. At a low magnification in the wild-type mouse (a), the reagent was completely excluded from the brain (arrow) and the spinal cord (arrowhead); in sharp contrast, in the Cld5 −/− mouse (a′), the CNS showed intense peroxidase activity. At a higher magnification, in the Cld5 −/− (b′ and c′) but not in the wild-type brain (b and c), the reagent infiltrated abundantly into the parenchyma. Bars: (a and a′) 2 mm; (b and b′) 40 μm; (c and c′) 10 μm.
Figure 6.

Time course of the biotinylation reagent leakage from the blood vessels in the Cld5 −/− brains. Primary amine-reactive biotinylation reagent (443 D) was perfused from the heart of resuscitated embryos. After a 1, 3, 5, or 10-min incubation, frozen sections from the brain were incubated with rhodamine-conjugated streptavidin to visualize the distribution of the injected biotinylation reagent. Bar, 50 μm.
If serum proteins extravasate freely in the brain, vasogenic edema should be induced. However, as mentioned in Fig. 2 b, there was no sign of the edema in the Cld5 −/− brain. We checked for the possible leakage of serum albumin (∼68 kD) from blood vessels in the Cld5 −/− brain (Liu, 1988; Vinores et al., 1989; Fig. 7 a). Interestingly, immunostaining with antialbumin polyclonal antibody (pAb) showed no leakage of serum albumin, which was completely retained in the blood vessels. Furthermore, we performed tracer experiments using microperoxidase (∼1.9 kD) as a tracer (Knothe Tate et al., 1998). As shown in Fig. 7 b, no leakage from the brain blood vessels was detected in the Cld5 −/− brain. These findings suggested that, in the Cld5 −/− brain, the BBB (i.e., the TJ barrier of endothelial cells) was loosened in a size-selective manner. If so, the absence of vasogenic edema in the Cld5 −/− brain can be explained. In Fig. 7 c, to confirm this speculation, we perfused a mixture of tetramethylrhodamine-conjugated dextran (∼10 kD) and a fluorescence dye for nuclear staining (Hoechst stain H 33258; 562 D) through the heart (Chang-Ling et al., 1992; Hu et al., 2000). After a 5-min incubation, in the wild-type brain, both dextran and H33258 were retained in blood vessels. H33258 stained only the nuclei of endothelial cells. Interestingly, in the Cld5 −/− brain, nuclei of not only endothelial cells but also surrounding neurons/glial cells were intensively stained by H33258, whereas dextran was detected only within blood vessels.
Figure 7.

Size-selective loosening of the BBB in Cld5 −/− mice. (a) Paraffin sections of the brain were de-paraffined and stained with rabbit antialbumin pAb, followed by incubation with HRP-conjugated anti–rabbit IgG pAb. Bound pAb was visualized by peroxidase activity. No leakage of endogenous serum albumin was observed in the Cld5 −/− brain. Bar, 40 μm. (b) Microperoxidase (∼1.9 kD) was injected into the heart of resuscitated embryos, and after a 5-min incubation, the brain was fixed. The localization of microperoxidase was visualized by peroxidase activity in frozen sections. No leakage of microperoxidase was observed in the Cld5 −/− brain. Bar, 40 μm. (c) A mixture of tetramethylrhodamine-conjugated dextran (∼10 kD) and a dye for nuclear staining (Hoechst stain H33258; 562 D) was perfused, and frozen sections were observed under a fluorescence microscope. In the wild-type brain, neither dextran (red) nor Hoechst dye (blue) extravasated, whereas in the Cld5 −/− brain, Hoechst dye, but not dextran, extravasated to stain the nuclei of surrounding neurons/glial cells. Bar, 30 μm.
Magnetic resonance imaging (MRI) analyses of the BBB impairment in Cld-5–deficient mice
Finally, we quantitatively examined impairment of the BBB in live Cld5 −/− mice using MRI with the longitudinal (T1) relaxation reagent gadolinium–diethylene triamine-N,N,N′,N″,N″-pentaacetic acid (Gd-DTPA; 742 D; Abbott et al., 1999; Caravan et al., 1999; Seo et al., 2002). We injected various dosages of Gd-DTPA into the heart of resuscitated 18.5-d embryos. The sagittal T1-weighted images of the wild-type brain clearly exhibited the existence of the BBB; the signal from most organs except the CNS was significantly enhanced by Gd-DTPA perfusion. In contrast, in the Cld5 −/− mice, the signals from all areas of the CNS were markedly enhanced, and the degree of enhancement depends on the dosage of injected Gd-DTPA (Fig. 8 a). In Fig. 8 b, we examined the relationship between the averaged T1 relaxation rate of water in the brain and the dosage of injected Gd-DTPA (Seo et al., 2002). On the assumption of a simple two-compartment exchange model (Fabry and Eisenstadt, 1978; Schwarzbauer et al., 1997), this relationship allowed us to estimate that the Gd-DTPA–accessible space per unit volume of the wild-type and Cld5 −/− brain was 1.2 ± 0.8% and 14.7 ± 4.2% (regression coefficient ± SD), respectively. This finding indicated that in the wild-type brain, Gd-DTPA was retained only within the blood vessels (∼1% of the total volume), and that in the Cld5 −/− brain, it extravasated and distributed diffusely in the extracellular space of neurons/glia cells (∼15% of the total volume). Therefore, it is safe to say that, quantitatively, the BBB against Gd-DTPA was severely affected in the Cld5 −/− brain.
Figure 8.
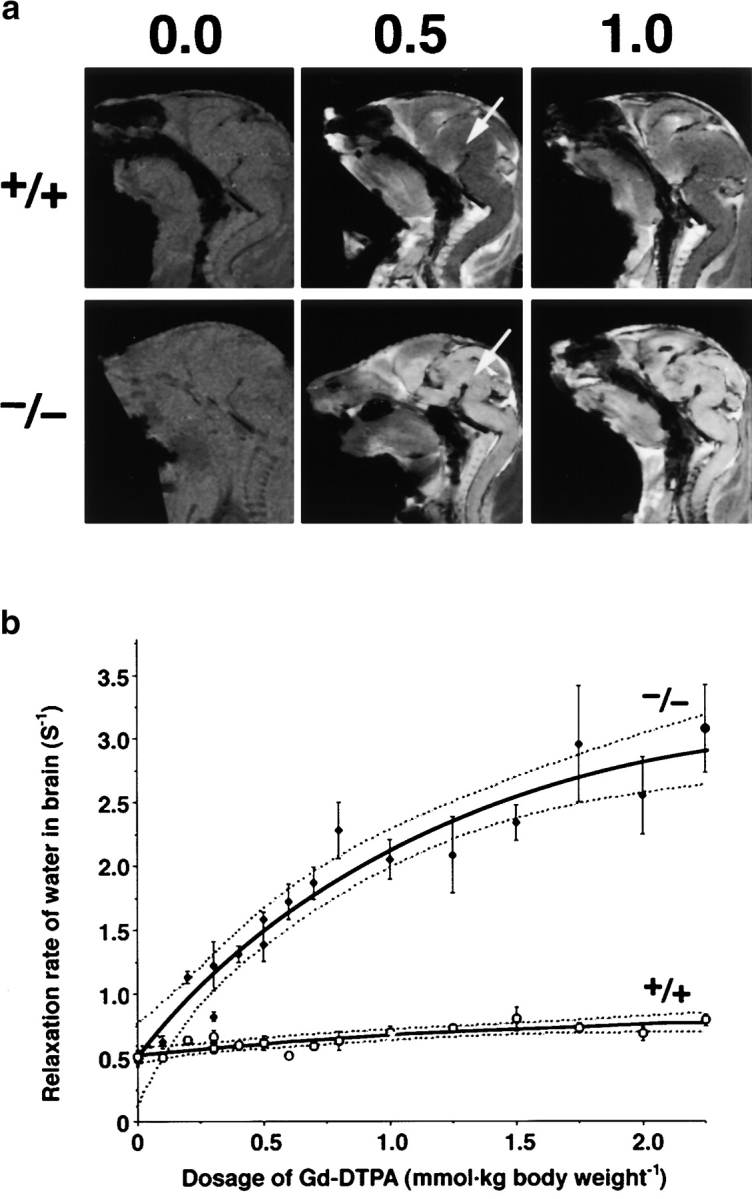
MRI of 18.5-d embryos. (a) Mid-sagittal T1-weighted images of the wild-type and Cld5 −/− brain after the transcardial injection of Gd-DTPA (0.0, 0.5, and 1.0 mmol/kg body wt). Due to shortening of the T1 relaxation time, the Gd-DTPA–accessible area was represented as a high signal intensity. In the wild-type mice, Gd-DTPA did not enter the extravascular space in the CNS, so the brain parenchyma (top middle, arrow) maintained a constant image intensity even at a high dose of Gd-DTPA. By contrast, in the Cld5 −/− brain, the image intensity in all areas of the CNS (bottom middle, arrow), including the spinal cord, was enhanced by Gd-DTPA, and the degree of enhancement depends on the dosage of injected Gd-DTPA. (b) The relationship between the relaxation rate of water (1/T1) in the brain and the dosage of injected Gd-DTPA. As 1/T1 of the brain, 1/T1 values of the five ROIs (156 × 156 × 750 mm) including the cortex, thalamus, hypothalamus, cerebellum, and pons were averaged. The bold and dotted lines are the results of fitting and 95% confidence limits, respectively. As explained in the text, on the assumption of a simple two-compartment exchange model (Fabry and Eisenstadt, 1978; Schwarzbauer et al., 1997), this relationship allowed us to estimate quantitatively the Gd-DTPA–accessible space per unit volume of the brain.
Discussion
Blood vessels are lined with a single layer of endothelial cellular sheets. These cellular sheets function as barriers to maintain the internal environment of blood vessels, but various materials must be selectively transported across these sheets. There are two pathways through which materials cross endothelial cellular sheets: the transcellular pathway including transcytosis through the cell and the paracellular pathway through TJs (for reviews see Spring, 1998; Tsukita et al., 2001). In the blood vessels in the CNS, the endothelial cellular sheets function as very tight barriers, constituting the so-called BBB (for reviews see Pardridge, 1998; Rubin and Staddon, 1999). For the establishment of the BBB, first, the transport of materials through transcellular pathways should be suppressed so completely that specialized transport systems are required in the plasma membranes of endothelial cells for the uptake of even small molecules such as glucose and amino acids by the CNS (for reviews see Pardridge, 1998, 2002; Edwards, 2001; Miller, 2002). Furthermore, when some materials are leaked into the parenchyma of the CNS, the multi-drug resistance transporter mdr1a (P-glycoprotein 1), which is concentrated in the endothelial cell plasma membranes, redistributes them out of the brain parenchyma into endothelial cells and, hence, back to the blood (Cordon-Cardo et al., 1989; Schinkel et al., 1994; Edwards, 2001). Second, the paracellular route also should be tightly sealed. TJs are markedly developed compared with those in endothelial cells in nonneuronal tissues (Reese and Karnovsky, 1967; Wolburg and Lippoldt, 2002). Taking these cellular bases for the BBB into consideration, TJs have been considered attractive targets for transient breakdown of the BBB in therapies for various CNS disorders.
We found that TJs in the brain blood vessels are primarily composed of at least two distinct species of Clds: Cld-5 and -12. Anti–Cld-1 pAb also stained the blood vessels in the wild-type brain as previously reported (Liebner et al., 2000), but this staining was still positive in the Cld-1–deficient mice (unpublished data). We examined the roles of Cld-5 in the BBB by generating Cld-5–deficient mice. In the Cld5 −/− brain, Cld-5 was simply removed from the TJs of endothelial cells, leaving morphologically normal blood vessels as well as the Cld-12–based TJs. However, in terms of the barrier function, these endothelial TJs showed a peculiar abnormality. Tracer experiments and MRI revealed that in these mice, the BBB is severely affected against small molecules (less than ∼800 D), but not larger molecules. In other words, the Cld-12–based TJs in Cld5 −/− brain blood vessels would function as a molecular sieve. They allow only small molecules (less than ∼800 D) to pass across TJs. Of course, this molecular mass cut-off (∼800 D) is tentative because the tracers used in this work have all a different chemical structure and differ in charge and hydrophilicity.
The size-selective loosening of TJs in Cld5 −/− brain blood vessels is consistent with the previous data on epithelial TJs. The TJs of MDCK I epithelial cells were primarily composed of Cld-1 and -4, and when Cld-4 was removed from these TJs using a Cld-4–binding peptide, the continuous Cld-1–based TJs still persisted, which leaked 4 kD/10 kD dextran, but not 40 kD dextran (Sonoda et al., 1999). Furthermore, we recently found that the TJs in the epidermis were also composed of Cld-1 and -4. In Cld-1–deficient mice, there still remained the continuous Cld-4–based TJs in the granular layer of the epidermis, and the layered organization of keratinocytes in the epidermis was not affected (Furuse et al., 2002). However, interestingly, the epidermal barrier against water was severely affected, resulting in the dehydration of newborn mice. In general, it is, thus, safe to say that, when TJs are composed of more than two distinct species of Clds, the removal of one Cld species changes the barrier function of TJs markedly while keeping their continuous structural integrity.
Therefore, in the Cld5 −/− brain, the existence of the Cld-12–based TJs would keep the structural integrity (and the polarity) of endothelial cells, showing no bleeding. Furthermore, the size selectivity of the Cld-12–based TJs did not allow most serum proteins to extravasate, resulting in no vasogenic edema. From the viewpoint of the drug delivery, the lack of bleeding and edema is very important and advantageous. Therefore, Cld-5 can be regarded as a potential target for developing a new drug delivery method for CNS disorders. Of course, it is possible that impairment of the BBB, even if temporary or in a size-selective manner, would be harmful to CNS activity to some extent. Indeed, Cld5 −/− mice died within 10 h of birth. Cld-5 is expressed in large amounts in all segments of the blood vessels in the brain, but it is also detected in some segments of the blood vessels of nonneural tissues such as the lung and kidney (Morita et al., 1999b). Various tissues were examined histologically in hematoxylin-eosin–stained sections of Cld5 −/− mice, but no significant abnormalities were detected (unpublished data). Therefore, at this moment it is not easy to discuss a causal sequence between BBB impairment and death in Cld5 −/− mice. To answer this question, as well as to better understand the basic physiology of the BBB, we should generate mice in which Cld-5 gene can be conditionally knocked out in the endothelial cells of the brain blood vessels in adult mice.
Materials and methods
Antibodies
Rat anti–PECAM-1 mAb (BD Biosciences), rat anti–mouse ZO-1 mAb (CHEMICON International, Inc.), rabbit anti–Cld-5 pAb (Zymed Laboratories), and rabbit anti–mouse albumin pAb (Inter-Cell Technology, Inc.) were purchased. Anti–Cld-12 pAb was raised as follows. A polypeptide corresponding to the COOH-terminal cytoplasmic domain of mouse Cld-12 (a cysteine residue was added at its NH2 terminus) was synthesized and coupled via the cysteine residue to keyhole limpet hemocyanin. This peptide was injected into rabbits as an antigen. Rabbit antisera were affinity-purified on nitrocellulose membranes with GST fusion proteins with Cld-12 before use.
Generation of Cld5 − /− mice
Two overlapping clones encoding mouse Cld-5 were obtained by screening a 129/Sv genomic library. Using them, the targeting vector was constructed as shown in Fig. 1 a. The diphthelia toxin A expression cassette (MC1pDT-A) was placed outside the 3′ arm of homology for negative selection. As shown in Fig. 1 a, only one exon covered the whole open reading frame of Cld-5. Thus, this targeting vector was designed to delete this exon by replacing with the pgk-neo cassette. J1 ES cells were electroporated with the targeting vector and selected for ∼9 d in the presence of G418. The G418-resistant colonies were removed and screened by Southern blotting with the 5′ and 3′ external probes (Fig. 1 a). When digested with EcoRI, correctly targeted ES clones were identified by an additional 4.7-kbp band together with the 8.4-kbp band of the wild-type allele with the 5′ probe, and by an additional 3.7-kbp band together with the 8.4-kb band of the wild-type allele with the 3′ probe. The targeted ES cells obtained were injected into C57BL/6 blastocysts, which were, in turn, transferred into BALB/c foster mothers to obtain chimeric mice. Male chimeras were mated with C57BL/6 females, and agouti offsprings were genotyped to confirm the germline transmission of the targeted allele. The littermates were genotyped by Southern blotting. Next, heterozygous mice were interbred to produce homozygous mice.
Immunostaining
For whole-mount staining, mouse 9.5-d embryos were killed. Samples were pretreated by microwaving in PBS for 20 s and fixed in 4% PFA/PBS for 30 min. They were dehydrated in methanol and bleached with 30% H202. Samples were then rehydrated, blocked with PBS-MT (0.2% Triton X-100 and 1% skimmed milk/PBS), and incubated overnight with rat anti–PECAM-1 mAb followed HRP-conjugated goat anti–rat IgG (CHEMICON International, Inc.). Next, they were washed with PBS-MT and PBS-T (0.2% Triton X-100/PBS) each for 5 h. Bound antibodies were visualized by incubating with 0.025% DAB, 0.08% NiCl2, and 30% H202 in PBS-T.
Immunofluorescence staining/peroxidase histochemistry for frozen and paraffin sections, and ultra-thin section electron microscopy were performed as previously described (Morita et al., 1999b).
Tracer experiments
Cld5 +/− intercross littermates were obtained by Caesarian section at embryonic day 18.5 and resuscitated. The following solutions were perfused from the left cardiac ventricle under a stereoscopic microscope using a low-pressure perfusion apparatus (Terumo) within 1 min; 5 μl/g body wt of 25 mg/ml microperoxidase (MP-11; 1,862 D; Sigma-Aldrich) in PBS containing 1 mM CaCl2 (Knothe Tate et al., 1998); 1 ml/g body wt of 2 mg/ml EZ-LinkTM Sulfo-NHS-Biotin (443 D; Pierce Chemical Co.) in PBS containing 1 mM CaCl2 (Chen et al., 1997; Furuse et al., 2002); and 1 ml/g body wt of 100 μg/ml Hoecht stain (H33258; 562 D; Calbiochem-Novabiochem) in PBS containing 1 mM CaCl2 with or without 1 mg/ml tetramethylrhodamine-conjugated lysine-fixable dextran (10 kD; Molecular Probes; Chang-Ling et al., 1992; Hu et al., 2000). 1–5 min after perfusion, the whole brain was removed, fixed with 3.7% formaldehyde, and frozen using liquid nitrogen. In the frozen sections, the distribution of microperoxidase, EZ-LinkTM Sulfo-NHS-Biotin, Hoecht stain, and dextran were examined as described previously (Chang-Ling et al., 1992; Chen et al., 1997; Knothe Tate et al., 1998; Hu et al., 2000; Furuse et al., 2002).
MRI
Mouse 18.5-d embryos were resuscitated and anesthetized with pentobarbital (50 μg/g body wt), and Gd-DTPA solution (<7 μl in total) was injected transcardially. The mice were placed in the head-up position on a polystyrene sledge, the position of the head was fixed with adhesive tape, and the sledge temperature was maintained at 34 ± 1°C using a warm air flow. 1H magnetic resonance images were obtained using an NMR spectrometer (7.05 T; model AMX-300wb; Bruker) with an active shielded gradient (micro2.5) and an 1H birdcage resonator (15-mm diam). The T1-weighted sagittal gradient-echo imaging (repeat time [TR] = 100 ms, echo-time [TE] = 4.4 ms, flip angle = 45°) was obtained with 78 μm in-plane resolution and 0.75-mm slice thickness. The T1 relaxation times were measured using an inversion recovery fast-imaging sequence (Haase et al., 1989) with a series of 16 detection pulses (12° flip angle), 10 inversion recovery delays (40–7,030 ms), 156-μm in-plane resolution, 0.75-mm slice thickness, 3,000-ms TR, 2.4-ms TE, and 2 accumulations. On the assumption of a two-compartment model with water exchange between Gd-DTPA–accessible space and nonaccessible space (Fabry and Eisenstadt, 1978; Schwarzbauer et al., 1997), the slow relaxation rate (RS) given by RS = 0.5 ((Rg + Rn + kn/f) − {[Rg − Rn + kn(1 − 2f)/f]2 + 4kn 2(1 − f)/f}0.5), where Rn is the intrinsic 1/T1 of water in the brain, kn is the rate constant of diffusive water influx from the Gd-DTPA–nonaccessible space into the accessible space, f and Rg are the volume fractions of water, and 1/T1 for water in the Gd-DTPA–accessible space, respectively. The Rg was estimated from the relaxivity of Gd-DTPA (Caravan et al., 1999), the dosage of the Gd-DTPA and the volume fraction of extracellular fluid.
Acknowledgments
We thank Drs. T. Noda and Y. Sugitani for technical advices in producing and analyzing Cld5 −/− mice, and Dr. T. Aoki for helpful discussions and encouragements.
This work was supported in part by a Grant-in-Aid for Cancer Research and a Grant-in-Aid for Scientific Research (A) from the Ministry of Education, Science and Culture of Japan to S. Tsukita, and by the Japan Society for the Promotion of Science Research for the Future Program to M. Furuse.
Footnotes
Abbreviations used in this paper: BBB, blood-brain barrier; Cld, claudin; CNS, central nervous system; ES, embryonic stem; Gd-DTPA, gadolinium–diethylene triamine-N,N,N′,N″,N″-pentaacetic acid; MRI, magnetic resonance imaging; pAb, polyclonal antibody; TJ, tight junction.
References
- Abbott, N.J., D.C. Chugani, G. Zaharchuk, and B.R. Rose. 1999. Delivery of imaging agents into brain. Adv. Drug Deliv. Rev. 37:253–277. [DOI] [PubMed] [Google Scholar]
- Anderson, J.M., and C.M. van Itallie. 1995. Tight junctions and the molecular basis for regulation of paracellular permeability. Am. J. Physiol. 269:G467–G475. [DOI] [PubMed] [Google Scholar]
- Balda, M.S., and K. Matter. 1998. Tight junctions. J. Cell Sci. 111:541–547. [DOI] [PubMed] [Google Scholar]
- Balda, M.S., J.A. Whitney, C. Flores, S. González, M. Cereijido, and K. Matter. 1996. Functional dissociation of paracellular permeability and transepithelial electrical resistance and disruption of the apical–basolateral intramembrane diffusion barrier by expression of a mutant tight junction membrane protein. J. Cell Biol. 134:1031–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caravan, P., J.J. Ilison, T.J. McMurry, and R.B. Lauffer. 1999. Gadolinium(III) chelates as MRI contrast agents: structure, dynamics, and applications. Chem. Rev. 99:2293–2352. [DOI] [PubMed] [Google Scholar]
- Chang-Ling, T., A.L. Neill, and N.H. Hunt. 1992. Early microvascular changes in murine cerebral malaria detected in retinal wholemounts. Am. J. Pathol. 140:1121–1130. [PMC free article] [PubMed] [Google Scholar]
- Chen, Y.-H., C. Merzdorf, D.L. Paul, and D.A. Goodenough. 1997. COOH terminus of occludin is required for tight junction barrier function in early Xenopus embryos. J. Cell Biol. 138:891–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordon-Cardo, C., J.P. O'Brien, D. Casals, L. Rittman-Grauer, J.L. Biedler, M.R. Melamed, and J.R. Bertino. 1989. Multidrug-resistance gene (P-glycoprotein) is expressed by endothelial cells at blood-barrier sites. Proc. Natl. Acad. Sci. USA. 86:695–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards, R.H. 2001. Drug delivery via the blood-brain barrier. Nat. Neurosci. 4:221–222. [DOI] [PubMed] [Google Scholar]
- Ehrlich, P. Ed. 1885. Das Sauerstoff-Bedurfnis des Organismus. Eine Farbenanalytishe Studie. Ph.D. thesis. Herschwarld, Berlin. 69–72.
- Fabry, M.E., and M. Eisenstadt. 1978. Water exchange across red cell membranes. Measurement by nuclear magnetic resonance T1, T2, and T12 hybrid relaxation. J. Membr. Biol. 42:375–398. [DOI] [PubMed] [Google Scholar]
- Farquhar, M.G., and G.E. Palade. 1965. Cell junctions in amphibian skin. J. Cell Biol. 26:263–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuse, M., T. Hirase, M. Itoh, A. Nagafuchi, S. Yonemura, S. Tsukita, and S. Tsukita. 1993. Occludin: a novel integral membrane protein localizing at tight junctions. J. Cell Biol. 123:1777–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuse, M., K. Fujita, T. Hiiragi, K. Fujimoto, and S. Tsukita. 1998. a. Claudin-1 and -2: novel integral membrane proteins localizing at tight junctions with no sequence similarity to occludin. J. Cell Biol. 141:1539–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuse, M., H. Sasaki, K. Fujimoto, and S. Tsukita. 1998. b. A single gene product, claudin-1 or -2, reconstitutes tight junction strands and recruits occludin in fibroblasts. J. Cell Biol. 143:391–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuse, M., H. Sasaki, and S. Tsukita. 1999. Manner of interaction of heterogeneous claudin species within and between tight junction strands. J. Cell Biol. 147:891–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuse, M., M. Hata, K. Furuse, Y. Yoshida, A. Haratake, Y. Sugitani, T. Noda, A. Kubo, and S. Tsukita. 2002. Claudin-based tight junctions are crucial for the mammalian epidermal barrier: a lesson from claudin-1–deficient mice. J. Cell Biol. 156:1099–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haase, A.D., Matthaei, R. Bartkowski, E. Duhmke, and D. Leibfritz. 1989. Inversion recovery snapshot FLASH MR imaging. J. Comput. Assist. Tomogr. 13:1036–1040. [DOI] [PubMed] [Google Scholar]
- Hu, P., J.D. Pollard, and T. Chan-Ling. 2000. Breakdown of the blood-retinal barrier induced by activated T cells of nonneural specificity. Am. J. Pathol. 156:1139–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh, M., H. Sasaki, M. Furuse, H. Ozaki, T. Kita, and S. Tsukita. 2001. Junctional adhesion molecule (JAM) binds to PAR-3: a possible mechanism for the recruitment of PAR-3 to tight junctions. J. Cell Biol. 154:491–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knothe Tate, M.L., P. Niederer, and U. Knothe. 1998. In vivo tracer transport through the lacunocanalicular system of rat bone in an environment devoid of mechanical loading. Bone. 22:107–117. [DOI] [PubMed] [Google Scholar]
- Liebner, S., U. Kniesel, H. Kalbacher, and H. Wolburg. 2000. Correlation of tight junction morphology with the expression of tight junction proteins in blood-brain barrier endothelial cells. Eur. J. Cell Biol. 79:707–717. [DOI] [PubMed] [Google Scholar]
- Liu, H.M. 1988. Neovasculature and blood-brain barrier in ischemic brain infarct. Acta Neuropathol. (Berl.). 75:422–426. [DOI] [PubMed] [Google Scholar]
- Martin-Padura, I., S. Lostaglio, M. Schneemann, L. Williams, M. Romano, P. Fruscella, C. Panzeri, A. Stoppacciaro, L. Ruco, A. Villa, et al. 1998. Junctional adhesion molecule, a novel member of the immunoglobulin superfamily that distributes at intercellular junctions and modulates monocyte transmigration. J. Cell Biol. 142:117–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, G. 2002. Drug targeting. Breaking down barriers. Science. 297:1116–1118. [DOI] [PubMed] [Google Scholar]
- Morita, K., M. Furuse, K. Fujimoto, and S. Tsukita. 1999. a. Claudin multigene family encoding four-transmembrane domain protein components of tight junction strands. Proc. Natl. Acad. Sci. USA. 96:511–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita, K., H. Sasaki, M. Furuse, and S. Tsukita. 1999. b. Endothelial claudin: claudin-5/TMVCF constitutes tight junction strands in endothelial cells. J. Cell Biol. 147:185–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardridge, W.M. 1998. Introduction to the Blood-Brain Barrier. Cambridge University Press, Cambridge. 486 pp.
- Pardridge, W.M. 2002. Drug and gene targeting to the brain with molecular Trojan horses. Nat. Rev. Drug Discov. 1:131–139. [DOI] [PubMed] [Google Scholar]
- Reese, T.S., and M.J. Karnovsky. 1967. Fine structural localization of a blood-brain barrier to exogenous peroxidase. J. Cell Biol. 34:207–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin, L.L., and J.M. Staddon. 1999. The cell biology of the blood-brain barrier. Annu. Rev. Neurosci. 22:11–28. [DOI] [PubMed] [Google Scholar]
- Saitou, M., K. Fujimoto, Y. Doi, M. Itoh, T. Fujimoto, M. Furuse, H. Takano, T. Noda, and S. Tsukita. 1998. Occludin-deficient embryonic stem cells can differentiate into polarized epithelial cells bearing tight junctions. J. Cell Biol. 141:397–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schinkel, A.H., J.J. Smit, O. van Tellingen, J.H. Beijnen, E. Wagenaar, L. van Deemter, C.A. Mol, M.A. van der Valk, E.C. Robanus-Maandag, H.P. te Riele, et al. 1994. Disruption of the mouse mdr1a P-glycoprotein gene leads to a deficiency in the blood-brain barrier and to increased sensitivity to drugs. Cell. 77:491–502. [DOI] [PubMed] [Google Scholar]
- Schneeberger, E.E., and R.D. Lynch. 1992. Structure, function, and regulation of cellular tight junctions. Am. J. Physiol. 262:L647–L661. [DOI] [PubMed] [Google Scholar]
- Schwarzbauer, C., S.P. Morrissey, R. Deichmann, C. Hillenbrand, J. Syha, H. Adolf, U. Noth, and A. Haase. 1997. Quantitative magnetic resonance imaging of capillary water permeability and regional blood volume with an intravascular MR contrast agent. Magn. Reson. Med. 37:769–777. [DOI] [PubMed] [Google Scholar]
- Seo, Y., A. Takamata, T. Ogino, H. Morita, S. Nakamura, and M. Murakami. 2002. Water permeability of capillaries in the subfornical organ of rats determined by Gd-DTPA2- enhanced 1H magnetic resonance imaging. J. Physiol. 545:217–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonoda, N., M. Furuse, H. Sasaki, S. Yonemura, J. Katahira, Y. Horiguchi, and S. Tsukita. 1999. Clostridium perfringens enterotoxin fragment removes specific claudins from tight junction strands: evidence for direct involvement of claudins in tight junction barrier. J. Cell Biol. 147:195–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spring, K. 1998. Routes and mechanism of fluid transport by epithelia. Annu. Rev. Physiol. 60:105–119. [DOI] [PubMed] [Google Scholar]
- Staehelin, L.A. 1974. Structure and function of intercellular junctions. Int. Rev. Cytol. 39:191–283. [DOI] [PubMed] [Google Scholar]
- Tsukita, S., M. Itoh, and M. Furuse. 1999. Structural and signaling molecules come together at tight junctions. Curr. Opin. Cell Biol. 11:628–633. [DOI] [PubMed] [Google Scholar]
- Tsukita, S., M. Furuse, and M. Itoh. 2001. Multi-functional strands in tight junctions. Nat. Rev. Mol. Cell Biol. 2:285–293. [DOI] [PubMed] [Google Scholar]
- Vinores, S.A., C. Gadegbeku, P.A. Campochiaro, and W.R. Green. 1989. Immunohistochemical localization of blood-retinal barrier breakdown in human diabetics. Am. J. Pathol. 134:231–235. [PMC free article] [PubMed] [Google Scholar]
- Wolburg, H., and A. Lippoldt. 2002. Tight junctions of the blood-brain barrier: development, composition and regulation. Vascul Pharmacol. 38:323–337. [DOI] [PubMed] [Google Scholar]