

Abstract
“In the long course of cell life on this earth it remained, for our age, for our generation, to receive the full ownership of our inheritance. We have entered the cell, the Mansion of our birth and started the inventory of our acquired wealth.” (Albert Claude, Nobel lecture, 1974)
Never before have Albert Claude's words been truer. Cell biologists now have at their disposal the entire inventory of genes in many organisms, and technologies that can enable the global interrogation of macromolecules and the structures they form. Indeed with the continued development of new high throughput technologies, such as expression arrays and mass spectrometry, the inventories that comprise cells appear within reach. But most certainly Claude had a loftier goal in mind. The challenge lying before us is to understand how these inventories work together as a system to bring about the life of the cell.
An approach to understanding the complexity of life that has emerged in concert with global high throughput approaches and the datasets that they generate is the systems biology approach. The goal of systems biology is to exploit these, and new, technologies to interrogate cells at multiple hierarchical levels of cellular organization (from molecules to modules to phenotypes) and to understand biological behaviors that emerge from the various interactions of a cell's many system elements. Thus, systems approaches hinge on combining multiparameter analyses with computational practices of systems engineering to develop dynamic system level models of cellular function. The vision is that these models will be necessary to understand how genetic and environmental perturbations cause disease, and to predict and ultimately prevent cellular dysfunction.
Like all good research, at the heart of systems biology is the tight coupling between experimentation, data analysis, and hypothesis generation. However, systems biology embodies three major concepts that make it unique. First, a discovery-based component employs high throughput data generation in an effort to define all the relevant elements of the system of interest and to quantitatively observe their activities in normal and perturbed cell states. This emphasis on genome-scale discovery complements the traditional emphasis on hypothesis-driven experimentation by leading to unanticipated findings. The second concept is the integration of multiple data types. This stems from the facts that system properties emerge from gene action and interaction at multiple molecular levels and that molecules act together to form modules serving specific functions that can be observed and quantified (Hartwell et al., 1999). This hierarchy of structure and function continues higher to measurable properties of cells and organisms. The integration of data collected at all of these levels is required for the formulation of quantitative system models, the third major concept. In the systems approach, biological responses are computationally analyzed, visualized, and modeled to generate hypotheses about the system properties of interest, which are then experimentally tested. Practically speaking, these hypotheses are often tested using classical approaches, but hypothesis testing can also take the form of monitoring global responses to specific perturbations. Through an iterative process, the model is thereby refined to bring it and the experimental results into close apposition.
Genome-scale data inventories
In recent years, several technologies have emerged to generate global datasets on the levels of gene expression, protein levels and modifications, molecular interactions, phenotypes, and genetic interactions. In most cases to date, yeast has been exploited for global interrogation; however, the completion of genome sequences of other organisms, including mouse and human, and the development of new approaches applicable to different eukaryotes support systems approaches in a wide range of models.
Control of gene expression.
Of the current genome-scale system measurement technologies, nucleic acid microarrays most closely approach the desired throughput and data quality required for systems biology. In addition to widely used technologies for quantifying global expression patterns, microarrays have recently been exploited to reveal chromatin targets for transcription factors. In this case, transcription factors containing an epitope tag are cross-linked to DNA and the complexes are immunopurified. The purified DNA is amplified, labeled, and hybridized to microarrays of the intergenic regions between adjacent open reading frames. The utility of this approach is dramatically exemplified by the recent identification of the chromatin regions bound by 106 yeast transcription factors (Lee et al., 2002). Together with expression arrays, these techniques have the potential to unravel the regulatory networks of yeast and the developmental programs of higher organisms.
Inventorying proteins.
Interestingly, mRNA expression profiles often do not reflect protein abundance or activities (Griffin et al., 2002). Thus to complement these data, it is essential to capture information on the protein status of cells. Here, it is desirable to determine, in a quantitative way, the inventory of all proteins present in a cell and to determine how normal cellular responses and experimentally directed perturbations affect protein abundance, posttranslational modifications, localizations, and turnover and synthesis rates, etc. Mainly through revolutionary advances in mass spectrometry (MS),* these challenges are becoming a reality (for reviews see Gygi and Aebersold, 2000; Aebersold and Mann, 2003). However, sample complexity, the wide range in abundance of proteins in biological systems, and the difficulty of deriving quantitative data are challenges inherent to this approach.
In addition to the well-known two-dimensional gel electrophoresis approaches, stable isotope labeling procedures can overcome some of the difficulties in quantifying proteins by MS. In these applications, proteins are labeled either metabolically or after isolation by stable isotopes. MS can then be performed on a mixture of the peptides derived from two different conditions and differentially labeled (with a heavy or light isotope). The ratio of signal derived from pairs of peptides differing by the masses of the incorporated isotopes can then be used to quantify the relative amounts of the proteins of interest in each original fraction (for reviews see Gygi and Aebersold, 2000; Aebersold and Mann, 2003).
This principle has been extended by chemically coupling an isotopically labeled affinity tag to specific reactive groups on the peptides. The first use of this strategy employed a thiol-reactive biotin-containing affinity tag (Gygi et al., 1999). Affinity purification of the tagged peptides (on avidin resin) first reduces the complexity of the sample and, by incorporating different isotopes into the tag, relative amounts of proteins can also be determined. Major efforts are currently underway to develop enrichment procedures or affinity-based reagents with chemistries specific to different posttranslational modifications. Most effort in this regard has been focused on phosphorylated and N-linked glycosylated peptides (McLachlin and Chait, 2001; Ficarro et al., 2002; Hirabayashi et al., 2002; Aebersold and Mann, 2003). These developments should enable one to measure not only the quantities of proteins, but also the relative amounts of specific posttranslational modifications and, by extension, activity states.
Protein localization.
Although it is currently not possible to inventory complete cells, organelles have been an attractive target for comprehensive proteomics studies. The first such organelle to be characterized in this way was the yeast spliceosome (Neubauer et al., 1997), but numerous other organelles have also been studied since that time. Because all subcellular fractions are contaminated to some extent with proteins from cellular compartments other than the one targeted, it is important to use additional techniques to define which proteins are bona fide constituents of the organelle of interest and which are transiently associated or contaminate the fraction. This was accomplished with the yeast nuclear pore complex by epitope tagging all suspected components and analyzing them individually by subcellular fractionation and in situ localization techniques (Rout et al., 2000). This is not easily done for larger, more dynamic organelles like the Golgi complex (Bell et al., 2001) or phagosome (Garin et al., 2001), but nevertheless, these approaches promise to contribute many new insights into the dynamics and biogenesis of organelles.
A complementary approach to defining the cellular localization of proteins has been undertaken by localizing epitope-tagged yeast proteins on a genome scale. Snyder's group has used an epitope containing transposable elements to randomly tag yeast genes by a method of shuttle mutagenesis. Each of the resulting tagged proteins could then be localized by immunofluorescence microscopy (Kumar et al., 2002).
Protein interactions.
The most common way to implicate a protein in a function is to identify physically interacting partners. Among the numerous techniques used to identify physical interactions between protein pairs, two-hybrid screens (Uetz et al., 2000; Ito et al., 2001) and protein pull-down assays (Gavin et al., 2002; Ho et al., 2002) have generated the largest datasets. The yeast two-hybrid and pull-down assays, together with the decades of acquired data on yeast proteins, have identified ∼15,000 interactions for ∼4,700 proteins (http://dip.doe-mbi.ucla.edu/dip/Stat.cgi). On a sobering note however, these mass-produced interaction data have relatively high error rates and provide no dynamic information, which will be of foremost importance to understanding these data. But, these are certainly powerful methods to identify the tens of thousands of interactions that define cellular interactomes, which arguably provide the most critical parameter for understanding new protein function.
An additional emerging proteomics technology is protein-based microarrays. In this approach, proteins are immobilized in array formats on derivatized glass slides, which are then used to identify proteins with specific binding properties or activities (for review see Kumar and Snyder, 2001). This approach has tremendous potential for assaying a library of proteins for interactions with specific ligands in high throughput on a small scale and has been used to identify proteins that have kinase activity or the ability to interact with specific antibodies or drugs. Such data types will be invaluable to understanding the roles of small molecules and metabolites, identifying peptide-binding domains, etc.
Phenotypes and genetic interactions.
The phenotypic impacts of single-gene perturbations can associate specific genes with specific cell properties. Almost all genes of the yeast genome have been systematically deleted by PCR-directed homologous recombination. This has been an outstanding resource for yeast researchers, who are interested in screening for phenotypes associated with their genes of interest. Each knockout strain is identifiable by “bar codes” flanking the deleted gene. Pooled yeast strains can then be grown together under defined conditions, and the pool can be quantitatively assayed for growth of each strain, revealing those that are either advantaged or disadvantaged by their gene loss (Shoemaker et al., 1996). As not all organisms are amenable to systematic knockout strategies, more complex model systems are being targeted by RNAi knockdown or random mutagenesis strategies. Indeed, RNAi may prove to be the most versatile tool for functional genomics studies of numerous multicellular organisms.
The phenotypic consequences of combined genetic perturbations reveal functional interactions that are not apparent from single-gene perturbations. In addition to extending the ability to associate specific genes with specific cellular processes, genetic interactions (e.g., epistasis and synthetic effects) allow the inference of the positions of gene products relative to the flow of information in the network. With the creation of the knockout library in yeast, it is possible to use robotics to systematically combine mutations and identify synthetic defects due to pairs of gene deletions (Tong et al., 2001). These methods have the ability to identify large classes of functionally interacting proteins in a rapid and systematic way.
Moving forward, a major frontier in systems biology is the development of high throughput quantitative assays of cell phenotypes. To create models that predict the cell biological effects of specific perturbations, we must have a well-developed understanding of how complex dynamic molecular networks determine measurable cell properties. This will require large amounts of cell property data and molecular data derived from cells subjected to many perturbations. High throughput image collection and analysis technologies, automated assays of growth, and real-time single-cell and single-molecule data collection will continue to be areas of increasing application and accelerating technological development.
Databases.
With the accumulation of seemingly endless lists of expression, interaction, localization, and phenotypic data from an increasing number of fully sequenced organisms, a major challenge for biologists is the ongoing assembly and organization of these data into databases that enable data integration. Currently databases, too numerous to list, have been assembled around different organisms, but they are not unified with respect to data organization, data types, etc. Standards of data quality, organization, and accessibility (through down loads or online queries) will greatly facilitate the ability of researchers to mine and analyze these large-scale datasets.
Insights from data integration and modeling
Systems biology attempts to exploit genome-wide datasets to achieve a new level of understanding and predictive power. However, although we can now generate long lists of proteins or genes from different types of high throughput expression or interaction data, a formidable challenge facing systems biology is integrating these disparate data into conceptual models of molecular function.
The first challenges come from the data themselves. By comparison to hand-crafted data, mass-produced data have high error rates; and second, the datasets are often too large for the human brain to integrate and model. Computational approaches can begin to remedy both of these issues. For example, microarrays can quantify the expression response of every gene in an organism across a number of different conditions. The data can then be analyzed by clustering tools that allow the classification of genes into groups reflecting common behaviors by comparatively analyzing the expression patterns across the various conditions. This process reveals trends of expression and counteracts and reveals individual gene fluctuations and experimental errors. The genes in each cluster group are often enriched in proteins of similar or related functions, enabling initial predictions for unfamiliar proteins. Furthermore, the integration of data from different sources is important for revealing biological themes and data significance. For example, there is a significant correlation between the coexpression of genes and the physical interactions among their encoded proteins (Ge et al., 2001). The integration of these data types can reinforce bona fide observations and weaken the effects of spurious data. Thus, full exploitation of these types of relationships can maximize the predictive value of integrated data. However, this raises an additional major challenge for systems biology. How do we integrate data to enable the visualization and understanding of the relationships among large-scale data from different sources? There are currently three general approaches in use that aim to address this issue: clustering methods, probabilistic methods, and graphical methods.
In the clustering methods, genes are first clustered based on one data type, and then a second data type is mapped onto the existing clusters. The visualization of this integration can be as simple as a color map of the superimposed data, where the position in a cluster represents one data type and the color of the gene or protein can indicate its identification from another data type. For example, Ge et al. (2001) clustered yeast genes based on expression patterns and color mapped protein interaction data onto pairs of clusters to visualize the correlation between gene expression and protein interaction.
Probabilistic methods typically express data as probabilities or Boolean states (true or false) and compute integrated probabilities that evaluate the likelihood that components present within two or more datasets are functionally related. Such methods have been applied to the classification and annotation of genes, and clusters of genes, from expression-profiling experiments. Smith et al. (2002) used the hypergeometric distribution to calculate P values for the overrepresentation of gene function categories (from a database) among clusters of genes from self-organizing map analysis of expression profiles. In this method, the probability of the observed coincidence of gene clusters and gene function categories is evaluated relative to the coincidence expected by random chance. Low P values suggest biological significance. In another example, Kumar et al. (2002) provided probabilistic predictions of the subcellular localization of all yeast proteins using a Bayesian method. Starting with default localization probabilities from experimental data, they sequentially updated these probabilities using various data sources, including gene expression and protein motifs.
The third general method expresses data in graphical form as vertices and edges (nodes and links). The vertices and edges of such graphs typically represent molecules and interactions, respectively. This intuitive method is essentially the same as that used commonly to represent molecular models in biology. Additional data can then be integrated by assigning them as additional attributes to each node or edge. Network graphs can thus encode and communicate these attributes of system elements in shape, color, position, and changes in these visual cues. For example, one could represent different molecule types with different node shapes, and different expression levels with color. Ideker et al. (2001), studying genes/proteins involved in galactose metabolism and the physical interactions among them, established the feasibility of this approach on a global scale. Tong et al. (2001) have extended graphical network analysis to data on genetic interactions.
Beyond visualization, graphs are amenable substrates for algorithmic analyses that incorporate the structure of the graph and the attributes assigned to graph elements. Subsequent graph visualizations can then represent both the input data and the derived data from such analyses. For example, Fig. 1 shows the identification of active subnetworks/pathways during galactose utilization in yeast (Ideker et al., 2002). Proteins implicated in the yeast cellular response to a shift to galactose metabolism are represented in a network graph as nodes. Protein–protein interactions are represented by edges connecting nodes, whereas directed edges (arrows) represent protein–DNA interactions. Simulated annealing methods were then used to identify connected groups of gene products whose genes show significant expression changes in response to the metabolic switch. These active subnetworks reveal unexpected connections between the gal module (denoted by GAL80) and other biomodules in the cell (see also Ideker et al., 2001). An increasing number of software packages for network analysis of this sort are available to visualize and analyze biological problems that can be formalized as graphs; examples include Cytoscape (http://www.cytoscape.org) and Osprey (http://biodata.mshri.on.ca/osprey).
Figure 1.
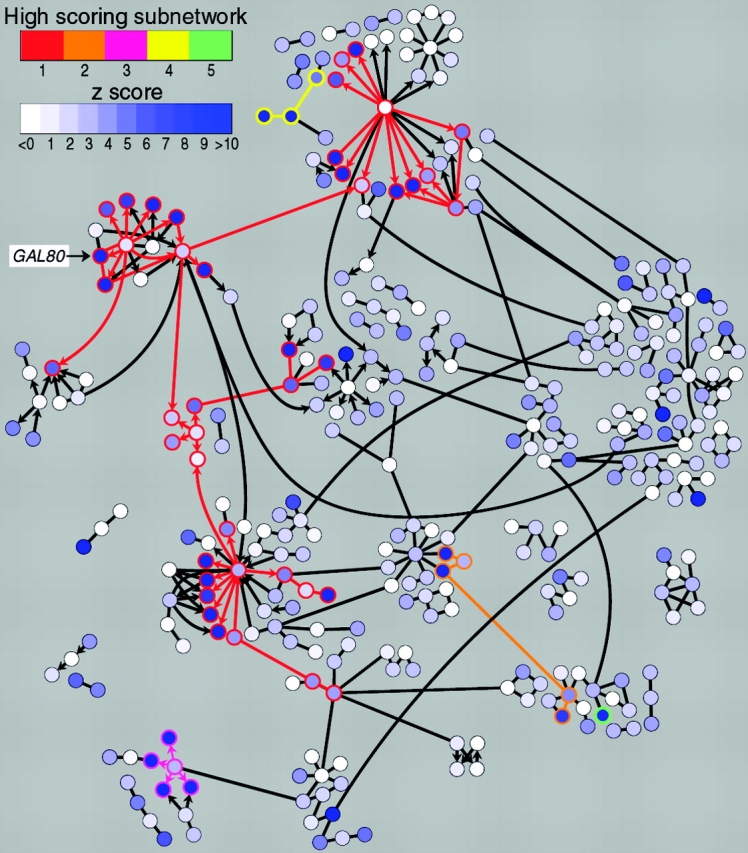
Identification of regulatory pathways using network graphing and simulated annealing methods. Shown is the network of interacting molecules implicated in galactose utilization in yeast. Each node (gene/protein) is colored based on the significance of its expression change (z-score) in an experiment comparing mRNA levels in a wild-type strain and a gal80 mutant. Higher z-scores indicate higher significance of change, either up or down. Simulated annealing methods were used to identify subnetworks (indicated by colored node borders and edges) with high aggregate z-scores. Note that the GAL module, including GAL80, is part of a high-scoring subnetwork. This figure was reprinted from Ideker et al., Discovering regulatory signalling circuits in molecular interaction networks, Bioinformatics, 2002, Vol. 18, pg. S233–S240 by permission of Oxford University Press.
Modeling.
Integrated genome-scale sets of diverse data present the possibility of modeling cell processes with global scope and potent hypothesis generation, that is, to make biological predictions from molecular data and identify likely molecular perturbations to control biology. The task requires bridging the gap between molecular and cellular behavior. This complex problem is quintessential systems biology and, as in system engineering, relies on simplifying the representation of thousands of interacting components through a hierarchy of complexity. Applied to biology, this provides a simplified way of integrating genome-wide data without losing sight of the contribution each molecule can make to the overall phenotype of the organism. Thus, it is desirable to classify collections of molecules that interact locally and temporally as biomodules, serving specific functions (Hartwell et al., 1999). Examples can include signal transduction pathways, metabolic pathways, or perhaps, on a larger scale, organelles. Biomodules, in turn, interact to form modular networks that specify cell biological properties.
Considerable recent effort has gone into identifying modular network organization within composite large-scale datasets generated as described above (e.g., Rives and Galitski, 2003). A major motivation for these studies is to abstract complex interaction data as simplified networks of connected structure/function modules. The activities and interactions of these modules, in turn, specify cell properties. Recognizing these relationships can aid in the design of molecular and genetic experiments and further global interrogation of the system (e.g., Ideker et al., 2001). Continued work in this area will likely aim to mathematically describe measured cell properties (phenotypes) as a function of the activities of modules, and module activity as a function of molecular activity. Quantification of modular and cellular activities can accelerate a convergence with efforts to simulate system behavior (Arkin, 2001; Davidson et al., 2002; Guet et al., 2002).
Although tremendous advances have enabled the inventorying of different levels of biological activities, the continued exploitation of systems biology approaches for cell biologists will require the development of high throughput technologies that enable us to assign quantitative measurements to cellular attributes that cell biologists currently describe in imprecise terms. As we move forward, data will increasingly be kinetic, spatially specific, and stochastic (e.g., transport, compartmentalization, and posttranslational states), which will, in turn, drive the evolution of network analyses and simulations. Furthermore, developments in bioinformatics hold the promise to extrapolate from model systems to humans to allow the indirect generation of predictions in humans from system-level insights in experimental organisms. Finally, it seems evident that the systems biology approaches pioneered in model systems increasingly will be applied directly to the prediction and prevention of human disease.
Acknowledgments
We thank Lee Hood, Ruedi Aebersold, Rick Rachubinski, and Mike Rout for critical reading of this manuscript and helpful discussions. We apologize to colleagues whose original research we were unable to cite due to constraints of article length.
T. Galitski is a recipient of a Burroughs Wellcome Fund Career Award in the Biomedical Sciences.
Footnotes
Abbreviation used in this paper: MS, mass spectrometry.
References
- Aebersold, R., and M. Mann. 2003. Mass spectrometry-based proteomics. Nature. 422:198–207. [DOI] [PubMed] [Google Scholar]
- Arkin, A.P. 2001. Synthetic cell biology. Curr. Opin. Biotechnol. 12:638–644. [DOI] [PubMed] [Google Scholar]
- Bell, A.W., M.A. Ward, W.P. Blackstock, H.N. Freeman, J.S. Choudhary, A.P. Lewis, D. Chotai, A. Fazel, J.N. Gushue, J. Paiement, et al. 2001. Proteomics characterization of abundant Golgi membrane proteins. J. Biol. Chem. 276:5152–5165. [DOI] [PubMed] [Google Scholar]
- Davidson, E.H., J.P. Rast, P. Oliveri, A. Ransick, C. Calestani, C.H. Yuh, T. Minokawa, G. Amore, V. Hinman, C. Arenas-Mena, et al. 2002. A genomic regulatory network for development. Science. 295:1669–1678. [DOI] [PubMed] [Google Scholar]
- Ficarro, S.B., M.L. McCleland, P.T. Stukenberg, D.J. Burke, M.M. Ross, J. Shabanowitz, D.F. Hunt, and F.M. White. 2002. Phosphoproteome analysis by mass spectrometry and its application to Saccharomyces cerevisiae. Nat. Biotechnol. 20:301–305. [DOI] [PubMed] [Google Scholar]
- Garin, J., R. Diez, S. Kieffer, J.F. Dermine, S. Duclos, E. Gagnon, R. Sadoul, C. Rondeau, and M. Desjardins. 2001. The phagosome proteome: insight into phagosome functions. J. Cell Biol. 152:165–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavin, A.C., M. Bosche, R. Krause, P. Grandi, M. Marzioch, A. Bauer, J. Schultz, J.M. Rick, A.M. Michon, C.M. Cruciat, et al. 2002. Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature. 415:141–147. [DOI] [PubMed] [Google Scholar]
- Ge, H., Z. Liu, G.M. Church, and M. Vidal. 2001. Correlation between transcriptome and interactome mapping data from Saccharomyces cerevisiae. Nat. Genet. 29:482–486. [DOI] [PubMed] [Google Scholar]
- Griffin, T.J., S.P. Gygi, T. Ideker, B. Rist, J. Eng, L. Hood, and R. Aebersold. 2002. Complementary profiling of gene expression at the transcriptome and proteome levels in Saccharomyces cerevisiae. Mol. Cell. Proteomics. 1:323–333. [DOI] [PubMed] [Google Scholar]
- Guet, C.C., M.B. Elowitz, W. Hsing, and S. Leibler. 2002. Combinatorial synthesis of genetic networks. Science. 296:1466–1470. [DOI] [PubMed] [Google Scholar]
- Gygi, S.P., and R. Aebersold. 2000. Mass spectrometry and proteomics. Curr. Opin. Chem. Biol. 4:489–494. [DOI] [PubMed] [Google Scholar]
- Gygi, S.P., B. Rist, S.A. Gerber, F. Turecek, M.H. Gelb, and R. Aebersold. 1999. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat. Biotechnol. 17:994–999. [DOI] [PubMed] [Google Scholar]
- Hartwell, L.H., J.J. Hopfield, S. Leibler, and A.W. Murray. 1999. From molecular to modular cell biology. Nature. 402:C47–C52. [DOI] [PubMed] [Google Scholar]
- Hirabayashi, J., T. Hashidate, Y. Arata, N. Nishi, T. Nakamura, M. Hirashima, T. Urashima, T. Oka, M. Futai, W.E. Muller, et al. 2002. Oligosaccharide specificity of galectins: a search by frontal affinity chromatography. Biochim. Biophys. Acta. 1572:232–254. [DOI] [PubMed] [Google Scholar]
- Ho, Y., A. Gruhler, A. Heilbut, G.D. Bader, L. Moore, S.L. Adams, A. Millar, P. Taylor, K. Bennett, K. Boutilier, et al. 2002. Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature. 415:180–183. [DOI] [PubMed] [Google Scholar]
- Ideker, T., V. Thorsson, J.A. Ranish, R. Christmas, J. Buhler, J.K. Eng, R. Bumgarner, D.R. Goodlett, R. Aebersold, and L. Hood. 2001. Integrated genomic and proteomic analyses of a systematically perturbed metabolic network. Science. 292:929–934. [DOI] [PubMed] [Google Scholar]
- Ideker, T., O. Ozier, B. Schwikowski, and A.F. Siegel. 2002. Discovering regulatory and signalling circuits in molecular interaction networks. Bioinformatics. 18(Suppl. 1):S233–S240. [DOI] [PubMed] [Google Scholar]
- Ito, T., T. Chiba, R. Ozawa, M. Yoshida, M. Hattori, and Y. Sakaki. 2001. A comprehensive two-hybrid analysis to explore the yeast protein interactome. Proc. Natl. Acad. Sci. USA. 98:4569–4574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, A., and M. Snyder. 2001. Emerging technologies in yeast genomics. Nat. Rev. Genet. 2:302–312. [DOI] [PubMed] [Google Scholar]
- Kumar, A., S. Agarwal, J.A. Heyman, S. Matson, M. Heidtman, S. Piccirillo, L. Umansky, A. Drawid, R. Jansen, Y. Liu, et al. 2002. Subcellular localization of the yeast proteome. Genes Dev. 16:707–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, T.I., N.J. Rinaldi, F. Robert, D.T. Odom, Z. Bar-Joseph, G.K. Gerber, N.M. Hannett, C.T. Harbison, C.M. Thompson, I. Simon, et al. 2002. Transcriptional regulatory networks in Saccharomyces cerevisiae. Science. 298:799–804. [DOI] [PubMed] [Google Scholar]
- McLachlin, D.T., and B.T. Chait. 2001. Analysis of phosphorylated proteins and peptides by mass spectrometry. Curr. Opin. Chem. Biol. 5:591–602. [DOI] [PubMed] [Google Scholar]
- Neubauer, G., A. Gottschalk, P. Fabrizio, B. Seraphin, R. Luhrmann, and M. Mann. 1997. Identification of the proteins of the yeast U1 small nuclear ribonucleoprotein complex by mass spectrometry. Proc. Natl. Acad. Sci. USA. 94:385–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rives, A.W., and T. Galitski. 2003. Modular organization of cellular networks. Proc. Natl. Acad. Sci. USA. 100:1128–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rout, M.P., J.D. Aitchison, A. Suprapto, K. Hjertaas, Y. Zhao, and B.T. Chait. 2000. The yeast nuclear pore complex: composition, architecture, and transport mechanism. J. Cell Biol. 148:635–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoemaker, D.D., D.A. Lashkari, D. Morris, M. Mittmann, and R.W. Davis. 1996. Quantitative phenotypic analysis of yeast deletion mutants using a highly parallel molecular bar-coding strategy. Nat. Genet. 14:450–456. [DOI] [PubMed] [Google Scholar]
- Smith, J.J., M. Marelli, R.H. Christmas, F.J. Vizeacoumar, D.J. Dilworth, T. Ideker, T. Galitski, K. Dimitrov, R.A. Rachubinski, and J.D. Aitchison. 2002. Transcriptome profiling to identify genes involved in peroxisome assembly and function. J. Cell Biol. 158:259–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong, A.H., M. Evangelista, A.B. Parsons, H. Xu, G.D. Bader, N. Page, M. Robinson, S. Raghibizadeh, C.W. Hogue, H. Bussey, et al. 2001. Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science. 294:2364–2368. [DOI] [PubMed] [Google Scholar]
- Uetz, P., L. Giot, G. Cagney, T.A. Mansfield, R.S. Judson, J.R. Knight, D. Lockshon, V. Narayan, M. Srinivasan, P. Pochart, et al. 2000. A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisiae. Nature. 403:623–627. [DOI] [PubMed] [Google Scholar]