

Abstract
Deletion of murine Smn exon 7, the most frequent mutation found in spinal muscular atrophy, has been directed to either both satellite cells, the muscle progenitor cells and fused myotubes, or fused myotubes only. When satellite cells were mutated, mutant mice develop severe myopathic process, progressive motor paralysis, and early death at 1 mo of age (severe mutant). Impaired muscle regeneration of severe mutants correlated with defect of myogenic precursor cells both in vitro and in vivo. In contrast, when satellite cells remained intact, mutant mice develop similar myopathic process but exhibit mild phenotype with median survival of 8 mo and motor performance similar to that of controls (mild mutant). High proportion of regenerating myofibers expressing SMN was observed in mild mutants compensating for progressive loss of mature myofibers within the first 6 mo of age. Then, in spite of normal contractile properties of myofibers, mild mutants develop reduction of muscle force and mass. Progressive decline of muscle regeneration process was no more able to counterbalance muscle degeneration leading to dramatic loss of myofibers. These data indicate that intact satellite cells remarkably improve the survival and motor performance of mutant mice suffering from chronic myopathy, and suggest a limited potential of satellite cells to regenerate skeletal muscle.
Keywords: SMN; muscle regeneration; myopathy; therapeutics; satellite cells
Introduction
Mature muscle fibers of mammalian skeletal muscle (myofibers) are multinucleate syncitia that arise from the fusion of mononucleate precursors or myoblasts. Adult skeletal muscle fibers display a remarkable capacity for regeneration in response to muscle injury including progressive myopathies. A population of precursor cells, termed satellite cells, located between the sarcolemma and the basal lamina of the muscle fibers, is responsible for this ability to regenerate (for review see Seale et al., 2001). Such mononucleate cells, normally quiescent, become activated after damage to proliferate and differentiate, leading to the formation of multinucleate myofibers either de novo or from preexisting muscle fibers. However, the therapeutic potential these cells might have for muscle repair in inherited myopathies remains to be determined.
Spinal muscular atrophy (SMA)* is a frequent recessive autosomal neuromuscular disorder characterized by degeneration of motor neurons associated with muscle paralysis and atrophy. Mutations of the survival of motor neuron gene (SMN1) are responsible for SMA (for review see Frugier et al., 2002). SMN is a ubiquitously expressed protein that has been involved in various processes including cytoplasmic assembly of snRNP into the spliceosome, pre-mRNA splicing, transcription, and metabolism of ribosomal RNA (for review see Paushkin et al., 2002). However, the molecular pathway linking SMN defect to SMA phenotype remains to be elucidated. Several strategies have been performed to generate mouse models of SMA (for review see Frugier et al., 2002). By using the Cre-LoxP system, deletion of murine Smn exon 7, the most frequent mutation found in SMA patients, has been directed to either neurons or skeletal muscle (neuronal or muscular mutants; Frugier et al., 2000, 2002; Cifuentes-Diaz et al., 2001, 2002).
To direct homozygous deletion of Smn to skeletal muscle, Cre recombinase has been placed under the control of the human α-skeletal actin gene promoter (HSA-Cre; Miniou et al., 1999). Muscular mutant mice carry the HSA-Cre transgene and a constitutive heterozygous deletion of Smn exon 7 (Smn Δ7), the other allele harboring two LoxP sequences flanking exon 7 (Smn F7). Such (HSA-Cre, Smn F7/Δ7) mice display a severe phenotype characterized by necrosis of muscle fibers associated with destabilization of the sarcolemma components (Cifuentes-Diaz et al., 2001). One striking observation was the marked reduced life span of Smn muscular mutant mice with median survival of 4 wk, whereas mice knockout for genes encoding components of the dystrophin–glycoprotein complex such as dystrophin (mdx mouse) develop a milder course of disease phenotype with an almost normal life span. Therefore, sarcolemma destabilization was not sufficient to explain muscle paralysis and death of muscular Smn mutant mice. In those mice, the low proportion of myocytes with central nuclei has suggested a weak regenerative process in response to necrosis (Cifuentes-Diaz et al., 2001).
Here, we investigated the influence of satellite cell activity in muscular mutant mice. As Cre-mediated deletion of Smn exon 7 was found to occur in fused myotubes but not in satellite cells, deleterious effect of the constitutive heterozygous deletion of Smn exon 7 (Smn Δ7) on muscle precursors was suspected. Death of 25% myogenic-committed cells carrying the (Smn Δ7/+) genotype was observed in vitro. To determine whether deleterious effect of Smn Δ7 allele on muscle precursor cells might be circumvented in vivo, mutant mice carrying the (HSA-Cre, Smn F7/F7) instead of (HSA-Cre, Smn F7/Δ7) genotype were generated. In (HSA-Cre, Smn F7/F7) mice, both Smn alleles are intact in satellite cells, then become deleted in multinucleate myotubes through Cre recombinase activity. The phenotype of (HSA-Cre, Smn F7/F7) mutant mice was tremendously attenuated in comparison with mice carrying the (HSA-Cre, Smn F7/Δ7) genotype with an increase of median survival from 1 to 8 mo. Similar muscular dystrophic changes were observed yet in both types of mutants, but active muscle regeneration process characterized (HSA-Cre, Smn F7/F7) mutant mice. Here, we present a functional study of this myopathic phenotype. The use of Cre recombinase transgenic line directing Smn deletion in differentiated myotubes (but not in satellite cells) allowed to generate a mouse model in which mature muscle fibers are mutated, resulting in necrosis, whereas satellite cells remain intact, leading to efficient muscle regeneration. For the first time, we demonstrate that intact satellite cells markedly improved the survival and motor performance of mutant mice suffering from chronic muscular necrosis through an active muscle regenerative process.
Results
Cre-mediated deletion of Smn occurs in fused myotubes, but not satellite cells
To determine the expression profile of the HSA-Cre recombinase transgene during myogenic differentiation, primary myogenic cultures were prepared by enzymatic disaggregation of forelimb and hindlimb muscles from wild-type or transgenic (HSA-Cre) newborn mice (Miniou et al., 1999). Cells isolated from skeletal muscle tissues contain a mixture of fibroblasts, myoblasts, adipocytes, and hematopoietic cells. However, the muscle-derived cells can be enriched using the preplate technique based on the differential adherence characteristic of primary muscle cells to dishes (Tassin et al., 1991; Rando and Blau, 1994; Qu et al., 1998). After two preplates, immunolabeling of desmin, the intermediate filament protein specific to myogenic cells, was observed in 95% of cells 3 d after myoblast purification (unpublished data). At that time, all desmin-positive cells were mononucleated, and immunolabeling of the Cre recombinase was negative in a total of 300 myogenic cells analyzed (Fig. 1) . At day 6, the ratio of differentiated myotubes containing two or more nuclei to desmin-positive mononucleate cells (i.e., fusion index) was determined by immunolabeling of desmin and DAPI staining of nuclei. Fusion index was 49.5% in a total of 300 cells analyzed. Immunolabeling of the Cre recombinase showed that 49.5% of cells were positive in a total of 300 cells analyzed, and all Cre-positive cells were multinucleated (Fig. 1). No Trypan blue uptake into myogenic committed cells was observed (unpublished data). These data indicate that the HSA-Cre recombinase transgene was expressed in multinucleate myotubes, but not in myoblasts, and was not toxic to myotubes.
Figure 1.
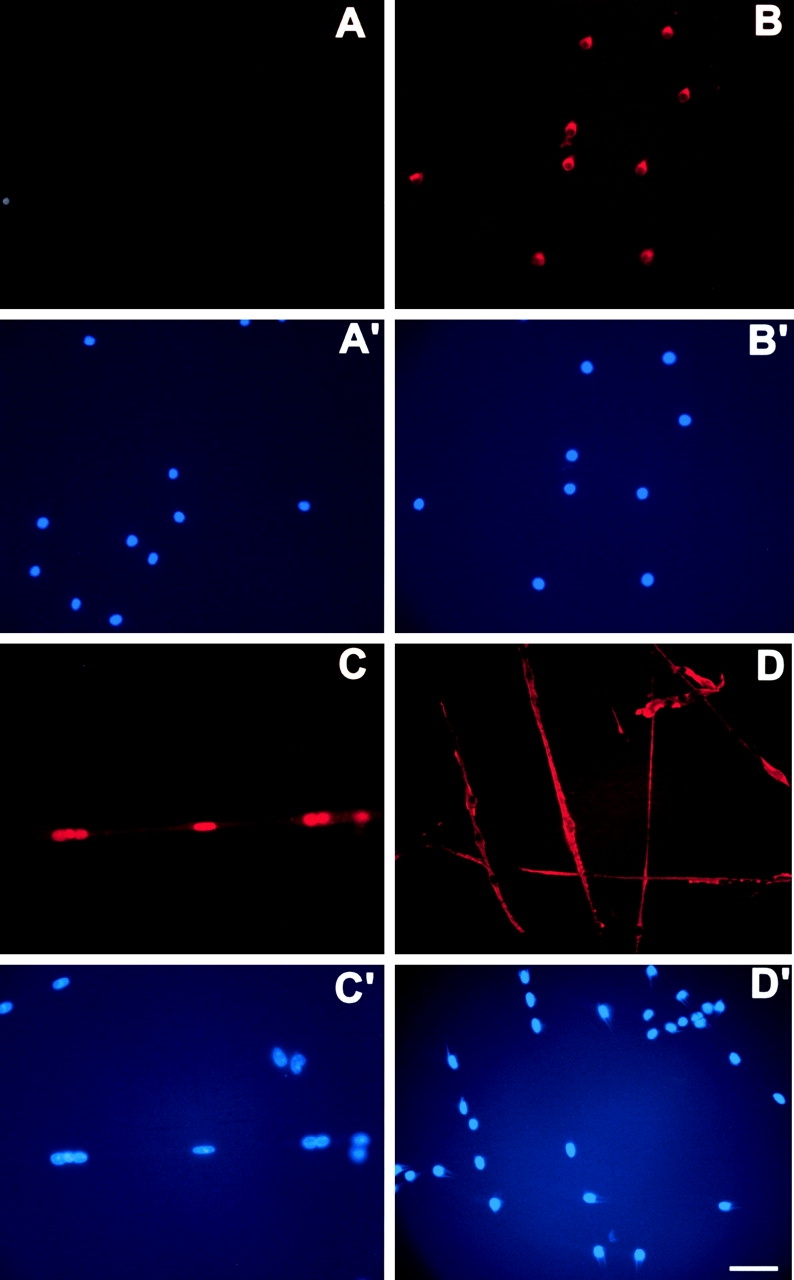
Cre recombinase and desmin expression in primary myogenic cultures. Immunolabeling of the Cre recombinase (A and C) or desmin (B and D) were performed 3 (A–B′) or 6 d after myoblast purification from (HSA-Cre) transgenic mice (C–D′). Nuclei were stained with DAPI (A′, B′, C′, and D′). Note the labeling of desmin (but not of Cre recombinase) in 3-d-old mononucleate myogenic cells, whereas in 6-d-old cells, Cre recombinase expression was observed in nuclei of fused myotubes. Bar, 40 μm.
To determine whether the Cre recombinase expression led to deletion of exon 7 from Smn F7 allele, primary myogenic cultures were performed from muscles dissected from (HSA-Cre, Smn F7/+) mice. PCR amplification analysis of DNA extracted from cells 0–8 d after myoblast purification was performed using primers flanking the Smn deleted genomic region (Fig. 2 A). No deletion of Smn exon 7 was observed at day 0, indicating that satellite cells or myoblasts are not Cre-mediated deleted in vivo. Smn exon 7 excision was observed from day 6 in agreement with Cre recombinase expression (Fig. 2 A).
Figure 2.

Genomic and protein analyses of Sm n in primary myogenic cultures. (A) Cre-mediated deletion of Smn exon 7 occurs 6 d after myoblast purification as determined by PCR amplification analysis of genomic DNA of (HSA-Cre, Smn F7/+) cells. DNA amplification of Cre recombinase transgene was used as internal positive control. (B) Immunoblot analysis of SMN in primary culture of skeletal muscles from control (Smn +/+) and (Smn Δ7/+) mice. Proteins were extracted from cells at day 0 (lanes 1 and 2), 3 (lanes 3 and 4), and 6 (lanes 5 and 6) after myoblast purification. Note the reduced amount of SMN in Smn Δ7/+ cells (lanes 2, 4, and 6) when compared with Smn +/+ (lanes 1, 3, and 5) and actin expression.
Heterozygous deletion of Smn exon 7 results in death of myogenic committed cells
To know whether constitutive heterozygous deletion of Smn exon 7 might have any effect on myogenic proliferation or differentiation, primary myogenic cultures were performed from newborn skeletal muscles of mice carrying the (Smn +/+) or (Smn Δ7/+) genotype. Immunoblot analysis was performed by using an mAb specific to the NH2 terminus of SMN, and revealed that SMN was highly expressed in myogenic committed cells including satellite cells or myoblasts from day 0 (Fig. 2 B). Reduced amount of SMN was observed in (Smn Δ7/+) cells when compared with (Smn +/+) cells, indicating that Smn Δ7 transcripts resulted in the absence of protein (Fig. 2 B). Proliferative capacity of myoblasts was determined by counting desmin-positive cells. Starting from 104 cells per plate at day 0, the number of desmin-positive cells increased similarly in both (Smn F7/+) and (Smn Δ7/+) genotypes between day 3 and day 6 after myoblast purification (Fig. 3 A). These data indicated that proliferation of myogenic cells was not affected by heterozygous deletion of Smn exon 7. However, death was observed in 16 and 25% of (Smn Δ7/+) cells in three independent experiments as determined by Trypan blue dye uptake into both myoblasts and fused myotubes at day 5 and day 6, respectively (Fig. 3 B). No Trypan blue dye uptake into (Smn +/+) or (Smn F7/+) cells was observed (Fig. 3 B, and unpublished data). TUNEL staining was negative in (Smn Δ7/+) cells, suggesting that cell death occurred through a nonapoptotic process (unpublished data).
Figure 3.


Kinetics of growth and cell death of myogenic committed cells from ( Smn F7/ + ), (Smn Δ7/+ ), and ( HSA-Cre, SmnF7/ + ) transgenic mice. (A) Starting from 104 cells per plate at day 0, number of living (curve) and dead cells (solid symbols) was evaluated from day 3 to day 6. Note that the kinetics of cell growth were similar in all genotypes analyzed. Dead cells were observed in both (Smn Δ7/+) and (HSA-Cre, Smn F7/+) cells with a significantly higher number in (Smn Δ7/+) than in (HSA-Cre, Smn F7/+). No cell death was observed in controls (Smn F7/+). (B) Trypan blue dye uptake into (Smn Δ7/+) myogenic cells including mononucleate (2, arrowhead) and multinucleate cells (3, arrow) 6 d after myoblast purification. No Trypan blue uptake was observed in 6-d-old (Smn F7/+) cells (1). Bar, 40 μm.
To determine whether Cre-mediated deletion of Smn led to similar deleterious effect on myogenic committed cells, primary cultures were performed from transgenic mice carrying the (HSA-Cre, Smn F7/+) genotype and compared with those carrying the (Smn Δ7/+) genotype. At day 5, death was noticed in 16% of (Smn Δ7/+) cells, but not in (HSA-Cre, Smn F7/+) cells. At day 6, death was observed in 11 and 25% of (HSA-Cre, Smn F7/+) and (Smn Δ7/+) cells, respectively (Fig. 3 A). The significantly higher proportion of dead cells in (Smn Δ7/+) than in (HSA-Cre, Smn F7/+, t test, P = 0.0025 at day 6) may be ascribed to either deleterious effect of Smn Δ7 involving both myoblasts and fused myotubes, or to a selective advantage of negative Cre recombinase (HSA-Cre, Smn F7/+) cells to grow. These data demonstrate that constitutive heterozygous deletion of Smn (Smn Δ7) led to a marked defect of myogenic differentiation with death occurring in 25% of myogenic committed cells.
Marked improvement of Smn mutant phenotype is associated with active muscle regeneration process
To assess whether deleterious effect of Smn Δ7 allele on muscle precursor cells could be circumvented in vivo, mutant mice carrying the (HSA-Cre, Smn F7/F7) instead of (HSA-Cre, Smn F7/Δ7) genotype were generated. In (HSA-Cre, Smn F7/F7) mice, both Smn alleles are intact in satellite cells, then Cre-mediated deleted in multinucleate myotubes. Surprisingly, although (HSA-Cre, Smn F7/Δ7) mice suffered from severe paralysis with a median survival of 33 d (n = 39; Cifuentes-Diaz et al., 2001), median survival of (HSA-Cre, Smn F7/F7) mice was 8 mo (n = 28) representing a eightfold increase in life span of mutant mice. A total of 24% of mice were still alive after 12 mo of age. In addition, (HSA-Cre, Smn F7/F7) mutant mice exhibited motor performance similar to that of control littermate (Smn F7/+) as determined by rotarod test. From 1 to 6 mo of age, 100% of (HSA-Cre, Smn F7/F7, n = 6) mutant mice were able to maintain their balance on the rod rotating at either 5 or 10 rpm for 7 min. For these reasons, (HSA-Cre, Smn F7/F7) mutant mice were called “mild” mutant compared with “severe” ones carrying the (HSA-Cre, Smn F7/Δ7) genotype (Fig. 4) .
Figure 4.

Phenotype of severe and mild mutant mice. Control littermate (A, Smn F7/+) and severe mutant mice (B, HSA-Cre, Smn F7/Δ7) at 1 mo of age. Note the posture of hindlimbs and dorsal kyphosis in severe mutant mice (B). (C and D) Mild mutant mice (HSA-Cre, Smn F7/F7) of 5 (C) or 11 mo (D). Note the marked hypotonia associated with kyphosis and reduction of muscle mass in 11-mo-old (but not in 5-mo-old) mutants.
To determine whether mild mutant mice developed muscular changes, histological examination of skeletal muscles including gastrocnemius, soleus, and quadriceps was performed by using hematoxylin and eosin staining. No obvious histological changes of muscle fibers were observed in 1-mo-old mild mutant mice when compared with control, except the presence of some rare necrotic fibers and myofibers with central nuclei (Fig. 5) . However, major changes were seen from 2 to 12 mo of age, including variability in fiber size, necrotic fibers, and regenerating myocytes with central nuclei (Fig. 5). These data indicate that mild mutant mice suffered from an active muscle necrosis regeneration process. Dystrophin and utrophin immunostaining on transverse muscle sections of 2-mo-old mild mutant mice showed abnormal expression of both proteins in a pattern similar to that previously observed in severe mutant mice (Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200210117/DC1; Cifuentes-Diaz et al., 2001). Therefore, in spite of the presence of severe myopathic process, the (HSA-Cre, Smn F7/F7) mutant mice display a much milder phenotype than that of (HSA-Cre, Smn F7/Δ7) mice.
Figure 5.
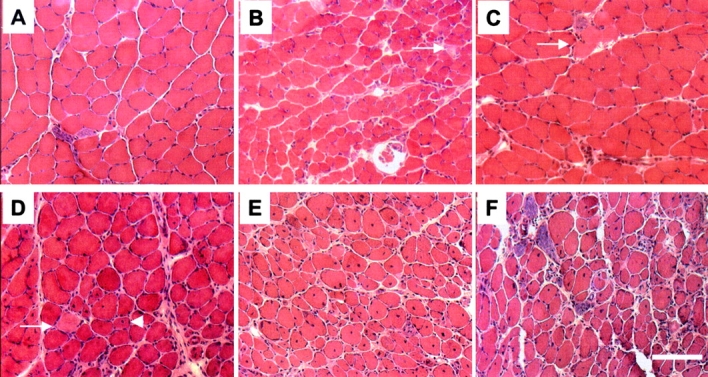
Hematoxylin and eosin staining of transverse sections of soleus from control, severe ( HSA-Cre, Smn F7/ Δ7 , and mild mutant mice (HSA-Cre, Smn F7/F7 ). At 1 mo of age, muscle histology of severe (B) or mild (C) mutants reveals necrotic fibers (arrow) and variation in fiber size (in severe mutant, only) when compared with control (A). At 2 mo of age (D), note the presence of necrotic fiber (arrow), numerous myofibers with central nuclei (arrowhead), and variation in fiber size in mild mutant mice. Marked morphological changes of skeletal muscle are observed in 6- (E) or 12-mo-old mild (F) mutant mice. Bar, 30 μm.
Heterozygous deletion of Smn present in all cell types of (HSA-Cre, Smn F7/Δ7), but not of (HSA-Cre, Smn F7/F7) mutant mice, might have a deleterious effect on the neighboring cells such as motor neurons. To know whether a neurogenic process, in addition to a myopathic one, was involved in the pathogenesis of severe mutants, electromyographic studies were performed in four 1-mo-old severe mutant (HSA-Cre, Smn F7/Δ7) and control mice. Distal motor latencies and compound muscle action potential (CMAP) amplitudes of gastrocnemius were recorded. Motor latencies of severe mutant mice (986 ± 172 ms) did not significantly differ from control mice (940 ± 134 ms, P = 0.68). CMAP amplitude can be subdivided in two parts, and the amplitude of the negative and positive waves was determined (Fournier, 2000). The amplitude of the negative wave of CMAP did not reveal any difference between severe mutant and control mice (35 ± 6 and 42 ± 7 mV, respectively; P = 0.21). In contrast, the positive wave of CMAP was significantly decreased in severe mutant mice (24 ± 7 mV) as compared with that of control mice of the same age (40 ± 7 mV, P = 0.02). The reduced amplitude of the positive wave strongly suggests that severe mutant mice suffer from a myopathic process, and the absence of giant motor unit as determined by the amplitude of the negative wave does not favor the hypothesis of a neurogenic effect as being responsible for difference in the phenotypic severity of mutant mice.
To determine whether the mild phenotype of (HSA-Cre, Smn F7/F7) mutant mice might be caused by change in Cre recombinase transgene expression, Cre-mediated deletion of Smn was evaluated in DNA extracted from skeletal muscle of (HSA-Cre, Smn F7/+) mice coming from littermate of mild or severe mutant mice. A similar ratio of Smn F7/Smn + was observed in both genetic backgrounds, indicating no change in Cre-mediated activity (Fig. S2). In addition, Cre-mediated deletion of Smn exon 7 was found to be similar in 1- or 12-mo-old (HSA-Cre, Smn F7/+) mice coming from littermate of mild mutants as determined by Smn F7/Smn + ratio (Fig. S2). Consistently, similar Cre recombinase expression was observed during life of mild mutant mice, as shown by immunolabeling experiments of Cre recombinase on transverse sections of gastrocnemius from 1- or 12-mo-old mild mutant mice (Fig. S3). Both experiments demonstrate that attenuation of the phenotype cannot be ascribed to reduced Cre recombinase activity. These results were further supported by Western blot analysis of SMN in skeletal muscle of mild and severe mutant mice. Similar reduction of SMN protein expression was observed in 20-d-old mild and severe mutant mice when compared with control littermate and actin expression (Fig. 6) . Interestingly, marked increase of SMN expression was observed in mild (but not in severe) mutant mice from 35 d of age with a level similar than that of control littermate of the same age and even higher at later stage (Fig. 6). SMN protein may be provided by cells that were not Cre-mediated deleted for Smn, and proliferating myogenic committed cells expressing high level of SMN might be regarded as strong candidates.
Figure 6.

Western blot analysis of SMN in skeletal muscles from control ( Smn F7/ + ), severe ( HSA-Cre, SmnF7/ Δ7 ), and mild mutant mice (HSA-Cre, SmnF7/F7). (A) At 20 d of age (left), the residual amount of SMN in mild (lane 3) or severe mice (lane 2) was similar. A marked increased amount of SMN was observed in 35-d-old mild (but not severe) mutant mice (right). (B and C) Note the marked decrease then increase of SMN expression in mild mutant mice from 20 d to 8 mo of age. The high proportion of regenerating myofibers in which Cre-mediated deletion of Smn has not yet occurred combined with the high expression of SMN in myogenic committed cells (see Fig. 2 B) lead to a high level of SMN in mild mutant mice.
To test this hypothesis, muscle regeneration was evaluated by determining the proportion of myofibers with central nuclei. Hematoxylin and eosin staining of transverse sections of the entire soleus was performed from three control, mild, or severe mutant mice. In mild mutant mice, the proportion of myofibers with central nuclei was increasing from 0.7% (mean of 6 out of 852, n = 3) at 1 mo, to 31% at 2 mo (mean of 256 out of 822, n = 3), reaching the value of 44% at 8 mo of age (mean of 278 out of 629, n = 3; Fig. 7) . In contrast, proportion of myofibers with centrally placed nuclei was <1% in both control (of 1- and 12-mo-old) and severe mutant mice (1-mo-old; Fig. 7). The significant increased proportion of regenerating myofibers from 1 to 2 mo (t test, P = 0.04) or to 8 mo of age (P < 0.01; Fig. 7) indicates the presence of an active muscle regeneration process in mild mutant mice. Significant loss of mature myofibers was observed in 2- and 8-mo-old mutant mice (566 ± 113 SD and 351 ± 162 SD, respectively; n = 3 in each group) when compared with 1-mo-old mild mutant mice (846 ± 94 SD, n = 3, P = 0.01; Fig. 7). However, the number of myofibers with central nuclei was able to compensate for the loss of mature myofibers of mild mutant mice from 2 to 6 mo of age, as total number of myofibers did not drop until 6 mo (Fig. 7).
Figure 7.

Myofiber number of the entire soleus of severe and mild mutant mice. Open columns indicate the number of myofibers without central nuclei, whereas gray columns indicate those with central nuclei in mild mutant mice. At each age, three mild mutant mice were analyzed, except at 6 mo (one animal). Hatched column indicates the mean of myofiber number in three severe mutant mice at 1 mo of age. Horizontal dotted line indicates the mean of myofiber number in three control mice analyzed at 1 and 12 mo of age. Standard deviations of the mean number of myofibers with (left bar) or without central nuclei (right bar) are given. Note that regenerating myofibers with central nuclei are able to compensate for loss of mature myofibers in 2- to 6-mo-old mild mutant mice, then reduction of both regenerating and mature myofibers was observed, leading to massive loss of muscle fibers in 12-mo-old mild mutant mice.
To compare the muscle regeneration capacity of severe mutants with that of mild mutant and control mice, myofiber degeneration was provoked by intramuscular injection of cardiotoxin in tibialis anterior of 3-wk-old control, severe, and mild mutant mice. As determined by hematoxylin and eosin staining on transverse sections, cardiotoxin induced myofiber necrosis within 1 d after injection (unpublished data). 8 d after cardiotoxin injection, muscle regeneration was quite similar in control and mild mutant mice, but severe mutant mice exhibited impaired muscle regeneration. Severe mutant mice displayed significant reduction in size of regenerating myofibers (Fig. 8) . Indeed, myofibers with central nuclei having size smaller than 900 μm2 represented 87% of regenerating myofibers in severe mutant mice (278 out of 320) compared with 53% (176 out of 331, χ2, P < 0.0001) in control or 46% in mild mutant mice (148 out of 323, χ2, P < 0.0001; Fig. 8). Early death of severe mutant mice (median survival of 33 d) did not allow us to determine whether skeletal muscle architecture was restored at a later stage.
Figure 8.
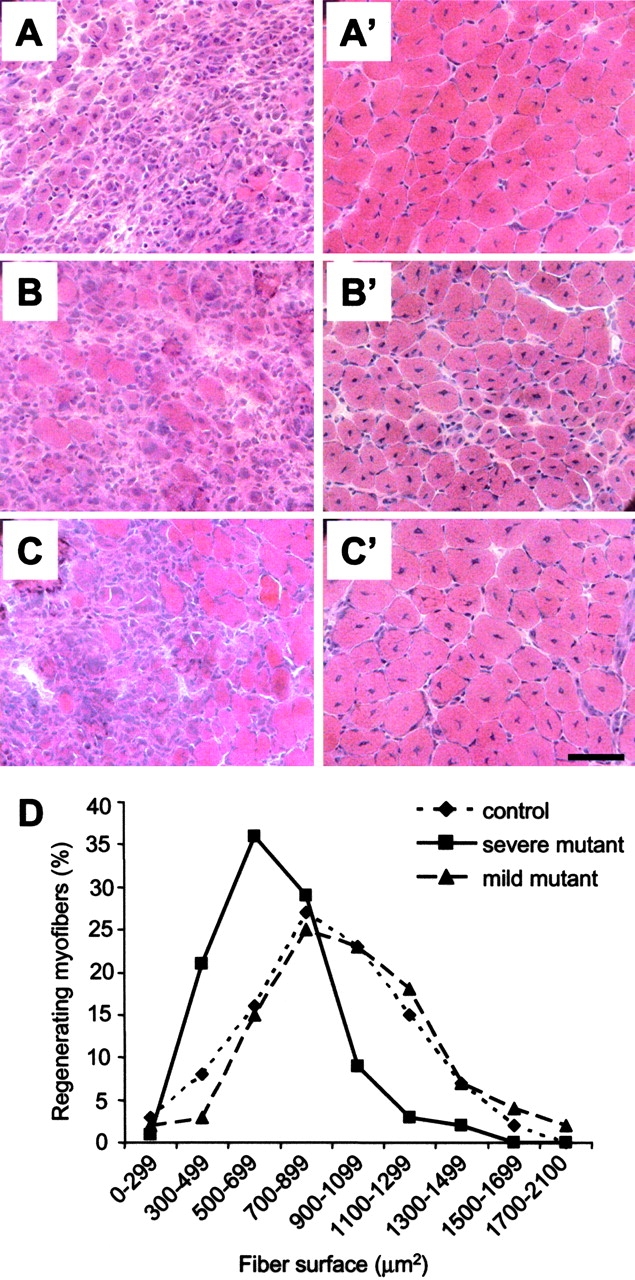
Skeletal muscle regeneration after cardiotoxin injection in control, severe, and mild mutant mice. (A–C′) At day 5 (A–C), necrotic myofibers are associated with cellular infiltration and regenerative myofibers in control (A), severe (B), and mild mutant mice (C). At day 8 (A′–C′), note the marked reduction in size of myofibers with central nuclei in severe mutants (B′) when compared with that of control (A′) or mild mutants (C′). Bar, 50 μm. (D) Distribution of regenerating myofiber surface as determined by hematoxylin and eosin staining on transverse sections of tibialis anterior 8 d after cardiotoxin injection. Note the marked change in myofiber surface profile of severe mutant mice with respect to that of either control or mild mutants (χ2, P < 0.0001). No significant difference was observed between control and mild mutant mice (χ2, P > 0.06).
To know whether the impairment of muscle regeneration in severe (but not in mild) mutant mice might be caused by defect of muscle progenitor cells in vivo, satellite cells were examined in control, severe, and mild mutant mice. Immunolabeling of Sca-1 and CD34 was performed on single muscle fibers isolated from collagenase-digested flexor digitorum brevis (FDB) of 1-mo-old control, severe, and mild mutant mice (Fig. 9) . Satellite cells were shown to express CD34, but not Sca-1, which further suggests that muscle-derived stem cells and satellite cells are distinct cell populations as recently described (Asakura et al., 2002). A significant reduction of CD34-expressing satellite cells per muscle fiber was observed in severe mutant mice (18 out of 35 muscle fibers, n = 3 mice) when compared with mild mutant mice (37 out of 36 muscle fibers, n = 3 mice, P < 0.02) or control mice (38 out of 50, n = 4 mice, P < 0.02). The average fiber lengths of severe mutant mice (318 μm ± 13 SEM, n = 102 muscle fibers) did not significantly differ from those of mild mutant mice (318 μm ± 13 SEM, n = 30, P = 0.98) or control mice (306 μm ± 12 SEM, n = 63, P = 0.45). These results indicate that differences in CD34-expressing satellite cell number were not caused by differences in average fiber length, and reveal a significant reduction of subpopulation of satellite cells expressing CD34 in severe (but not in mild) mutant mice.
Figure 9.

Satellite cells on freshly isolated muscle fibers from control, severe, and mild mutant mice. Immunolabeling of CD34 was performed on isolated muscle fibers from FDB of control (A–A′′), severe (B–B′′), and mild mutant mice (C–C′′). Reduced number of CD34-expressing satellite cells was observed in severe (but not mild) mutants. Satellite cell nuclei and myonuclei were stained with DAPI. Satellite cells do not express Sca-1 on isolated wild-type muscle fibers (D–D′′). Bar, 50 μm.
Progressive motor defect of mild Smn mutant mice correlates with reduction of both regenerating and mature muscle fibers
Mild mutant mice did not display any detectable motor defect within the first 6 mo of age, then developed progressive motor weakness as determined by rotarod test. At 8 mo of age, mutant mice (n = 6) were no more able to maintain their balance on the rod rotating at 5 rpm for 1 min, whereas control mice succeeded the test for 7 min. Motor defect was associated with the appearance of severe kyphosis and atrophy of muscle masses (Fig. 4).
To determine whether the progressive decline in motor performance of mild mutant mice was of myogenic or neurogenic origin, electromyographic studies were performed in three 11-mo-old control and mutant mice. Motor latencies of CMAP of mild mutants (838 ± 159 ms) did not significantly differ from control mice (772 ± 16 ms, P > 0.3). In mild mutant mice, the negative wave amplitude was not statistically different from control mice (34 ± 5 and 32 ± 6 mV, respectively; P > 0.3). In contrast, positive wave amplitude was significantly decreased in mutant mice (17 mV ± 2.1 SD) as compared with that of controls of the same age (27.4 mV ± 5 SD, t test, P < 0.03). Furthermore, Cre-mediated deletion of Smn was observed in skeletal muscles, but not in other tissues including spinal cord or sciatic nerve of 8-mo-old mutant mice (Fig. S4). These data indicated that the progressive motor defect observed in mild mutant mice was of myogenic origin only.
Mechanical properties of muscle were investigated on isolated extensor digitorum longus (EDL), a fast-twitch muscle, from 8-mo-old control and mild mutant mice. Marked reduction of the muscle weight from 15.9 ± 0.6 mg in controls to 10.1 ± 0.6 mg in mutants (P < 0.001, n = 5) was first noted. The amplitude and kinetic characteristics of short isometric tetanus showed a marked reduction in isometric force (Table I). However, when isometric forces were corrected for the muscle cross section (the “normalized” force, mN.cm−2), no difference between mutant and control mice was observed, suggesting that reduction of isometric force was a direct consequence of muscle mass reduction. The rate of force development was markedly slowed down in mutants and the difference remained, yet attenuated, when this rate was expressed as a percentage of the tetanic force developed. Isolated muscles were also submitted to a series of eccentric contractions where forced lengthening was imposed during maximal contraction. In five EDLs from mild Smn mutants, an average force drop of 15% (± 7, SD), which remained within the range of drops observed in age-matched controls, was observed (Table I; Deconinck et al., 1997). The cytosolic concentration of Ca2+ at rest was found to be quite similar in both mutant (24.5 nM ± 4.0, SD, n = 8) and control mice (28.9 nM ± 7.4, SD, n = 4). The passive influx of Ca2+ from the external medium was estimated by measuring the inward flux of Mn2+ ions (Merritt et al., 1989). It amounted to 2.35%.min−1 (± 1.23, SD, n = 8) in fibers from mild mutants and 4.0%.min−1 (± 2.19, SD, n = 4) in controls. These values fall within the normal ranges described previously (De Backer et al., 2002), and the difference between control and mutant mice was not significant (P > 0.1). Altogether, these data indicate that mild Smn mutant mice suffer from degenerative process of myofibers that affects neither contractile properties nor Ca2+ homeostasis. Consistently, mutant mice exhibited moderate elevation of serum creatine kinase activity (1,160 U/l ± 330 SD, n = 18) when compared with control littermates of the same age (476 U/l ± 220 SD, n = 12, P = 0.02). Therefore, reduction of muscle force and mass was not caused by defect of contractile properties of myofibers.
Table I. Amplitude and kinetic characteristics of isometric tetanic contraction of EDL.
| Mechanical parameters | Mild Smn mutants n = 5, SEM |
Controls n = 4, SEM |
P t test |
|---|---|---|---|
| Isometric force (mN) | 182 ± 19 | 323 ± 5 | <0.001 |
| Normalized isometric force (mN.cm−2) | 106 ± 13 | 122 ± 6 | n.s. |
| Rate of force development (mN.ms−1) | 4.8 ± 0.5 | 11.6 ± 0.3 | <0.001 |
| Rate of percentage of force development (% mN.ms−1) |
2.6 ± 0.1 | 3.6 ± 0.1 | <0.001 |
| Time for half relaxation (ms) | 65 ± 6.6 | 53 ± 7.5 | n.s. |
n.s., not significant.
Hematoxylin and eosin staining of skeletal muscles of 8- or 12-mo-old mild mutant mice including gastrocnemius, soleus, and biceps brachii showed major reduction of muscle size associated with myopathic changes including myofiber necrosis, variation in size of myofibers with centrally placed nuclei, and proliferative interstitial connective tissue (Fig. 5). To determine whether atrophy of muscle mass was caused by loss of muscle fibers, total myofiber number of the entire soleus was evaluated. Progressive reduction of myofiber number was observed in 8-mo-old mutant mice (629 ± 226 SD, n = 3) and become dramatic at 12 mo (361 ± 142 SD, n = 3) when compared with that of control mice of the same age (811 ± 122 SD, n = 3, 55% reduction, t test, P = 0.01; Fig. 7). To know whether the loss of muscle fibers was due to a progressive decline of muscle regeneration process in response to chronic necrosis, the total number of regenerating myofibers with central nuclei was evaluated in the same tissue samples. A dramatic decreased number of myofibers with central nuclei was observed in the entire soleus of 12-mo-old mutant mice (78 ± 56 SD, n = 3) when compared with that of 8-mo-old mutant mice (278 ± 88 SD, 72% reduction, P = 0.03; Fig. 7). These results suggest that progressive defect of muscle regeneration process associated with chronic skeletal muscle necrosis are responsible for loss of muscle fibers leading to progressive motor defect of mild mutant mice.
Discussion
Studying SMA mouse models and skeletal muscle cells or biopsies from SMA patients suggested that SMA phenotype not only results from motoneuron impairment, but also from constitutive skeletal muscle defects (Henderson et al., 1987; Braun et al., 1995; Cifuentes-Diaz et al., 2001). Although SMN was reported to be present in cytoplasmic dotlike structures in both myoblasts in vitro and skeletal muscle fibers of mice from postnatal day 1 to day 15 (Burlet et al., 1998; Fan and Simard, 2002), its role in myogenic cell survival has never been explored. Primary myogenic cultures from newborn mice revealed that SMN is highly expressed in myoblasts and fused myotubes. A twofold decrease in SMN expression results in death of 25% of myogenic committed cells including myoblasts and fused myotubes through a nonapoptotic process. These data indicate a marked dosage effect of SMN on muscle precursor cell survival in vitro. The availability of a cellular model derived from skeletal muscle affected in vivo in SMA should contribute hereafter to a better knowledge of SMA pathogenesis through investigation of molecular mechanisms underlying muscle cell death.
Smn mutant mice were generated using the Cre-loxP system that directs Smn exon 7 deletion not only in a given tissue, but also at a given time depending on the Cre recombinase transgene expression (Miniou et al., 1999; Frugier et al., 2000; Cifuentes-Diaz et al., 2001, 2002). The Cre recombinase expression controlled by the HSA promoter was observed in post-mitotic fused myotubes, but not in myoblasts both in vitro and in vivo. This system enabled us to induce a deletion of Smn exon 7 restricted to fused myotubes leading to necrosis process of myofibers while satellite cells remain nondeleted. Regenerative capacity of intact satellite cells in response to damage of mature muscle fibers can thus be evaluated. (HSA-Cre, Smn F7/F7) mutant mice, in which the satellite cells are not deleted for Smn, exhibit mild phenotype with median survival of 8 mo and motor performance similar to that of controls within the first 6 mo of age, despite severe myopathic process. In contrast, mutant mice carrying the (HSA-Cre, Smn F7/Δ7), in which satellite cells are heterozygously deleted for Smn, develop acute necrosis process of muscle fibers, progressive motor paralysis, and early death at 1 mo of age (Cifuentes-Diaz et al., 2001). Cre recombinase activity was similar in both types of mutants. We cannot exclude the hypothesis that the heterozygous deletion of Smn present in all cell types of severe (but not of mild) mutant mice might have a deleterious effect on the neighboring cells. However, neither morphological changes of motor neurons nor skeletal muscle denervation were observed in severe muscular mutant mice (Cifuentes-Diaz et al., 2001 and the present paper). Our data strongly suggest that the difference in phenotypic expression of mutant mice was of myogenic origin only.
The major difference in muscle morphology between severe and mild mutant mice consists of the appearance of an active muscle regeneration process in mild (but not severe) mutants. High proportion of regenerating myofibers is associated with marked reexpression of SMN in skeletal muscle of mild (but not severe) mutants, which likely arises from proliferative nondeleted satellite cells. These results strongly suggest that the partial rescue of the mild mutant mice comes from an improved capacity of muscle regeneration from intact satellite cells in response to similar damage of mature muscle fibers. Consistently, impaired muscle repair was observed in severe (but not mild) mutant mice in response to muscle injury by cardiotoxin, growth of regenerating myofibers being markedly deficient. Impairment of muscle regeneration in severe mutant mice can be ascribed to defect of myogenic precursor survival in vitro and reduced number of CD34-expressing satellite cells in vivo. Several reports support the evidence that satellite cells are heterogeneous in nature with respect, for example, to the expression of CD34, Myf5, and Sca-1 (Beauchamp et al., 2000; Qu-Petersen et al., 2002). Characterizing subpopulation(s) of cells and their derivatives dying in mutants should provide insight into the nature of precursor cells committed to efficient muscle regeneration.
Mild mutant mice exhibit progressive motor defect from 6 mo of age associated with severe reduction of muscle mass. This correlates with a marked reduction of total muscle force, as shown in vivo by the rotarod test or in vitro by measuring the tetanic force of isolated muscles. However, surprisingly, the contractile properties of the remaining muscle fibers seem unaffected as they develop normal force per unit cross section area and show no abnormal susceptibility to high mechanical stress. The reduction of the rate of force development is most likely the consequence of the marked reduction of the fast myosin isoform transcripts, although direct correlation remains to be determined (unpublished data). Intracellular homeostasis of Ca2+ ions also seems unaffected, and serum creatine kinase is moderately elevated. These data clearly differentiate the mild Smn mutant myopathy from a typical form of dystrophinopathy such as found in mdx mice (for review see Gillis, 1999).
Marked reduction of muscle force correlates with a dramatic reduction of both regenerating and mature muscle fibers. These data suggest a progressive decline in capacity of myogenic committed cells to regenerate muscle fibers in response to chronic necrosis in mild mutant mice. mdx mice lacking dystrophin, a model for muscular dystrophy and regeneration, develop neither motor defect of limbs nor reduction of muscle mass within the first year of life (Carnwath and Shotton, 1987; Coulton et al., 1988). This apparent paradox could be related to a progressive defect of skeletal muscle regeneration in Smn (but not in mdx) mutant mice. In 12-mo-old mild Smn mutant mice, dramatic decreased number of both regenerating and mature muscle fibers was observed in the entire soleus (mean of 72 out of a total of 361 ± 142 SD, n = 3). In contrast, a high number of regenerating myofibers was able to compensate for loss of mature myofibers in mdx mice of the same age, leading to a total number of myofibers similar to that of control mice (mean of 555 out of a total of 1048 ± 117 SD, n = 2; unpublished data). Although satellite cells are not deleted for Smn in mild mutant mice, Cre-mediated deletion that occurs in fused myotubes could lead to a progressive impairment of the muscle regeneration process. Alternatively, proliferation potential of satellite cells may be limited, as suggested by studies of human muscular dystrophies (for review see Cossu and Mavilio, 2000). Because satellite cells carry the conditional mutation, they undergo the same fate of degeneration as soon as they differentiate into mature muscle fibers. One can hypothesize that short and repeated cycles of myofiber degeneration and regeneration in Smn (but not in mdx) mutant mice could reveal a limited capacity of satellite cells to regenerate skeletal muscle. Evaluating the amplitude and kinetics of degeneration process in both mdx and Smn mutant mice should allow us to clarify this point.
The ability of myoblasts to become incorporated into skeletal muscle during regeneration is exploited through myoblast transplantation, a potential muscle cell–mediated therapeutic approach for human progressive muscular dystrophies (for reviews see Cossu and Mavilio, 2000; Partridge, 2000; Seale et al., 2001; Burton and Davies, 2002). Transplantation of muscle precursor cells into mdx model has shown that normal myoblasts can participate in muscle regeneration, resulting in dystrophin expression in the host muscle (Huard et al., 1994; Beauchamp et al., 1999; Qu-Petersen et al., 2002). However, it remains difficult to determine whether this local rescue could lead to any functional improvement, and the therapeutic potential that muscle progenitor cells might have remains unclear. By using a transgenic mouse line expressing the Cre recombinase in differentiated skeletal muscle, Cohn et al. (2002) demonstrated that maintenance of self-renewing potential of skeletal muscle can prevent the development of severe dystrophic alterations resulting from dystroglycan disruption. For the first time, our data proved that intact satellite cells remarkably improved the survival and motor performance of mutant mice, despite the presence of severe myopathic process. These results provide strong support to the view that use of muscle progenitor cells should represent a rational and efficient therapeutic strategy in muscular dystrophies. Smn mutant mice should represent valuable tools for evaluating the therapeutic benefits of cellular transplantation approaches.
Materials and methods
Mice
(HSA-Cre, Smn F7/F7) or (HSA-Cre, Smn F7/Δ7) mutant mice were generated by crossing homozygous (Smn F7/F7) mice with mice carrying the Cre recombinase transgene (HSA-Cre) and the (Smn F7/+) or (Smn Δ7/+) genotype, respectively. (HSA-Cre, Smn F7/+), (HSA-Cre, Smn Δ7/+), (HSA-Cre, Smn +/+), and (Smn Δ7/+) mice were maintained on C57BL/6J genetic background. (Smn F7/+) or (Smn +/+) animals from littermate were used as controls. The genomic organization of the Smn locus carrying either constitutive or Cre-mediated deletion of Smn exon 7 is identical as described previously by Frugier et al. (2000). Animals were genotyped by PCR amplification of DNA extracted from tail biopsies (Miniou et al., 1999; Frugier et al., 2000). All animal procedures were performed in accordance with institutional guidelines (agreement no. A91–228–2 and 3429).
Serum level of creatine kinase activity
Quantitative determination of creatine kinase activity of serum of control (Smn F7/+) or mutant mice (HSA-Cre, Smn F7/F7) was performed as described previously (Cifuentes-Diaz et al., 2001).
Primary muscle cultures
Primary muscle cultures were prepared from newborn mice (3–5 d of age) using a modified version of a previously described preplate technique (Tassin et al., 1991; Rando and Blau, 1994). The muscle tissue was enzymatically dissociated in 0.25% trypsin without EDTA (Life Technologies) in an equal volume of PBS at 37°C for 15 min. Cells were resuspended in proliferation medium (PM) consisting of Dulbecco's modified Eagle's medium supplemented with 10% horse serum (HS), 10% FBS, 1.25% chick embryo extract, and 1% penicillin-streptomycin (GIBCO BRL). Myoblasts were enriched by preplate technique on noncoated plastic dishes for 1 h. The nonadherent cells were then collected and transferred to new dishes for 4 h. The nonadherent cells were collected, counted, and plated (104 cells per dish) on 35-mm gelatin-coated dishes (1% gelatin; Sigma-Aldrich). Cells were grown in PM at 37°C in 7.5% CO2. For Trypan blue dye labeling, cells were trypsinized (0.25% trypsin with 2 mM EDTA; GIBCO BRL), then an equal volume of Trypan blue dye was added (0.4% Trypan blue stain; GIBCO BRL). Trypan blue uptake into cells was also performed directly on dishes. Cells were washed in PBS, incubated in an equal volume of PBS and 0.4% Trypan blue stain for 1 min, and were then directly observed under a microscope (Axiophot; Carl Zeiss MicroImaging, Inc.). For each analysis, independent experiments were performed from skeletal muscles of at least three mice of each phenotype: wild type, (Smn F7/+), (HSA-Cre), (Smn Δ7/+), or (HSA-Cre, Smn F7/+).
Single muscle fiber isolation
Single muscle fibers were isolated from the entire FDB muscle including tendons of three 1-mo-old control, severe, and mild mutant mice according to Rosenblatt et al. (1995). Muscles were digested with 0.2% collagenase type IV (Sigma-Aldrich) for 30 min at 37°C under O2. The muscle was then transferred to 35-mm collagen-coated dishes containing 2 ml Krebs and triturated using fire-polished pipettes to gently disaggregate the muscle fibers. Muscle fibers were incubated for 2 h in a 37°C humidified incubator with 5% CO2 to allow adhesion to the substratum. The average fiber lengths were evaluated by using an imaging densitometer (model GS710; Bio-Rad Laboratories). For immunolabeling of CD34 and Sca-1, isolated muscle fibers were fixed with 4% PFA in PBS for 5 min at RT, permeabilized with 0.03% Triton X-100 in PBS, and were then incubated with 10% normal goat serum for 1 h. Antibodies were incubated at 37°C for 1 h in concentrations as follows: FITC-coupled rat anti–mouse CD34 mAb (1:100, RAM34; BD Biosciences) and biotin anti–mouse Sca-1 antibody (1:200; BD Biosciences) revealed by Cy3-conjugated streptavidin (1:500; US Biological). The nuclei were stained with DAPI.
Cardiotoxin-induced muscle injury
Mice were deeply anesthetized with 100 mg/kg ketamine and 10 mg/kg xylazine. Cardiotoxin (50 μl of 10 μM diluted in PBS; Latoxan) was injected in tibialis anterior of three 3-wk-old control, severe, and mild mutant mice (Garry et al., 1997). The tibialis anterior muscles were harvested 1, 5, and 8 d after the cardiotoxin injection and frozen in cold isopentane. 10-μm transverse sections performed each 100 μm were stained with hematoxylin and eosin. Surface of myofibers with central nuclei was evaluated on serial transverse sections 8 d after cardiotoxin injection using Q-Fluoro software (Leica). 53% of myofibers with central nuclei had size <900 μm2 in control mice. The proportion of regenerating myofibers <900 μm2 was then determined in severe and mild mutant mice and compared with that of control mice by using a χ2 test for statistical analysis.
Histological and immunolabeling experiments of muscle tissues and cultures
10-μm transverse sections of isopentane-frozen skeletal muscles including gastrocnemius, soleus, quadriceps, and biceps brachii were stained with hematoxylin and eosin. To evaluate the total number of muscle fibers with or without central nuclei, the entire soleus was dissected and serial sections were performed every 400 μm. Counting was made after hematoxylin and eosin staining, and the highest number of myofibers per muscle section was retained for statistical analysis. Three (HSA-Cre, Smn F7/F7) mutant mice of 1, 2, 8, and 12 mo of age, one of 6 mo, and three 1-mo-old (HSA-Cre, Smn F7/Δ7) mutant mice were included in this work. Three (Smn F7/+) mice of 1 and 12 mo of age were used as controls.
Double immunostaining of dystrophin and utrophin was performed as described previously (Cifuentes-Diaz et al., 2001). For desmin immunolabeling of culture muscle cells, cells were fixed in cold methanol for 1 min at 4°C, incubated in 10% horse serum for 1 h at RT, and were then incubated with desmin mAb (1:500, DE-B5; CHEMICON International). After washing, cells were incubated with rhodamine-coupled secondary antibody (1:5,000; Jackson ImmunoResearch Laboratories), then mounted in Vectashield® with DAPI (Vector Laboratories). For Cre recombinase immunolabeling, the same procedure was applied by using Cre recombinase mAb (1:200, 2D8; Euromedex). Preparations were observed under a fluorescence microscope (Axiophot; Carl Zeiss MicroImaging, Inc.).
Determination of Cre recombinase activity by DNA analysis
Tissues were incubated overnight at 55°C in 0.1 M Tris-HCl, 5 mM EDTA, pH 8.0 0.2% SDS, and 0.2 M NaCl supplemented with 0.2 mg/ml of proteinase K. After phenol/chloroform extraction, genomic DNA was resuspended in Tris EDTA 10:1 buffer. PCR amplification of Smn exon 4 using primers flanking this exon was used as internal positive control (Frugier et al., 2000). Detection of the Smn Δ7 allele was performed by PCR amplification using primers pHR5 and GS8 (Frugier et al., 2000). The wild-type and Smn F7 alleles were simultaneously amplified by PCR using primers ex7sou1 and GS8 (Frugier et al., 2000). The efficiency of excision was estimated by comparing the intensity of the band amplified from the Smn F7 allele with that from the wild-type allele (Smn +). Ratio of Smn F7/Smn + was evaluated by scanning of PCR amplification products with an imaging densitometer (model GS710; Bio-Rad Laboratories) and analyzed with Quantity One software (Bio-Rad Laboratories).
Immunoblotting experiments
Frozen skeletal muscle (quadriceps or gastrocnemius) of three control and mutant mice were rapidly frozen and crushed in liquid nitrogen using a mortar and pestle. The pulverized muscle samples were transferred into 5 vol of a buffer containing 25 mM sodium phosphate, pH 7.2, 5 mM EDTA, and 1% SDS supplemented with protease inhibitor cocktail (Sigma-Aldrich), and boiled for 5 min. Protein sample (25–50 μg) was mixed with an equal volume of 2× Laemmli buffer, electrophoresed, then transferred. For SMN and actin immunodetection, anti-SMN and anti-actin mAb were used (1:5,000, Transduction laboratories; 1:10,000, CHEMICON International, respectively). Frozen muscle cell pellets were lysed into the same extraction buffer.
Rotarod test
Testing began at 15 d of age and animals were studied every 4 d, then once a week after 1 mo of age. Rotarod was performed as described previously (Cifuentes-Diaz et al., 2002).
Electromyographic studies
After deep anaesthetization, CMAP of gastrocnemius of three 11-mo-old mild mutants, four 1-mo-old severe mutants, and three control mice of the same ages were elicited after supramaximal stimulation of the sciatic nerve using an electromyographic apparatus (Keypoint 2; Medtronic). Latency and peak-to-peak amplitude were recorded and analyzed.
Muscle mechanics
Animals were deeply anaesthetized with ketamine and xylazine in order to preserve muscle perfusion during dissection of the EDL and the FDB. The Animal Ethics Committee of the Catholic University of Louvain (Brussels, Belgium) has approved this protocol. Electrical stimuli and isometric force of isolated EDL were sampled through an RTI-8 15 AD converter and digitized as described previously (Deconinck et al., 1997). A series of twitches and 0.3 s tetani were applied, in order to determine the length at which isometric tetanus force was maximal. After the recording of representative isometric twitch and tetanus, a 5-min rest was imposed, followed by the eccentric contraction protocol (Deconinck et al., 1997). This consisted of a series of 6 isometric tetani of 0.35 s duration in which a 7% lengthening of the muscle was applied, at a rate of 1 fiber length.s−1, when maximal force had been developed, i.e., after 0.15 s. Tetani were separated by 5 min rest during which the initial muscle length was restored. The isometric force drop (in %) is the decrease of the isometric force from the first to the sixth eccentric contraction.
Cytosolic Ca2+ concentration and Ca2+ influx in single FDB fibers
The experimental protocol has been previously described in detail (De Backer et al., 2002). In short, FDB muscles were submitted to collagenase digestion, then single fibers were loaded with the cell-permeant Ca2+ indicator Fura-PE3-AM. Cytosolic [Ca2+] was estimated by recording the intensity of fluorescence (Grynkiewicz et al., 1985; De Backer et al., 2002). Then, fibers were changed to a Ca2+-free Krebs solution containing 500 μM MnCl2. Influx of Mn2+ into fibers loaded with Fura-PE3 quenches the fluorescence of the dye and the quenching rate reflects the influx rate of Ca2+ ions (Merritt et al., 1989). Results were expressed as percent decrease of Fura-PE3 fluorescence per minute (in short: %.min−1).
Online supplemental material
Fig. S1 shows the immunostaining of sarcolemma components (dystrophin and utrophin) in mild mutant mice. Fig. S2 shows the characterization of Cre recombinase activity in mild and severe genetic backgrounds. Fig. S3 shows the immunolabeling of Cre recombinase on tissue sections of 1- and 12-mo-old mild mutant mice. Fig. S4 shows that Cre-mediated deletion of Smn exon 7 is restricted to skeletal muscle in mild mutant mice. Online supplemental material available at http://www.jcb.org/cgi/content/full/jcb.200210117/DC1.
Acknowledgments
We are very grateful to René-Marc Mège for helpful suggestions with primary myogenic cultures and Suse Barbosa and Gillian Butler-Browne for advice in cardiotoxin muscle injury. We thank Michel Fardeau (Institut de Myologie) for fruitful discussion, and M.H. Moreau and G. François (Centre Hospitalier Sud Francilien) for helpful assistance.
This work was supported by grants to J. Melki from INSERM (no. 690), the Association Française contre les Myopathies (no. 8DLO7E), Families of SMA (no. MEL111999-13102), Andrew's Buddies (no. LF002F), the Fondation pour la Recherche Médicale (no. 1000390-01), Aventis (no. 98277), the Conseil Regional d'Ile de France (no. E1534), GENOPOLE, and from the Fondation Bettencourt Schueller. J.-M. Gillis was supported by a grant from the Concerted Action (no. 00/05-260, Belgium).
The online version of this article includes supplemental material.
S. Nicole's present address is INSERM, U-546, Faculté de Médecine Pitié-Salpétriêre, 75013 Paris, France.
Footnotes
Abbreviations used in this paper: CMAP, compound muscle action potential; EDL, extensor digitorum longus, FDB, flexor digitorum brevis; HSA, human α-skeletal actin; SMA, spinal muscular atrophy; SMN, survival of motor neuron gene.
References
- Asakura, A., P. Seale, A. Girgis-Gabardo, and M.A. Rudnicki. 2002. Myogenic specification of side population cells in skeletal muscle. J. Cell Biol. 159:123–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beauchamp, J.R., J.E. Morgan, C.N. Pagel, and T.A. Partridge. 1999. Dynamics of myoblast transplantation reveal a discrete minority of precursors with stem cell-like properties as the myogenic source. J. Cell Biol. 144:1113–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beauchamp, J.R., L. Heslop, D.S. Yu, S. Tajbakhsh, R.G. Kelly, A. Wernig, M.E. Buckingham, T.A. Partridge, and P.S. Zammit. 2000. Expression of CD34 and Myf5 defines the majority of quiescent adult skeletal muscle satellite cells. J. Cell Biol. 155:1221–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun, S., B. Croizat, M.C. Lagrange, J.M. Warter, and P. Poindron. 1995. Constitutive muscular abnormalities in culture in spinal muscular atrophy. Lancet. 345:694–695. [DOI] [PubMed] [Google Scholar]
- Burlet, P., C. Huber, S. Bertrandy, M.A. Ludosky, I. Zwaenepoel, O. Clermont, J. Roume, A.L. Delezoide, J. Cartaud, A. Munnich, and S. Lefebvre. 1998. The distribution of SMN protein complex in human fetal tissues and its alteration in spinal muscular atrophy. Hum. Mol. Genet. 7:1927–1933. [DOI] [PubMed] [Google Scholar]
- Burton, E.A., and K.E. Davies. 2002. Muscular dystrophy: reason for optimism? Cell. 108:5–8. [DOI] [PubMed] [Google Scholar]
- Carnwath, J.W., and D.M. Shotton. 1987. Muscular dystrophy in the mdx mouse: histopathology of the soleus and extensor digitorum longus muscles. J. Neurol. Sci. 80:39–54. [DOI] [PubMed] [Google Scholar]
- Cifuentes-Diaz, C., T. Frugier, F.D. Tiziano, E. Lacene, N. Roblot, V. Joshi, M.H. Moreau, and J. Melki. 2001. Deletion of murine SMN exon 7 directed to skeletal muscle leads to severe muscular dystrophy. J. Cell Biol. 152:1107–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cifuentes-Diaz, C., S. Nicole, M.E. Velasco, C. Borra-Cebrian, C. Panozzo, T. Frugier, G. Millet, N. Roblot, V. Joshi, and J. Melki. 2002. Neurofilament accumulation at the motor endplate and lack of axonal sprouting in a spinal muscular atrophy mouse model. Hum. Mol. Genet. 11:1439–1447. [DOI] [PubMed] [Google Scholar]
- Cohn, R.D., M.D. Henry, D.E. Michele, R., Barresi, F. Saito, S.A. Moore, J.D. Flanagan, M.W. Skwarchuk, M.E. Robbins, J.R. Mendell, R.A. Williamson, and K.P. Campbell. 2002. Disruption of DAG1 in differentiated skeletal muscle reveals a role for dystroglycan in muscle regeneration. Cell. 110:639–648. [DOI] [PubMed] [Google Scholar]
- Cossu, G., and F. Mavilio. 2000. Myogenic stem cells for the therapy of primary myopathies: wishful thinking or therapeutic perspective? J. Clin. Invest. 105:1669–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulton, G.R., J.E. Morgan, T.A. Partridge, and J.C. Sloper. 1988. The mdx mouse skeletal muscle myopathy: I. A histological, morphometric and biochemical investigation. Neuropathol. Appl. Neurobiol. 14:53–70. [DOI] [PubMed] [Google Scholar]
- De Backer, F., C. Vandebrouck, P. Gailly, and J.M. Gillis. 2002. Long-term study of Ca(2+) homeostasis and of survival in collagenase-isolated muscle fibres from normal and mdx mice. J. Physiol. 542:855–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deconinck, N., J. Tinsley, F. De Backer, R. Fisher, D. Kahn, S. Phelps, K. Davies, and J.M. Gillis. 1997. Expression of truncated utrophin leads to major functional improvements in dystrophin-deficient muscles of mice. Nat. Med. 3:1216–1221. [DOI] [PubMed] [Google Scholar]
- Fan, L., and L.R. Simard. 2002. Survival motor neuron (SMN) protein: role in neurite outgrowth and neuromuscular maturation during neuronal differentiation and development. Hum. Mol. Genet. 11:1605–1614. [DOI] [PubMed] [Google Scholar]
- Fournier, E. 2000. Étude et sémiologie de la conduction nerveuse motrice. In Examen Electromyographique et Étude de la Conduction Nerveuse. 2nd ed. Ed. Médicale Internationale, Cachan, France. 161–171.
- Frugier, T., F.D. Tiziano, C. Cifuentes-Diaz, P. Miniou, N. Roblot, A. Dierich, M. Le Meur, and J. Melki. 2000. Nuclear targeting defect of SMN lacking the C-terminus in a mouse model of spinal muscular atrophy. Hum. Mol. Genet. 9:849–858. [DOI] [PubMed] [Google Scholar]
- Frugier, T., S. Nicole, C. Cifuentes-Diaz, and J. Melki. 2002. The molecular bases of spinal muscular atrophy. Curr. Opin. Genet. Dev. 12:294–298. [DOI] [PubMed] [Google Scholar]
- Garry, D.J., Q. Yang, R. Bassel-Duby, and R.S. Williams. 1997. Persistent expression of MNF identifies myogenic stem cells in postnatal muscles. Dev. Biol. 188:280–294. [DOI] [PubMed] [Google Scholar]
- Gillis, J.M. 1999. Understanding dystrophinopathies: an inventory of the structural and functional consequences of the absence of dystrophin in muscles of the mdx mouse. J. Muscle Res. Cell Motil. 20:605–625. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz, G., M. Poenie, and R.Y. Tsien. 1985. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 260:3440–3450. [PubMed] [Google Scholar]
- Henderson, C.E., S.L. Hauser, M. Huchet, F. Dessi, F. Hentati, T. Taguchi, J.P. Changeux, and M. Fardeau. 1987. Extracts of muscle biopsies from patients with spinal muscular atrophies inhibit neurite outgrowth from spinal neurons. Neurology. 37:1361–1364. [DOI] [PubMed] [Google Scholar]
- Huard, J., S. Verreault, R. Roy, M. Tremblay, and J.P. Tremblay. 1994. High efficiency of muscle regeneration after human myoblast clone transplantation in SCID mice. J. Clin. Invest. 93:586–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merritt, J.E., R. Jacob, and T.J. Hallam. 1989. Use of manganese to discriminate between calcium influx and mobilization from internal stores in stimulated human neutrophils. J. Biol. Chem. 264:1522–1527. [PubMed] [Google Scholar]
- Miniou, P., D. Tiziano, T. Frugier, N. Roblot, M. Le Meur, and J. Melki. 1999. Gene targeting restricted to mouse striated muscle lineage. Nucleic Acids Res. 27:e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partridge, T. 2000. The current status of myoblast transfer. Neurol. Sci. 21:S939–S942. [DOI] [PubMed] [Google Scholar]
- Paushkin, S., A.K. Gubitz, S. Massenet, and G. Dreyfuss. 2002. The SMN complex, an assemblyosome of ribonucleoproteins. Curr. Opin. Cell Biol. 14:305–312. [DOI] [PubMed] [Google Scholar]
- Qu, Z., L. Balkir, J.C. van Deutekom, P.D. Robbins, R. Pruchnic, and J. Huard. 1998. Development of approaches to improve cell survival in myoblast transfer therapy. J. Cell Biol. 142:1257–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu-Petersen, Z., B. Deasy, R. Jankowski, M. Ikezawa, J. Cummins, R. Pruchnic, J. Mytinger, B. Cao, C. Gates, A. Wernig, and J. Huard. 2002. Identification of a novel population of muscle stem cells in mice: potential for muscle regeneration. J. Cell Biol. 157:851–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rando, T.A., and H.M. Blau. 1994. Primary mouse myoblast purification, characterization, and transplantation for cell-mediated gene therapy. J. Cell Biol. 125:1275–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenblatt, J.D., A.I. Lunt, D.J. Parry, and T.A. Partridge. 1995. Culturing satellite cells from living single muscle fiber explants. In Vitro Cell. Dev. Biol. Anim. 31:773–779. [DOI] [PubMed] [Google Scholar]
- Seale, P., A. Asakura, and M.A. Rudnicki. 2001. The potential of muscle stem cells. Dev. Cell. 1:333–342. [DOI] [PubMed] [Google Scholar]
- Tassin, A.M., R.M. Mège, D. Goudou, M.E. Edelman, and F. Rieger. 1991. Modulation of expression and cell surface distribution of N-CAM during myogenesis in vitro. Neurochem. Int. 18:97–106. [DOI] [PubMed] [Google Scholar]