

Abstract
Signal transduction by reactive oxygen species (ROS; “redox signaling”) has recently come into focus in cellular biology studies. The signaling properties of ROS are largely due to the reversible oxidation of redox-sensitive target proteins, and especially of protein tyrosine phosphatases, whose activity is dependent on the redox state of a low pKa active site cysteine. A variety of mitogenic signals, including those released by receptor tyrosine kinase (RTKs) ligands and oncogenic H-Ras, involve as a critical downstream event the intracellular generation of ROS. Signaling by integrins is also essential for the growth of most cell types and is constantly integrated with growth factor signaling. We provide here evidence that intracellular ROS are generated after integrin engagement and that these oxidant intermediates are necessary for integrin signaling during fibroblast adhesion and spreading. Moreover, we propose a synergistic action of integrins and RTKs for redox signaling. Integrin-induced ROS are required to oxidize/inhibit the low molecular weight phosphotyrosine phosphatase, thereby preventing the enzyme from dephosphorylating and inactivating FAK. Accordingly, FAK phosphorylation and other downstream events, including MAPK phosphorylation, Src phosphorylation, focal adhesion formation, and cell spreading, are all significantly attenuated by inhibition of redox signaling. Hence, we have outlined a redox circuitry whereby, upon cell adhesion, oxidative inhibition of a protein tyrosine phosphatase promotes the phosphorylation/activation and the downstream signaling of FAK and, as a final event, cell adhesion and spreading onto fibronectin.
Keywords: reactive oxygen species; integrin-mediated cell adhesion; Rac; LMW-PTP; focal adhesion kinase
Introduction
Although superoxide anions (O2.−) and hydrogen peroxide (H2O2) are generally considered to be toxic by-products of respiration, recent evidence suggests that the production of these reactive oxygen species (ROS)* might be an integral component of membrane receptor signaling. In mammalian cells, a variety of extracellular stimuli have been shown recently to induce a transient increase in the intracellular concentration of ROS, and specific inhibition of the ROS generation results in a complete blockage of stimulant-dependent signaling (Gulati et al., 2001). Recently, important observations on the role of ROS as physiological regulators of intracellular signaling cascades activated by growth factors through their tyrosine kinase receptors have shed new light on the possible mechanisms underlying the growth regulatory and tumor-promoting activity of oxygen species and on the antiproliferative and antitumoral action of some antioxidant agents. A growing body of evidence indicates in fact that growth factor–induced oxygen species are necessary for optimal downstream propagation of mitogenic and antiapoptotic signals through mechanism which are still not understood completely.
The downstream effect of ROS production is the more or less reversible oxidation of proteins (Finkel, 2001). Thiols, by virtue of their ability to be reversibly oxidized, are recognized as key targets of oxidative stress. Redox-sensitive proteins, which include protein tyrosine phosphatases (PTPs) as the active site cysteine, are the target of specific oxidation by various oxidants, including H2O2, and this modification can be reversed by intracellular reducing agents (Xu et al., 2002). The reversible oxidation of PTPs family member was first demonstrated for PTP1B (Lee et al., 1998) during EGF signaling and then for low molecular weight PTP (LMW-PTP) (Chiarugi et al., 2001), and Src homology phosphatase (SHP)-2 (Meng et al., 2002) during PDGF stimulation. The inhibition exerted by ROS on PTPs helps the propagation of receptor tyrosine kinase (RTK) signals mediated by protein tyrosine phosphorylation, generally associated with the proliferative stimulus (Chiarugi et al., 2002).
The small GTPase Rac-1 has a key role in the regulation of cell growth and orchestrates cytoskeletal changes and gene expression in response to mitogenic cues from both soluble growth factors and ECM proteins. Although a function for Rac proteins in regulating the formation of ROS during the phagocyte respiratory burst has been long recognized, it is a recent notion that transient expression of constitutively activated forms of the small GTP-binding proteins Ras or Rac-1 in nonphagocytic cells also leads to a significant increase in intracellular ROS, suggesting that the family of Ras-related small GTP-binding proteins may function as general regulators of the intracellular redox state (Sundaresan et al., 1996). More importantly, Rac-1 mediates the transient rise of ROS observed after H-Ras expression or after cell stimulation by either growth factors or cytokines in a variety of cell types (Irani and Goldschmidt-Clermont, 1998). There are several recent studies on the role of Rac in the generation of ROS for both NADPH and arachidonic acid (AA)–dependent oxidases, although many details of the signaling pathway are still remaining unclear. These findings indicate that phospholipase A2 and subsequent AA metabolism by 5-lipoxygenase (LOX) act as downstream mediators in a Rac-signaling pathway leading to the generation of ROS (Woo et al., 2000). Moreover, in neutrophils the activity of the NADPH oxidase system is regulated by the small GTP-binding protein Rac-2 (Knaus et al., 1991), whereas in macrophages the NADPH oxidase appears to be regulated by Rac-1 (Abo et al., 1991).
In spite of the growing attention toward the mechanisms of intracellular signaling by adhesion molecules and the modality of signal integration between integrins and RTK receptors, a direct role for ROS in cell response to ECM proteins has not been directly addressed so far. Given the known role of small GTP-binding proteins in the signal transduction of integrin-mediated cell adhesion and in driving the production of intracellular ROS, we reasoned that ROS may be produced during cell adhesion, thus functioning as second messengers.
In this report, we provide evidence that ROS take a role in integrin signaling. Moreover, the production of ROS during integrin receptor engagement is dramatically more pronounced than during growth factor administration. In suggesting a role for oxidant species in integrin signaling, we propose that the production of ROS during cell adhesion leads to an up-regulation of FAK through the reversible oxidation of the LMW-PTP.
Results
Integrin-mediated cell adhesion generates a transient increase of ROS
A growing list of recent reports have demonstrated rapid and significant increases in intracellular ROS after growth factor or cytokine stimulation. Analysis of cells in culture has demonstrated that a variety of ligands, including PDGF, EGF, angiotensin II, and a host of cytokines, all trigger the rapid production of intracellular ROS (Sundaresan et al., 1995; Lo et al., 1996; Bae et al., 1997; Zafari et al., 1998). Integrin engagement by ECM proteins activates several intracellular cascades, which are similar to and largely integrated with those triggered by RTKs. As a main role in ligand-induced ROS production that has been ascribed to the small GTPase Rac, we reasoned that phenomena such as cell adhesion and cytoskeleton organization into spreading could trigger the production of intracellular ROS in response to Rac activation. To evaluate the production of hydrogen peroxide during cell adhesion, NIH-3T3 fibroblasts were presuspended for 30 min and then seeded for the indicated times onto plastic dishes. The production of hydrogen peroxide has been reported in Fig. 1 A (right). We observed a dramatic increase (up to 10-fold) in oxidants production starting at 15 min, peaking at 45 min, and slowly decreasing thereafter. Such an increase in intracellular oxidants, revealed by the redox-sensitive fluorescent dye 2′,7′-dichlorofluorescein diacetate (DCF-DA), is most likely due to hydrogen peroxide to which this probe is selectively sensitive.
Figure 1.




Intracellular ROS level during cell adhesion. (A) 106 cells were serum starved for 24 h before detaching and maintained in suspension with gentle agitation for 30 min at 37°C. Cells were then kept in suspension during treatment with 30 ng/ml of PDGF-BB or either kept in suspension or plated on fibronectin precoated dishes for the indicated times. Hydrogen peroxide production was evaluated with DCF-DA. (B) Cells were treated as in A except for the analysis of preadherent cells, which have not been detached for presuspension but left on plastic dishes for 24 h in serum starvation. After 30 min of presuspension, cells have been either treated in suspension with 30 ng/ml of PDGF-BB (S) or seeded onto fibronectin-treated dishes simultaneously with PDGF-BB treatment (A) for the indicated times. In parallel, preadherent cells have been stimulated with 30 ng/ml of PDGF-BB (preA). Hydrogen peroxide was evaluated with DCF-DA. (C) Cells are treated as in A and then were kept in suspension (Susp.) or seeded onto plastic culture dishes precoated with polylysine (PL) or fibronectin (FN) for the indicated times. Hydrogen peroxide was evaluated with DCF-DA. (D) Cells were treated as in A except that they are maintained in suspension during treatment with anti–α5-integrin monoclonal antibodies (mAb). These data, normalized per protein content, are obtained by at least three independent experiments. Values are mean ± SD of triplicate samples.
We also attempted to determine the relative contribution on ROS production of cell adhesion to ECM and of soluble growth factors. For this purpose, we compared exposure of suspended NIH-3T3 cells to PDGF with adhesion on fibronectin-coated dishes in a time course experiment. The oxidative response to integrin engagement appeared much higher in amplitude and different in kinetic with a peak at 45 min instead of 10 min (Fig. 1 A, left).
Since cell adhesion is associated with increased generation of intracellular peroxide, we next tested whether integrin redox signaling also has a part in cell oxidative response to soluble growth factors. ROS production elicited by PDGF was evaluated in suspended cells and in cells preadhered from 24 h (the general condition in which growth factor–elicited ROS induction has been reported previously; Sundaresan et al., 1995; Bae et al., 1997) and in cells in which adhesion and PDGF stimulation are concomitant. The results indicate that hydrogen peroxide production is much more pronounced in adherent than in suspended cells, in particular when integrin and PDGF receptors stimulation is simultaneous, suggesting a synergistic action of integrins and PDGF receptors for redox signaling (Fig. 1 B). Together, these findings indicate that the redox signaling by PDGF is largely anchorage- and integrin-dependent but also suggest that oxidative responses induced by cell adhesion and soluble growth factors are qualitatively and quantitatively different and are likely mediated by partially distinct mechanisms.
To analyze the reliance of cell adhesion–dependent hydrogen peroxide production upon integrin receptor engagement, we seeded presuspended NIH-3T3 cells onto either fibronectin or polylysine pretreated dishes. The production of hydrogen peroxide is reported in Fig. 1 C. The findings indicate that stimulation of integrin receptors is likely responsible for the intracellular production of ROS, since fibronectin treatment is extremely more effective than polylysine in the production of H2O2. The relevance of the engagement of integrin receptors in ROS production was further confirmed by the treatment of suspended cells with anti–α5-integrin–stimulating antibodies (Fig. 1 D). Together, these observations indicate that integrin clustering, rather than physical interaction with a solid substrate, induces intracellular production of hydrogen peroxide during cell adhesion.
To identify the source of intracellular ROS generated in response to cell adhesion, we tried to block the production of oxidants using selective inhibitors (Fig. 2). We used 5 μM diphenyl-iodide (DPI) to block NADPH oxidase, 10 μM nordihydroguaiaretic acid (NDGA) to block LOX, and 5 μM rotenone to inhibit mitochondrial superoxide production (Werner and Werb, 2002). The results exclude the involvement of mitochondrial respiratory chain in the production of ROS during cell adhesion, since rotenone is ineffective in preventing the rise of ROS, and indicate a major involvement of LOX and partially of NADPH oxidase in ROS production, since NDGA and moderately DPI impair the generation of hydrogen peroxide in response to cell adhesion.
Figure 2.

The source of ROS during cell adhesion. (A) 106 cells were serum starved for 24 h before detaching and maintained in suspension with or without 5 μM DPI to block NADPH oxidase, 10 μM NDGA to block 5-LOX, or 5 μM rotenone (Rot) to block mitochondrial superoxide production. Then cells were kept in suspension or seeded on fibronectin-treated dishes. Hydrogen peroxide was evaluated with DCF-DA and normalized per protein content. Values are mean ± SD of triplicate samples.
Integrin-induced ROS production requires functional Rac-1
The small GTPase Rac-1 is a central component of the signaling machinery downstream of adhesion molecules (del Pozo et al., 2000; Brakebusch et al., 2002). Moreover, Rac-1 mediates ligand-dependent generation of ROS in a variety of physiological settings (Knaus et al., 1991; Cool et al., 1998; Moldovan et al., 1999; Diebold and Bokoch, 2001; Brakebusch et al., 2002). Interestingly, both LOX and NADPH oxidases, two oxygen radical sources, are reportedly modulated by this GTPase. To assess the potential involvement of Rac-1 in the oxidative events associated with cell adhesion, we first determined whether Rac-1 is activated in NIH-3T3 cells allowed to adhere after presuspension. In agreement with previous reports (del Pozo et al., 2000), we observed an early accumulation of GTP-bound (i.e., active) Rac-1 upon cell plating on fibronectin with a kinetic comparable with the generation of oxygen species (Fig. 3 A). A causal link between Rac activation and adhesion-induced ROS was further demonstrated by overexpressing constitutively active Rac-1 mutant (RacQL) or the corresponding dominant-negative form (RacN17) of the GTPase (Fig. 3 B). As shown in Fig. 3 C, Rac-1–overexpressing cells plated on fibronectin produce significantly more hydrogen peroxide than cells in suspension. ROS production in adherent cells requires functional Rac-1, since it is nearly completely abolished in cells harboring the dominant-negative mutant RacV12N17. These data reinforce the idea that Rac-1 lies downstream of adhesion molecules in the redox cascade triggered by cell–ECM interaction.
Figure 3.



Involvement of Rac-1 in adhesion-dependent redox signaling. (A) Rac-1 activation during fibroblast adhesion to fibronectin. NIH-3T3 cells were kept in suspension for 2 h in serum-free HBSS. After medium renewal, aliquots of 5 × 105 cells were directly lysed or plated onto fibronectin-coated dishes for the indicated times. Adherent cells were then lysed and processed as described in Materials and methods. PAK-bound (GTP-loaded; top) and total (bottom) Rac-1 were quantified by anti Rac-1 Western blotting on GST-PAK pull downs and total cell lysates, respectively. (B) Expression of RacQL or RacN17 mutants in NIH-3T3 fibroblasts. NIH-3T3 were infected for 24 h with the indicated retroviral supernatants. Protein expression was verified by Western blot after 48 h of recovery. After anti-Rac immunoblotting, the filter was reprobed with an antiactin antiserum to control for equal protein loading in all the lanes. (C) Mock- and Rac-infected cells were serum starved for 24 h before detaching and maintained in suspension with gentle agitation for 30 min at 37°C. Cells were then kept in suspension or plated on fibronectin precoated dishes for 45 min. Hydrogen peroxide production was evaluated with DCF-DA and normalized per protein content. The infection efficiency in this experiment was ∼60%. Values are mean ± SD of triplicate samples.
ROS are mediators of cell adhesion
Stimulation of rat vascular smooth muscle cells with PDGF transiently increases the intracellular concentration of hydrogen peroxide. This increase could be blunted by cotreatment with chemical antioxidants, thus suggesting that H2O2 may act as a signal-transducing molecule during PDGF treatment (Sundaresan et al., 1995). To clarify the role of hydrogen peroxide produced during integrin-mediated cell adhesion, we treated presuspended NIH-3T3 cells with 5 μM NDGA or 10 μM DPI before seeding them onto fibronectin-coated dishes to selectively block the integrin-mediated ROS production from the LOX or NADPH oxidases, respectively. Fig. 4 A shows that both NDGA and DPI are effective in inhibiting both cell adhesion and cytoskeletal organization. In fact, cells treated with inhibitors are round shaped, more brilliant, and do not show any cytoskeletal organization. To confirm the key role of oxidants during cell adhesion and spreading, we attempted to scavenge the hydrogen peroxide produced during adhesion by using N-acetylcysteine (NAC), a known cell-permeant antioxidant. The results (Fig. 4 B) demonstrate that the removal of oxidant species by NAC administration dramatically delays cell attachment on ECM proteins and spreading. Together these findings suggest that the ROS produced after integrin receptor engagement during cell adhesion and spreading are essential molecules, very likely acting as signal-transducing messengers.
Figure 4.


ROS are mediators of cell adhesion. (A) 105 cells were serum starved for 24 h before detaching and maintained in suspension for 30 min at 37° with or without 5 μM DPI or 10 μM NDGA. Cells were then kept in suspension or seeded onto fibronectin-treated dishes for 60 min. Photographs of PFA-fixed cells were taken in a phase–contrast microscope. (B) The cells are treated as in A except that they are pretreated with 20 μM NAC (instead of NDGA or DPI) for 30 min in suspension. These experiments are representative of at least three others with similar results.
The redox regulation of LMW-PTP during cell adhesion
Redox-sensitive proteins, including PTPs as the active site cysteine, are the target of specific oxidation by ROS (Xu et al., 2002). This modification can be reversed by intracellular reducing agents. The reversible oxidation of PTP family members was demonstrated for PTP1B (Lee et al., 1998), LMW-PTP (Caselli et al., 1998), PTEN (Lee et al., 2002), SHP-2 (Meng et al., 2002), and cdc25C (Savitsky and Finkel, 2002), although a redox regulation during physiological conditions has been reported only for PTP1B during EGF signaling (Lee et al., 1998) and for LMW-PTP and SHP-2 during PDGF stimulation (Chiarugi et al., 2001; Meng et al., 2002). LMW-PTP is a cytosolic tyrosine phosphatase involved in the regulation of cell proliferation through dephosphorylation of PDGF receptor and of cytoskeletal organization and focal adhesion (FA) formation through the dephosphorylation of p190RhoGAP and FAK, respectively (Chiarugi et al., 1995, 2000; Raugei et al., 2002; Rigacci et al., 2002). It has been reported that fibroblasts overexpressing LMW-PTP display a significantly decreased number of FAs and increased cell motility (Chiarugi et al., 1998; Rigacci et al., 2002). To verify the involvement of LMW-PTP in adhesion to ECM proteins, we overexpressed the phosphatase in NIH-3T3 murine fibroblasts, and we analyzed the ability of these cells to adhere onto fibronectin in a time course experiment (Fig. 5). Generation of ROS in response to adhesion was not affected by LMW-PTP overexpression, and the levels of α5-integrins do not differ among all LMW-PTP–expressing clones or mock-transfected cells (unpublished data). However, in keeping with an inhibitory role of this phosphatase on integrin signaling, overexpression of LMW-PTP was associated with a significant delay in cell adhesion and spreading, which closely resembles the defect observed in parental cells treated with antioxidants (Fig. 4, A and B). This is consistent with the idea that LMW-PTP is a major target for redox signaling by integrins and suggests that elimination of ROS during cell–ECM interaction may lead to a functional up-regulation of LMW-PTP activity, which in turn inhibits FA formation and cell adhesion.
Figure 5.

LMW-PTP down-regulates cell adhesion to fibronectin. 3 × 104 mock-transfected or wtLMW-PTP–overexpressing cells were seeded in serum-depleted medium, for the indicated times, onto fibronectin pretreated dishes. Photographs of PFA-fixed cells were taken in a phase–contrast microscope. This experiment is representative of at least three others with similar results.
Because of the involvement of LMW-PTP in cell adhesion regulation and its redox sensitivity (Chiarugi et al., 2001), we hypothesized that this enzyme may represent a target for oxidative signaling by integrins, and we analyzed the redox state of LMW-PTP during cell adhesion to ECM proteins. Presuspended NIH-3T3 cells overexpressing LMW-PTP were seeded onto fibronectin-coated dishes or kept in suspension for the indicated times and then lysed in RIPA buffer containing 5 μM of 5′-iodoacetamidofluorescein (5′-F-IAA). Fluorescein-carboxymethylation is a method to tag proteins containing low pKa cysteine residues (Wu et al., 1998) that we have already applied to study the oxidation state of LMW-PTP in vivo (Chiarugi et al., 2001), demonstrating a specificity of 5′-F-IAA for LMW-PTP active site-reduced cysteines. LMW-PTP was then immunoprecipitated from lysates, and an antifluorescein immunoblot was performed. The result shows that LMW-PTP is strongly oxidized during cell adhesion (Fig. 6 A). Moreover, in a parallel experiment we quantitated the PTP activity of immunoprecipitated LMW-PTP in suspended and adherent cells by an enzymatic assay on p-paranitro phenyl-phosphase (PNPP). An aliquot of the samples was used in an anti–LMW-PTP immunoblot for normalization. The ratio between enzymatic activity and LMW-PTP content of the immunoprecipitates is reported in Fig. 6 B. The results demonstrate that during cell adhesion the oxidation of the phosphatase is accompanied by its enzymatic inactivation followed by a complete rescue of its catalytic activity. On the contrary, no variation of LMW-PTP activity is observed in cells kept in suspension. Interestingly, the variation of enzymatic activity of LMW-PTP follows the kinetic of the production of ROS due to cell adhesion. We note that the redox-dependent inhibition of ectopic LMW-PTP is not complete, reaching 70% of the total enzyme. Nevertheless, the residual activity of overexpressed LMW-PTP is sufficient to affect cell adhesion and spreading (Fig. 5). In addition, we confirm that the redox behavior of endogenous LMW-PTP is identical to the ectopically expressed LMW-PTP (Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200211118/DC1).
Figure 6.




LMW-PTP oxidation during cell adhesion. (A) 106 NIH-3T3 cells were serum starved for 24 h before detaching and maintained in suspension for 30 min at 37°. Cells were then kept in suspension or seeded onto fibronectin-treated dishes for 30 and 60 min. Cells were then lysed in RIPA buffer and treated with 5′-F-IAA. LMW-PTP was immunoprecipitated, and an antifluorescein immunoblotting was performed. The blot was then stripped and reprobed with anti–LMW-PTP antibodies for normalization. (B) 106 NIH-3T3 cells overexpressing wtLMW-PTP were treated as in A except that LMW-PTP was immunoprecipitated without 5′-IAF labeling, and a PTP activity assay was performed using PNPP as substrate. LMW-PTP activity is shown in U/mg. (C) 106 NIH-3T3 cells overexpressing wtLMW-PTP were treated as in B except that they were pretreated for 16 h with or without BSO in order to inhibit the synthesis of glutathione. (D) 106 NIH-3T3 cells overexpressing wtLMW-PTP were treated as in B except that they were pretreated for 30 min with or without 5 μM DPI or 10 μM NDGA in order to inhibit the synthesis of ROS. These data are representative of at least three independent experiments.
We have demonstrated previously that a GSH-dependent reduction process is involved in the rescue of enzymatic activity of the oxidized LMW-PTP after PDGF treatment (Chiarugi et al., 2001). To analyze the GSH dependence of the recovery of LMW-PTP activity during integrin-mediated cell adhesion, we blocked the glutathione-dependent cellular system of oxidized protein reduction by using an inhibitor of the γ-glutamyl–cysteine-synthetase, butionine-sulphoximide (BSO) during cell adhesion. The cellular glutathione concentration was decreased by 90% in a 24-h pretreatment of NIH-3T3 cells with BSO (unpublished data). Cells were pretreated for 16 h with or without 25 mM BSO, and the LMW-PTP activity in the immunoprecipitates was quantitated in suspended and adherent cells. The results (Fig. 6 C) show that the treatment with BSO causes an impairment of LMW-PTP reactivation, indicating that the intracellular reduced glutathione takes a central role in the recovery of LMW-PTP activity after cell adhesion to ECM.
Finally, in order to discover the source of ROS that inactivate/oxidize LMW-PTP, we selectively blocked ROS production by NADPH oxidase with DPI and/or by LOX with NDGA before assaying LMW-PTP activity in immunoprecipitates. Cells were pretreated with the inhibitors for 30 min in suspension, and then cell adhesion on fibronectin was allowed. LMW-PTP immunoprecipitates were assayed for phosphatase activity and the results, shown in Fig. 6 D, confirm that the intracellular oxidase system, which is mainly involved in LMW-PTP inactivation during cell adhesion, is likely LOX, although NADPH oxidase appears to retain a secondary role.
ROS control FAK activation through the redox regulation of LMW-PTP
It has been demonstrated recently that LMW-PTP associates with and dephosphorylates p125FAK, interfering with cell motility and spreading. Moreover, LMW-PTP overexpression significantly decreases the number of FAs in adherent cells (Rigacci et al., 2002). To analyze the role of cell adhesion–dependent ROS production during FA formation, we explored the activation of p125FAK through an artificial block of ROS production. Presuspended cells, either kept in suspension or seeded onto fibronectin-coated dishes, were treated with NDGA or DPI. p125FAK activation was assessed by antiphosphotyrosine immunoblot of anti-FAK immunoprecipitates (Fig. 7 A). The blot was then reprobed with anti-p125FAK antibodies for normalization, and densitometric analysis was performed. The results prove that phosphorylation of FAK is dependent on ROS generation since NDGA and DPI leads to an impairment of FAK activation. In addition, we analyzed the effect of antioxidants on the activation of MAPKs and p60 Src tyrosine kinase, other well-known events of downstream FAK activation. Our results (Fig. 7, B and C) confirm the central role of ROS in the transduction signals, i.e., MAPK and Src activation, starting from FAK when cytoskeleton rearrangement takes place.
Figure 7.

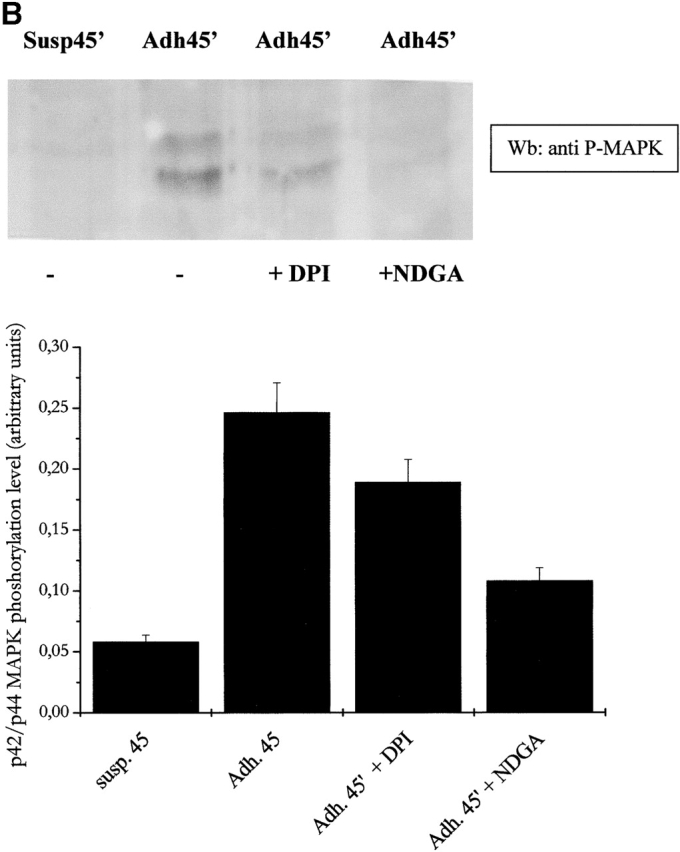

ROS produced by cell adhesion affects FAK activation and downstream signals. (A) FAK activation analysis: 106 NIH-3T3 cells overexpressing wtLMW-PTP were serum starved for 24 h before detaching and maintained in suspension for 30 min at 37°C with or without 5 μM DPI or 10 μM NDGA. Then cells were either kept in suspension or seeded onto fibronectin-treated dishes for 45 min. Cells were then lysed in RIPA buffer, p125FAK was immunoprecipitated, and an antiphosphotyrosine immunoblotting was performed. The blot was then stripped and reprobed with anti-p125FAK antibodies for normalization by densitometric analysis. The bottom plot reports the activation ratio of p125FAK. (B and C) MAPK and Src kinase activation analysis: cells were treated as in A except that 10 μg of total lysates in RIPA buffer was used for an anti–phospho-MAPK immunoblot. The blot was then stripped and reprobed with anti-MAPK antibodies for normalization by densitometric analysis. The bottom plot reports the net activation of MAPK (B) or Src kinase (C). These data are representative of at least three independent experiments.
To clarify if the presence of oxidant messenger molecules could affect the association between LMW-PTP and FAK, we immunoprecipitated LMW-PTP from NDGA pretreated cells during cell adhesion to fibronectin. Anti-FAK immunoblotting showed that the association between the two molecules is severely impaired during adhesion with respect to suspension. In addition, a central role of the adhesion-dependent ROS production in the abrogation of LMW-PTP/FAK association is strongly suggested by antioxidant treatment (Fig. 8 A). The equalization of LMW-PTP content in immunoprecipitates was achieved by anti–LMW-PTP immunoblot (Fig. 8 B). On the basis of these data, we suggest that the oxidation of LMW-PTP during cell adhesion impairs the ability of the phosphatase to bind its substrate, namely FAK. As a direct consequence, an enhancement of FAK tyrosine phosphorylation level occurs and cell attachment through FAs takes place.
Figure 8.


ROS produced by cell adhesion affects LMW-PTP binding to p125FAK. (A) 106 NIH-3T3 cells overexpressing wtLMW-PTP were serum starved for 24 h before detaching and maintained in suspension for 30 min at 37°C with or without 10 μM NDGA. Then, cells were either kept in suspension or seeded onto fibronectin-treated dishes for 45 min. Cells were then lysed in RIPA buffer, and LMW-PTP was immunoprecipitated, and an anti-p125FAK immunoblotting was performed. The blot was then stripped and reprobed with anti–LMW-PTP antibodies for normalization (B). These data are representative of at least three independent experiments.
Discussion
Data presented here lead to two major conclusions: (a) ROS have a major role in the signaling cascade triggered by integrins during cell–ECM interaction; and (b) modulation of integrin signaling and cell adhesion through FA formation by ROS is mediated, at least in part, by an up-regulation of FAK through an oxidative inhibition of LMW-PTP.
Our contribution to the general idea of a key role of ROS during cell proliferation is related to the proposal that ROS, in particular those produced in a Rac-1–dependent fashion, play a major role in the propagation of intracellular signals triggered by integrins. Given the recent observation that ROS are required for the growth factor–induced tyrosine phosphorylation of proteins, we investigated whether these oxidant molecules are produced and could play a role in integrin-mediated cell adhesion. First, we have shown that during cell adhesion to extracellular matrix proteins, a rise in intracellular ROS occurs. The increase in ROS generation is large (∼10-fold) and dependent on integrin receptor engagement (Fig. 1 A) as indicated by the growth of ROS during cell adhesion to fibronectin but not to polylysine and by the response elicited by anti–α5-integrin antibodies (Fig. 1, C and D).
An important issue raised by our experiments deals with the relative contribution of growth factors and adhesion molecules to redox signaling. Since ROS elicited by PDGF in suspended fibroblasts are significantly less pronounced than those triggered by PDGF during simultaneous integrin receptor engagement (Fig. 1 B), we conclude that the frequently reported generation of ROS by growth factors (Sundaresan et al., 1995; Bae et al., 1997; Zafari et al., 1998; Finkel, 2001) is likely an anchorage-dependent phenomenon. In addition, the relative contribution to ROS production of integrin receptor engagement and growth factor administration is unbalanced, being higher than oxidants produced during cell–ECM interaction with respect to those produced in response to growth factors (Fig. 1 A). By extension, we suggest that oxidative signals during fibroblast stimulation by mitogens may be combinatory in nature and reflect an integrated activity of both RTKs and adhesion molecules.
Besides mitochondrial respiratory chain malfunction, there are several potential intracellular sources of ROS, including NADPH oxidase and LOX, which catalyze the synthesis of superoxide anion rapidly converted in hydrogen peroxide (Wientjes and Segal, 1995; Bae et al., 1997; Finkel, 2001; Pani et al., 2001). Herein, we rule out that integrins engage ROS molecules through mitochondrial pathways, and we suggest the involvement of cytosolic oxidases, mainly of LOX and only marginally of NADPH oxidase (Fig. 2). Hence, although several reports have indicated the source of growth factor–induced ROS in NADPH-dependent, membrane-bound oxidases (Bae et al., 1997; Zafari et al., 1998; Chiarugi et al., 2001; Finkel, 2001), we propose that cytosolic lipoxygenases account for most oxidative events triggered by integrins. Although the molecular basis of this difference is yet to be elucidated, the different source of ROS may account for the differences in both intensity and the kinetic observed when redox signals elicited by PDGF and cell adhesion were compared.
Moreover, we report that Rac takes a specific role in the production of ROS after cell adhesion (Fig. 3). It has been reported that ROS production in growth factor–stimulated nonphagocytic cells appears to require the participation of Rac (Kim et al., 1997; Cool et al., 1998). Although in part speculative, we propose the idea that PDGF-elicited ROS production is greatly dependent on cell anchorage due to the limited capacity of growth factors to activate Rac-1 in the absence of integrin engagement (del Pozo et al., 2000). In this view, Rac may to play a central role in the integration of the combinatorial oxidative signals owing to RTKs and adhesion molecules.
Many data suggest that hydrogen peroxide acts as a signal-transducing molecule during growth factor signaling (Sundaresan et al., 1995; Bae et al., 1997; Zafari et al., 1998; Chiarugi et al., 2001; Colavitti et al., 2002). Our data emphasize that ROS behave as messenger molecules during integrin receptor signaling and growth factor ones. In fact, both the specific blockage of oxidant production by cytosolic oxidases with DPI or NDGA treatment or the scavenge of the ROS produced after cell adhesion with NAC treatment cause a great inhibition or delay of cell adhesion and spreading (Fig. 4). These data are in keeping with a central role of hydrogen peroxide in the cytoskeleton rearrangement that drives cells toward contacts with ECM proteins and completion of cytoskeletal architecture, since it is also indicated by the kinetic of ROS production, with a peak at 45 min in strict concomitance with the stabilization of FAs.
There is increasing evidence that oxidative stress or redox-dependent protein modifications modulate early events of signal transduction for cell growth and death (Pani et al., 2000b; Finkel, 2001). One of the effects of these redox signals may be the inactivation of PTPs through the oxidation of critical sulfydryl groups (Xu et al., 2002). All PTPs contain an essential cysteine residue (pK a 4.7–5.4) in the signature active site motif that exists as a thiolate anion at neutral pH (Zhang et al., 1992). The active site cysteine is the target of specific oxidation by various oxidants, including H2O2, and this modification can be reversed by incubation with thiol compounds such as dithiothreitol and reduced glutathione (Caselli et al., 1998; Lee et al., 1998, 2002; Meng et al., 2002). These observations suggest that PTPs might undergo H2O2-dependent inactivation in cells, resulting in a shift in the equilibrium with PTKs toward protein phosphorylation. The reversible oxidation has been demonstrated for PTP1B during EGF signaling and for LMW-PTP and SHP-2 during PDGF stimulation (Bae et al., 1997; Chiarugi et al., 2001; Meng et al., 2002). Among these phosphatases, we selected LMW-PTP for its role in cell adhesion, since it has been reported that this phosphatase is involved in cell motility control and in down-regulation of FA formation (Chiarugi et al., 1998; Chiarugi et al., 2000; Rigacci et al., 2002). We report herein that LMW-PTP overexpression causes a delay in cell adhesion to ECM proteins, confirming a negative function of LMW-PTP on FA development and cytoskeleton organization (Fig. 5). We demonstrated that LMW-PTP is oxidized during cell adhesion and that this oxidation is followed by a transient inactivation of the phosphatase enzymatic activity (Fig. 6, A and B). After the removal of the oxidative burst following adhesion, LMW-PTP totally rescues its catalytic activity, due to intracellular reduced glutathione, in agreement with our previously reported findings on the central role of glutathione in the redox regulation of LMW-PTP after PDGF receptor hydrogen peroxide production (Chiarugi et al., 2001). Finally, LMW-PTP oxidation after integrin receptors engagement is mainly due to the activation of LOX, in keeping with the source of ROS during cell adhesion (Fig. 6). The finding that phosphatase inhibition during cell–ECM interaction is transient and reversible by intracellular reductants may also have important functional implications. It is in fact an emerging concept that cell adhesion is a dynamic process, closely related to cell motility. Although phosphatase inhibition promotes cell adhesion, phosphatase recovery may be essential to local detachment and directional migration in response to integrin signaling. Although partially speculative, this idea is consistent with previous reports of decreased cell motility in dominant-negative LMW-PTP–expressing cells (Chiarugi et al., 1998; Rigacci et al., 2002) and also with the need of PTEN phosphatase activity for cellular chemotaxis (Iijima and Devreotes, 2002).
It has been reported that LMW-PTP is able to down-regulate the formation of FA structures by dephosphorylating p125FAK (Rigacci et al., 2002). FAK is a nonreceptor protein tyrosine kinase involved in signal transduction from integrin-enriched FA sites, mediating cell contact with the extracellular matrix. Multiple protein–protein interaction sites allow FAK to associate with adapters and structural proteins, allowing for the modulation of MAPKs, stress-activated protein kinases, and small GTPases activity. FAK-enhanced signals have been shown to mediate the survival of anchorage-dependent cells and are critical for efficient cell migration in response to growth factor receptor and integrin stimulation (Brakebusch et al., 2002; Hauck et al., 2002). Herein we reported that ROS produced after cell adhesion behave as positive regulators for FAK activation, since the blockage of their synthesis greatly reduced the activation of the kinase. Actually, the ROS produced by 5-LOX are mainly responsible for FAK activation, leaving NADPH oxidase with a marginal role (Fig. 7 A). The key role of cell adhesion–dependent ROS increase is further stressed by the analysis of FAK downstream pathways. In fact, both MAPK and Src kinase are dramatically down-regulated when cells are treated with antioxidants (Fig. 7, B and C). Finally, we reported that the oxidation of LMW-PTP during cell adhesion is accompanied by a disruption of the interaction between the phosphatase and FAK (Fig. 8). The peculiarity of LMW-PTP redox regulation is the ability to rescue its catalytic activity by virtue of an intramolecular disulphur bond between two vicinal cysteines (Caselli et al., 1998; Chiarugi et al., 2001). These two cysteines are both in the catalytic site: Cys12 forms the transient cysteinyl-phosphate intermediate, whereas Cys17 cooperates with Arg18 for the binding of the phosphate moiety of the substrate (Cirri et al., 1993 ). We suppose that the oxidation of both these cysteines to form an intramolecular disulfide affects the ability of the protein to bind the substrate. Hence, during oxidative conditions LMW-PTP is not only oxidized and enzymatically inactivated but it is no more able to bind its natural substrate. With respect to the simple enzyme inhibition, this additional mechanism could further delay FAK dephosphorylation upon enzyme recovery or, more importantly, may prevent phosphotyrosine residues to be occupied and functionally sequestered by the inactive phosphatase, thus permitting these phosphorylated residues to signal through binding of SH2 domain–containing proteins, i.e., Src kinase, Grb2 adaptor, and so on. Whether this phenomenon is restricted to the LMW-PTP–FAK interaction or applies to other phosphatase-substrate pairs is still to be determined.
On the basis of our data, we propose a model of redox regulation of FA formation in which ROS play a key role in the transduction of the signals engaged by cell adhesion through inhibiting LMW-PTP and allowing the activation of p125 FAK. The requirement for ROS in the integrin signaling cascade upstream of FAK may account for the inhibition of cell adhesion and spreading observed in antioxidant-treated cells; however, a direct effect of oxidants on different targets cannot be excluded at the moment.
Finally, we stress that our findings open new avenues for pharmacological intervention in anchorage-independent cell transformation. In fact, malignant cells are often anchorage-independent for their growth, and this property directly correlates with their metastatic potential (Brakebusch et al., 2002). Loss of anchorage dependence is frequently due to a deregulated activation of the signaling pathway normally triggered by integrins. In particular, excess ROS production associated with cell transformation by H-Ras (Irani and Goldschmidt-Clermont, 1998) or c-Myc (Tanaka et al., 2002) could release costimulatory signals which are normally triggered by cell–ECM interaction. In line with this view, Rat-1 cells overexpressing either active Rac-1 or oncogenic R12 Ras display anchorage-independent growth, which is dramatically inhibited by ROS scavengers (unpublished data).
Materials and methods
Materials
Unless specified, all reagents were obtained from Sigma-Aldrich. NIH-3T3 and C2C12 cells were from American Type Culture Collection; PDGF-BB was from Peprotech; all antibodies were from Transduction Laboratories except the activating monoclonal anti-α5 integrin antibodies that are gifts from Dr. Bosco Chan (University of Western Ontario, London, Canada), and those against c-Myc tag that were from Santa Cruz Biotechnology, Inc.; DCF-DA, 5′-iodoacetamide-fluoresceinated (5′-IAF), and antifluorescein antibodies were obtained from Molecular Probes.
Cell culture and protein overexpression
NIH-3T3 cells were cultured in DME with 10% FCS in 5% CO2 humidified atmosphere. 10 μg of pRcCMV-wtLMW-PTP (Chiarugi et al., 1995) were stably transfected in NIH-3T3 cells using the calcium phosphate method. For cell infections, retroviral constructs expressing the RacQL and RacN17 cDNA in the pLPC/Puro backbone were generated and transfected by calcium phosphate coprecipitation in the 293T Phoenix packaging cell line (a gift from Dr. Scott Lowe, Cold Spring Harbor Laboratories, Cold Spring Harbor, NY). Supernatants were collected every 4–6 h and overlayed onto NIH-3T3 cells. After 24 h of infection, cells were left to recover for 48 h before analyses.
Cell adhesion
106 cells were serum starved for 24 h before detaching with 0.25% trypsin for 1 min. Trypsin was blocked with 0.2 mg/ml soybean trypsin inhibitor, and cells were resuspended in 2 ml/10-cm dish of fresh medium, maintained in suspension for 30 min at 37°C, and then directly seeded onto precoated dishes treated overnight with 10 μg/ml human fibronectin or 10 μg/ml poly-d-lysine in PBS. Control cells were kept in suspension by plating them onto dishes pretreated with 1 mg/ml of BSA in culture medium, thus preventing adhesion to the dish.
Cell adhesion assay
Cells were serum starved for 24 h, and then 3 × 104 cells were seeded onto serum-depleted medium for the indicated times in a 24-well dish precoated with 10 μg/ml human fibronectin. Cells were fixed in 1 ml of 0.25% PFA and photographs were taken with a phase–contrast microscope (Nikon).
Immunoprecipitation and Western blot analysis
106 cells were lysed for 20 min on ice in 500 μl of complete RIPA lysis buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1% Nonidet P-40, 2 mM EGTA, 1 mM sodium orthovanadate, 1 mM phenyl-methanesulphonyl fluoride, 10 μg/ml aprotinin, 10 μg/ml leupeptin). Lysates were clarified by centrifugation and immunoprecipitated for 4 h at 4°C with 1–2 μg of the specific antibodies. Immune complexes were collected on protein A Sepharose, separated by SDS-PAGE, and transferred onto nitrocellulose. Immunoblots were incubated in 3% BSA, 10 mM Tris/HCl, pH 7.5, 1 mM EDTA, and 0.1% Tween-20 for 1 h at room temperature, probed first with specific antibodies and then with secondary antibodies. Quantity-One software (Bio-Rad Laboratories) was used to perform quantitative analyses.
In vivo 5′-F-IAA labeling
As reported in Wu et al. (1998), cells were lysed in RIPA buffer at pH 7.5, and 5 μM 5′-F-IAA was added. The lysates were labeled 10 min at 37°C and then were immunoprecipitated with anti–LMW-PTP antibodies. The redox state was evidenced by antifluorescein immunoblot.
LMW-PTP activity assay
The tyrosine phosphatase activity was measured as reported previously (Bucciantini et al., 1999). Briefly, cells were lysed in RIPA buffer, and LMW-PTP was immunoprecipitated from lysates. Immunoprecipitates were then resuspended in 100 μl of 0.1 M sodium acetate, pH 5.5, 10 mM EDTA. Phosphatase activity assay was performed adding 100 μl of 10 mM PNPP at 37°C for 1 h. The production of p-nitrophenol was measured colorimetrically at 410 nm. The results were normalized on the basis of LMW-PTP content analyzed by anti–LMW-PTP immunoblot.
MAPK and Src activation
1.5 × 105 cells were serum starved for 24 h before detachment. After a 30-min presuspension treatment, cells were seeded on fibronectin-coated dishes for different times and then lysed in RIPA buffer. 20 μg of lysates were used for anti–phospho-ERK1/2 or anti–phospho-Src immunoblots. The data were normalized by anti-MAPK or anti-Src immunoblot.
Assay of intracellular H2O2
Intracellular production of H2O2 was assayed as described previously (Pani et al., 2000a). 5 min before the end of incubation time, adherent or suspended cells were treated with 5 μg/ml DCF-DA. After PBS washing, adherent cells were lysed in 1 ml of RIPA buffer and analyzed immediately by fluorescence spectrophotometric analysis at 510 nm. Data have been normalized on total protein content.
Determination of Rac-1 activity
NIH-3T3 cells were kept in suspension for 2 h in serum-free medium. After medium renewal, aliquots of 5 × 105 cells were directly lysed or plated onto fibronectin-coated dishes for the indicated times before lysis in RIPA buffer. Rac-GTP was quantified in precleared protein lysates according to Sander et al. (1998). Briefly, lysates were incubated with 5–10 μg of PAK-CRIB-GST fusion protein absorbed on glutathione-Sepharose beads. Immunoreactive Rac-1 precipitated by PAK-GST was then quantified by anti-Rac Western blot analysis.
Online supplemental material
Western blot, immunoprecipitation, and 5′-IAF labelling are used in Fig. S1 to show the redox regulation of endogenous LMW-PTP. Fig. S1 and the techniques used in Fig. S1 are available at http://www.jcb.org/cgi/content/full/jcb.200211118/DC1.
Supplemental Material
Acknowledgments
The authors wish to thank Dr. Barbara Bedogni for preparing the pLPC/RacQL and pLPC/RacN17 constructs, and Dr. Bosco Chan (University of Western Ontario, London, Canada) for generously providing the anti–mouse alpha-5 mAb. We thank Prof. P. Cirri and Dr. Barbara Bedogni for helpful discussions.
This work was supported by the Italian Association for Cancer Research, the Ministero della Università e Ricerca Scientifica e Tecnologica (MIUR-PRIN 2002 to G. Ramponi), in part by grants from the Consorzio Interuniversitario Biotecnologie (to G. Ramponi) and the Cassa di Risparmio di Firenze (to P. Chiarugi), and by Consiglio Nazionale delle Ricerche, Target project on Biotechnology (to S. Borrello).
P. Chiarugi and G. Pani contributed equally to this work.
The online version of this article includes supplemental material.
Footnotes
Abbreviations used in this paper: AA, arachidonic acid; BSO, butionine sulphoximide; DCA-DA, 2′,7′-dichlorofluorescein diacetate; DPI, diphenyl iodide; FA, focal adhesion; 5′-F-IAA, 5′-iodoacetamidofluorescein; LMW-PTP, low molecular weight PTP; LOX, 5-lipoxygenase; NAC, N-acetyl-cysteine; NDGA, nordihydroguaiaretic acid; PNPP, p-paranitro phenyl-phosphase; PTP, phosphotyrosine protein phosphatase; ROS, reactive oxygen species; SHP, Src homology phosphatase.
References
- Abo, A., E. Pick, A. Hall, N. Totty, C.G. Teahan, and A.W. Segal. 1991. Activation of the NADPH oxidase involves the small GTP-binding protein p21rac1. Nature. 353:668–670. [DOI] [PubMed] [Google Scholar]
- Bae, Y.S., S.W. Kang, M.S. Seo, I.C. Baines, E. Tekle, P.B. Chock, and S.G. Rhee. 1997. Epidermal growth factor (EGF)-induced generation of hydrogen peroxide. Role in EGF receptor-mediated tyrosine phosphorylation. J. Biol. Chem. 272:217–221. [PubMed] [Google Scholar]
- Brakebusch, C., D. Bouvard, F. Stanchi, T. Sakai, and R. Fassler. 2002. Integrins in invasive growth. J. Clin. Invest. 109:999–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucciantini, M., P. Chiarugi, P. Cirri, L. Taddei, M. Stefani, G. Raugei, P. Nordlund, and G. Ramponi. 1999. The low Mr phosphotyrosine protein phosphatase behaves differently when phosphorylated at Tyr131 or Tyr132 by Src kinase. FEBS Lett. 456:73–78. [DOI] [PubMed] [Google Scholar]
- Caselli, A., R. Marzocchini, G. Camici, G. Manao, G. Moneti, G. Pieraccini, and G. Ramponi. 1998. The inactivation mechanism of low molecular weight phosphotyrosine-protein phosphatase by H2O2. J. Biol. Chem. 273:32554–32560. [DOI] [PubMed] [Google Scholar]
- Chiarugi, P., P. Cirri, G. Raugei, G. Camici, F. Dolfi, A. Berti, and G. Ramponi. 1995. PDGF receptor as a specific in vivo target for low M(r) phosphotyrosine protein phosphatase. FEBS Lett. 372:49–53. [DOI] [PubMed] [Google Scholar]
- Chiarugi, P., P. Cirri, F. Marra, G. Raugei, T. Fiaschi, G. Camici, G. Manao, R.G. Romanelli, and G. Ramponi. 1998. The Src and signal transducers and activators of transcription pathways as specific targets for low molecular weight phosphotyrosine-protein phosphatase in platelet-derived growth factor signaling. J. Biol. Chem. 273:6776–6785. [DOI] [PubMed] [Google Scholar]
- Chiarugi, P., P. Cirri, L. Taddei, E. Giannoni, G. Camici, G. Manao, G. Raugei, and G. Ramponi. 2000. The low M(r) protein-tyrosine phosphatase is involved in Rho-mediated cytoskeleton rearrangement after integrin and platelet-derived growth factor stimulation. J. Biol. Chem. 275:4640–4646. [DOI] [PubMed] [Google Scholar]
- Chiarugi, P., T. Fiaschi, M.L. Taddei, D. Talini, E. Giannoni, G. Raugei, and G. Ramponi. 2001. Two vicinal cysteines confer a peculiar redox regulation to low molecular weight protein tyrosine phosphatase in response to platelet-derived growth factor receptor stimulation. J. Biol. Chem. 276:33478–33487. [DOI] [PubMed] [Google Scholar]
- Chiarugi, P., P. Cirri, M.L. Taddei, D. Talini, L. Doria, T. Fiaschi, F. Buricchi, E. Giannoni, G. Camici, G. Raugei, and G. Ramponi. 2002. New perspectives in PDGF receptor downregulation: the main role of phosphotyrosine phosphatases. J. Cell Sci. 115:2219–2232. [DOI] [PubMed] [Google Scholar]
- Cirri, P., P. Chiarugi, G. Camici, G. Manao, G. Raugei, G. Cappugi, and G. Ramponi. 1993. The role of Cys12, Cys17 and Arg18 in the catalytic mechanism of low-M(r) cytosolic phosphotyrosine protein phosphatase. Eur. J. Biochem. 214:647–657. [DOI] [PubMed] [Google Scholar]
- Colavitti, R., G. Pani, B. Bedogni, R. Anzevino, S. Borrello, J. Waltenberger, and T. Galeotti. 2002. Reactive oxygen species as downstream mediators of angiogenic signaling by vascular endothelial growth factor receptor-2/KDR. J. Biol. Chem. 277:3101–3108. [DOI] [PubMed] [Google Scholar]
- Cool, R.H., E. Merten, C. Theiss, and H. Acker. 1998. Rac1, and not Rac2, is involved in the regulation of the intracellular hydrogen peroxide level in HepG2 cells. Biochem. J. 332:5–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diebold, B.A., and G.M. Bokoch. 2001. Molecular basis for Rac2 regulation of phagocyte NADPH oxidase. Nat. Immunol. 2:211–215. [DOI] [PubMed] [Google Scholar]
- del Pozo, M.A., L.S. Price, N.B. Alderson, X.D. Ren, and M.A. Schwartz. 2000. Adhesion to the extracellular matrix regulates the coupling of the small GTPase Rac to its effector PAK. EMBO J. 19:2008–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkel, T. 2001. Reactive oxygen species and signal transduction. IUBMB Life. 52:3–6. [DOI] [PubMed] [Google Scholar]
- Gulati, P., P.C. Klohn, H. Krug, M. Gottlicher, B. Markova, F.D. Bohmer, and P. Herrlich. 2001. Redox regulation in mammalian signal transduction. IUBMB Life. 52:25–28. [DOI] [PubMed] [Google Scholar]
- Hauck, C.R., D.A. Hsia, and D.D. Schlaepfer. 2002. The focal adhesion kinase—a regulator of cell migration and invasion. IUBMB Life. 53:115–119. [DOI] [PubMed] [Google Scholar]
- Iijima, M., and P. Devreotes. 2002. Tumor suppressor PTEN mediates sensing of chemoattractant gradients. Cell. 109:599–610. [DOI] [PubMed] [Google Scholar]
- Irani, K., and P.J. Goldschmidt-Clermont. 1998. Ras, superoxide and signal transduction. Biochem. Pharmacol. 55:1339–1346. [DOI] [PubMed] [Google Scholar]
- Kim, B.C., C.J. Lim, and J.H. Kim. 1997. Arachidonic acid, a principal product of Rac-activated phospholipase A2, stimulates c-fos serum response element via Rho-dependent mechanism. FEBS Lett. 415:325–328. [DOI] [PubMed] [Google Scholar]
- Knaus, U.G., P.G. Heyworth, T. Evans, J.T. Curnutte, and G.M. Bokoch. 1991. Regulation of phagocyte oxygen radical production by the GTP-binding protein Rac 2. Science. 254:1512–1515. [DOI] [PubMed] [Google Scholar]
- Lee, S.R., K.S. Kwon, S.R. Kim, and S.G. Rhee. 1998. Reversible inactivation of protein-tyrosine phosphatase 1B in A431 cells stimulated with epidermal growth factor. J. Biol. Chem. 273:15366–15372. [DOI] [PubMed] [Google Scholar]
- Lee, S.R., K.S. Yang, J. Kwon, C. Lee, W. Jeong, and S.G. Rhee. 2002. Reversible inactivation of the tumor suppressor PTEN by H2O2. J. Biol. Chem. 277:20336–20342. [DOI] [PubMed] [Google Scholar]
- Lo, Y.Y., J.M. Wong, and T.F. Cruz. 1996. Reactive oxygen species mediate cytokine activation of c-Jun NH2-terminal kinases. J. Biol. Chem. 271:15703–15707. [DOI] [PubMed] [Google Scholar]
- Meng, T.C., T. Fukada, and N.K. Tonks. 2002. Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo. Mol. Cell. 9:387–399. [DOI] [PubMed] [Google Scholar]
- Moldovan, L., K. Irani, N.I. Moldovan, T. Finkel, and P.J. Goldschmidt-Clermont. 1999. The actin cytoskeleton reorganization induced by Rac1 requires the production of superoxide. Antiox. Redox. Signal. 1:29–43. [DOI] [PubMed] [Google Scholar]
- Pani, G., R. Colavitti, B. Bedogni, R. Anzevino, S. Borrello, and T. Galeotti. 2000. a. A redox signaling mechanism for density-dependent inhibition of cell growth. J. Biol. Chem. 275:38891–38899. [DOI] [PubMed] [Google Scholar]
- Pani, G., B. Bedogni, R. Anzevino, R. Colavitti, B. Palazzotti, S. Borrello, and T. Galeotti. 2000. b. Deregulated manganese superoxide dismutase expression and resistance to oxidative injury in p53-deficient cells. Cancer Res. 60:4654–4660. [PubMed] [Google Scholar]
- Pani, G., B. Bedogni, R. Colavitti, R. Anzevino, S. Borrello, and T. Galeotti. 2001. Cell compartmentalization in redox signaling. IUBMB Life. 52:7–16. [DOI] [PubMed] [Google Scholar]
- Raugei, G., G. Ramponi, and P. Chiarugi. 2002. Low molecular weight protein tyrosine phosphatases: small, but smart. Cell. Mol. Life Sci. 59:941–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigacci, S., E. Rovida, S.P. Dello, and A. Berti. 2002. Low Mr phosphotyrosine protein phosphatase associates and dephosphorylates p125 focal adhesion kinase, interfering with cell motility and spreading. J. Biol. Chem. 277:41631–41636. [DOI] [PubMed] [Google Scholar]
- Sander, E.E., S. van Delft, J.P. ten Klooster, T. Reid, R.A. van der Kammen, F. Michiels, and J.G. Collard. 1998. Matrix-dependent Tiam1/Rac signaling in epithelial cells promotes either cell–cell adhesion or cell migration and is regulated by phosphatidylinositol 3-kinase. J. Cell Biol. 143:1385–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savitsky, P.A., and T. Finkel. 2002. Redox regulation of Cdc25C. J. Biol. Chem. 277:20535–20540. [DOI] [PubMed] [Google Scholar]
- Sundaresan, M., Z.X. Yu, V.J. Ferrans, K. Irani, and T. Finkel. 1995. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science. 270:296–299. [DOI] [PubMed] [Google Scholar]
- Sundaresan, M., Z.X. Yu, V.J. Ferrans, D.J. Sulciner, J.S. Gutkind, K. Irani, P.J. Goldschmidt-Clermont, and T. Finkel. 1996. Regulation of reactive-oxygen-species generation in fibroblasts by Rac1. Biochem. J. 318:379–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka, H., I. Matsumura, S. Ezoe, Y. Satoh, T. Sakamaki, C. Albanese, T. Machii, R.G. Pestell, and Y. Kanakura. 2002. E2F1 and c-Myc potentiate apoptosis through inhibition of NF-kappaB activity that facilitates MnSOD-mediated ROS elimination. Mol. Cell. 9:1017–1029. [DOI] [PubMed] [Google Scholar]
- Werner, E., and Z. Werb. 2002. Integrins engage mitochondrial function for signal transduction by a mechanism dependent on Rho GTPases. J. Cell Biol. 158:357–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wientjes, F.B., and A.W. Segal. 1995. NADPH oxidase and the respiratory burst. Semin. Cell Biol. 6:357–365. [DOI] [PubMed] [Google Scholar]
- Woo, C.H., Y.W. Eom, M.H. Yoo, H.J. You, H.J. Han, W.K. Song, Y.J. Yoo, J.S. Chun, and J.H. Kim. 2000. Tumor necrosis factor-alpha generates reactive oxygen species via a cytosolic phospholipase A2-linked cascade. J. Biol. Chem. 275:32357–32362. [DOI] [PubMed] [Google Scholar]
- Wu, Y., K.S. Kwon, and S.G. Rhee. 1998. Probing cellular protein targets of H2O2 with fluorescein-conjugated iodoacetamide and antibodies to fluorescein. FEBS Lett. 440:111–115. [DOI] [PubMed] [Google Scholar]
- Xu, D., I.I. Rovira, and T. Finkel. 2002. Oxidants painting the cysteine chapel: redox regulation of PTPs. Dev. Cell. 2:251–252. [DOI] [PubMed] [Google Scholar]
- Zafari, A.M., M. Ushio-Fukai, M. Akers, Q. Yin, A. Shah, D.G. Harrison, W.R. Taylor, and K.K. Griendling. 1998. Role of NADH/NADPH oxidase-derived H2O2 in angiotensin II-induced vascular hypertrophy. Hypertension. 32:488–495. [DOI] [PubMed] [Google Scholar]
- Zhang, Z.Y., J.P. Davis, and R.L. Van Etten. 1992. Covalent modification and active site-directed inactivation of a low molecular weight phosphotyrosyl protein phosphatase. Biochemistry. 31:1701–1711. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.