

Abstract
Signals transduced by kinases depend on the extent and duration of substrate phosphorylation. We generated genetically encoded fluorescent reporters for PKC activity that reversibly respond to stimuli activating PKC. Specifically, phosphorylation of the reporter expressed in mammalian cells causes changes in fluorescence resonance energy transfer (FRET), allowing real time imaging of phosphorylation resulting from PKC activation. Targeting of the reporter to the plasma membrane, where PKC is activated, reveals oscillatory phosphorylation in HeLa cells in response to histamine. Each oscillation in substrate phosphorylation follows a calcium oscillation with a lag of ∼10 s. Novel FRET-based reporters for PKC translocation, phosphoinositide bisphosphate conversion to IP3, and diacylglycerol show that in HeLa cells the oscillatory phosphorylations correlate with Ca2+-controlled translocation of conventional PKC to the membrane without oscillations of PLC activity or diacylglycerol. However, in MDCK cells stimulated with ATP, PLC and diacylglycerol fluctuate together with Ca2+ and phosphorylation. Thus, specificity of PKC signaling depends on the local second messenger-controlled equilibrium between kinase and phosphatase activities to result in strict calcium-controlled temporal regulation of substrate phosphorylation.
Keywords: calcium; fluorescence resonance energy transfer; oscillation; phosphatase; PKC
Introduction
Dynamic interplay between phosphorylation and dephosphorylation critically controls cellular function. This is particularly evident in signal transduction, where the extent and duration of substrate phosphorylation dictates the nature of downstream signaling. Perturbation of the balance between phosphorylation and dephosphorylation can alter a cell's fate, from triggering apoptosis to promoting proliferation.
Since its discovery over two decades ago as the “phorbol ester receptor” (Nishizuka, 1984), PKC has been known to play a key role in maintaining the balance between normal growth and transformation (Nishizuka, 1995). Indeed, the tumor-promoting properties of phorbol esters exemplify the cellular consequences of tipping the balance of cellular signals excessively toward phosphorylation. PKC function in cells is exquisitely controlled by three major mechanisms: phosphorylation, required for catalytic competence, membrane targeting, required for conformational activation, and protein–protein interactions, which poise the enzyme at specific intracellular locations (Mellor and Parker, 1998; Newton, 2002b). Thus, PKC activity is dependent on the spatiotemporal context of the enzyme, its substrates, and its activating cofactors. Pertubation of any of these mechanisms disrupts cell function by altering the degree of substrate phosphorylation.
There are 10 mammalian PKC isozymes that share a highly homologous catalytic domain, conferring relatively low substrate selectivity between family members (Nishikawa et al., 1997). All isozymes contain an autoinhibitory pseudosubstrate sequence in the regulatory moiety that allosterically regulates access of substrates to the active site. In addition, the regulatory moiety contains one or two membrane-targeting modules: the C1 domain, which binds diacylglycerol (DAG)* and phorbol esters, and the C2 domain, which binds Ca2+. Conventional PKC isozymes (α, βI, βII, and γ) have functional C1 and C2 domains and are thus stimulated by DAG and calcium. Novel PKC isozymes (δ, ɛ, and ζ) have a functional C1 domain but nonligand binding C2 domain and are stimulated by DAG but insensitive to calcium. Atypical PKC isozymes (ζ and ι/λ) have a nonligand binding C1 domain and no C2 domain and are thus not regulated by either of these second messengers. Additional isozyme-specific regulation is provided by protein–protein interactions that tether PKC near its substrates (Mochly-Rosen, 1995; Jaken and Parker, 2000). Because PKC is activated through many different receptor types that couple to phospholipid hydrolysis with or without calcium release, there is potential for a wide range of amplitude and duration of PKC signals.
The translocation of PKC from the cytosol to the membrane has served as the hallmark for PKC activation since the seminal discovery that phorbol esters cause PKC to redistribute from the cytosol to the membrane (Kraft et al., 1982; Kraft and Anderson, 1983). Recent advances have taken advantage of GFP to visualize translocation of PKC to membranes upon generation of DAG and calcium in living cells. (Sakai et al., 1997; Oancea and Meyer, 1998; Shirai et al., 1998b). These studies have revealed a wealth of information on the kinetics and localization of PKC inside the cell. It is not clear, however, to what extent PKC translocation corresponds to PKC activation and substrate phosphorylation.
The study of complex signaling systems in living cells has become possible with advances in fluorescent reporters. Signaling events can be monitored by either fluorometric chemical chelators such as the calcium dyes (Tsien, 1989) or genetically encoded fluorescent proteins engineered to change either subcellular location or fluorescent properties upon sensing cellular signals. Numerous examples now exist of fusions of fluorescent proteins to either translocating signaling domains such as C1, C2, and PH domains (Oancea et al., 1998; Gray et al., 1999) or proteins that contain these domains (Sakai et al., 1997; Gijon et al., 1999). Additionally, a growing number of reporters have been designed that alter fluorescence resonance energy transfer (FRET) between fluorescent proteins (Miyawaki and Tsien, 2000) or the intrinsic fluorescent properties of a fluorescent protein (Llopis et al., 1998; Nagai et al., 2001). Reporters based on FRET between fluorescent proteins have several advantages: unlike small molecule reporters, they are genetically encodable, and unlike translocation indicators, they require simple optics and software to analyze. Additionally, a FRET reporter may be pretargeted to subcellular sites if translocation is not an integral part of the readout. Thus, much information can be gleaned from living cells, provided that such reporters do not significantly perturb cell function (for example, by buffering of cell signals resulting from reporter overexpression) and provided reporter specificity is maintained in cells.
Recent work has shown that FRET reporters for kinase activity are a viable method to probe phosphorylation in live cells (Ting et al., 2001; Zhang et al., 2001; Sato et al., 2002). We developed a reporter for phosphorylation by PKC that we call C kinase activity reporter (CKAR) and used this in conjunction with calcium-sensing fluorophores and novel FRET-based translocation assays to test the spatiotemporal properties of the PKC signal pathway. We find a surprisingly high temporal fidelity of signaling restricted to the plasma membrane, controlled by the temporally limiting (i.e., fastest changing) second messenger calcium, and dependent on a strict coupling of kinase and phosphatase activities.
Results
Design and characterization of CKAR
A reporter for PKC-mediated phosphorylation, CKAR, was constructed analogously to a previously described reporter (A kinase activity reporter [AKAR]) for protein A (Zhang et al., 2001). CKAR is composed of monomeric CFP (mCFP) and monomeric YFP (mYFP) (Zacharias et al., 2002) flanking a PKC substrate sequence tethered by a flexible linker to an FHA2 phosphothreonine-binding domain from the yeast checkpoint protein rad53p (Durocher et al., 2000) (Fig. 1 A). Phosphorylation of the PKC substrate sequence triggers an intramolecular clamp with the FHA2 module, causing a conformational change that alters the amount of FRET from CFP to YFP (Fig. 1 A). This change in FRET provides a continuous nondestructive fluorometric readout for CKAR phosphorylation.
Figure 1.
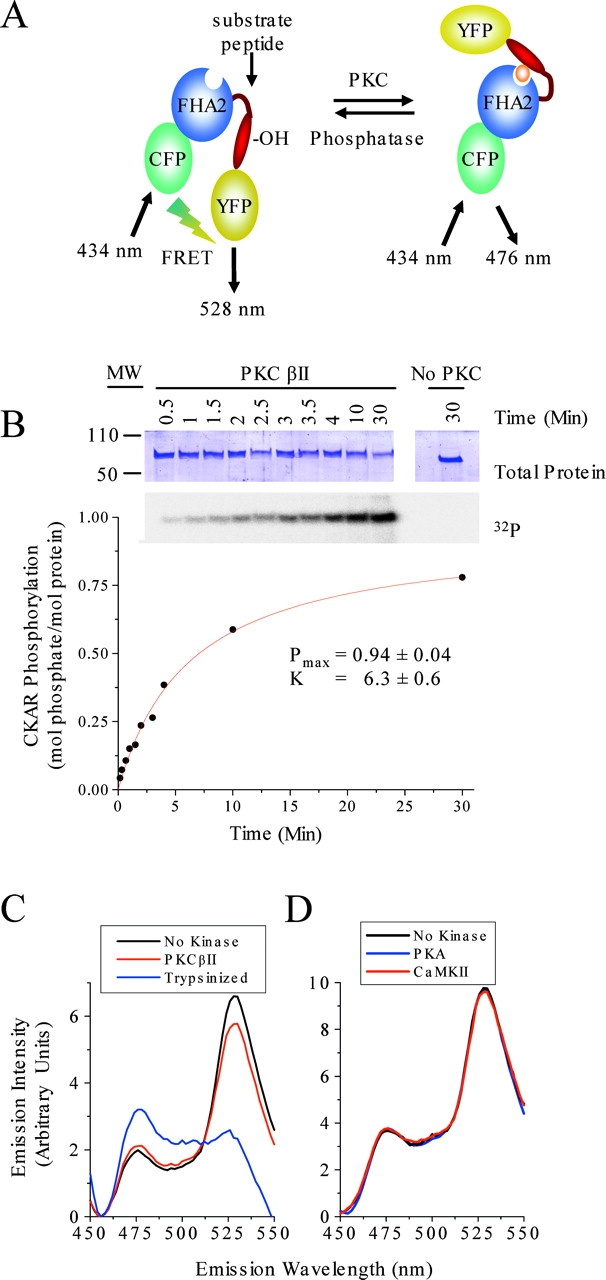
Generation of CKAR. (A) CKAR is comprised of mCFP, the FHA2 domain of Rad53p, a PKC substrate sequence, and mYFP. The substrate sequence, when phosphorylated, binds the FHA2 phospho-peptide–binding domain. This conformational change results in a change in FRET, reversible by phosphatases. (B) CKAR is stoichiometrically phosphorylated by PKC in vitro. Time course of 32P incorporation into nickel-purified histidine-tagged CKAR measured by scintillation counts of excised Coomassie blue–stained bands. (Inset) Coomassie blue–stained purified CKAR (top) and 32P autoradiography (bottom). (C) Emission spectra of CKAR incubated for 30 min at 30°C with and without purified PKCβII. Excitation at 434 nm resulted in a CFP emission peak (476 nm) and YFP emission peak caused by FRET from CFP (528 nm). PKC phosphorylation resulted in decreased intensity at 528 nm and increased intensity at 476 nm, consistent with a decrease in FRET. Incubation of CKAR with trypsin for 30 min at 30°C destroyed the YFP emission, demonstrating that FRET was caused by intramolecular energy transfer. (D) Incubation with active calmodulin-dependent kinase II (CaMKII) or cAMP-dependent kinase (PKA) resulted in no change in FRET.
In designing CKAR, we focused on finding both an appropriate phosphopeptide-binding domain and a specific substrate sequence. We switched from the 14-3-3τ phosphoserine-binding domain used in AKAR (Zhang et al., 2001) to FHA2 for two reasons. 14-3-3τ is an obligate dimer, whereas FHA2 is monomeric. Also, AKAR was a poor substrate for phosphatases in living cells, perhaps because 14-3-3τ binds phosphoserine too tightly and protects it from phosphatases, whereas AKAR analogs with FHA1 could be rapidly dephosphorylated upon cessation of PKA activity (unpublished data). The dissociation constants of FHA1 and FHA2 for their respective optimal peptides are 530 nM and 10 μM (Durocher et al., 2000), whereas 14-3-3 binds its optimal peptide with a dissociation constant of 56 nM (Yaffe et al., 1997) The modest affinity of FHA2 for phosphothreonine makes CKAR reversible and allows CKAR to monitor the ongoing balance between PKC and phosphatases. We designed a specific PKC substrate sequence that would also bind FHA2. This sequence was designed de novo based on information from oriented peptide library screens that have determined optimal substrate sequences for all PKC isoforms (Songyang et al., 1994; Nishikawa et al., 1997). We designed the substrate sequence based on three criteria: (1) consensus phosphorylation sequence for PKC, (2) minimal susceptibility to other basophilic kinases (Yaffe et al., 2001), and (3) determinants that favor FHA2 binding. We decided upon the sequence GGSGGRFRRFQTLKIKAKAGGSGG, where the underlined region is the substrate sequence, flanked by a flexible linker sequence GGSGG. This sequence is predicted to be an excellent substrate for all PKC isoforms but suboptimal for all other kinases (Nishikawa et al., 1997; Yaffe et al., 2001).
CKAR expressed in bacterial cells as a 6xHis-tagged fusion construct was purified and tested for substrate specificity in vitro. Fig. 1 B shows that CKAR was stoichiometrically phosphorylated by PKC in a standard phosphorylation assay using recombinant PKC βII. In contrast, CKAR was not phosphorylated by either CaMKII or PKA using standard assays for these two kinases (unpublished data), even though these two kinases are predicted to be the most likely alternatives to phosphorylate CKAR (Yaffe et al., 2001).
Emission spectra of CKAR at the CFP excitation maximum (434 nm) before and after phosphorylation by PKC (30 min) show a decrease of YFP emission (528 nm) and concomitant increase in CFP emission (476 nm) upon phosphorylation (Fig. 1 C). Trypsinolysis of CKAR to cleave CFP from YFP resulted in a dramatic loss of YFP emission, confirming that the YFP emission peak in intact CKAR is caused by intramolecular FRET (Fig. 1 C, blue line). In contrast, emission spectra of CKAR before and after incubation in phosphorylation reactions with active PKA and CaMKII showed no change (Fig. 1 D). Thus, specific phosphorylation of CKAR by PKC results in a decrease in FRET. The absolute amounts of FRET can be estimated from Fig. 1 C to be 38% before and 34% after phosphorylation, assuming that emission at 476 nm arises solely from the CFP donor and that FRET is negligible after trypsinolysis.
CKAR reports PKC activity in live cells
We next explored whether CKAR could function as a reporter for PKC in live cells. Fig. 2 A shows that CKAR expressed in MDCK cells showed a decrease in FRET upon activation of PKC with phorbol dibutyrate (PDBu). Note that the data in Fig. 2 A are plotted as the ratio of cyan fluorescence (which increases as FRET decreases) to yellow emission (which decreases as FRET decreases). This decrease in FRET was rapidly reversed by the specific PKC inhibitor Gö6983. Treatment of MDCK cells with vehicle (DMSO) and then forskolin to activate PKA did not alter the FRET ratio, indicating that the sensor does not respond to PKA (Fig. 2 B, red line). Similarly, no response was observed when cells were first treated with Go6983 to inhibit PKC and then treated with thapsigargin to stimulate CaMKII by calcium release (Fig. 2 B, green line). These experiments performed in HeLa cells yielded the same results (unpublished data). Thus, CKAR senses PKC but not PKA or CaMKII activation in live cells. We calibrated fluorescence intensity to protein concentration by imaging pure fluorescent protein of known concentration to obtain a standard curve (unpublished data). Based on this calibration, we estimate CKAR concentration in cells to be 0.5–2 μM except where otherwise specified. This is well within the range of concentration of endogenous PKC substrates, which can be as high as 20 μM for abundant proteins such as MARCKS (Wang et al., 2002).
Figure 2.
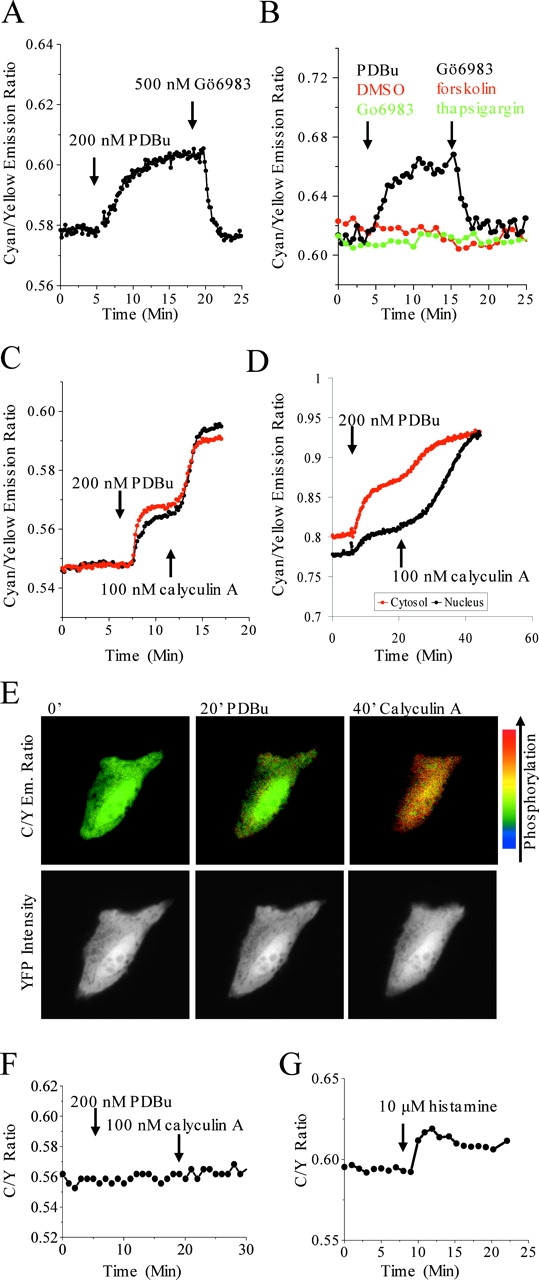
CKAR is a specific, reversible reporter for PKC activation in live cells. (A) CKAR expressed in HeLa cells is phosphorylated upon stimulation of PKC with 200 nM PDBu. This is reversed by addition of 500 nM Go6983, a specific inhibitor of PKC. (B) CKAR phosphorylation is specific for PKC. (Black) PKC is activated by 200 nM PDBu and inhibited by 1 μM Go6983. (Red) Neither DMSO (vehicle for PDBu) nor 10 μM forskolin cause a change in FRET. (Green) Preinhibition of PKC (1 μM Go6983) does not change basal FRET, which remains unchanged by release of intracellular calcium by thapsigargin to stimulate other calcium-sensitive kinases such as CaMKII. (C) Supramaximal stimulation of PKC with 200 nM PDBu results in stable phosphorylation of CKAR, but inhibition of phosphatases with 100 nM calyculin A results in additional phosphorylation. Data are from two cells in the same field of view. (D) Phosphorylation of CKAR in the cytosol (red) and nucleus (black) reveal preferential cytosolic phosphorylation after PDBu but greater and uniform phosphorylation after calyculin A treatment. (E) Images corresponding to Fig. 2 D show phosphorylation (red shift of pseudocolored FRET ratio image, top) of CKAR after PDBu (20') and PDBu and calyculin (40'). YFP intensity images (bottom) indicate no change in CKAR localization over the course of the experiment. (F) Mutation of the threonine phosphoacceptor in the designed PKC substrate of CKAR precludes FRET changes in response to either 200 nM PDBu or 100 nM calyculin. (G) CKAR is also sensitive to receptor-mediated activation of PKC. 10 μM histamine resulted in rapid phosphorylation of CKAR in HeLa cells. All data are representative of at least three experiments.
We next tested whether full activation of PKC results in complete phosphorylation of CKAR or if cellular phosphatases counteract PKC activity to maintain a significant pool of unphosphorylated substrate. To maximally activate PKC, we treated MDCK cells with 200 nM phorbol esters. This resulted in a robust change in FRET (Fig. 2 C; red and black lines are from two separate cells). Surprisingly, addition of the phosphatase inhibitor calyculin A (100 nM) to cells already treated with 200 nM PDBu resulted in an even larger increase in phosphorylation, indicating that maximal PKC activation does not saturate CKAR unless phosphatases are inhibited. This phosphorylation is compartmentalized: PDBu results in preferential phosphorylation of CKAR in the cytosol over the nucleus, the further increase in phosphorylation after calyculin A results in uniform phosphorylation throughout the cell (Fig. 2 D). Pseudocolored images of this FRET change are shown in Fig. 2 E. Importantly, similar results were observed with CKAR expression ranging from 0.8 to 8 μM (unpublished data), indicating that substrate buffering of PKC activity does not greatly alter CKAR responses at the concentrations used in this work.
To confirm that changes in FRET result from phosphorylation of CKAR at the intended amino acid, we changed the phosphoacceptor threonine in the PKC substrate sequence to alanine. Fig. 2 F shows that this variant does not respond to either PDBu or calyculin A, consistent with the designed site of phosphorylation.
We next explored whether CKAR was sensitive enough to respond to physiological activation of PKC. Fig. 2 G shows that histamine treatment of HeLa cells resulted in a small but reproducible change in FRET.
Membrane-tethered CKAR detects oscillations in substrate phosphorylation
We reasoned that because PKC activity is highest at membranes, a membrane-targeted CKAR would be a better substrate for PKC in intact cells. Thus, we fused the 10 NH2-terminal residues of Lyn kinase to CKAR. This sequence contains signals for myristoylation and palmitoylation (Zacharias et al., 2002), effectively targeting CKAR to the plasma membrane (Fig. 3 A). Fig. 3 B reveals that this myristoylated and palmitoylated (MyrPalm)–CKAR undergoes a FRET change upon treatment of HeLa cells with PDBu similar to that observed for cytosolic CKAR. However, unlike cytosolic CKAR, the PDBu-induced FRET change of the membrane-tethered CKAR was only modestly sensitive to calyculin A (Fig. 3 B compared with Fig. 2 C). Thus, tethering CKAR at the site of action of PKC alters the phosphorylation/dephosphorylation equilibrium to favor phosphorylation.
Figure 3.
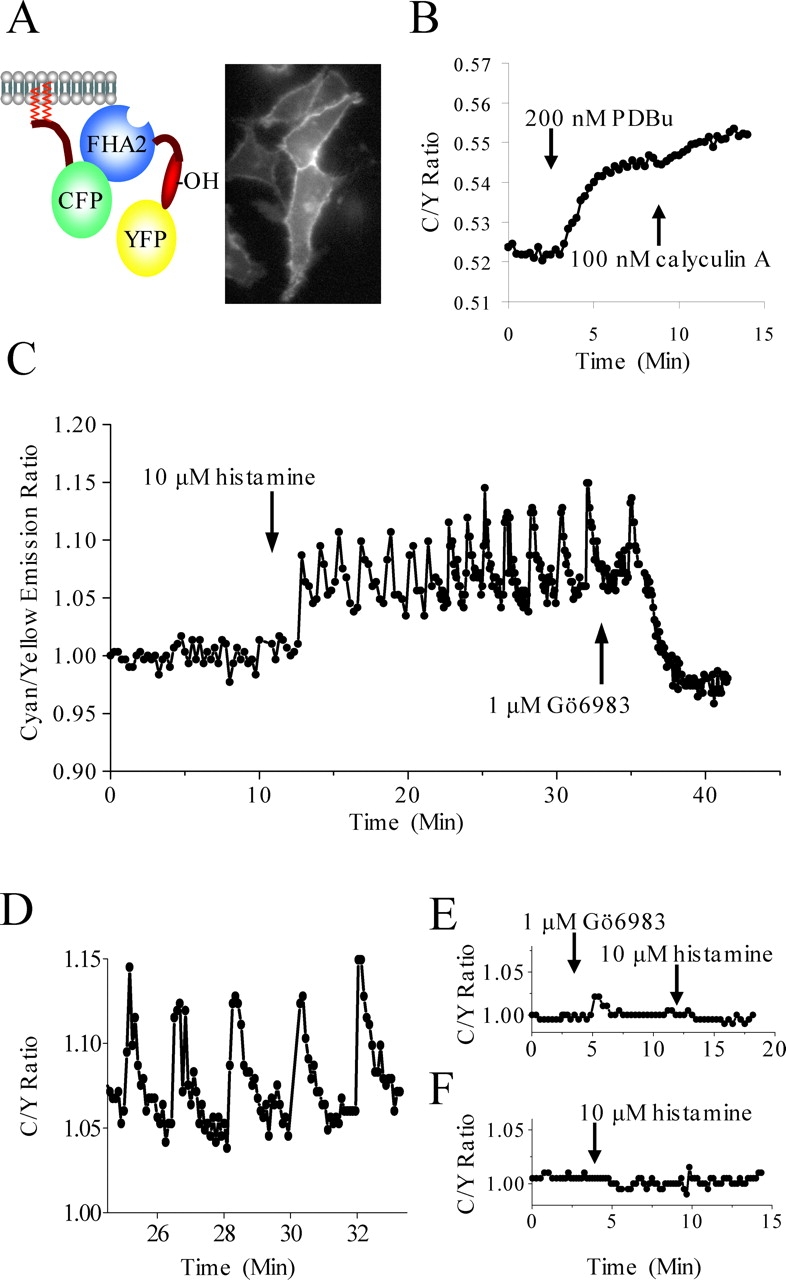
Targeting CKAR to plasma membrane. (A) CKAR was targeted to plasma membrane by fusion of the 10 amino acid NH2 terminus of the kinase Lyn to the NH2 terminus of CKAR, encoding myristoylation and palmitoylation. (Inset) An image of MyrPalm-CKAR expressed in HeLa cells showing effective targeting of CKAR to the plasma membrane. (B) Supramaximal stimulation of PKC with 200 nM PDBu results in nearly complete phosphorylation of CKAR, since inhibition of phosphatases with 100 nM calyculin A results in only slight additional phosphorylation. (C) MyrPalm-CKAR phosphorylation oscillates after 10 μM histamine and is inhibited by 1 μM Gö6983. (D) Expanded time scale of Fig. 3 C. (E) Pretreatment with 1 μM Gö6983 prevents MyrPalm-CKAR phosphorylation by 10 μM histamine. (F) Mutation of the threonine phosphoacceptor to alanine (T413A) makes MyrPalm-CKAR unresponsive to 10 μM histamine. All data are representative of at least three experiments. Oscillatory phosphorylation, while highly variable, was detected in 10–20% of cells studied, observed in over 30 cells in more than 12 different experiments.
We next examined the effect of receptor-mediated activation of PKC on membrane-tethered CKAR. Fig. 3 C shows that in some cells histamine stimulation of HeLa cells triggered a sustained series of transient phosphorylations. These were terminated after 20 min by inhibition of PKC with 1 μM Gö6983. Each phosphorylation “spike” displayed a rapid rise followed by a slower decline (Fig. 3 D, expanded time scale of C), consistent with a burst of kinase activity followed by dephosphorylation to a baseline equilibrium between kinase and phosphatase activities. Oscillations were extremely variable in amplitude, period, and duration, and the data shown are representative of sustained, regular oscillations seen in a minority of cells (5–10%). Either inhibition of PKC with 1 μM Gö6983 (Fig. 3 E) or mutation of the phosphoacceptor threonine to alanine (Fig. 3 F) prevented a FRET change upon stimulation with 10 μM histamine, consistent with accurate reporting of phosphorylation by MyrPalm-CKAR.
The striking rhythmicity of histamine-induced oscillations in substrate phosphorylation led us to explore whether they reflected the well-characterized histamine-evoked calcium oscillations in HeLa cells. We measured histamine-evoked phosphorylation and Ca2+ changes simultaneously with MyrPalm-CKAR and the Ca2+ indicator Fura red. Fig. 4 A shows that the oscillations in CKAR phosphorylation (black line) are phase locked with Ca2+ oscillations (red line). Quantitative analysis of the peaks reveals that phosphorylation lags ∼10–15 s behind Ca2+ elevations (Fig. 4 B). To eliminate the possibility of spectral overlap from Fura red causing illusory FRET changes in these experiments, we measured histamine-induced calcium spikes with Fura red in cells expressing the Thr to Ala mutant MyrPalm-CKAR and saw almost no overlap of Fura red signal into the cyan and yellow emission channels (Fig. 4 C). Although oscillations are highly variable in HeLa cells, we noted that MyrPalm-CKAR phosphorylation was always coupled to Ca2+ (examined in at least 15 cells from eight separate experiments), whether the Ca2+ increases were regular, sustained oscillations, or brief, irregular transients.
Figure 4.
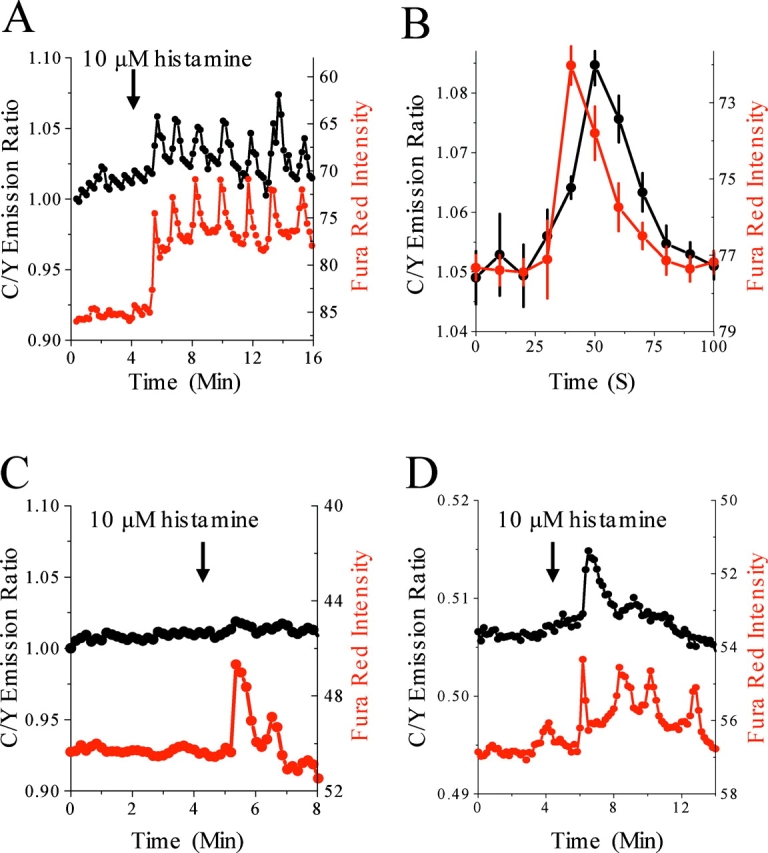
MyrPalm-CKAR oscillatory phosphorylation corresponds to calcium oscillations. (A) Calcium (Fura red intensity, red) and MyrPalm-CKAR phosphorylation (CFP-YFP FRET, black) show that MyrPalm-CKAR phosphorylation corresponds directly to calcium transients. (B) Averaging the calcium and phosphorylation peaks in A illustrates a consistent lag of 10–20 s in MyrPalm-CKAR phosphorylation after initiation of calcium transients. The time of each Fura red intensity spike was normalized and the Fura red intensities and FRET ratios averaged for each image acquisition surrounding that fixed time. (C) Histamine stimulation of HeLa cells expressing MyrPalm-CKAR T413A shows that FRET changes are almost entirely independent of spectral overlap from Fura red signals and instead depend on the phosphoacceptor T413 in the PKC substrate of MyrPalm-CKAR. (D) Histamine stimulation resulting in calcium oscillations does not result in oscillatory phosphorylation of cytosolic CKAR. All data are representative of at least three experiments. Phase-locked calcium and phosphorylation oscillations have been noted in 15 different cells from eight experiments.
In contrast to the oscillatory phosphorylation response of membrane-tethered MyrPalm-CKAR, the data in Fig. 2 F showed that response of cytosolic CKAR is not oscillatory. To confirm this result, we simultaneously measured CKAR phosphorylation and Ca2+ oscillations within the same cell. Fig. 4 D shows that calcium-controlled oscillatory PKC phosphorylation does not propagate through the cytosol (Fig. 4 D). Thus, the balance between PKC and phosphatases oscillates only at the membrane, where PKC is most active.
Correlation of activity with translocation
The preceding results show phase-locked oscillations of PKC activity and calcium release. We next addressed whether this Ca2+-controlled activity reflected Ca2+-controlled membrane association or whether it reflected Ca2+-controlled changes in membrane affinity independent of dissociation of PKC from the membrane. (Oancea and Meyer, 1998; Nalefski and Newton, 2001). To measure translocation in HeLa cells, we developed a novel FRET assay that does not require complex optics such as total internal reflection fluorescence (Codazzi et al., 2001) or subjective discrimination of membrane and cytosol regions in acquired images (Oancea and Meyer, 1998). We fused the NH2-terminus of Lyn kinase to mCFP to encode for myristoylation and palmitoylation (MyrPalm-mCFP) and coexpressed this with a PKC that had mYFP fused at both NH2- and COOH-termini (Fig. 5 A). When PKC translocates from the cytosol to the membrane upon generation of calcium and DAG, the average distance between CFP and YFP decreases, causing increased FRET. YFP was fused to both ends of PKC to increase the probability of FRET upon PKC translocation. This method allows for whole cell monitoring of PKC translocation to the site of MyrPalm-CFP. Fig. 5 B shows that histamine stimulation of HeLa cells results in repetitive transient translocations (black line) that are phase locked with calcium oscillations imaged with the fluorescent dye Fura red (red line), consistent with a visible redistribution of mYFP-tagged PKC to membrane (unpublished data). Oscillatory translocation was observed in the absence of Fura red, indicating that FRET and not spectral overlap from Fura red is responsible for the increase in measured yellow to cyan emission ratio (unpublished data).
Figure 5.

Histamine induces oscillations of PKC translocation. (A) YFP fused to both termini of PKC functions as a FRET acceptor for CFP fused to the myristoylation and palmitoylation sequence from Lyn (MyrPalm-CFP). Translocation of YFP-PKC-YFP to a membrane containing MyrPalm-CFP results in increased FRET. (B) Addition of 10 μM histamine to HeLa cells causes rapid, oscillating translocation of YFP-PKC-YFP to plasma membrane (black) corresponding to oscillating calcium transients imaged by Fura red in the same cell (red). Data are representative of three experiments.
Ca2+ oscillations are uncoupled from diacylglycerol production
To explore whether DAG oscillates in phase with calcium, we designed a probe for PLC activity. The most direct readout for DAG is translocation of the DAG-binding C1 domain (Oancea and Meyer, 1998). Unfortunately, GFP-tagged C1 domains from PKCβ, δ, or γ are insoluble in HeLa cells and do not translocate effectively (unpublished data). Instead, we took advantage of methodologies to detect IP3 as a measure of PLC activity. As shown previously (Hirose et al., 1999; van der Wal et al., 2001), the PH domain of PLCδ, which binds phosphoinositide bisphosphate (PIP2), translocates from the membrane to the cytosol upon PLC activation and cleavage of PIP2. We adopted a novel strategy to measure translocation of this PH domain that reports translocation from membrane to cytosol upon PLC activation. Fusion of both CFP and YFP to the PH domain of PLCδ (cyan/yellow PH domain reporter [CYPHR]) allows us to measure both intramolecular and intermolecular FRET. Although changes in intramolecular FRET are unpredictable, intermolecular FRET depends on the concentration of fluorophore: because the effective concentration of fluorophore is lower when diffusing in three-dimensional cytosol than when diffusing in a two-dimensional membrane, the dilution experienced by the PH domain upon PLC activation is reported as a decrease in FRET (Fig. 6 A). Decreased FRET corresponded to visible translocation from membrane to cytosol (unpublished data).
Figure 6.

Translocation of the PH domain of PLCδ reported by FRET reveals constant PLC activity during calcium oscillations. (A) CYPHR, a CFP-PHδ-YFP fusion construct, reports PLC activity by reduced intermolecular FRET upon translocation from membrane. The PH domain of PLCδ1, which binds PIP2 in the plasma membrane, translocates to cytosol upon PIP2 hydrolysis. Translocation from membrane (two dimensions) to cytosol (three dimensions) results in a decreased effective concentration and hence lower intermolecular FRET. (B) CYPHR reports constant PLC activity (black) during calcium oscillations (red). Data are representative of four experiments.
CYPHR expressed in HeLa cells reports a sustained PLC activity during histamine-evoked calcium oscillations (Fig. 6 B), implying a calcium-induced calcium release (CICR) mechanism of oscillation instead of dynamic uncoupling. This is consistent with earlier reports of the insensitivity of histamine-evoked calcium oscillations in HeLa cells to IP3 concentration (Sauve et al., 1991) and contrasts with systems showing phase-locked oscillations of IP3 and calcium (Hirose et al., 1999; Nash et al., 2001). Indeed, a GFP-tagged PH domain exhibits oscillatory translocation in phase with ATP-evoked calcium oscillations in MDCK cells, an example of the dynamic uncoupling mechanism of oscillating PLC activity (Hirose et al., 1999). Consistent with these findings, MyrPalm-CKAR, DAG reporter, (DAGR; identical to CYPHR but with a PKCβ C1 domain replacing the PH domain), and CYPHR report transient responses of phosphorylation, DAG, and PLC activity phase locked with calcium transients in MDCK cells (Fig. 7, A–C). DAGR visibly translocated from cytosol to membrane upon ATP stimulation or PDBu addition to MDCK cells (unpublished data), consistent with its characterization as a DAG sensor (Oancea et al., 1998) This differs markedly from histamine-evoked oscillations of phosphorylation in HeLa cells in which PLC activity reported by CYPHR is independent of calcium transients. We conclude that histamine-evoked PKC responses in HeLa and ATP-evoked PKC responses in MDCK cell represent two distinct mechanisms for temporal control of PKC activation.
Figure 7.
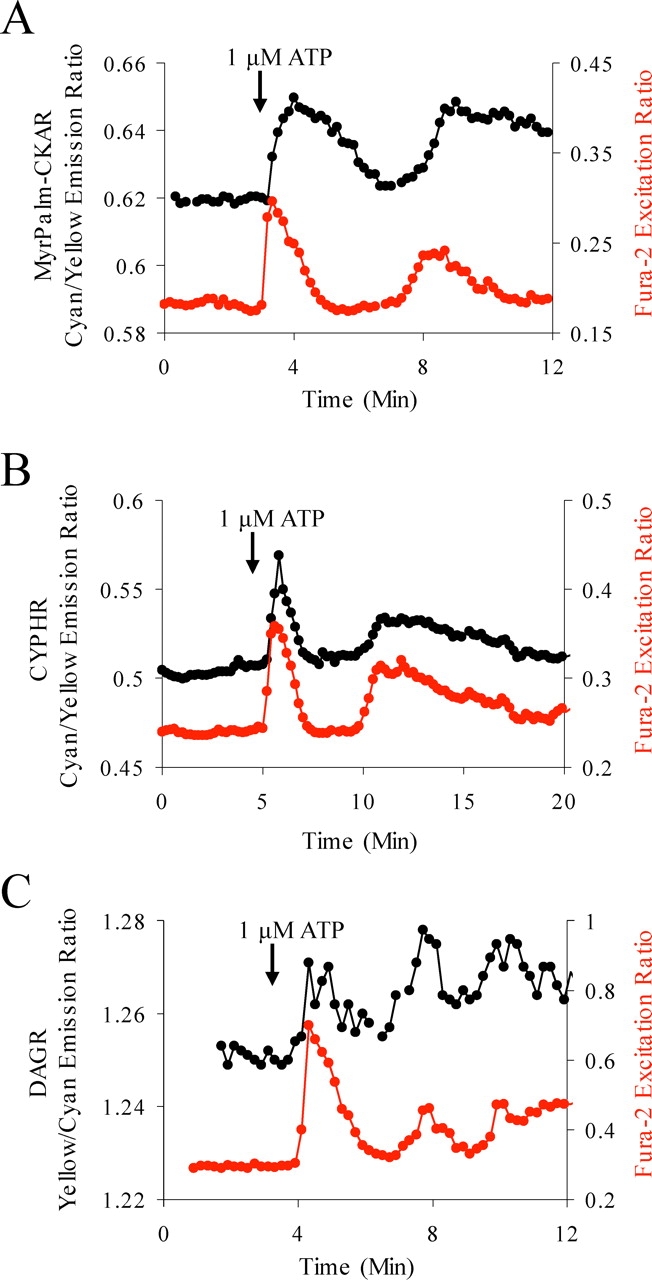
PLC controls oscillations of PKC activity in MDCK cells. 10 μM ATP induces calcium transients phase locked with responses of MyrPalm-CKAR (reports phosphorylation) (A), CYPHR (reports PLC activity) (B), and DAGR (reports DAG) (C). All data are representative of three to five experiments.
Discussion
The foregoing results underscore the importance of spatiotemporal control of second messengers and their signaling systems. Specifically, using a genetically engineered reporter for monitoring the activity of endogenous PKC, we show that substrate phosphorylation is under the dynamic control of second messenger levels, which directly regulate the intrinsic activity of PKC, and cellular phosphatase activity, which rapidly terminates signaling by PKC. We show that in HeLa and MDCK cells, histamine and ATP trigger sustained oscillatory phosphorylation of membrane-tethered reporter and that these oscillations are phase locked with Ca2+ oscillations.
CKAR: specific reporter for the dynamic interplay between phosphorylation and dephosphorylation of PKC substrates
CKAR was successfully designed to be a specific, reversible reporter of phosphorylation by PKC (Figs. 1 and 2). Specificity was achieved by de novo design of a substrate sequence using information from oriented peptide library screens identifying determinants that dictate kinase substrate specificity (Yaffe et al., 2001). Reversibility was achieved by choosing an FHA2 domain to bind phosphothreonine with modest affinity (10 μM) (Durocher et al., 2000).
PKC translocation, CYPHR, and DAGR
We also devised new FRET-based reporters for PKC translocation, PIP2 to IP3 hydrolysis, and diacylglycerol. These reporters not only translocate to and from the plasma membrane but also alter the ratio of yellow to cyan emissions in response to the appropriate signals. These indicators may have significant value in flow cytometry, complex tissues, or high throughput screening, where quantitative monitoring of translocation of a GFP-tagged C1 domain, PKC, or PH domain to the plasma membrane is difficult.
Subcellular specificities of CKAR phosphorylation
We found that CKAR is only partially phosphorylated in vivo when localized to the cytosol, even with maximal activation of PKC by phorbol esters (Fig. 2 C). Presumably, this phosphorylation arises from diffusional collision of CKAR with PKC at the membrane, where it becomes phosphorylated and then diffuses back into the cytosol. The lower level of phosphorylation seen in the nucleus (Fig. 2, D and E) indicates a different balance of kinase and phosphatase there, either through impaired accessibility to active PKC or higher nuclear phosphatase activity. This is consistent with nuclear exclusion noted for most PKC isoforms (Shirai et al., 1998a; Kajimoto et al., 2001). In contrast, targeting CKAR to the plasma membrane, the site of activation of PKC, results in nearly complete phosphorylation of the reporter as revealed by the minimal effect of phosphatase inhibition on the phosphorylation of membrane-tethered CKAR (Fig. 3 B). This likely reflects the increased probability of phosphorylating a substrate colocalized with PKC. These data indicate a spatial gradient in the balance between kinase and phosphatase more favorable for phosphorylation at the plasma membrane than in the cytosol, and more favorable in the cytosol than in the nucleus. Such radial gradients were predicted some time ago (Brown and Kholodenko, 1999) but were not observable until the advent of genetically encoded phosphorylation reporters. It is interesting in this regard that not only is there an abundance of kinases tethered to their substrates (Jaken and Parker, 2000), but also there is a growing number of scaffolds that tether both a phosphatase and a kinase in the same complex (Coghlan et al., 1995; Westphal et al., 1998).
Calcium oscillations dictate substrate phosphorylation by conventional PKCs
HeLa cells, like many electrically nonexcitable cell types, undergo repetitive cycles of calcium release and reuptake upon stimulation with some agonists, resulting in periodic spikes of free intracellular calcium. Conventional isoforms of PKC can translocate in phase with these spikes, leading to an oscillatory translocation, ostensibly producing bursts of PKC activity (Oancea and Meyer, 1998; Tanimura et al., 2002). However, it was not known if PKC-mediated substrate phosphorylation and phosphatase activity were kinetically competent to result in distinct spikes of phosphorylation during individual calcium oscillations. We find that when CKAR is targeted to plasma membrane by acylation (Fig. 3), regular, pulsatile phosphorylation oscillations occur in phase with calcium oscillations (Fig. 4).
Oscillations in PKC activity that are phase locked with calcium oscillations can occur either through a predominantly calcium-mediated activation of conventional PKC isozymes or through concurrent oscillations in DAG and calcium. Although the first of these possibilities is consistent with any model of calcium oscillations, the second is inconsistent with CICR (Harootunian et al., 1991; Thomas et al., 1996; Nash et al., 2001), which postulates no feedback of calcium to PLC activity and thus does not predict nor account for oscillations of DAG and IP3. Instead, a model of concurrently oscillating calcium and DAG implies an oscillator mechanism in dynamic uncoupling of receptor occupation from PLC activity (Harootunian et al., 1991; Hirose et al., 1999). These distinct modes of calcium oscillation are proposed to constitute disparate signals transduced by the same PLC-mediated pathway (Nash et al., 2001).
Previous work has shown oscillatory conventional PKC translocation during both CICR (Oancea and Meyer, 1998) and dynamic uncoupling (Codazzi et al., 2001) mediated calcium oscillations as evidenced by sustained and oscillating DAG, respectively. Although we are unable to measure DAG directly in HeLa cells, our results are consistent with a CICR mechanism of calcium oscillation since PLC activity does not oscillate (Fig. 6), in agreement with previous results (Sauve et al., 1991) and inconsistent with oscillations of DAG. We nevertheless noted oscillatory PKC translocation, consistent with a requirement for both calcium and DAG for maximal membrane binding (Fig. 5); active PKC at membranes at least partially translocates to cytosol upon calcium reuptake, since DAG alone cannot maintain full PKC membrane affinity. It is important to note that translocation does not directly reflect activation, since weakly membrane-bound PKC is not fully active (Orr and Newton, 1992; Newton and Keranen, 1994; Johnson et al., 2000). Thus, in contrast to previous work showing such calcium-controlled PKC activity, we now have a direct readout of PKC substrate phosphorylation. The revelation of oscillatory substrate phosphorylation indicates a high temporal fidelity for PKC signals: changes in PKC activation are rapidly transduced to substrate phosphorylation or dephosphorylation by a close coupling of PKC and phosphatases. This temporal specificity is evident only when CKAR is targeted to membranes, since a cytosolic CKAR does not experience oscillatory phosphorylation even in the presence of calcium oscillations (Fig. 6 D). This is most likely because PKC activity is membrane limited, allowing for enhanced phosphorylation of a membrane-bound substrate; a cytosolic substrate must rely on diffusion to membrane-bound PKC to become phosphorylated. Only a tight spatial coupling of kinase, phosphatase, and substrate allows such remarkably high temporal fidelity of phosphorylation signals.
An intriguing corollary of this result is the possibility of temporal PKC isoform specificity. In both CICR and dynamic uncoupling, DAG is present when cytosolic calcium is elevated, so conventional PKC oscillations are independent of DAG. In contrast, novel PKC isoforms, unresponsive to calcium, may differentiate between sustained and oscillatory DAG. Thus, novel PKCs are equipped to discriminate CICR from dynamic uncoupling. However, without the advantage of a calcium-enhanced search for DAG (Nalefski and Newton, 2001), it is unknown if novel PKC's respond fast enough to maintain temporal fidelity of DAG oscillations. It will be important to test each PKC isoform for oscillatory translocation in both CICR and dynamic uncoupling calcium oscillations to illuminate differences in the temporal signals of individual PKC isoforms.
It is widely accepted that the outcome of PKC activity depends on the amount and duration of activation, but the importance of oscillatory phosphorylation is unclear. Such oscillations may be a mechanism of encoding signal specificity, or they may be a necessary consequence of calcium oscillations, unrelated to effects downstream of PKC. However, calcium oscillations increase both the sensitivity and specificity of calcium signals (De Koninck and Schulman, 1998; Dolmetsch et al., 1998; Li et al., 1998), setting precedent for such temporal decoding by the cell. As tools and techniques become available to better identify and assay the phosphorylation of physiological substrates of PKC, it will be important to evaluate if the same is true of oscillatory phosphorylation.
Molecular mechanism for Ca2+-controlled PKC activation
Fig. 8 shows a model for the molecular control of PKC activity by second messengers based on previous studies (Newton, 2001) that accounts for the Ca2+-controlled oscillations in substrate phosphorylation found in histamine-stimulated HeLa cells and PLC-controlled oscillations found in MDCK cells. In the absence of lipid hydrolysis, PKC is localized primarily to the cytosol (Fig. 8, 1) where it is maintained in an inactive conformation by the pseudosubstrate (green rectangle) which occupies the substrate-binding cavity, sterically blocking substrate phosphorylation. In vitro studies show that this autoinhibited species does have modest basal activity (an order of magnitude less than maximal cofactor-stimulated activity), so cellular phosphatase activity likely plays a key role in rapidly abolishing any basal phosphorylation. This species of PKC bounces on and off the membrane by a diffusion-controlled mechanism but is not retained on the membrane in the absence of the appropriate second messengers. Generation of Ca2+ and DAG through receptor-mediated mechanisms results in the recruitment of PKC to the membrane by the following mechanism: Ca2+ binds the C2 domain of cytosolic PKC (Fig. 8, 2), altering the electrostatics of this module so that upon the next encounter with the membrane the C2 domain binds anionic lipids and PKC is tethered to the membrane by a low affinity interaction (Fig. 8, 3). This species of PKC then diffuses in the two-dimensional plane of the membrane until it finds DAG, which then binds the C1 domain, resulting in a high affinity interaction of PKC with the membrane (Fig. 8, 4). The energy provided by engaging both these modules on the membrane releases the basic pseudosubstrate from the acidic substrate-binding cavity, allowing substrate binding and phosphorylation. Loss of either membrane tether through a decrease in either Ca2+ or DAG results in a decrease in the affinity of PKC for membranes (Fig. 8, 5). If this decrease in affinity is significant, PKC is released from the membrane; however, it is also possible that it is retained at the membrane but via only one of the membrane-targeting modules. Importantly, loss of one anchor results in loss of the binding energy required to maintain the pseudosubstrate away from the active site, and the enzyme regains the autoinhibited conformation. This pause in PKC activity allows phosphatase activity to dominate, resulting in substrate dephosphorylation. Elevation of Ca2+ allows the C2 domain to engage on the membrane again, favoring release of the pseudosubstrate to allow renewed substrate phosphorylation. Thus, the oscillations in PKC activity observed arise from altering the equilibrium of the pseudosubstrate blocking the substrate-binding cavity. This molecular rearrangement is controlled by how tightly the C1 and C2 domains are engaged on the membrane, events which, in turn, are controlled by the levels of DAG and Ca2+, respectively. As a coincidence detector for these messengers, PKC is temporally controlled by whichever messenger is regulated the most rapidly. In the case of histamine treatment of HeLa cells, Ca2+ oscillations and not DAG oscillations dictate this allosteric regulation of PKC, whereas in ATP-stimulated MDCK cells, Ca2+ and DAG oscillate together.
Figure 8.

A model of the temporal control of PKC signals. Inactive, cytosolic PKC is autoinhibited by a pseudosubstrate (1). Phospholipase activation results in DAG formation and calcium release: calcium binds cytosolic PKC (2), imparting weak membrane affinity (3). Calcium-bound PKC rapidly binds DAG in the membrane, resulting in maximal membrane affinity and full PKC activity by removal of the pseudosubstrate from the active site (4). A drop in either calcium or DG results in decreased membrane affinity and reinhibition by the pseudosubstrate (5). This coincidence detection by PKC defines a signaling nexus in the cell, whereby strict temporal fidelity of phosphorylation is dictated by rapid turnover of either second messenger and counterbalancing phosphatase activity.
Conclusion
In this study, we show that PKC-mediated phosphorylation signals depend on the spatiotemporal context of second messengers, kinase, substrate, and phosphatase. Specifically, we find that the phosphorylation state of the reporter CKAR reflects coincidence of the second messengers calcium and DAG that together activate PKC. Tight spatial coupling of kinase and phosphatase activities allows for highly dynamic phosphorylation: when CKAR is tethered to membranes, its phosphorylation is phase locked with calcium oscillations in both HeLa and MDCK cells, independent of constant (HeLa) or oscillating (MDCK) DAG generation. This temporal fidelity of the PKC signal pathway, from second messenger to substrate phosphorylation, allows precise control of cellular regulation by creating high temporal bandwidth for PKC signals and is likely to be of critical importance to many cellular processes.
Materials and methods
CKAR construction
CKAR was generated in the mammalian expression vector pcDNA3.1(+) (Invitrogen). CFP was amplified by PCR from a plasmid template to encode a HindIII restriction site followed by a consensus initiation site for translation (CGCCACC) (Kozak, 1987) before the initiating ATG of CFP, and a KpnI restriction site at the 3′ end instead of a terminating codon. FHA2, a gift from Michael Yaffe (Massachusetts Institute of Technology, Cambridge, MA) was amplified by PCR to include KpnI and BamHI restriction sites at the 5′ and 3′ ends, respectively. Citrine, our preferred version of YFP (Griesbeck et al., 2001), was amplified by PCR to include a 5′ BamHI followed by the PKC substrate sequence and a 3′ XbaI following the terminating codon. These pieces were cloned into pcDNA3.1 and confirmed by sequencing.
A parallel construct was made including the mutation A206K in both CFP (mCFP) and citrine (mYFP) to reduce the intrinsic homoaffinity of all GFPs (Zacharias et al., 2002) and preclude intermolecular FRET by CFP-mYFP dimerization. This construct was shown to function as well as the original CKAR and so was used for all experiments. For in vitro experiments, CKAR was amplified to include a 5′ BglII site and a 3′ SalI site and cloned into pRSETB (Invitrogen) cut from BamHI to XhoI. MyrPalm-CKAR was generated by the addition of a HindIII restriction site, a consensus translational initiation site, and the 5′ 30 bp of Lyn kinase to the 5′ end of CKAR. This sequence encodes for myristoylation and palmitoylation, shown previously to be sufficient to target a protein to the plasma membrane (Zacharias et al., 2002).
Construction of other plasmids
MyrPalm-CFP was made by PCR with the same 5′ primer as used for MyrPalm-CKAR and a 3′ primer to the COOH terminus of CFP with an Xba site and cloned into pcDNA3.1(+). YFP-PKCβII-YFP was made by PCR of mYFP from HindII to KpnI, rat PKCβII from KpnI to XhoI, and YFP from XhoI to XbaI, cloned sequentially into pcDNA3.1(+). CYPHR was generated by PCR of CFP from HindIII to KpnI, the PH domain of murine PLCδ1 (aa 10–140) from KpnI to XhoI, and mYFP from XhoI to XbaI cloned sequentially into pcDNA3.1(+).DAGR was similarly created by PCR of the C1A and C1B domains of rat PKCβII (aa 37–152) from KpnI to XhoI and substitution for the PH domain in CYPHR.
Protein expression, kinase assays, and in vitro spectroscopy
CKAR in the pRSETB vector was transformed into BL21 Gold Escherichia coli (Stratagene) and purified by nickel chelation chromatography as described previously (Miyawaki and Tsien, 2000). Protein yield was estimated by CFP absorption at 434 nm. Fluorescence emission spectra were measured with an excitation wavelength of 434 nm.
For kinase assays, CKAR (125 nmol) was incubated with purified PKCβII (1 U μL−1), PKA (5 U μL−1), or CamKII (5 U μL−1) with appropriate buffers and cofactors including [γ-32P]ATP (100 μM, 10 μCi) as described earlier for PKC (Newton, 2002a) or in the manufacturers' instructions for PKA and CamKII (New England Biolabs, Inc.) for varying amounts of time. Proteins were separated by SDS-PAGE, stained with Coomassie blue, bands corresponding to CKAR excised, and 32P incorporation measured by scintillation counting. Fluorescence emission scans were obtained from samples phosphorylated in parallel with unradiolabeled ATP with excitation at 434 nm to excite CFP.
Cell culture
HeLa and MDCK cells were plated onto sterilized glass coverslips in 35-mm dishes and grown in DME containing 10% FBS at 37°C with 5% CO2. At 60–80% confluency, cells were transfected with either Superfect or Polyfect (QIAGEN) and allowed to grow for 12–24 h posttransfection before imaging. For calcium imaging experiments, 0.2–1.0 μM Fura red/AM or Fura-2/AM (Molecular Probes) was loaded into cells for 30 min, washed once with PBS, and incubated for 30–60 min in fresh media before imaging.
Cell imaging
Cells were washed once with HBSS and imaged in the dark at room temperature. Images were acquired on a Zeiss Axiovert microscope (Carl Zeiss MicroImaging, Inc.) with a cooled charge-coupled device camera (Photometrics) controlled by MetaFluor 3.0 software (Universal Imaging, Corp). CFP, FRET, and Fura red images were obtained through a 440DF20 excitation filter, a 455DRLP dichroic mirror, and three separate emission filters (480DF30 for CFP, 535DF25 for FRET, and 653DF95 for Fura red). Citrine intensity was imaged through a 495DF10 excitation filter, 455DRLP dichroic mirror, and 535DF25 emission filter. Excitation and emission filters were switched in filter wheels (Lambda 10–2; Sutter). Integration times were varied between 200 and 500 ms to optimize signal and minimize photobleaching. Optical filters were obtained from Chroma Technologies and Omega Optical. Because Fura red emission overlaps Citrine/FRET emission, care was taken to limit the amount of Fura red loaded. Measurements were restricted to cells in which CKAR expression was high enough to be unaffected by changes in Fura red intensity.
Acknowledgments
This work was supported by the National Institutes of Health (grants P01 DK54441 and GM43154 to A.C. Newton, 2T32 GM07752 to J.D. Violin, and NS27177 to R.Y. Tsien), Alliance for Cellular Signaling (grant GM62114 to J. Zhang and R.Y. Tsien), and the Howard Hughes Medical Institute (to R.Y. Tsien).
Footnotes
Abbreviations used in this paper: AKAR, A kinase activity reporter; CICR, calcium-induced calcium release; CKAR, C kinase activity reporter; CYPHR, cyan/yellow PH domain reporter; DAG, diacylglycerol; DAGR, DAG receptor; FRET, fluorescence resonance energy transfer; IP3, inositol 1,4,5-trisphosphate; mCFP, monomeric CFP; mYFP, monomeric YFP; MyrPalm, myristoylated and palmitoylated; PDBu, phorbol dibutyrate;PIP2, phosphoinositide bisphosphate.
References
- Brown, G.C., and B.N. Kholodenko. 1999. Spatial gradients of cellular phospho-proteins. FEBS Lett. 457:452–454. [DOI] [PubMed] [Google Scholar]
- Codazzi, F., M.N. Teruel, and T. Meyer. 2001. Control of astrocyte Ca(2+) oscillations and waves by oscillating translocation and activation of protein kinase C. Curr. Biol. 11:1089–1097. [DOI] [PubMed] [Google Scholar]
- Coghlan, V.M., B.A. Perrino, M. Howard, L.K. Langeberg, J.B. Hicks, W.M. Gallatin, and J.D. Scott. 1995. Association of protein kinase A and protein phosphatase 2B with a common anchoring protein. Science. 267:108–111. [DOI] [PubMed] [Google Scholar]
- De Koninck, P., and H. Schulman. 1998. Sensitivity of CaM kinase II to the frequency of Ca2+ oscillations. Science. 279:227–230. [DOI] [PubMed] [Google Scholar]
- Dolmetsch, R.E., K. Xu, and R.S. Lewis. 1998. Calcium oscillations increase the efficiency and specificity of gene expression. Nature. 392:933–936. [DOI] [PubMed] [Google Scholar]
- Durocher, D., I.A. Taylor, D. Sarbassova, L.F. Haire, S.L. Westcott, S.P. Jackson, S.J. Smerdon, and M.B. Yaffe. 2000. The molecular basis of FHA domain:phosphopeptide binding specificity and implications for phospho-dependent signaling mechanisms. Mol. Cell. 6:1169–1182. [DOI] [PubMed] [Google Scholar]
- Gijon, M.A., D.M. Spencer, A.L. Kaiser, and C.C. Leslie. 1999. Role of phosphorylation sites and the C2 domain in regulation of cytosolic phospholipase A2. J. Cell Biol. 145:1219–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray, A., J. Van Der Kaay, and C.P. Downes. 1999. The pleckstrin homology domains of protein kinase B and GRP1 (general receptor for phosphoinositides-1) are sensitive and selective probes for the cellular detection of phosphatidylinositol 3,4-bisphosphate and/or phosphatidylinositol 3,4,5-trisphosphate in vivo. Biochem. J. 344(Pt 3):929–936. [PMC free article] [PubMed] [Google Scholar]
- Griesbeck, O., G.S. Baird, R.E. Campbell, D.A. Zacharias, and R.Y. Tsien. 2001. Reducing the environmental sensitivity of yellow fluorescent protein. Mechanism and applications. J. Biol. Chem. 276:29188–29194. [DOI] [PubMed] [Google Scholar]
- Harootunian, A.T., J.P. Kao, S. Paranjape, and R.Y. Tsien. 1991. Generation of calcium oscillations in fibroblasts by positive feedback between calcium and IP3. Science. 251:75–78. [DOI] [PubMed] [Google Scholar]
- Hirose, K., S. Kadowaki, M. Tanabe, H. Takeshima, and M. Iino. 1999. Spatiotemporal dynamics of inositol 1,4,5-trisphosphate that underlies complex Ca2+ mobilization patterns. Science. 284:1527–1530. [DOI] [PubMed] [Google Scholar]
- Jaken, S., and P.J. Parker. 2000. Protein kinase C binding partners. Bioessays. 22:245–254. [DOI] [PubMed] [Google Scholar]
- Johnson, J.E., J. Giorgione, and A.C. Newton. 2000. The C1 and C2 domains of protein kinase C are independent membrane targeting modules, with specificity for phosphatidylserine conferred by the C1 domain. Biochemistry. 39:11360–11369. [DOI] [PubMed] [Google Scholar]
- Kajimoto, T., S. Ohmori, Y. Shirai, N. Sakai, and N. Saito. 2001. Subtype-specific translocation of the delta subtype of protein kinase C and its activation by tyrosine phosphorylation induced by ceramide in HeLa cells. Mol. Cell. Biol. 21:1769–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak, M. 1987. An analysis of 5′-noncoding sequences from 699 vertebrate messenger RNAs. Nucleic Acids Res. 15:8125–8148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft, A.S., and W.B. Anderson. 1983. Phorbol esters increase the amount of Ca2+, phospholipid-dependent protein kinase associated with plasma membrane. Nature. 301:621–623. [DOI] [PubMed] [Google Scholar]
- Kraft, A.S., W.B. Anderson, H.L. Cooper, and J.J. Sando. 1982. Decrease in cytosolic calcium/phospholipid-dependent protein kinase activity following phorbol ester treatment of EL4 thymoma cells. J. Biol. Chem. 257:13193–13196. [PubMed] [Google Scholar]
- Li, W., J. Llopis, M. Whitney, G. Zlokarnik, and R.Y. Tsien. 1998. Cell-permeant caged InsP3 ester shows that Ca2+ spike frequency can optimize gene expression. Nature. 392:936–941. [DOI] [PubMed] [Google Scholar]
- Llopis, J., J.M. McCaffery, A. Miyawaki, M.G. Farquhar, and R.Y. Tsien. 1998. Measurement of cytosolic, mitochondrial, and Golgi pH in single living cells with green fluorescent proteins. Proc. Natl. Acad. Sci. USA. 95:6803–6808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellor, H., and P.J. Parker. 1998. The extended protein kinase C superfamily. Biochem. J. 332(Pt 2):281–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyawaki, A., and R.Y. Tsien. 2000. Monitoring protein conformations and interactions by fluorescence resonance energy transfer between mutants of green fluorescent protein. Methods Enzymol. 327:472–500. [DOI] [PubMed] [Google Scholar]
- Mochly-Rosen, D. 1995. Localization of protein kinases by anchoring proteins: a theme in signal transduction. Science. 268:247–251. [DOI] [PubMed] [Google Scholar]
- Nagai, T., A. Sawano, E.S. Park, and A. Miyawaki. 2001. Circularly permuted green fluorescent proteins engineered to sense Ca2+. Proc. Natl. Acad. Sci. USA. 98:3197–3202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalefski, E.A., and A.C. Newton. 2001. Membrane binding kinetics of protein kinase C betaII mediated by the C2 domain. Biochemistry. 40:13216–13229. [DOI] [PubMed] [Google Scholar]
- Nash, M.S., K.W. Young, R.A. Challiss, and S.R. Nahorski. 2001. Intracellular signalling. Receptor-specific messenger oscillations. Nature. 413:381–382. [DOI] [PubMed] [Google Scholar]
- Newton, A.C. 2001. Protein kinase C: structural and spatial regulation by phosphorylation, cofactors, and macromolecular interactions. Chem. Rev. 101:2353–2364. [DOI] [PubMed] [Google Scholar]
- Newton, A.C. 2002. a. Analyzing protein kinase C activation. Methods Enzymol. 345:499–506. [DOI] [PubMed] [Google Scholar]
- Newton, A.C. 2002. b. Regulation of the ABC kinases by phosphorylation: protein kinase C as a paradigm. Biochem J. 370(Pt 2):361–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton, A.C., and L.M. Keranen. 1994. Phosphatidyl-L-serine is necessary for protein kinase C's high-affinity interaction with diacylglycerol-containing membranes. Biochemistry. 33:6651–6658. [DOI] [PubMed] [Google Scholar]
- Nishikawa, K., A. Toker, F.J. Johannes, Z. Songyang, and L.C. Cantley. 1997. Determination of the specific substrate sequence motifs of protein kinase C isozymes. J. Biol. Chem. 272:952–960. [DOI] [PubMed] [Google Scholar]
- Nishizuka, Y. 1984. The role of protein kinase C in cell surface signal transduction and tumour promotion. Nature. 308:693–698. [DOI] [PubMed] [Google Scholar]
- Nishizuka, Y. 1995. Protein kinase C and lipid signaling for sustained cellular responses. FASEB J. 9:484–496. [PubMed] [Google Scholar]
- Oancea, E., and T. Meyer. 1998. Protein kinase C as a molecular machine for decoding calcium and diacylglycerol signals. Cell. 95:307–318. [DOI] [PubMed] [Google Scholar]
- Oancea, E., M.N. Teruel, A.F. Quest, and T. Meyer. 1998. Green fluorescent protein (GFP)-tagged cysteine-rich domains from protein kinase C as fluorescent indicators for diacylglycerol signaling in living cells. J. Cell Biol. 140:485–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr, J.W., and A.C. Newton. 1992. Interaction of protein kinase C with phosphatidylserine. 1. Cooperativity in lipid binding. Biochemistry. 31:4661–4667. [DOI] [PubMed] [Google Scholar]
- Sakai, N., K. Sasaki, N. Ikegaki, Y. Shirai, Y. Ono, and N. Saito. 1997. Direct visualization of the translocation of the gamma-subspecies of protein kinase C in living cells using fusion proteins with green fluorescent protein. J. Cell Biol. 139:1465–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato, M., T. Ozawa, K. Inukai, T. Asano, and Y. Umezawa. 2002. Fluorescent indicators for imaging protein phosphorylation in single living cells. Nat. Biotechnol. 20:287–294. [DOI] [PubMed] [Google Scholar]
- Sauve, R., A. Diarra, M. Chahine, C. Simoneau, N. Morier, and G. Roy. 1991. Ca2+ oscillations induced by histamine H1 receptor stimulation in HeLa cells: Fura-2 and patch clamp analysis. Cell Calcium. 12:165–176. [DOI] [PubMed] [Google Scholar]
- Shirai, Y., K. Kashiwagi, K. Yagi, N. Sakai, and N. Saito. 1998. a. Distinct effects of fatty acids on translocation of gamma- and epsilon-subspecies of protein kinase C. J. Cell Biol. 143:511–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirai, Y., N. Sakai, and N. Saito. 1998. b. Subspecies-specific targeting mechanism of protein kinase C. Jpn. J. Pharmacol. 78:411–417. [DOI] [PubMed] [Google Scholar]
- Songyang, Z., S. Blechner, N. Hoagland, M.F. Hoekstra, H. Piwnica-Worms, and L.C. Cantley. 1994. Use of an oriented peptide library to determine the optimal substrates of protein kinases. Curr. Biol. 4:973–982. [DOI] [PubMed] [Google Scholar]
- Tanimura, A., A. Nezu, T. Morita, N. Hashimoto, and Y. Tojyo. 2002. Interplay between calcium, diacylglycerol, and phosphorylation in the spatial and temporal regulation of PKCalpha-GFP. J. Biol. Chem. 277:29054–29062. [DOI] [PubMed] [Google Scholar]
- Thomas, A.P., G.S. Bird, G. Hajnoczky, L.D. Robb-Gaspers, and J.W. Putney, Jr. 1996. Spatial and temporal aspects of cellular calcium signaling. FASEB J. 10:1505–1517. [PubMed] [Google Scholar]
- Ting, A.Y., K.H. Kain, R.L. Klemke, and R.Y. Tsien. 2001. Genetically encoded fluorescent reporters of protein tyrosine kinase activities in living cells. Proc. Natl. Acad. Sci. USA. 98:15003–15008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsien, R.Y. 1989. Fluorescent indicators of ion concentrations. Methods Cell Biol. 30:127–156. [DOI] [PubMed] [Google Scholar]
- van der Wal, J., R. Habets, P. Varnai, T. Balla, and K. Jalink. 2001. Monitoring agonist-induced phospholipase C activation in live cells by fluorescence resonance energy transfer. J. Biol. Chem. 276:15337–15344. [DOI] [PubMed] [Google Scholar]
- Wang, J., A. Gambhir, G. Hangyas-Mihalyne, D. Murray, U. Golebiewska, and S. McLaughlin. 2002. Lateral sequestration of phosphatidylinositol 4,5-bisphosphate by the basic effector domain of myristoylated alanine-rich C kinase substrate is due to nonspecific electrostatic interactions. J. Biol. Chem. 277:34401–34412. [DOI] [PubMed] [Google Scholar]
- Westphal, R.S., K.A. Anderson, A.R. Means, and B.E. Wadzinski. 1998. A signaling complex of Ca2+-calmodulin-dependent protein kinase IV and protein phosphatase 2A. Science. 280:1258–1261. [DOI] [PubMed] [Google Scholar]
- Yaffe, M.B., K. Rittinger, S. Volinia, P.R. Caron, A. Aitken, H. Leffers, S.J. Gamblin, S.J. Smerdon, and L.C. Cantley. 1997. The structural basis for 14-3-3:phosphopeptide binding specificity. Cell. 91:961–971. [DOI] [PubMed] [Google Scholar]
- Yaffe, M.B., G.G. Leparc, J. Lai, T. Obata, S. Volinia, and L.C. Cantley. 2001. A motif-based profile scanning approach for genome-wide prediction of signaling pathways. Nat. Biotechnol. 19:348–353. [DOI] [PubMed] [Google Scholar]
- Zacharias, D.A., J.D. Violin, A.C. Newton, and R.Y. Tsien. 2002. Partitioning of lipid-modified monomeric GFPs into membrane microdomains of live cells. Science. 296:913–916. [DOI] [PubMed] [Google Scholar]
- Zhang, J., Y. Ma, S.S. Taylor, and R.Y. Tsien. 2001. Genetically encoded reporters of protein kinase A activity reveal impact of substrate tethering. Proc. Natl. Acad. Sci. USA. 98:14997–15002. [DOI] [PMC free article] [PubMed] [Google Scholar]