

Abstract
Agrin is a motor neuron–derived factor that directs formation of the postsynaptic apparatus of the neuromuscular junction. Agrin is also expressed in the brain, raising the possibility that it might serve a related function at neuron–neuron synapses. Previously, we identified an agrin signaling pathway in central nervous system (CNS) neurons, establishing the existence of a neural receptor that mediates responses to agrin. As a step toward identifying this agrin receptor, we have characterized the minimal domains in agrin that bind and activate it. Structures required for agrin signaling in CNS neurons are contained within a 20-kD COOH-terminal fragment of the protein. Agrin signaling is independent of alternative splicing at the z site, but requires sequences that flank it because their deletion results in a 15-kD fragment that acts as an agrin antagonist. Thus, distinct regions within agrin are responsible for receptor binding and activation. Using the minimal agrin fragments as affinity probes, we also studied the expression of the agrin receptor on CNS neurons. Our results show that both agrin and its receptor are concentrated at neuron–neuron synapses. These data support the hypothesis that agrin plays a role in formation and/or function of CNS synapses.
Keywords: α-dystroglycan; agrin; agrin receptor; synapse; neuromuscular junction; neuron
Introduction
The hypothesis that agrin mediates the motor neuron–induced accumulation of acetylcholine receptors (AChRs)* at the neuromuscular junction is now widely accepted (for review see Hoch, 1999). Although a complete account of how agrin triggers such changes in AChR distribution has yet to be established, much of what we know about this process has been learned from structural studies of agrin and the proteins with which it interacts.
Native agrin is an ∼400-kD heparan sulfate proteoglycan assembled on an ∼200-kD polypeptide backbone characterized by multiple cysteine-rich domains homologous to other ECMs and secreted proteins (Fig. 1). Structural determinants required for agrin's AChR clustering activity are localized to its 95-kD COOH-terminal half distinguished by the presence of four EGF repeats and three laminin-like G domains (Ferns et al., 1993). Relatively little is known about the properties of the EGF repeats, but functions of the G domains are better understood. For example, α-dystroglycan, a component of the dystrophin-associated glycoprotein complex expressed on the surface membranes of skeletal muscle fibers, binds to a region within the G1/G2 domain (Gesemann et al., 1996; Hopf and Hoch, 1996). However, neither the G1 nor G2 domains are required for agrin's AChR clustering activity, which instead is a property of a 21-kD COOH-terminal region consisting of the G3 domain and the alternatively spliced z site (Gesemann et al., 1995, 1996). The receptor that mediates agrin-induced clustering of AChRs in muscle is a complex consisting of a muscle-specific kinase (MuSK) and an as yet to be identified myotube-associated specificity component (MASC; Glass et al., 1996). Thus, structural elements sufficient to bind and activate the MuSK–MASC receptor are present within 21 kD of agrin's COOH terminus. However, the observation that the AChR clustering activity of the 21-kD fragment is markedly lower than the 95-kD fragment (Gesemann et al., 1995) suggests the G1/G2 domains and/or EGF repeats also contribute to agrin's activity, either by stabilizing the structure of the active site or binding to a component on the muscle cell surface.
Figure 1.

The structure of agrin and agrin deletion constructs used in this paper. Alternate transcriptional start sites give rise to short and long NH2-terminal (SN, LN) forms of agrin. Agrin's polypeptide chain is characterized by numerous cysteine-rich repeats similar to follistatin (F), laminin B (LB), EGF (E), and laminin A G domains (G). Two serine/threonine-rich regions (S/T), consensus glycosaminoglycans side-chain attachment sites (lollipops), and sites of alternative splicing (X, Y, Z) are also shown. Horizontal bars indicate location of binding sites for various cell surface and ECM molecules. The minimal region required for agrin's AChR clustering activity is also shown. Agrin deletion constructs C-Ag95z0/8 and C-AgΔ20 included an NH2-terminal signal peptide for expression in mammalian cells and 4 amino acid insert at the y site. All deletion constructs included COOH-terminal myc (m) and polyhistidine (H) epitope tags.
Many populations of neurons in brain also express agrin, and several lines of evidence support a role for agrin in neuronal synaptogenesis analogous to that at the neuromuscular junction. Agrin is concentrated at neuron–neuron synapses (Mann and Kröger, 1996; Koulen et al., 1999; Gingras and Ferns, 2001), and suppressing agrin expression in cultured hippocampal neurons with antisense oligonucleotides inhibits accumulation of neurotransmitter receptors and other postsynaptic markers at synaptic sites (Ferreira, 1999; Böse et al., 2000). However, other papers have suggested alternate or additional neural functions for agrin. Consistent with a role as a “stop and differentiation” signal (Campagna et al., 1995), blocking agrin expression in cultured hippocampal neurons alters rates of axonal and dendritic elongation (Mantych and Ferreira, 2001) and inhibits differentiation of presynaptic terminals (Böse et al., 2000). In fact, the recent finding that agrin-deficient neurons exhibit altered responses to transient changes in cytoplasmic Ca2+ suggests the potential to modulate a wide range of processes in brain (Hilgenberg et al., 2002).
Little is known about the receptor(s) that might mediate agrin's effects in neurons. However, using the immediate early gene c-fos as a reporter, we have identified an agrin-dependent signaling pathway in central nervous system (CNS) neurons, providing evidence that a neuronal receptor for agrin does exist (Hilgenberg et al., 1999). Agrin signaling in neurons shares several biochemical similarities with that in muscle, including concentration dependence, requirement for extracellular Ca2+, and inhibition by heparin. However, unlike AChR clustering, which is exquisitely sensitive to splicing at the z site (Ferns et al., 1992; Ruegg et al., 1992; Gesemann et al., 1995), agrin z+ and z− isoforms are equally potent activators of the agrin signal pathway in neurons (Hilgenberg et al., 1999). To learn more about the nature of the neuronal receptor for agrin, we analyzed the functional properties of deletion mutants derived from the 95-kD COOH-terminal fragment used in our original works. Our results provide evidence for a novel neuronal receptor for agrin concentrated at synapses formed between CNS neurons.
Results
Agrin signaling in neurons is independent of splicing at the z site
Our initial characterization of the agrin signal transduction pathway in CNS neurons demonstrated its inability to discriminate between “active” z+ and “inactive” z− agrin isoforms (Hilgenberg et al., 1999). However, these works used alternatively spliced variants of the 95-kD COOH-terminal region of rat agrin (rC-Agz0/8), and were limited by the fact that only indirect estimates of agrin concentration could be made, leaving open the possibility that some difference in the specific activities of alternatively spliced isoforms might have gone undetected. To address this issue directly, new 95-kD mouse agrin constructs (Fig. 1; C-Ag95z0/8) were assembled in the pSecTag2 expression vector (Invitrogen), incorporating COOH-terminal myc and polyhistidine epitope tags, permitting purification and detection of the expressed protein and accurate concentration measurement to be made (see Materials and methods). Because the vast majority of agrin molecules expressed in brain include the 4 amino acid exon at the y site (Hoch et al., 1993; Li et al., 1997), all agrin constructs included the y4 exon, and only the properties of z-site variants were examined.
Rat rC-Agz0 and rC-Agz8 induce a neuron-specific increase in Fos expression (Hilgenberg et al., 1999). To confirm the properties of the corresponding mouse constructs, 12-d-old cortical cultures were treated with 1 nM purified mouse C-Ag95z0 or C-Ag95z8, and were then double labeled with antibodies against Fos and either microtubule-associated protein 2 (MAP2) or glial fibrillary acidic protein (GFAP) to identify neurons and glial cells, respectively. Consistent with our previous results, treatment with either C-Ag95z0 or C-Ag95z8 caused a marked increase in Fos expression in neurons, but not nonneuronal cells (Fig. 2). Although differences in the level of Fos expression between neurons were apparent, virtually all neurons (>90%) responded to the C-Ag95z0/8 treatment. In contrast, treatment with a similar concentration of prostate serum antigen control protein expressed in the same vector had no effect on Fos levels in either neurons or glia (Fig. 2). In light of these results, we conclude that neither the myc epitope nor polyhistidine tags induce c-fos, nor do they affect the ability of the C-Ag95z0/8 sequences to do so.
Figure 2.
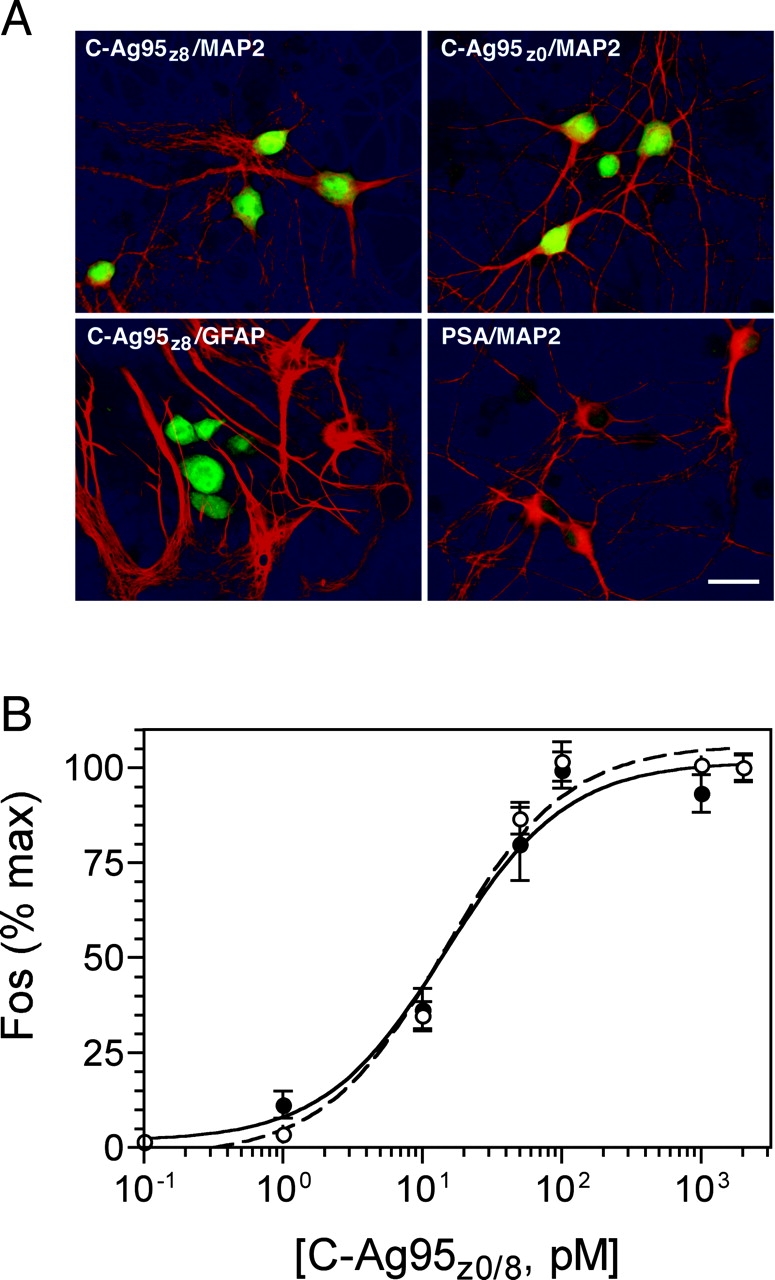
C-Ag95z8 and C-Ag95z0 induce expression of c-fos in cultured cortical neurons. (A) 12-d-old cortical cultures were treated for 10 min with either C-Ag95z8 or C-Ag95z0, followed by double labeling with antibodies for Fos (fluorescein channel) and either MAP2 or GFAP (rhodamine channel). Cell bodies and nuclei of MAP2-positive neurons were intensely labeled for Fos in cultures treated with either C-Ag95z8 or C-Ag95z0. In contrast, only basal levels of Fos expression were observed in GFAP-positive nonneuronal cells. Induction of c-fos was agrin specific in that no detectable increase in Fos was apparent in cultures treated with prostate serum antigen control protein. Bar, 20 μm. (B) Cultures were incubated for 10 min in C-Ag95z8 (open circles, broken line) or C-Ag95z0 (filled circles, solid line), and levels of Fos expression were determined by in situ enzyme-linked assay as described in Materials and methods. Both agrin constructs induced essentially identical concentration-dependent and saturable increases in c-fos expression. Data were normalized to maximal level of Fos expression in each experiment. Each point represents the mean of triplicate determinations from three independent experiments ± SEM. Curves were fit by single-site nonlinear regression model R2 ≥ 0.94. Values from mock-treated sister wells have been subtracted.
Next, we determined the specific activity of each z-site isoform using an in situ enzyme-linked immunoassay (Hilgenberg et al., 1999) to examine the concentration dependence of c-fos induction by C-Ag95z0 and C-Ag95z8. As shown in Fig. 2 B, both agrin isoforms induced c-fos in a concentration-dependent and saturable fashion. Fos expression curves were well fit by a single-site nonlinear regression model (R2 ≥ 0.94) predicting EC50 values of 11.93 ± 0.44 pM for C-Ag95z0 and 12.67 ± 0.58 pM for C-Ag95z8 (mean ± SEM). Similar EC50 values have also been reported for agrin-induced clustering of AChR in muscle, but in contrast to the >1,000-fold difference in AChR clustering activity between isoforms (Ferns et al., 1992; Ruegg et al., 1992; Gesemann et al., 1995), the c-fos–inducing activity of C-Ag95z8 and C-Ag95z0 in neurons is the same.
The 20-kD COOH-terminal portion of agrin is necessary and sufficient for signaling in neurons
As a first step in localizing the structural domains responsible for signaling in neurons, we took advantage of knowledge gained from previous structural analyses of agrin function in muscle (Hoch et al., 1994; Gesemann et al., 1995, 1996) and divided C-Ag95z0/8 into two fragments. The first, an 75-kD NH2-terminal fragment (Fig. 1; C-AgΔ20), which includes the 4 amino acid exon at the y site, has been shown to mediate agrin binding to α-dystroglycan (Gesemann et al., 1996; Hopf and Hoch, 1996). The remaining 20-kD COOH-terminal fragment contains the alternatively spliced z site (Fig. 1; C-Ag20z0 or C-Ag20z8), and is homologous to the minimal agrin fragment able to induce clustering of AChR in cultured muscle cells (Gesemann et al., 1995).
Treatment with C-AgΔ20, at a concentration equivalent to either a near saturating (50 pM) or supersaturating (5 nM) amount of C-Ag95z0/8, had no effect on Fos expression in cortical cultures, suggesting that the active domain is not present within the C-AgΔ20 region (Fig. 3 A). Agrin binding to α-dystroglycan has been shown to modulate agrin-induced AChR clustering in muscle, and we considered the possibility of a similar function for α-dystroglycan signaling in neurons. However, coincubation with either an equal or 100-fold molar excess of C-AgΔ20 had no effect on Fos expression induced by either the C-Ag95z0 or C-Ag95z8 isoforms (Fig. 3 A). Together, these data suggest that domains present within C-AgΔ20 are neither required for, nor modulate, agrin induction of c-fos in neurons.
Figure 3.

The 20-kD COOH-terminal region of agrin is necessary and sufficient for induction of c-fos. (A) Cortical neurons were treated for 10 min with C-AgΔ20 alone or in the presence of 50 pM C-Ag95z8 or C-Ag95z0. Neither a subsaturating (50 pM) nor supersaturating (50 nM) concentration of C-AgΔ20 induced expression of c-fos, nor were they able to modulate the activity of the larger fragments. (B) In contrast, cortical cultures exhibited a concentration-dependent and saturable increase in response to C-Ag20z8 (open circles, broken line) or C-Ag20z0 (solid circles, solid line) similar to that seen with the C-Ag95z8/0 fragments. Data were normalized to the level of Fos expression induced by 50 pM C-Ag95z8 alone in A and maximal level of Fos expression in B. Each data point shows the mean of triplicate determinations from three independent experiments ± SEM. Curves were fit by single-site nonlinear regression model R2 ≥ 0.97. Background levels of Fos expression from mock-treated sister cultures have been subtracted.
The C-Ag20 fragments were both potent inducers of c-fos. As with C-Ag95z0/8, c-fos induction by C-Ag20z0 and C-Ag20z8 was concentration-dependent and saturable (Fig. 3 B). In fact, the EC50 values obtained for the 20-kD fragments (C-Ag20z0, 13.33 ± 0.26 pM; C-Ag20z8, 11.25 ± 2.88 pM) were indistinguishable from each other and from those of the C-Ag95z0/8 isoforms. In light of these observations, we conclude that the structural domains that mediate agrin induction of c-fos are contained within the C-Ag20z0 fragment.
Sequences flanking the z site are critical for agrin signaling
Agrin's AChR clustering activity is regulated by alternative splicing at the z site. However, the observation that a peptide of the 8 amino acid alternatively spliced insert is itself inactive (Gesemann et al., 1995) suggests that domains important for agrin's bioactivity in muscle include not only the z site, but also amino acids that flank it. Because inclusion of alternatively spliced exons at the z site has no effect on agrin activity in neurons, we sought to determine the role of sequences surrounding the z site. To address this question, we deleted 37 amino acids from the NH2 terminus of C-Ag20z0 to the border of the G3 domain, giving rise to a 15-kD COOH-terminal fragment, C-Ag15 (Fig. 1).
Treatment with C-Ag15 alone had no effect on the levels of Fos expression, even when present at a concentration fivefold above saturation for C-Ag20z0/8 (unpublished data). However, when added together with C-Ag95z8, C-Ag15 appeared to inhibit the c-fos–inducing activity of the larger agrin fragment. To examine this effect in more detail, C-Ag15 dose–response studies were performed in the presence of a near saturating (50 pM) concentration of C-Ag95z8. Increasing concentrations of C-Ag15 inhibited the c-fos–inducing activity of C-Ag95z8 (Fig. 4 A). The curve was well fit by a nonlinear regression model for single-site competition (R2 = 0.95) predicting an IC50 of 64.45 ± 10.94 pM and close to a 1:1 agonist:antagonist molar ratio. Similar results were also obtained for competition against 50 pM C-Ag95z0 and the C-Ag20z0/8 isoforms (unpublished data).
Figure 4.

The 15-kD COOH-terminal region of agrin is a cell- specific competitive inhibitor of C-Ag95z8 induction of c-fos. (A) Cortical cultures were incubated in 50 pM C-Ag95z8 in the presence of different concentrations of C-Ag15. C-Ag15 inhibited C-Ag95z8 induction of c-fos in a concentration-dependent manner well described by a single-site competition model (R2 = 0.95) with an IC50 of 64.5 pM; close to a 1:1 agonist:antagonist molar ratio. Data were normalized to percentage of Fos expression induced by 50 pM C-Ag95z8 alone, and represent the mean of triplicate determinations from three independent experiments ± SEM. Background levels of Fos expression have been subtracted. (B) To test the cell specificity of C-Ag15 inhibition, hippocampal (Hip) or cerebellar (Cer) neurons and chick muscle fibers (Mus) were incubated in 50 pM C-Ag95z8 alone (open bars) or in the presence (filled bars) of 1 nM C-Ag15. Levels of Fos expression were expressed as fold change over mock-treated sister cultures such that a value of 1 indicates no change. C-Ag15 completely inhibited C-Ag95z8-induced expression of Fos in hippocampal and cerebellar neurons, but not in muscle. (C) The effect of C-Ag15 on C-Ag95z8-induced AChR aggregation was also tested. Cultured chick myotubes were incubated overnight in 50 pM C-Ag95z8 alone (−) or in the presence (+) of 1 nM C-Ag15, and AChR clusters were labeled with rhodamine-conjugated α-bungarotoxin. AChR clusters were counted blind with respect to treatment in five random fields/well and expressed as the ratio of clusters in mock-treated sister cultures. Consistent with the results of the Fos expression analysis, C-Ag15 had no effect on C-Ag95z8-induced AChR clustering. Bars in B and C represent the mean ± SEM of triplicate wells from four independent experiments.
To learn whether inhibition of agrin signaling by C-Ag15 might extend to other neurons or even muscle, mouse hippocampal and cerebellar neurons or chick skeletal myotubes were incubated with 50 pM C-Ag95z8 alone or in the presence of 1 nM C-Ag15, a concentration that blocks activity in cortical neurons. Treatment with C-Ag95z8 triggered a robust increase in Fos levels in both populations of neurons and in muscle compared with control sister cultures receiving vehicle (Fig. 4 B). However, co-incubation with C-Ag15 inhibited Fos expression in the neurons, but had no effect on muscle. Because agrin's bioactivity in muscle is normally measured in terms of its AChR clustering activity, we also examined the ability of C-Ag15 to antagonize agrin-induced clustering of AChR. Chick myotubes were incubated overnight with 50 pM C-Ag95z8 alone or with 1 nM C-Ag15, AChR labeled with rhodamine-conjugated α-bungarotoxin, and the number of AChR clusters were determined. In line with the results of the Fos assay, C-Ag15 had no effect on the AChR clustering activity of C-Ag95z8 (Fig. 4 C). Based on the results of these works, it appears that C-Ag15 is a specific antagonist for the neuronal receptor for agrin.
C-Ag20z0/8 rescues an agrin-deficient neuronal phenotype
Although induction of c-fos is a convenient reporter facilitating biochemical characterization of agrin signaling, its significance in terms of agrin function in CNS neurons is unknown. Recently, we have provided evidence that agrin plays an important role in neural differentiation by showing that agrin-deficient neurons exhibit reduced responses to glutamate both in cell culture and in vivo (Hilgenberg et al., 2002). Therefore, to learn more about the role of the 20-kD COOH-terminal region in agrin function, we tested the ability of the C-Ag20 isoforms to rescue the agrin deficiency.
When grown in normal media, levels of glutamate-induced Fos expression in agrin-deficient neurons were reduced by ∼70% compared with wild type. However, consistent with our earlier findings, supplementing the growth media for 2 d with a saturating concentration of either C-Ag95z8 or C-Ag95z0 significantly increased the response of agrin-deficient neurons close to wild-type levels (Fig. 5 A). Similar results were obtained when agrin-deficient neurons were grown in the presence of the C-Ag20z0/8 fragments. Compared with agrin-deficient neurons in vehicle-supplemented control media, treatment with either 5 nM C-Ag20z8 or C-Ag20z0 resulted in a significant 2.2-fold increase in glutamate response. Thus, not only do the C-Ag20z0/8 isoforms exhibit full activity in terms of their ability to induce c-fos, they are also competent to reverse the physiological deficit resulting from loss of expression of full-length agrin in the agrin-deficient cultures.
Figure 5.

The 20-kD COOH-terminal region of agrin is the minimal fragment sufficient to rescue an agrin-deficient phenotype. (A) 10-d-old wild-type (filled bars) or agrin-deficient (open bars) cultures were grown for 2 d in media supplemented to 5 nM with the indicated agrin fragment. Cultures were challenged for 5 min with 100 μM glutamate as described previously (Hilgenberg et al., 2002), and the levels of Fos expression were determined. Data were normalized to levels of glutamate-induced Fos expression in mock-treated wild-type cultures. Compared with mock, glutamate responses of homozygous agrin-deficient neurons were rescued to near wild-type levels by either the long C-Ag95z8/0 or short C-Ag20z8/0 fragments (***, P ≤ 0.0005; **, P ≤ 0.002; *, P ≤ 0.02; paired t test). (B) To test the ability of C-Ag15 to antagonize agrin action, 10-d-old agrin-deficient neurons were maintained for 2 d in the presence of 5 nM C-Ag95z0 alone or in combination with 10 nM C-Ag15, and neuronal responses to glutamate were determined as above. Treatment with C-Ag15 significantly (*, P ≤ 0.004; paired t test) inhibited rescue of the agrin-deficient response to glutamate by C-Ag95z0.
Despite its apparent effectiveness as an antagonist of agrin-induced expression of c-fos, growth in media supplemented with up to 10 nM C-Ag15 had no effect on glutamate-induced expression of c-fos in either wild-type or heterozygous agrin-deficient neurons (unpublished data). This suggests that C-Ag15 is unable to block signaling by native agrin. However, the concentration of endogenous agrin in our cultures is unknown, and may exceed the maximal concentration of C-Ag15 we were able to use, especially if agrin is present as high density clusters on neuronal surface membranes (see next section and Results). As an alternative approach to examining the functional properties of the C-Ag15 fragment, we tested its ability to inhibit rescue of the agrin-deficient phenotype by C-Ag95z0/8. Inclusion of 10 nM C-Ag15 in the growth medium significantly reduced the efficacy of 5 nM C-Ag95z0, blocking slightly more than half (54%) of the rescue normally achieved by C-Ag95z0 alone (Fig. 5 B). Similar results were also seen in single experiments using either C-Ag95z8 or C-Ag20z8 (unpublished data). Based on these observations we conclude that, under the appropriate conditions, C-Ag15 can inhibit agrin function in CNS neurons.
Agrin receptors are concentrated at neuron–neuron synapses
The works described so far provide evidence for a neuron-specific agrin receptor, and identify a minimal fragment of agrin capable of activating it. Next, we sought to establish the cellular distribution of the neuronal receptor for agrin using the short agrin fragments as affinity ligands. Live cortical cultures were incubated in C-Ag20z8, C-Ag20z0, or C-Ag15, and were then labeled with an antibody against the polyhistidine epitope tag to visualize bound agrin. A similar pattern of labeling was evident for all three agrin fragments and appeared as bright puncta scattered over neuronal somata and neurites (Fig. 6). Labeling was specific in that nonneuronal cells were not labeled (unpublished data), and little or no labeling was evident when either the agrin fragments were omitted or a β-galactosidase–myc–6His vector control protein was used in their place (Fig. 6). More significantly, binding of all three agrin fragments was completely blocked in cultures labeled in the presence of a large excess of rC-Ag with both “neural” rC-Agy4z8 and “inactive” rC-Agy0z0 appearing equally effective at blocking binding of either C-Ag20z isoform or C-Ag15. Precise estimates of the concentration dependence of the competition between rC-Ag and the short mouse agrin fragments are difficult to obtain using immunofluorescence. However, NIH Image analysis of the mean pixel intensities obtained from fixed exposure photomicrographs of random fields revealed labeling to be reduced by ∼30% in cultures coincubated with a 1:1 molar ratio of C-Ag20z8 to rC-Agy4z8, and barely detectable at 1:100 (unpublished data). Together, these results provided strong evidence that the bioactivity of the different agrin isoforms reported here is mediated through binding to a single population of agrin receptors expressed on cortical neuron cell membranes.
Figure 6.
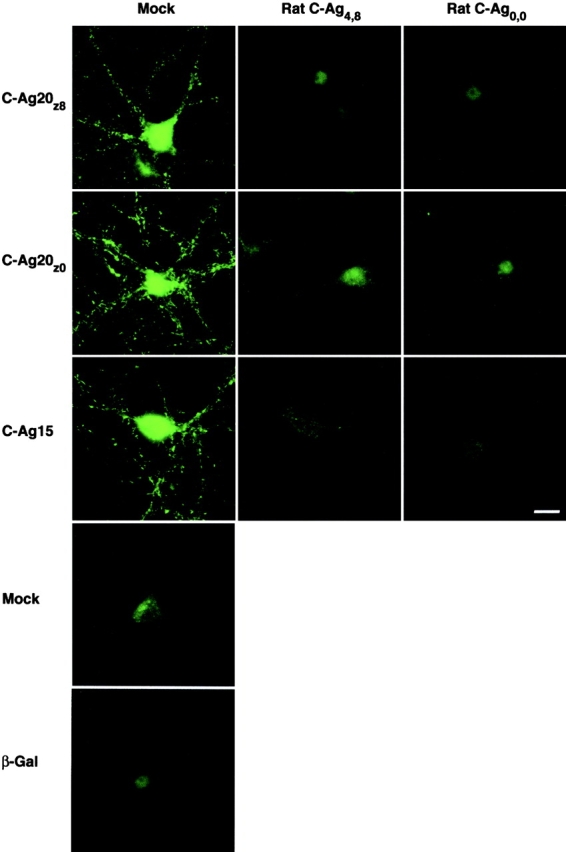
Agrin receptor expression in nerve cell membranes. Live cortical neurons were incubated in C-Ag20z8, C-Ag20z0, or C-Ag15, either alone or in the presence of mock-conditioned medium or a 500-fold molar excess of the active (Rat C-Ag4,8) or inactive (Rat C-Ag0,0) isoform of rat agrin. Immunostaining with an anti-polyhistidine antibody reveals binding of the short agrin fragments to receptors distributed in numerous small clusters on neuron cell bodies and neurites; patches of agrin receptors outside the focal plane contribute to the diffuse staining evident in some neuron cell bodies. Consistent with a single class of agrin receptors, each fragment shows a similar pattern of binding that can be blocked by either isoform of rat agrin. The ability of rC-Ag to block the short agrin fragments is also strong evidence that binding is specific. In addition, no labeling was observed when the short agrin fragments were omitted or replaced by β-galactosidase (β-Gal) as a control for vector-specific sequences. Bar, 20 μm.
Previous papers have shown that agrin is present at neuron–neuron synapses in peripheral ganglia and retina (Mann and Kröger, 1996; Koulen et al., 1999; Gingras and Ferns, 2001). The punctate labeling observed with the short COOH-terminal agrin fragments suggested that receptors for agrin might also be synaptic. To test this hypothesis, we first determined the distribution of endogenous agrin on cultured cortical neurons by labeling live cultures with RαAg-1, a pan-specific anti-agrin serum, followed by fixation and labeling with a second antibody against the synaptic vesicle protein synaptophysin. Agrin immunostaining was distributed in patches on the neuronal cell bodies and neurites, and colocalized with synaptophysin-positive nerve terminals (Fig. 7). Although some variation in the intensity of the immunostaining was evident, few (if any) agrin-positive/synaptophysin-negative or synaptophysin-positive/agrin-negative patches were observed. Similar results were also obtained using an anti-agrin mAb (MAB5204; CHEMICON International, Inc.) and rabbit anti-synaptophysin antiserum (unpublished data). Therefore, we conclude that agrin is specifically localized at synapses formed between cultured cortical neurons.
Figure 7.

Agrin and agrin receptors are colocalized at synaptic contacts. The subcellular distribution of endogenous agrin and agrin receptors on cultured cortical neurons was determined by labeling with either an anti-agrin serum, RαAg-1, or short agrin fragment, followed by fixation and incubation with an antibody to synaptophysin to identify nerve terminals by labeling synaptic vesicles. Both agrin and the agrin receptor were present at virtually all synaptophysin-positive nerve terminals (arrowheads), evidence that agrin and its receptor are colocalized at synaptic sites. Nerve terminal staining was specific, and was not observed in control cultures labeled with an agrin receptor probe in the absence of the anti-synaptophysin antibody. Bar, 20 μm.
Next, to learn whether agrin receptors also exhibit a synaptic pattern of expression, a parallel work was performed in which live cortical cultures were labeled with C-Ag20z8, C-Ag20z0, or C-Ag15 as before, followed by fixation and staining for synaptophysin. Irrespective of the agrin probe used, agrin receptor clusters exhibited a high degree of colocalization with the synaptic vesicle marker. Staining for synaptophysin was specific; nonneuronal cells were not labeled by the synaptophysin antibody (unpublished data), and nerve terminal staining was not observed in cultures treated with C-Ag20z0 or other short fragments in the absence of the anti-synaptophysin antibody (Fig. 7). Together with the agrin immunostaining, the results of these studies show that both agrin and its receptor are concentrated at neuron–neuron synapses.
Discussion
Several papers have provided evidence that agrin plays a role in differentiation of CNS neurons. In particular, using the immediate early gene c-fos as a reporter, we have characterized an agrin-signaling pathway that influences development of neuronal responses to excitatory neurotransmitters and depolarization (Hilgenberg et al., 1999, 2002). To learn more about the receptor mediating agrin's function in brain, we have analyzed the structural features of agrin required for activation of its signaling pathway. Our results confirm agrin's activity in CNS neurons is independent of alternative splicing at the z site, and demonstrate further that agrin's functional domains reside within 20 kD of its COOH terminus. Also, we show that the receptor mediating these agrin-dependent signals is colocalized with agrin at neuron–neuron synapses. Here, we discuss the implications of our findings for agrin function and the identity of its receptor in CNS neurons.
Initial attempts to characterize molecules that might mediate agrin-induced clustering of AChR identified α-dystroglycan as a major agrin-binding protein in muscle cell membranes (Campanelli et al., 1994; Gee et al., 1994). Although it now seems unlikely to represent the “functional” agrin receptor, evidence suggests that α-dystroglycan still plays an important role in consolidation and maintenance of AChR clusters once formed (Jacobson et al., 1998, 2001; Heathcote et al., 2000). Dystroglycan is also found in mammalian brain, where its expression and synaptic localization have suggested a role in synaptic function (Tian et al., 1996; Zaccaria et al., 2001; Lévi et al., 2002). Interestingly, several biochemical properties of agrin induction of c-fos in CNS neurons, including Ca2+ dependence, inhibition by heparin, and ability to bind z+ and z− isoforms (Hilgenberg et al., 1999), are consistent with agrin binding to α-dystroglycan (Sugiyama et al., 1994; Gesemann et al., 1996). High affinity binding of agrin to α-dystroglycan is mediated by a region that includes the first two laminin G domains (Gesemann et al., 1996; Hopf and Hoch, 1996). However, C-AgΔ20, which contains the G1 and G2 domains, neither induced c-fos nor inhibited the activity of the larger fragments. Conversely, both C-Ag20z0/8 isoforms, lacking domains for high affinity binding to α-dystroglycan, induced c-fos and rescued the agrin-deficient phenotype with a similar efficacy as their 95-kD C-Agz0/8 counterparts. Although these results do not rule out a role for α-dystroglycan mediating other aspects of agrin signaling, α-dystroglycan is unlikely to be a component of the agrin receptor mediating the responses measured here.
Also, we considered the possibility that the MuSK–MASC complex might mediate agrin signaling in neurons. Both neural and muscle signaling pathways are activated by picomolar concentrations of agrin, are inhibited by heparin, and are Ca2+ dependent (Hilgenberg et al., 1999). Even more striking is the fact that the same minimal agrin fragment is sufficient to activate the MuSK–MASC complex (Gesemann et al., 1995) and neuronal receptor; evidence that significant structural homology exists between the two receptors. However, despite these similarities, we believe the neuronal and muscle receptors are distinct. MuSK expression has not been detected in either embryonic or adult mammalian brain (Valenzuela et al., 1995). Moreover, although a critical determinant of agrin's AChR clustering activity in muscle (Ferns et al., 1992; Ruegg et al., 1992; Gesemann et al., 1995), alternative splicing at the z site has no effect on agrin's bioactivity assayed here, even in the case of the smallest active fragment where variations in structure might be expected to exert the greatest effect. The finding that the C-Ag20z0/8 fragments are fully active also contrasts with observations in muscle where the AChR clustering activity of the corresponding fragment is >100-fold lower than the 95-kD C-Ag polypeptide from which it was derived (Gesemann et al., 1995). Together with the inability of C-Ag15 to antagonize agrin induction of either c-fos or AChR clustering in muscle, these data point to a fundamental difference in the receptors that catalyze the agrin response in nerve and muscle cells.
The inhibition of c-fos induction by C-Ag15, its ability to block rescue of the agrin-deficient phenotype, distribution of binding sites, and ability of different agrin isoforms to block binding to them argue that C-Ag15 and the active agrin polypeptide fragments compete for a common binding site on the neuronal receptor for agrin. How then might C-Ag15 bind to the receptor but not activate it? One possibility is that deletion of the 5-kD NH2-terminal region may have disordered secondary or tertiary structures in the remaining G3 and COOH-terminal domain necessary for signaling. However, that such a structural change could occur without affecting the apparent affinity of C-Ag15 binding to the receptor seems improbable. Alternatively, activation of the receptor may require agrin binding to two sites, with the 5-kD NH2-terminal region of C-Ag20z0/8 targeted to the second site. The agrin receptor in muscle is made up of two components (MuSK and MASC) although agrin appears only to bind directly to MASC (Glass et al., 1996). Whereas a similar two-component model of the neuronal receptor for agrin would be consistent with our data, the fact that C-Ag15 has no effect on agrin's bioactivity in muscle suggests that, although functionally homologous to MASC, the agrin-binding component of the neuronal receptor would be structurally distinct.
Blocking agrin expression with antisense oligonucleotides inhibits synapse formation and alters synaptic function in CNS neurons (Ferreira, 1999; Böse et al., 2000). The ability of rC-Ag95z8 (but not rC-Ag95z0) to rescue changes induced by the antisense oligonucleotides suggests these effects are mediated by a loss of isoform-specific signals associated with agrin's COOH-terminal domains (Ji et al., 1998; Böse et al., 2000). These observations contrast to our own findings that, in terms of their ability to induce c-fos and rescue an agrin mutant phenotype, the bioactivity of even the shortest alternatively spliced active fragments are the same. One possible explanation for this apparent inconsistency is that different domains within the 95-kD COOH-terminal region of agrin might mediate distinct responses through specific receptors. In this regard, it is interesting to note that a recent paper showed that in addition to α-dystroglycan/heparin binding, the 95-kD COOH-terminal region of agrin contains three other sites that interact with neuronal cell surface receptors (Burgess et al., 2002). Two of these sites are targeted to integrins, one of which modulates agrin's AChR clustering activity, whereas the receptor for the third site, located within 20 kD of agrin's COOH terminus, has yet to be identified. It will be interesting, in light of these observations, to determine whether C-Ag15 inhibition is limited to the bioassays used here, or whether it extends to other agrin-dependent neuronal responses.
Agrin induces expression of c-fos in CNS neurons, and mutation of the agrin gene decreases neuronal responses to excitatory neurotransmitters (Hilgenberg et al., 2002). Although neurons express multiple molecules capable of binding agrin, several lines of evidence support the conclusion that the agrin-binding sites visualized using the affinity probes are the receptors responsible for the physiologic responses to agrin reported here and earlier (Hilgenberg et al., 1999). First, the apparent affinities and potency of the various agrin fragments, in terms of their c-fos–inducing activity or ability to rescue the agrin-deficient phenotype, are the same. Moreover, the ability of the smallest agrin fragment, C-Ag15, to inhibit both c-fos induction and rescue of the agrin-deficient phenotype is strong evidence for a common receptor mediating both activities. A similar profile was also apparent for the binding properties of the agrin fragments when used to visualize the distribution of the agrin receptors. Consistent with the biochemical studies, neurons were clearly labeled using agrin concentrations in the picomolar range irrespective of z site composition. Most importantly, the distribution of high density binding sites for C-Ag15 and their sensitivity to blockade by rC-Agz0/8 was indistinguishable from that observed with either C-Ag20z0/8 constructs.
Previous papers have shown that agrin is concentrated at cholinergic synapses formed between cultured sympathetic neurons (Gingras and Ferns, 2001) and GABAergic synapses in the retina (Mann and Kröger, 1996; Koulen et al., 1999). Cultured cortical neurons receive both glutamatergic and GABAergic inputs (Li et al., 1997), and because no evidence of synaptic contacts lacking agrin was found, we can extend these findings to include glutamatergic synapses as well. A similarly high correlation between agrin receptors and nerve terminals was also evident, suggesting that agrin and its receptor colocalize at synaptic sites. Together, these observations suggest that regardless of neurotransmitter phenotype, synapses are likely to be significant sites of agrin action. Consistent with these conclusions, agrin has been implicated in regulating a number of pre- and postsynaptic properties of cholinergic, glutamatergic, and GABAergic neurons (Ferreira, 1999; Böse et al., 2000; Gingras et al., 2002). Higher resolution techniques than those used here will be required to determine the agrin receptor's subcellular distribution. However, if all agrin receptors are presynaptic, then a long-range retrograde signal would be needed to account for agrin's ability to rapidly induce c-fos. Therefore, it seems likely that at least some agrin receptors, for example, those at or near the neuronal soma, are postsynaptic.
At the neuromuscular junction, agrin is anchored to the synaptic basal lamina by a laminin-binding domain present within its NH2 terminus (Denzer et al., 1995). However, the mechanism controlling agrin's spatial organization in neurons, and in particular, its concentration at synaptic sites, is unknown. Neuron–neuron synapses lack a basal lamina; moreover, the short NH2-terminal form of agrin (SN-agrin) expressed in CNS neurons not only lacks the laminin-binding domain, but consistent with a type II membrane protein, is oriented such that its NH2 terminus is cytoplasmic (Burgess et al., 2000; Neumann et al., 2001). That signals embedded within this cytoplasmic domain might play a role in agrin's subcellular localization also seems unlikely. Fusion of the SN-agrin NH2 terminus to YFP directs YFP to neuronal membranes, but the pattern of expression is uniform rather than concentrated at synaptic sites (Burgess et al., 2002). In light of these findings, agrin's spatial distribution would seem to be defined by interactions between its extracellular domains and candidate neuronal cell surface agrin-binding proteins such as neural cell adhesion molecules, integrins, and α-dystroglycan. Clearly, the high affinity and synapse-specific pattern of expression of the neuronal receptor for agrin are also consistent with the possibility that, in addition to signal transduction, it could play a role sculpting agrin's spatial distribution. Future studies comparing the time course of agrin and agrin receptor expression and any effect C-Ag15 might have on agrin's distribution will be helpful in testing this hypothesis.
Materials and methods
Tissue culture
Mouse cortical cultures were prepared from newborn or 1-d-old ICR strain mice (Harlan) as described previously (Hilgenberg et al., 1999). For the first 24 h after plating, cells were maintained in neural basal medium (NBM) plus B27 supplements (Invitrogen), and in nonneuronal cell–conditioned NBM (cNBM) plus B27 thereafter, at 37°C in humidified 5% CO2 atmosphere. To further reduce proliferation of nonneuronal cells, cultures on glass coverslips used for histology were treated with 5 μM 5-fluoro-2-deoxyuridine (Sigma-Aldrich) 3–4 d after plating. Hippocampal and cerebellar cultures were prepared in a similar manner. Experiments were performed on 10–14-d-old cultures.
Agrin-deficient neuron cultures were prepared from cortices of embryonic d 18–19 fetuses resulting from matings between mice heterozygous for a mutation in the agrin gene (Gautam et al., 1996). Cultures were prepared and genotyped as described previously (Li et al., 1999), and were maintained as above.
Chick muscle cultures were prepared from pectoral muscles of 10–11-d-old White Leghorn chick embryos as described previously (Hilgenberg et al., 1999). Experiments were performed on 4–6-d-old cultures.
Expression constructs
Parent constructs, C-Ag95z0 and C-Ag95z8 (Fig. 1), encoding the soluble 95-kD COOH terminus of mouse agrin, were generated from cDNA prepared by RT-PCR of adult mouse cortex RNA using the F95/R95 primer pair (Table I) subcloned into the pGEM-T (Promega) shuttle vector and transformed into JM109-competent bacteria. Individual ampicillin-resistant colonies were picked, and C-Ag95z0- and C-Ag95z8-containing clones were identified by PCR analysis using primers F24/B2 flanking the z site. After double digestion of the BamHI and EcoRV l linker sites contained within the F95 and R95 primers, agrin inserts were gel purified and ligated into the pSecTag2B expression vector (Invitrogen) in frame with a COOH-terminal myc epitope and 6× polyhistidine tag.
Table I. PCR primers.
| Construct | Primer | Sequence | Nucleotide |
|---|---|---|---|
| C-Agy4z0/8 | F95 | TAGGATCCACCGCCAGTATTGACCGA | 1104–1121 |
| R95 | TAGATATCAGAGTGGGGCAGGGTCTT | 6678–6663 | |
| C-Ag Δ20 | RΔ20 | TAGATATCGTCCGCCCATCAAAGGCC | 4585–4568 |
| C-Ag20z0/8 | F20 | AATGGATCCTCGGACCTACATCG | 4581–4593 |
| R20 | TTCGAATTCAGAGTGGGGCAGGG | 6679–6666 | |
| C-Ag15 | F15 | AATGGATCCGTGGATTGGCAAGGC | 5750–5764 |
Engineered BamHI and EcoRV restrictions sites, in forward and reversed primers, respectively, are underlined. Nucleotide positions are numbered according to Rupp et al. (1992).
C-AgΔ20 lacking the 20-kD COOH-terminal region of C-Agz0/8 was prepared by PCR amplification of the C-Ag95z8 pGEM-T template using F95 and the reverse primer RΔ20 that includes a 3′ EcoRV site. DNA from the PCR reaction was digested with BamHI and EcoRV, gel purified, and ligated into pSecTag2B.
A similar strategy was used to generate C-Ag20z0/8 and C-Ag15 constructs by PCR amplification of the appropriate pGEM-T C-Agz0/8 template using either F20 (for C-Ag20z0/8) or F15 (for C-Ag15) in combination with R20. However, for more efficient expression, aliquots of the PCR reaction were ligated into the inducible bacterial expression vector pTrcHis2 (Invitrogen).
Expression and quantitation of recombinant agrin
pSecTag2B agrin vector DNA encoding either C-Agz0/8 or C-AgΔ20 was transfected into HEK 293T cells using LipofectAMINE™ (Invitrogen) according to the manufacturer's directions. Controls were either sham transfected with LipofectAMINE™ alone or with control vector encoding prostate-specific antigen (pSecTag2-PSA). Agrin constructs in the pTrcHis2 expression vector were maintained in the JM109 bacteria. The plasmid pTrcHis2-lacZ encoding β-galactosidase was expressed as a control.
Polyhistidine-tagged agrin fragments were purified from conditioned media and bacterial extracts using the Talon™ (CLONTECH Laboratories, Inc.) metal affinity resin eluted with 200–500 mM imidazole (Sigma-Aldrich) according to the manufacturer's instructions. The identity of the isolated fragments was confirmed by immunoblot analysis using RαAg-1, a rabbit antiserum raised against a synthetic peptide corresponding to amino acids 1862–1895 conserved in all isoforms of mouse agrin. For some experiments, the elution buffer was removed by dialysis against PBS or 20 mM Tris and 250 mM NaCl, pH 8.0.
The molar concentration of each fragment was determined by comparison to a C-Ag95z0 standard prepared as follows: HEK 293T cells were transfected with pSecTag2B-C-Agz0, and were then transferred to 80% methionine-free DME containing 100 μCi/ml [35S]methionine. C-Ag95z0 present in the medium was purified over a Talon™ metal affinity resin column, and the apparent molar concentration was determined by counting aliquots of the column eluate in a scintillation counter (model LS7500; Beckman Coulter). A small correction factor was applied to account for the fraction of counts incorporated into C-Ag95z0 (≥90%) versus total counts determined by phosphorimager analysis (Molecular Dynamics, Inc.) of an aliquot of the eluate separated on an 8% SDS-PAGE gel.
The concentration of the other agrin fragments was determined by comparison to a 35S-labeled C-Ag95z0 standard in immunoblots probed with a mouse anti-myc antibody (Invitrogen) and 125I-labeled anti–mouse second antibody (Amersham Biosciences). The amount of 125I bound to both the standard and unknown was determined by phosphorimager analysis and, after correcting for the contribution of the [35S]methionine in the standard, the concentration of the unknown was determined from the standard curve.
Soluble 95-kD COOH-terminal fragments of rat agrin (rC-Agy4z8/y0z0) were harvested in media conditioned by transiently transfected COS-7 cells (Hilgenberg et al., 1999) and dialyzed against PBS. The concentration of the rC-Ag was estimated by comparison to a mouse agrin standard in the c-fos induction assay and by immunoblot analysis with RαAg-1.
Quantitative analysis of Fos expression
Fos expression in cortical cultures was measured by in situ enzyme-linked immunoassay (Hilgenberg et al., 1999). In brief, 11–14-d-old neuronal cultures were treated for 10 min with agrin or other agent diluted in NBM or PBS, then washed in cNBM and returned to the incubator for 2 h. Cultures were rinsed in PBS, fixed in ice cold 4% PFA, and blocked in PBS containing 0.1% Triton X-100 and 4% BSA (PBSTB) before being incubated in a primary rabbit antibody against Fos (Ab-2; Oncogene Research Products) and secondary goat antibody against mouse conjugated to alkaline phosphatase (Southern Biotechnology Associates, Inc.). The level of Fos expression was determined by monitoring conversion of p-nitrophenyl phosphate to a soluble yellow reaction product at 405 nm.
AChR clustering assay
4–6-d-old myotubes were treated with agrin overnight followed by incubation with 20 nM rhodamine-conjugated α-bungarotoxin (Molecular Probes, Inc.) in culture medium for 1 h at 37°C. Cells were then fixed in 4% PFA in PBS, washed in PBS, and viewed at 200× under epifluorescent illumination on a microscope (Optiphot-2; Nikon). For each well, the mean number of AChR clusters/field was determined from counts obtained from five random fields. All counts were performed blind with respect to treatment. To facilitate comparison between experiments, the number of AChR clusters/field was normalized to the cluster density of control cultures treated with vehicle alone.
Fos immunohistochemistry
10–14-d-old cortical neurons, grown on glass coverslips, were treated with agrin, fixed, and blocked as for the Fos in situ enzyme-linked immunoassay. Cells were double labeled overnight at 4°C with Ab-2 (1:200) together with either a mouse monoclonal antibody against MAP2 (SMI-52; 1:400; Sternberger Monoclonals), or GFAP (G-A-5; 1:1,000; Sigma-Aldrich) in PBSTB to identify neurons or glial cells, respectively. Bound antibodies were visualized by incubation for 2 h at RT in a mixture of fluorescein-conjugated goat anti–rabbit and Texas red–labeled goat anti–mouse secondary antibodies (Vector Laboratories) diluted 1:200 in PBSTB. Coverslips were washed in PBS, mounted in Fluoromount™ (Southern Biotechnology Associates, Inc.), and examined using epifluorescent illumination.
Agrin immunohistochemistry
Live cortical cultures on glass coverslips were incubated for 30 min at 37°C in RαAg-1 diluted 1:500 in NBM. Cultures were then washed in NBM, fixed and blocked as described for the Fos in situ enzyme-linked immunoassay, and were then washed and incubated overnight at 4°C with the anti-synaptophysin antibody, SVP38 (Sigma-Aldrich), diluted 1:400 in PBS containing 4% BSA (PBSB). Primary antibodies were visualized by double labeling with fluorescein-conjugated goat anti–rabbit and Texas red–labeled anti–mouse antibodies as above.
Agrin receptor immunohistochemistry
The distribution of agrin receptors was studied using agrin deletion fragments as affinity probes. Neurons, plated on glass coverslips, were washed briefly (1–2 min) in cold PBS containing 10 mM EDTA followed by a second wash in cold PBS alone before incubation for 15 min at 4°C with 1 pM recombinant mouse agrin in NBM. In some experiments, labeling with mouse agrin was performed in the presence of various concentrations of rat rC-Ag95y4z8 or rC-Agy0z0. Control cultures were treated with vector control protein or an equivalent volume of vehicle in which the recombinant agrin was dissolved. Cells were then washed in cold NBM, fixed for 40 min on ice in 4% PFA in PBS, then washed in PBS and blocked for 1 h in PBSB. Recombinant agrin was detected through the COOH-terminal 6× polyhistidine tag using an anti-his antibody (1:500; Invitrogen). In experiments where cells were double labeled for synaptophysin, the anti-synaptophysin antiserum (1:400) was also added at this step. Incubations were performed overnight at 4°C in PBSB, followed by labeling with fluorescein-conjugated goat anti–mouse and Texas red–labeled anti–rabbit antibodies as described in the previous paragraphs. In some experiments, the amount of mouse agrin bound was estimated by digital photomicrography using the public domain NIH Image program (http://rsb.info.nih.gov/nih-image/).
Acknowledgments
The authors thank Betty Sicaeros for technical assistance and Dr. Diane O'Dowd for critical reading of the manuscript.
This work was supported by National Institutes of Health grant NS33213 to M.A. Smith.
Footnotes
Abbreviations used in this paper: AChR, acetylcholine receptor; CNS, central nervous system; GFAP, glial fibrillary acidic protein; MAP2, microtubule-associated protein 2; MASC, myotube-associated specificity component; MuSK, muscle-specific kinase; NBM, neural basal medium.
References
- Böse, C.M., D. Qiu, A. Bergamaschi, B. Gravante, M. Bossi, A. Villa, F. Rupp, and A. Malgaroli. 2000. Agrin controls synaptic differentiation in hippocampal neurons. J. Neurosci. 20:9086–9095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess, R.W., W.C. Skarnes, and J.R. Sanes. 2000. Agrin isoforms with distinct amino termini: differential expression, localization, and function. J. Cell Biol. 151:41–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess, R.W., D.K. Dickman, L. Nunez, D.J. Glass, and J.R. Sanes. 2002. Mapping sites responsible for interactions of agrin with neurons. J. Neurochem. 83:271–284. [DOI] [PubMed] [Google Scholar]
- Campagna, J.A., M. Ruegg, and J.L. Bixby. 1995. Agrin is a differentiation-inducing “stop signal” for motorneurons in vitro. Neuron. 15:1365–1374. [DOI] [PubMed] [Google Scholar]
- Campanelli, J.T., S.L. Roberds, K.P. Campbell, and R.H. Scheller. 1994. A role for dystrophin-associated glycoproteins and utrophin in agrin-induced AChR clustering. Cell. 77:663–674. [DOI] [PubMed] [Google Scholar]
- Denzer, A.J., M. Gesemann, B. Schumacher, and M.A. Ruegg. 1995. An amino-terminal extension is required for the secretion of chick agrin and its binding to extracellular matrix. J. Cell Biol. 131:1547–1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferns, M., W. Hoch, J.T. Campanelli, F. Rupp, Z.W. Hall, and R.H. Scheller. 1992. RNA splicing regulates agrin-mediated acetylcholine receptor clustering activity on cultured myotubes. Neuron. 8:1079–1086. [DOI] [PubMed] [Google Scholar]
- Ferns, M.J., J.T. Campanelli, W. Hoch, R.H. Scheller, and Z. Hall. 1993. The ability of agrin to cluster AChRs depends on alternative splicing and on cell surface proteoglycans. Neuron. 11:491–502. [DOI] [PubMed] [Google Scholar]
- Ferreira, A. 1999. Abnormal synapse formation in agrin-depleted hippocampal neurons. J. Cell Sci. 112:4729–4738. [DOI] [PubMed] [Google Scholar]
- Gautam, M., P.G. Noakes, L. Moscoso, F. Rupp, R.H. Scheller, J.P. Merlie, and J.R. Sanes. 1996. Defective neuromuscular synaptogenesis in agrin-deficient mutant mice. Cell. 85:525–536. [DOI] [PubMed] [Google Scholar]
- Gee, S.H., F. Montanaro, M.H. Lindenbaum, and S. Carbonetto. 1994. Dystroglycan-α, a dystrophin-associated glycoprotein, is a functional agrin receptor. Cell. 77:675–686. [DOI] [PubMed] [Google Scholar]
- Gesemann, M., A.J. Denzer, and M.A. Ruegg. 1995. Acetylcholine receptor-aggregating activity of agrin isoforms and mapping of the active site. J. Cell Biol. 128:625–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gesemann, M., V. Cavalli, A.J. Denzer, A. Brancaccio, B. Schumacher, and M.A. Ruegg. 1996. Alternative splicing of agrin alters its binding to heparin, dystroglycan, and the putative agrin receptor. Neuron. 16:755–767. [DOI] [PubMed] [Google Scholar]
- Gingras, J., and M. Ferns. 2001. Expression and localization of agrin during sympathetic synapse formation in vitro. J. Neurobiol. 48:228–242. [DOI] [PubMed] [Google Scholar]
- Gingras, J., S. Rassadi, E. Cooper, and M. Ferns. 2002. Agrin plays an organizing role in the formation of sympathetic synapses. J. Cell Biol. 158:1109–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass, D.J., D.C. Bowen, T.N. Stitt, C. Radziejewski, J. Bruno, T.E. Ryan, D.R. Gies, S. Shah, K. Mattson, S.J. Burden, et al. 1996. Agrin acts via a MuSK receptor complex. Cell. 85:513–524. [DOI] [PubMed] [Google Scholar]
- Heathcote, R.D., J.M. Ekman, K.P. Campbell, and E.W. Godfrey. 2000. Dystroglycan overexpression in vivo alters acetylcholine receptor aggregation at the neuromuscular junction. Dev. Biol. 227:595–605. [DOI] [PubMed] [Google Scholar]
- Hilgenberg, L.G.W., K.D. Ho, D. Lee, D. O'Dowd, and M.A. Smith. 2002. Agrin regulates neuronal responses to excitatory neurotransmitters in vitro and in vivo. Mol. Cell. Neurosci. 19:97–110. [DOI] [PubMed] [Google Scholar]
- Hilgenberg, L.G.W., C.L. Hoover, and M.A. Smith. 1999. Evidence of an agrin receptor in cortical neurons. J. Neurosci. 19:7384–7393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoch, W. 1999. Formation of the neuromuscular junction. Agrin and its unusual receptors. Eur. J. Biochem. 265:1–10. [DOI] [PubMed] [Google Scholar]
- Hoch, W., M. Ferns, J.T. Campanelli, Z.W. Hall, and R.H. Scheller. 1993. Developmental regulation of highly active alternatively spliced forms of agrin. Neuron. 11:479–490. [DOI] [PubMed] [Google Scholar]
- Hoch, W., J.T. Campanelli, S. Harrison, and R.H. Scheller. 1994. Structural domains of agrin required for clustering of nicotinic acetylcholine receptors. EMBO J. 13:2814–2821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopf, C., and W. Hoch. 1996. Agrin binding to α-dystroglycan. J. Biol. Chem. 271:5231–5236. [DOI] [PubMed] [Google Scholar]
- Jacobson, C., F. Montanaro, M. Lindenbaum, S. Carbonetto, and M. Ferns. 1998. α-Dystroglycan functions in acetylcholine receptor aggregation but is not a coreceptor for agrin-MuSK signaling. J. Neurosci. 18:6340–6348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson, C., P.D. Cote, S.G. Rossi, R.L. Rotundo, and S. Carbonetto. 2001. The dystroglycan complex is necessary for stabilization of acetylcholine receptor clusters at neuromuscular junctions and formation of the synaptic basement membrane. J. Cell Biol. 152:435–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji, R.-R., C.M. Böse, C. Lesuisse, D. Qiu, J.C. Huang, Q. Zhang, and F. Rupp. 1998. Specific agrin isoforms induce cAMP response element binding protein phosphorylation in hippocampal neurons. J. Neurosci. 18:9695–9702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koulen, P., L.S. Honig, E.L. Fletcher, and S. Kröger. 1999. Expression, distribution and ultrastructural localization of the synapse-organizing molecule agrin in the mature avian retina. Eur. J. Neurosci. 11:4188–4196. [DOI] [PubMed] [Google Scholar]
- Lévi, S., R.M. Grady, M.D. Henry, K.P. Campbell, J.R. Sanes, and A.M. Craig. 2002. Dystroglycan is selectively associated with inhibitory GABAergic synapses but is dispensable for their differentiation. J. Neurosci. 22:4274–4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Z., J.L. Massengill, D.K. O'Dowd, and M.A. Smith. 1997. Agrin gene expression in mouse somatosensory cortical neurons during development in vivo and in cell culture. Neuroscience. 79:191–201. [DOI] [PubMed] [Google Scholar]
- Li, Z., L.G.W. Hilgenberg, D.K. O'Dowd, and M.A. Smith. 1999. Formation of functional synaptic connections between cultured cortical neurons from agrin-deficient mice. J. Neurobiol. 39:547–557. [DOI] [PubMed] [Google Scholar]
- Mann, S., and S. Kröger. 1996. Agrin is synthesized by retinal cells and colocalizes with gephyrin. Mol. Cell. Neurosci. 8:1–13. [DOI] [PubMed] [Google Scholar]
- Mantych, K.B., and A. Ferreira. 2001. Agrin differentially regulates the rates of axonal and dendritic elongation in cultured hippocampal neurons. J. Neurosci. 21:6802–6809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann, F.R., G. Bittcher, M. Annies, B. Shumacher, S. Kröger, and M.A. Ruegg. 2001. An alternative amino-terminus expressed in the central nervous system converts agrin to a type II transmembrane protein. Mol. Cell. Neurosci. 17:208–225. [DOI] [PubMed] [Google Scholar]
- Ruegg, M.A., K.W.K. Tsim, S.E. Horton, S. Kröger, and U.J. McMahan. 1992. The agrin gene codes for a family of basal lamina proteins that differ in function and distribution. Neuron. 8:691–699. [DOI] [PubMed] [Google Scholar]
- Rupp, F., T.H. Özçelik, M. Linial, K. Peterson, U. Francke, and R. Scheller. 1992. Structure and chromosomal localization of the mammalian agrin gene. J. Neurosci. 12:3535–3544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiyama, J., D.C. Bowen, and Z.W. Hall. 1994. Dystroglycan binds nerve and muscle agrin. Neuron. 13:103–115. [DOI] [PubMed] [Google Scholar]
- Tian, M., C. Jacobson, S.H. Gee, K.P. Campbell, S. Carbonetto, and M. Jucker. 1996. Dystroglycan in the cerebellum is a laminin alpha 2-chain binding protein at the glial-vascular interface and is expressed in Purkinje cells. Eur. J. Neurosci. 8:2739–2747. [DOI] [PubMed] [Google Scholar]
- Valenzuela, D.M., T.N. Stitt, P.S. DiStefano, E. Rojas, K. Mattsson, D.L. Compton, L. Nunez, J.S. Park, J.L. Stark, D.R. Gies, et al. 1995. Receptor tyrosine kinase specific for the skeletal muscle lineage: expression in embryonic muscle, at the neuromuscular junction, and after injury. Neuron. 15:573–584. [DOI] [PubMed] [Google Scholar]
- Zaccaria, M.L., F. Di Tommaso, A. Brancaccio, P. Paggi, and T.C. Petrucci. 2001. Dystroglycan distribution in adult mouse brain: a light and electron microscopy study. Neuroscience. 104:311–324. [DOI] [PubMed] [Google Scholar]