

Abstract
Vascular endothelial growth factor (VEGF-A) is a major regulator of blood vessel formation and function. It controls several processes in endothelial cells, such as proliferation, survival, and migration, but it is not known how these are coordinately regulated to result in more complex morphogenetic events, such as tubular sprouting, fusion, and network formation. We show here that VEGF-A controls angiogenic sprouting in the early postnatal retina by guiding filopodial extension from specialized endothelial cells situated at the tips of the vascular sprouts. The tip cells respond to VEGF-A only by guided migration; the proliferative response to VEGF-A occurs in the sprout stalks. These two cellular responses are both mediated by agonistic activity of VEGF-A on VEGF receptor 2. Whereas tip cell migration depends on a gradient of VEGF-A, proliferation is regulated by its concentration. Thus, vessel patterning during retinal angiogenesis depends on the balance between two different qualities of the extracellular VEGF-A distribution, which regulate distinct cellular responses in defined populations of endothelial cells.
Keywords: VEGF; endothelial cell; filopodia; astrocyte; migration; proliferation
Introduction
The development of branched tubular organs like the vascular system, lung, kidney, and many glandular tissues poses several fundamental biological questions. What determines the cellular architecture of tubes and how do new branches arise? What controls the size of a new branch and the direction of its outgrowth? How do branches fuse to form a continuous network?
The most pervasive vertebrate tubular organ, the vasculature, is first assembled from scattered precursor cells that shape blood islands, which fuse to create the first primitive plexus of vessels (Risau and Flamme, 1995). Subsequently, enlargement and remodeling of the plexus, involving sprouting, splitting, and regression of branches, shape hierarchical vascular patterns that allow directional blood flow. These patterns become precisely adapted to organ anatomy and physiology, hence they differ extensively between organs.
Principally, at least two different mechanisms may lead to organ-specific vascular patterns. First, the formation of a primary vascular network may be a random process followed by specific branch regression. This “vascular pruning” represents a major mechanism of vascular remodeling and is likely regulated at the level of endothelial cell survival, which depends on vascular endothelial growth factor (VEGF-A)* and unidentified signals from surrounding vascular smooth muscle cells or pericytes (Benjamin et al., 1999). Second, angiogenic sprouting and fusion may be a guided process, leading to specific primary vascular patterns. Such angiogenic guidance is mainly inferred by the seemingly nonrandom angiogenic sprouting in the developing central nervous system (CNS), for example, in the mammalian retina, where a vascular plexus initially forms superimposed on a preexisting astrocyte plexus (Stone and Dreher, 1987; Fruttiger et al., 1996).
Precision guidance of specialized cells is involved in the formation of other pervasive organ systems. Axonal guidance by attractive and repulsive forces is well established, and also the formation of the insect tracheal system, which is both structurally and functionally analogous to the vertebrate vasculature, relies on guidance of cells and subcellular processes along predefined tracks (for review see Zelzer and Shilo, 2000). Certain molecules with a role in axon and/or tracheal guidance have also been implicated in vascular morphogenesis (for review see Shima and Mailhos, 2000). For angiogenesis, however, the functions of these and other angiogenic modulators remain ill defined.
The concept of precision guidance requires a sensor that relays external signals into specific cell behavior. In axonal guidance, this is provided by a specialized tip structure, the growth cone. Also, the guidance of Drosophila tracheal branches depends on specialized sensor cells situated at the sprouting tips. These tip cells are unique in morphology and gene expression and appear to respond to guidance cues conferring positional information (Samakovlis et al., 1996). Both the growth cone and the tracheal tip cells use dynamic filopodia to sense guidance cues in their surroundings and to migrate (Kater and Rehder, 1995; Ribeiro et al., 2002). There is evidence from several studies that endothelial sprouts can also extend multiple filopodia at their distal tips (Bär and Wolff, 1972; Marin-Padilla, 1985 and earlier literature cited therein), indicating that growing vascular sprouts are endowed with specialized tip structures with potential functions in guidance and migration. These descriptions have received surprisingly little attention, and with few recent exceptions (Dorrell et al., 2002; Ruhrberg et al., 2002) they go unnoticed in today's concepts of vascular development. Importantly, the numerous pro- and antiangiogenic factors discovered during the past 15 yr have not been studied in relation to endothelial tip cells and their filopodia, and in particular, the possibility that endothelial tip cells may respond specifically to such factors has not been explored.
By analyzing mice lacking heparin-binding VEGF-A isoforms, we have recently provided evidence that the spatial distribution of secreted VEGF-A is critical for the balance between capillary branching and growth in vessel size (Ruhrberg et al., 2002). Here, we have used several genetic and pharmacological gain and loss of function approaches to show that different modes of VEGF-A distribution in the extracellular space independently guide tip cell migration and control proliferation in stalk cells. Collectively, our data explain how the pattern of cellular expression and extracellular distribution of a single growth factor shapes vascular patterns during angiogenic sprouting by regulating different events in defined subpopulations of endothelial cells.
Results
We focused our studies of developmental angiogenesis on the early postnatal mouse retina, which develops a stereotypical vascular pattern in a well-defined sequence of events (Fig. 1). Simultaneous vascular sprouting at the periphery and remodeling at the center (observable, for example, at postnatal day [P]5), allows the study of different aspects of vessel formation, maturation, and specialization in a single preparation. Retinas are ideal structures to visualize using whole-mount immunostaining and in-situ hybridization techniques, coupled with high resolution three-dimensional imaging by confocal laser scanning microscopy. We studied retinas from various mice between birth (P0) and P14. During this time, spreading of the inner vascular plexus proceeds from the optic disc to the peripheral margin. From approximately P6, vascular branches also extend from the inner plexus into the retina to form the outer plexuses (Fig. 1, P8, arrows).
Figure 1.
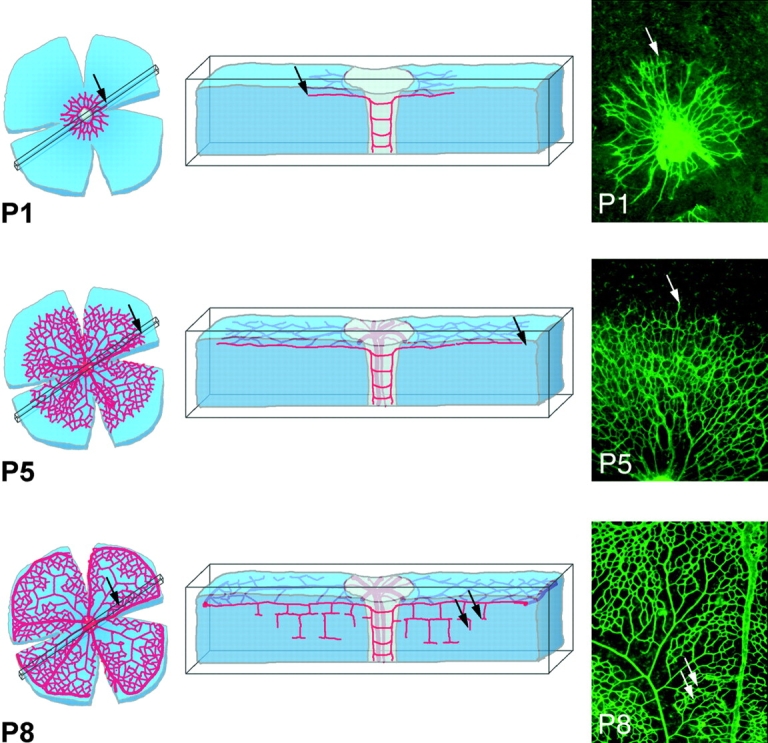
Schematic presentation of retina development as a model system for investigation of angiogenic sprouting in the CNS. Corresponding top view micrographs of whole mount isolectin- labeled specimen are shown to the right. The top view displays the primary plexus in the fiber layer of the retina. Sprouting occurs toward the periphery in the primary plexus (P1 and P5, arrows) and subsequently into deeper layers (P8, arrows), where again branching and fusion leads to plexus formation.
Characterization of the endothelial tip cell
High resolution imaging of isolectin B4–stained retinas revealed that the endothelial cells at the tips of vascular sprouts extended long filopodia (Fig. 2, a–d and f). In the retina, this was most evident at the edge of the expanding inner vascular plexus (Fig. 2, a and b), at sites of sprouting into and within the deeper retinal layers (Fig. 2 c), and at prospective fusion sites in the central, remodeling zone (Fig. 2 d, arrows). Endothelial filopodia were uniform in thickness (∼100 nm) but of variable length, with the longest extending >100 μm. Staining of nuclei in combination with isolectin B4, vascular endothelial (VE) cadherin, and fibronectin (Fig. 2, a and e) revealed that the sprouting tip consisted of a single, highly polarized endothelial cell, hereafter referred to as the tip cell. The endothelial identity of this cell was further confirmed by staining for platelet–endothelial cell adhesion molecule (PECAM)-1, endomucin (unpublished data), and VEGF receptor (VEGFR)2 (see Fig. 5, d–f). Therefore, we consider the tip cell the leader in the phalanx of endothelial cells constituting a vascular sprout. Accordingly, trailing cells are referred to as stalk cells. Double staining for endothelial markers and nuclei revealed that the extension of long filopodia is largely restricted to the tip cells. Staining of the actin cytoskeleton by phalloidin conjugates highlighted the tip cells and in particular their leading edge and filopodia (Fig. 2 f). Tip cells were also distinguished from stalk cells by their strong expression of PDGF-B mRNA (Fig. 2 g) and VEGFR2 mRNA and protein (see Fig. 5, d–f), implying that tip cells have a gene expression profile that is distinct from the stalk cells. PDGF-B has been shown previously to play an essential role in the recruitment of pericytes to new vessels (Lindahl et al., 1997). The VEGFR2 expression in tip cells is further discussed below. Vascular perfusion with labeled dextran demonstrated that the vascular lumen extends up to but not into the tip cells (Fig. 2 h). Together, these characteristics suggest that the tip cells are distinct and functionally specialized microvascular endothelial cells. Their morphology and localization in the sprout is schematically illustrated in Fig. 2 i. Tip cells were not unique to the retina but were present in other parts of the developing mouse CNS harboring active angiogenesis (unpublished data).
Figure 2.
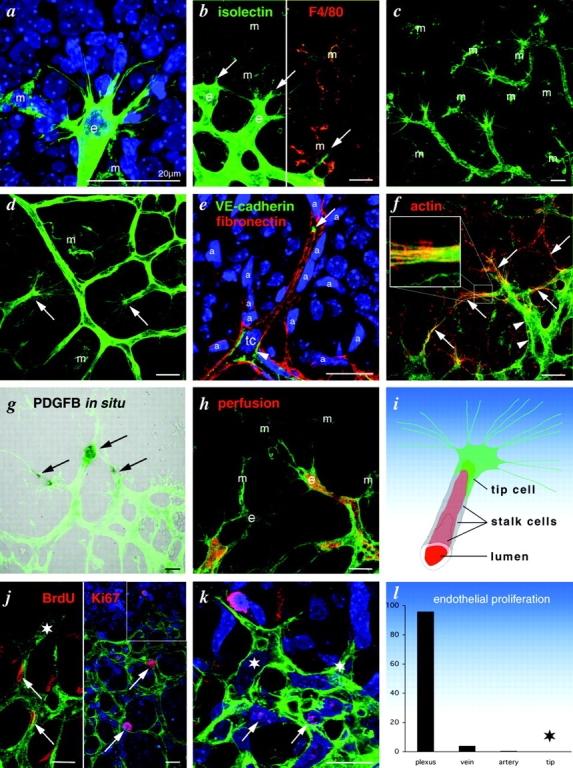
Characterization of the endothelial tip cell. Isolectin staining is shown in green and nuclei staining is shown in blue. e, endothelium; m, macrophages/microglia. (a) High magnification confocal micrograph showing typical filopodia extension at the leading edge of the primary retinal plexus. Note the single endothelial nucleus. (b) Double labeling for isolectin and macrophage marker F4/80 distinguishes between endothelium (e, only green) and macrophages (m, green and red). Arrows point to sprouting tips. (c) Filopodia at sprouting tips in the deeper retinal plexus, and (d) at remodeling sites (arrows) in the primary plexus. (e) High magnification of a sprouting tip triple labeled for VE-cadherin (cell borders and junctions), fibronectin (astrocytes and vessel basement membrane), and nuclei. Note focal filopodia extension at the tip (arrow), whereas following cells extend no filopodia (arrowheads). (f) Filopodia are highly enriched in actin filaments (red and green, arrows and blow up inset), whereas endothelial cells without filopodia show little actin (green, arrowheads). (g) PDGFB in situ hybridization reveals tip cell–specific gene expression (arrows). (h) Dextran perfusion (false colored in red) revealing endothelial lumina. Note lumen-free tip cell to the left. (i) Graphic illustration of tip cell and stalk cell terminology. (j) Overview micrograph visualizing endothelial proliferation in the plexus (arrows) but not in the tip region (box, magnified in k). Note lack of Ki67 labeling or Brdu labeling in tip cells (stars), whereas stalk cells and plexus cells near the tip label weakly (arrows). (l) Distribution of proliferating endothelial cells given as percent of the total. 10 retinas labeled for Ki67 were analyzed. Note lack of tip cell proliferation (star). Bars, 20 μm.
Figure 5.

Illustration of tip cell guidance toward VEGF sources and of VEGFR expression on endothelial filopodia. (a–c) Confocal laser scanning micrographs of VEGF-A in situ hybridization (black signal) combined with double labeling for isolectin and GFAP. (a) Overview illustrating strong VEGF-A expression ahead of the vascular plexus and very low behind the leading edge. (b) Higher magnification showing astrocytic VEGF-A expression (black and red overlap) and strong down-regulation in astrocytes covered by the primary plexus. (c) Tip cell filopodia orientate toward and along VEGF-A–expressing astrocytes. (d) VEGFR2 in situ hybridization identifies strongest expression in the tip cells (arrows). (e and f) VEGFR2 is prominent on tip cell filopodia (arrowheads). (e) Rat mAb labeling using tyramide enhancer kit on cryosection, and (f) goat polyclonal antibody on whole mount labeling together with isolectin.
To determine whether vascular sprouting in the CNS involved expansion of the endothelial population from the tip or the stalk, we performed double labeling of BrdU or the proliferation marker Ki-67, and isolectin B4. We did not observe proliferating tip cells in either 10 P5–P7 retinas labeled with Ki-67 or an additional 10 labeled with BrdU (Fig. 2, j–l). In contrast, proliferation was abundant in stalk cells and in the immature capillary plexus but occurred also in veins and to a lesser extent in arteries (Fig. 2 l; unpublished data). This suggests that tip cells are nonproliferative; proliferation in the spreading plexus occurs in the sprout stalks and further back in the remodeling plexus.
The abundance of filopodia on tip cells is indicative of an active migratory phenotype. To study more directly the dynamics of tip cell behavior in real time, we used an organ culture model of vessel sprouting adapted from the rat aortic ring model (Nicosia and Ottinetti, 1990) in which all external stimuli such as TPA, VEGF, or bFGF were omitted. These modifications resulted in a system that is entirely self driven, characterized by rapid growth of sprouts, which form lumens and are enveloped by mural cells (unpublished data). The tips of the sprouts were composed of highly migratory cells with numerous filopodia- and lamellipodia-like processes (Fig. 3 a and see Fig. 6 i). Time-lapse recordings revealed that protrusion and retraction of lamellipodia from these tip cells was a highly dynamic process with single endothelial cells being retained at the tip of the sprout (Fig. 3 a). Endothelial proliferation was conspicuous in stalk cells (Fig. 3 c); however, we did not observe tip cell mitosis using Ki-67 or phospho-histone staining. Thus, with respect to the functional polarization in the sprout, angiogenesis in the aortic ring assay mimics retinal angiogenesis.
Figure 3.
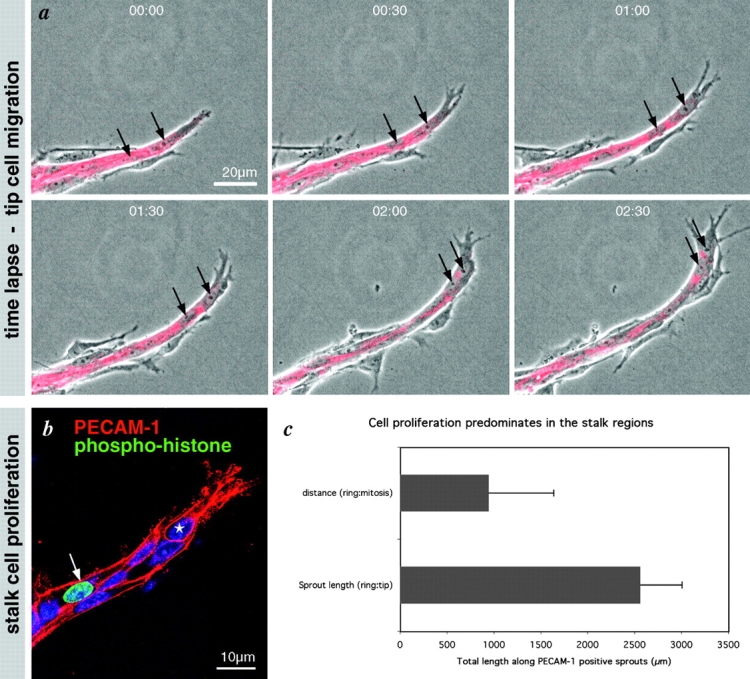
Tip cell migration and stalk cell proliferation in the aortic ring sprouting model. (a) Selected sequence from a time-lapse movie focusing on a single sprout prelabeled for PECAM-1 (red). Note lamellipodia protrusions and continued migration of the leading cell (arrows). (b) Representative image of a single sprout labeled for PECAM-1 (red) and mitosis (phospho-histone, green) and nuclei (Blue). Note that proliferation is confined to the stalk; tip cells do not proliferate (see star and panel c). Total sprout length from the ring margin to the tip of each sprout was measured in a single ring quadrant from each of the three time points (days 5, 7, and 9). The distance from the ring to mitotic endothelial cells along these same sprouts was also measured. The histograms represent the mean of the total length of PECAM-positive sprouts over the 3 d versus the mean length distance to endothelial cell mitosis.
Figure 6.
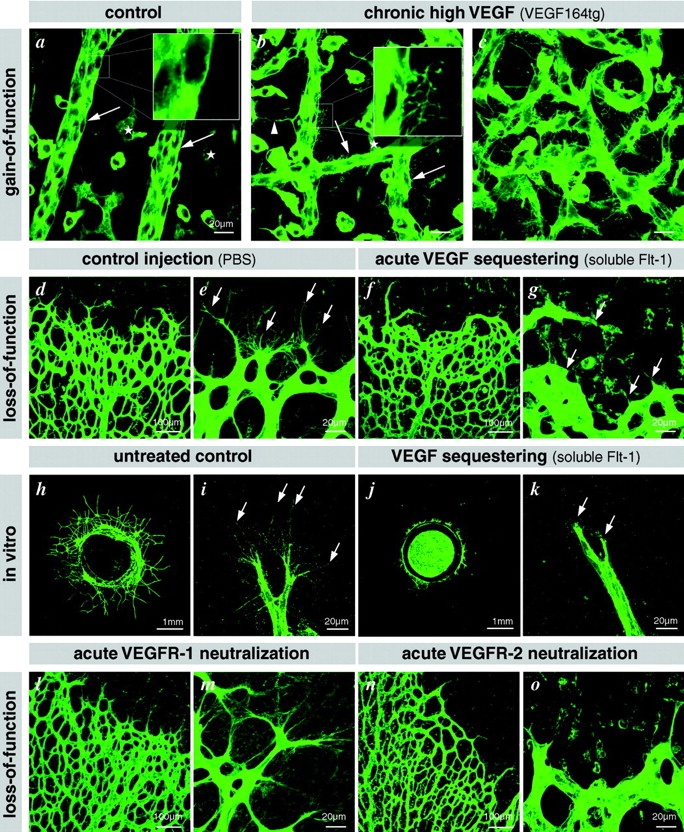
Illustration of filopodia induction in hyaloid vessels of VEGF164tg. (a) Wild-type littermate showing normal smooth surface of the hyaloid vessels (arrows) lying on the inner surface of the retina. Filopodia are only present in the intraretinal vascular plexus (asterisks). (b and c) Hyaloid vessels in VEGF164tg are studded with filopodia (arrows and inset). Bundles of filopodia are involved in sprouting and fusion (arrowhead), leading to an aberrant hyperfused vascular structure. Bars, 20 μm. VEGF is necessary for filopodia extension: (d–g) acute sequestering of VEGF by intraocular injection of soluble Flt-1–IgG chimeric protein leads to filopodia retraction already after 6 h (earliest time point investigated). (h–k) VEGF sequestering inhibits tip cell filopodia in the aortic ring assay. (l–o) Acute neutralization of VEGFR2 (n and o) but not VEGFR1 (l and m) leads to retraction of tip cell filopodia.
Tip cell filopodia extend on VEGF-producing astrocytes
The inner retinal vascular plexus develops in close association with a preexisting layer of astrocytes (Stone and Dreher, 1987; Fruttiger, 2002). We confirmed that the retinal vascular plexus initially forms super imposed on the astrocyte network that remodels after contact with the vasculature (Fig. 4 a). These two plexuses subsequently dissociate in the remodeling zone, indicating a transient importance of their close interaction in the peripheral sprouting region. All tip cells were closely attached to astrocytes and stretched most of their filopodia along the astrocyte cell bodies and processes (Fig. 4 b, filled arrowheads). Filopodia without apparent astrocyte contact were consistently shorter and undulating (Fig. 4 b, open arrowheads).
Figure 4.
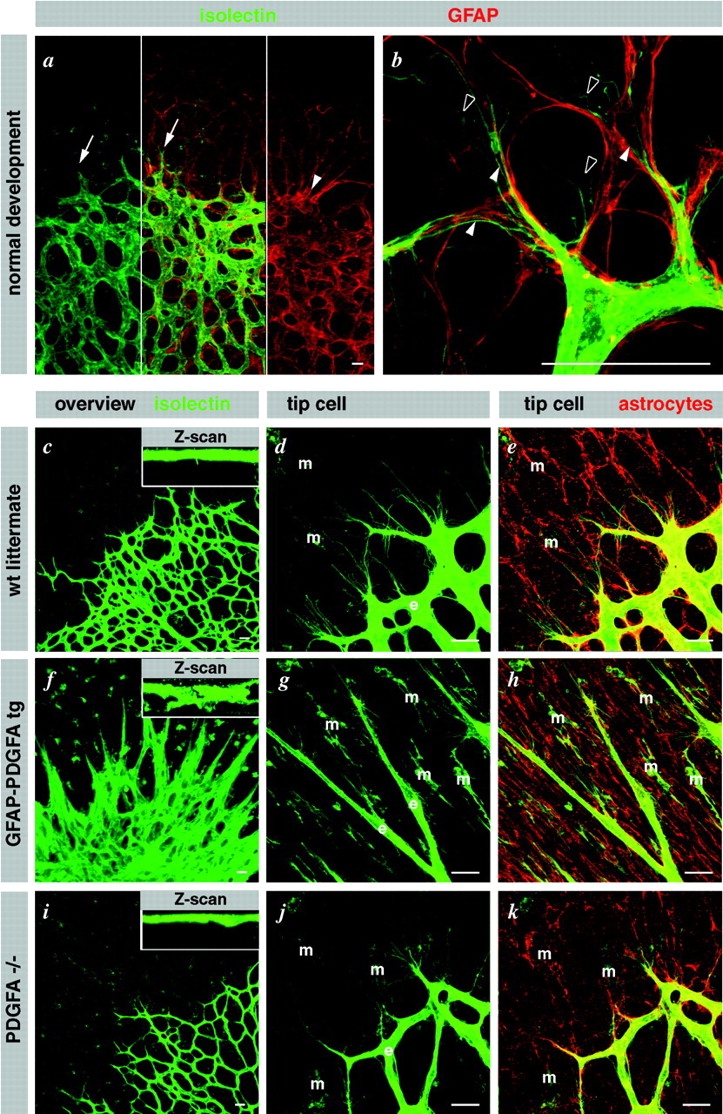
Astrocytes guide endothelial tip cell filopodia. (a) Over view micrograph, displaying overlap of vascular and astrocytic network. Note that the leading edge of the vascular plexus (arrows) is clearly visible in the astrocytic pattern (arrowhead). (b) High magnification illustrating alignment of tip cell filopodia with astrocytic processes (filled arrowheads). Note undulating appearance of filopodia that lack astrocytic contact (open arrowheads). (c–k) Illustration of the dominant effect of astrocytes on the retinal vascular plexus. Vessel density is regulated by astrocyte density (c). Wild-type overview displaying a single layer of vessels in alignment with the astrocytic network (d and e). (f) Supernumerous astrocytes in GFAP-PDGFA transgenics lead to multilayered blood vessels (see also z scan inset). Tip cells truthfully follow the aberrantly dense astrocytic cords (g and h). (i–k) PDGFA deficiency leads to a sparse vascular network as a result of a sparse astrocytic network. m, macrophages; e, endothelial cells. Bars, 20 μm.
These observations suggest that an astrocyte scaffold guides the extension of tip cell filopodia. Previous studies have implied that astrocytes are dominant in retinal vessel pattern formation (Fruttiger et al., 1996). To analyze if changes in the architecture of the astrocyte plexus were dominant also with regard to the guidance of tip cell filopodia, we studied transgenic mice in which the growth factor PDGF-A was expressed under the astrocyte-specific glial fibrillary acidic protein (GFAP) promoter (Fruttiger et al., 2000). Since astrocytes strongly express the PDGF-A receptor (PDGFRα) (Fruttiger, 2002), autocrine mitogenic stimulation results in super-numerous astrocytes intertwining into a dense network of radially oriented bundles (Fig. 4 h). The endothelial tip cells and their filopodia oriented along these abnormal astrocyte bundles (Fig. 4, g and h), leading to the formation of a super imposed multilayered vascular network (Fig. 4 f). PDGF-A knockout mice developed a sparser network of astrocytes and vessels, however, with retained association between tip cell filopodia and the astrocyte network (Fig. 4, i–k). Together, the effects of astrocyte hyper- or hypoplasia on retinal vascular patterning suggest that astrocytes provide the principal cues for guidance of endothelial tip cells and their filopodia.
Previous studies have shown that astrocytes express VEGF-A in CNS angiogenesis during developmental and pathological processes (Pierce et al., 1995; Stone et al., 1995; Provis et al., 1997). To map VEGF-A expression to specific cell types, we performed VEGF-A in situ hybridization in combination with isolectin/GFAP double staining. VEGF-A mRNA expression only occurred in GFAP-positive retinal astrocytes (Fig. 5, a–c), with the strongest signals located in astrocytes at the leading edge and immediately ahead of the plexus. VEGF-A mRNA was also detected in astrocytes further back in the plexus surrounding veins and capillaries but not surrounding arteries (unpublished data).
VEGF-A stimulates tip cell filopodia
Since tip cell filopodia extended along VEGF-A mRNA-positive astrocytes, we asked whether ectopic VEGF-A could induce filopodial extension from endothelial cells. Support for this idea came from examination of the hyaloid vasculature in transgenic mice overexpressing VEGF from the lens-specific αA-crystallin promoter. (The generation and basic characterization of these mice will be reported elsewhere [unpublished data].) Hyaloid arteries extend from the optic disc and ramify around the lens supporting its early development, and ultimately these regress postnatally. The hyaloid arteries are normally straight vessels with smooth abluminal surfaces and few branch and fusion points; however, those in αA-crystallin–VEGF164 transgenics were densely covered with abluminal filopodia. These hyaloid vessels were also aberrantly fused into chaotic vascular networks (Fig. 6, a–c).These observations indicate that VEGF overexpression is sufficient to induce continuing filopodial extension, vascular sprouting, and hyperfusion in vessels in which these processes normally have ceased. However, these studies do not discriminate between the possibilities that filopodial extension is directly and dynamically regulated by VEGF or whether it is inherent to a tip cell phenotype induced by VEGF. To address these alternatives, we acutely deprived the growing retinal vasculature of VEGF-A by injecting a soluble VEGFR1 extracellular domain–Fc fusion protein (soluble Flt) into the eyes of P5 mice. Soluble Flt has a high affinity for VEGF-A and acts as a potent extracellular VEGF-A trap. 6 h after injection, most tip cell filopodia were completely retracted in the sprouting region (Fig. 6, d–g). To assess the dynamics of VEGF-A–regulated tip cell process extensions and migration, we employed the aortic ring assay. Filopodial retraction was widespread after addition of soluble Flt but not after treatment with other Fc fusion proteins (Fig. 6, h–k). Time-lapse videomicroscopy also showed that the protrusive activity of tip cell lamellipodia was impaired in conjunction with a block in tip cell migration and sprout elongation (Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200302047/DC1).
VEGF stimulates tip cell filopodia via VEGFR2
VEGF-A binds to and signals through two receptors, VEGFR1 (Flt-1) and -2 (Flk-1) (Ferrara, 1999), which are coexpressed in angiogenic endothelium. In the retina, VEGFR2 in situ hybridization highlighted tip cells, but there was also significant signal in the stalk (Fig. 5 d). However, VEGFR1 mRNA expression was similar in both tip and stalk cells (Fruttiger, 2002). VEGFR2 protein staining was prominent on tip cell filopodia (Fig. 5, e and f).
The intensity of VEGFR2 immunostaining on the filopodia suggested that this receptor is implicated in VEGF-mediated filopodia protrusion. To test this hypothesis, neutralizing VEGFR2-specific antibodies were injected into the eyes of P5 mice, and the retinas were removed for examination 6 h later. The tip cell filopodia were retracted, and plexus spreading was inhibited (Fig. 6, n and o), suggesting that VEGFR2-mediated signaling is necessary for tip cell filopodial extension. A neutralizing VEGFR1 antibody had no effect on tip cell morphology or their filopodia (Fig. 6, l and m). We conclude that the extension and maintenance of tip cell filopodia depends on VEGF-A signaling via VEGFR2.
Spatially restricted VEGF guides tip cell filopodia
We next asked if VEGF is directly involved in the guidance of filopodia along the astrocyte scaffold. Considering their length and display of VEGFR2 protein, the tip cell filopodia may be capable of sensing VEGF-A at considerable distance from the cell soma. However, it is also possible that VEGF-A stimulates random protrusion of filopodia and that other factors (e.g., extracellular matrix) subsequently guide the filopodia. Direct guidance by VEGF-A would require the existence of precisely shaped extracellular gradients or deposits of VEGF-A protein, which could be sensed by the filopodia. We showed recently that the heparin-binding isoforms VEGF164 and 188 are required for the establishment of steep extracellular VEGF gradients in the mouse embryonic hindbrain (Ruhrberg et al., 2002). Therefore, we asked if heparin-binding VEGF-A isoforms had a similar role in the retina. RT-PCR indicated that VEGF164 is the dominant isoform expressed in the developing retina followed by 120, 144, and 188 (Fig. 7 a). Immunolabeling showed that extracellular VEGF-A is distributed mainly along the astrocyte tracks in developing wild-type retinas (Fig. 7 b). In contrast, mouse mutants expressing only VEGF120 (120/120 mice) lacked a distinctive astrocytic association of extracellular VEGF; instead VEGF120 is distributed more diffusely in the retina (Fig. 7 c). This finding is consistent with the shallow gradient of VEGF protein around the hindbrain midline demonstrated previously in 120/120 mice (Ruhrberg et al., 2002).
Figure 7.
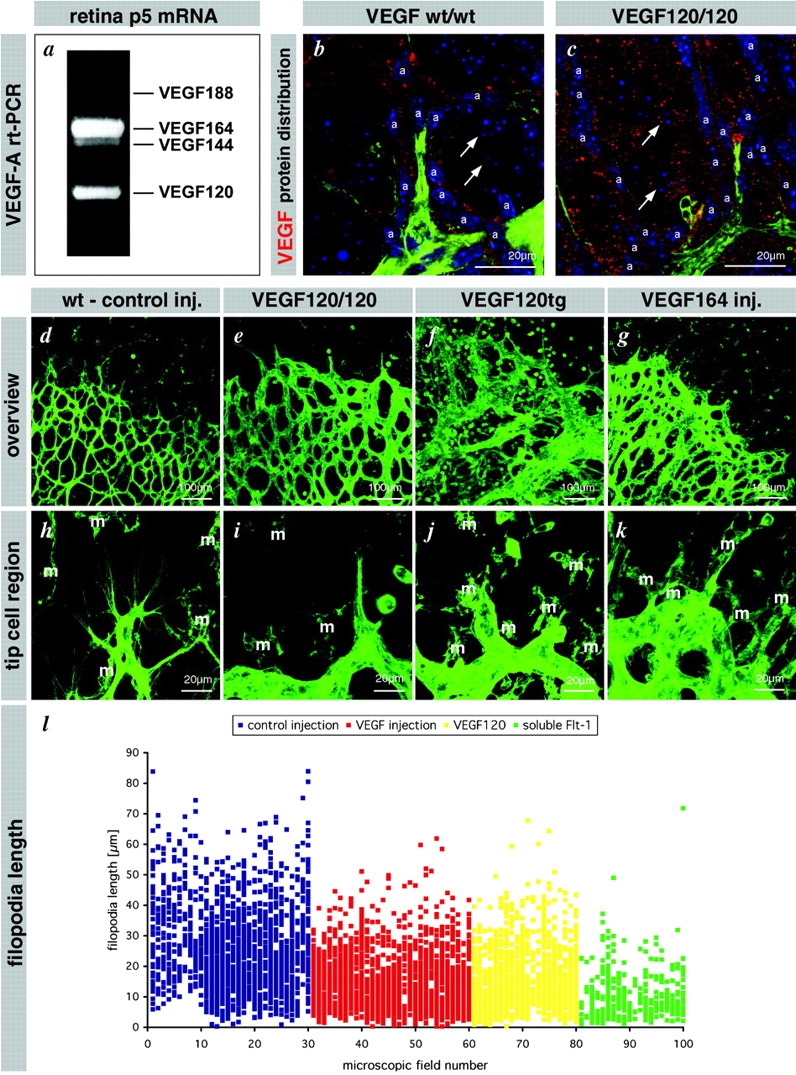
VEGF gradients shaped by heparin-binding isoforms are necessary for directed tip cell filopodia extension. (a) RT-PCR analysis detects four VEGF isoforms in the retina at P5. The predominant form is VEGF 164 that binds to heparan sulfate proteoglycan. (b) VEGF protein is located in the vicinity of astrocytes in wild-type retinas (a). (c) VEGF 120 protein distributes further away from the astrocytes and can be found around ganglion cells (arrows). (d–l) Flattening of the VEGF gradient leads to filopodia shortening, misorientation (see also Fig. 8) and reduced migration (see also Fig. 9, j and k). (d and h) Control injection overview and tip cell filopodia detail. (e and i) VEGF120/120 retinas display reduced filopodia length and sprouting but increased vessel diameter. Filopodia also extend in aberrant directions (see also Fig. 9). (f and j) VEGF120 transgenes expressing VEGF120 from the lens-specific crystallin promoter. Note massive enlargement of vessel diameter and chaotic sprouting and fusion but little sprouting toward the periphery. Filopodia length is also reduced (not quantified). (g and k) Intraocular injection of VEGF leads to a similar situation as seen in the VEGF120/120 mice. Reduced sprouting and filopodia length but ongoing proliferation (data not depicted) leads to enlarged vessel diameter. Note that in all situations the flattening of VEGF gradients leads to inhibition of filopodia extension and migration. Additional misorientation is documented in Fig. 9. (l) Quantification of filopodia length in the control situation, two situations of flattened VEGF gradient, however with difference in overall concentration, and one situation with acute VEGF sequestering. Each dot represents one filopodium. Data are collected from at least five different retinas for each group.
Normally, the tip cell filopodia extend directionally in a narrow plane defined by the astrocytic network (Fig. 7, d and h). Such filopodia also exist in VEGF120/120 mice, but they were fewer in number and shorter than in the wild-type retinas (Fig. 7, e, i, and l). In addition, VEGF120/120 mutant tip cells extended filopodia also in several directions, i.e., upward (toward the inner limiting membrane (ILM), and backward (peripheral to central) (Fig. 8, a and b). Excessive filopodia extending from stalk cells were also seen. Filopodia that were not oriented centroperipherally and parallel to the astrocytic network were generally undulating and lacked clear spatial orientation (Fig. 8 b). In conclusion, the wide extracellular distribution of VEGF-A in 120/120 retinas correlated with loss of tip cell filopodial polarity.
Figure 8.

Disturbance of VEGF gradients leads to misguidance of tip cell filopodia. Representative illustrations of the tip cell in wild-type (a), VEGF120/120 (b), and VEGF120tg (c) retinas. Left hand panels (a1–c1) show confocal images focused around the ILM, middle panels (a2–c2) show extended depth of focus ranging from the ILM to the fiber layer, and right hand panels (a3–c3) show schematic z sections. Black bars on the right indicate the level and depth of focus in panels a1–c1 and a2–c2. (a) Wild-type vessels extend filopodia strictly in the plane of the astrocytes, thus giving only a faint shadow in the level of the ILM (a1, arrow). Extended depth of focus reveals smooth surface of the stalk cells (a2, arrowheads). Orientation and steepness of wild-type VEGF gradient is indicated by a white arrowhead in a3. (b) VEGF120/120 tip cells show a characteristic loss of polarization leading to ectopic filopodia (blow up in inset) that leave the astrocytic plane (b1, arrows) and protrude also from the stalk cells (b2, arrowheads). This is likely to be caused by a flattened VEGF gradient (b3, arrowhead). (c) VEGF120tg tip cells display the most drastic loss of polarization once they leave the retinal tissue proper (c1). Note the sprout is extending in the plane of the ILM. Filopodia contacting the ILM possess characteristic swellings at the ends (inset). Sprouts within the retina also display loss of polarization characterized by ectopic filopodia (c1 and c2, arrowheads). Vertical arrowhead in c3 indicates ectopic VEGF 120 expression from the lens. Bar, 20 μm.
In addition to a change in distribution of extracellular VEGF-A, the remaining VEGF-A protein in VEGF120/120 mice lacks VEGF164 and 188–specific COOH-terminal sequences. This may affect signaling in ways that are critical for guidance. For example, VEGF164 (but not 120) binds to the axonal guidance receptor neuropilin-1, which enhances signaling via VEGFR2 in endothelial cells (Soker et al., 1998). In an attempt to distinguish between the possibilities of isoform-specific signaling and extracellular VEGF gradients as alternative mechanisms of filopodial guidance, we compared the VEGF120/120 retinas with retinas from transgenic mice that overexpress specific VEGF isoforms and from mice injected intraocularly with VEGF164. When overexpressed from the lens-specific αA-crystallin promoter, each of the VEGF120, 164, or 188 isoforms led to abnormal filopodial guidance, including ectopic filopodia and a shortening of the remaining astrocyte-associated filopodia, similar to the situation in VEGF120/120 mice (Fig. 7, f and j; unpublished data). Direct injection of VEGF164 had a similar effect (Fig. 7, g, k, and l). The most severe misguidance, which also resulted in entire tip cells extending across the ILM and into the vitreous, was seen in αA-crystallin–VEGF120 mice (Fig. 8 c), probably reflecting chronic exposure to high concentrations of VEGF (unpublished data).
In summary, aberrantly oriented filopodia were seen in all situations in which disruption of a normal extracellular VEGF gradient occurred. However, aberrant filopodia orientation was not associated with the presence or absence of specific VEGF isoforms. Thus, a properly shaped extracellular pattern of VEGF-A distribution, rather than specific VEGF-A isoforms or concentrations, is necessary for the correct guidance of tip cell filopodia.
Tip cell migration depends on the distribution, whereas stalk cell proliferation depends on the concentration of VEGFR2 agonistic activity
In addition to the sensor role, filopodia are known to exert a motor function. Abnormal filopodial guidance would then correlate with altered or decreased tip cell migration. As a measure of directed tip cell migration, we assessed the peripheral spreading of the inner retinal plexus in the various situations of altered VEGF distribution. We observed slower spreading in 120/120 and αA-crystallin–VEGF mice and in response to direct intraocular injections of VEGF-A (unpublished data; see also Fig. 9, j and k).
Figure 9.
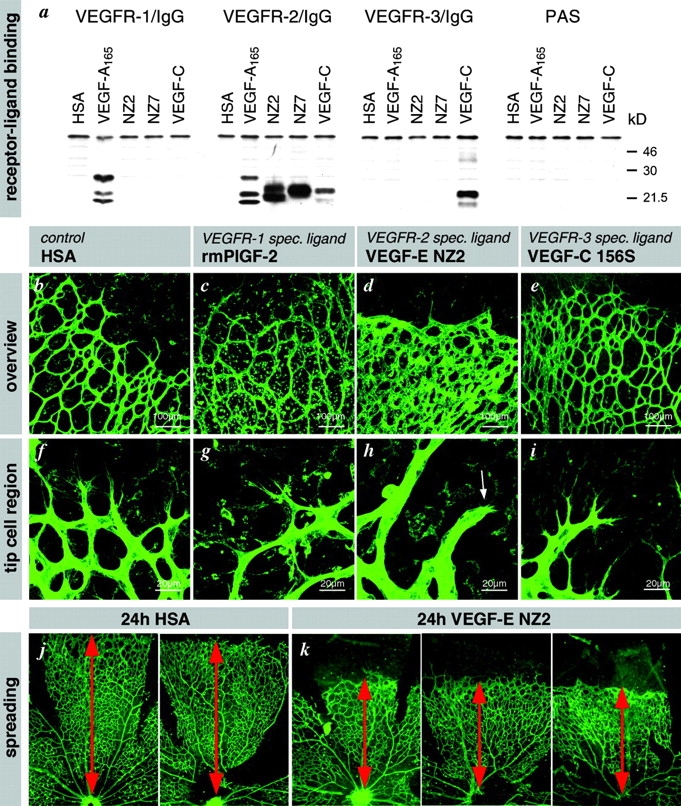
Receptor specificity. (a) Binding of VEGF-E to VEGFR–IgG fusion proteins. VEGFR–IgG fusion proteins were incubated with purified histidine-tagged growth factors and precipitated with protein A sepharose. After reducing SDS-PAGE and Western blotting, receptor-bound growth factors were detected with pentahistidine antibodies. Both forms of VEGF-E (NZ2 and NZ7) show significant interaction only with VEGFR-2, whereas VEGF-A interacts additionally with VEGFR-1 and VEGF-C additionally with VEGFR-3. (b–i) Intraocular injection of VEGFR–specific ligands at 1 μg/μl concentrations. Retinas were fixed after 24 h. (b) Human serum albumin injection has no effect on patterning or filopodia (f). (c) VEGFR1-specific ligand leads to macrophage recruitment (round cells) but has no effect on patterning or tip cell filopodia (g). (d) VEGFR2-specific ligand mimics VEGF-A injection and VEGF120/120 retinas (compare with Fig. 7, e and g). Note the lack of sprouting but increased vessel diameter and density. Only few very short filopodia are present (h, arrow). (e) VEGFR-3–specific ligand VEGF-C156S had no effect on patterning or tip cell filopodia (i). (j and k) Spreading of the vascular plexus over the retina is a measure of migration. Under all conditions of disturbed VEGF gradients, the spreading of the vasculature toward the periphery is significantly reduced. Examples are shown for VEGF-E injection. Overview pictures were taken on P6. Spreading distance as measured from the optic disc is indicated by red doubled arrow.
The observation that a disturbed VEGF-A gradient inhibits both filopodial extension and peripheral endothelial spreading in the retina suggests that directed tip cell migration depends on the ability of the tip cell to distinguish receptor signaling arising at the tips of filopodia from signals arising at the cell soma. To address the VEGFR dependence of this ability, we used a panel of VEGFR-specific ligands. Placenta growth factor (PlGF) and VEGF-B are selective ligands for VEGFR1 (Eriksson and Alitalo, 1999). No endogenous mammalian VEGF is selective for VEGFR2; however, the orf virus–encoded VEGF-E protein shows selective high affinity binding to VEGFR2 (Ogawa et al., 1998; Wise et al., 1999). We produced recombinant VEGF-E protein and characterized its receptor-binding properties. This protein bound and activated the VEGFR2 protein selectively (Fig. 9 a). VEGF-E injection into the eye led to dramatic inhibition of peripheral plexus spreading (Fig. 9, j and k) and to associated shortening of tip cell filopodia (Fig. 9, d and h). It also led to protrusion of short, ectopic filopodia from stalk cells (unpublished data). In contrast, injections of PlGF had no significant effect on spreading or filopodial extension, but it apparently had other biological effects, resulting in massive influx of macrophages (Fig. 9, c and g). A third VEGFR, VEGFR3, is mainly expressed in lymphatic endothelium in late embryogenesis and postnatal life but also plays a role in early blood vessel development (Dumont et al., 1998). After intraocular injections of the VEGFR3-selective ligand VEGF-C156S (Joukov et al., 1998), there was a lack of detectable effects on tip cell filopodial extensions in the developing retina (Fig. 9, e and i). Based on the selective and specific retinal responses to VEGF-E, we conclude that retinal tip cell migration depends on an intact gradient of VEGFR2 agonistic activity.
Although ectopic VEGFR2 agonists inhibited peripheral spreading, they increased proliferation widely in the retinal vascular plexus. This was observed in αA-crystallin–VEGF transgenics (Fig. 7 f) and after injection of VEGF-A (Fig. 7 g; unpublished data) or VEGF-E (Fig. 9 d), where the number of endothelial cells per vessel length, the size of the vessels, and the plexus density were increased. Together, these observations demonstrate that in the developing retina tip cell migration and stalk cell proliferation are independently controlled phenomena that depend on VEGF-A stimulation of VEGFR2. However, whereas tip cell migration depends on the extracellular VEGF-A distribution pattern (this study), stalk cell proliferation appears to depend on the actual VEGF-A concentration (this study; unpublished data).
Discussion
We have identified a mechanism by which retinal angiogenesis is guided. In response to VEGF-A, specialized endothelial cells situated at the tips of retinal vascular sprouts extend long filopodia. Extracellular VEGF-A gradients guide these filopodia, leading to directed migration of tip cells. These effects are mediated via VEGFR2. VEGF-A reaching cells situated in the vascular stalks also stimulates their proliferation through the same receptor. The activation of VEGFR2 is therefore interpreted differently by tip and stalk cells. The reason for this is currently unclear, and insight would require additional information about the molecular differences between the two cell types. Nevertheless, our study highlights the crucial importance of a balance between the tip and stalk cell responses to VEGF-A. The coordination between the two responses demands for a meticulous control of extracellular VEGF-A distribution. This control utilizes cell type (astrocyte), environmental (oxygen), and transcriptional (splicing) dependent mechanisms. An accurate balance between migration of the tip cell and proliferation of the stalk cells is only achieved when the correct relationship between VEGF-A gradient and concentration occurs as a consequence of these combined control mechanisms. This provides an elegant illustration of how a single growth factor may exert intricate control of a complex morphogenetic process. Together with our recent demonstration that heparin-binding VEGF isoforms are required to shape extracellular VEGF gradients and primary vascular patterns (Ruhrberg et al., 2002), the present data provides initial evidence for the development of a mechanistic understanding of sprouting angiogenesis. This concept may also extend to pathological angiogenesis in which the formation of chaotic and dysfunctional networks (for example, in growing tumors and VEGF gene/protein therapy) results from uncontrolled VEGF expression (Carmeliet, 2000; Carmeliet and Jain, 2000).
The tip cell filopodia
Filopodia fulfill at least three different functions in a variety of cell types: intercellular communication, cell migration, and cell adhesion (Wood and Martin, 2002). Specialized filopodia may solely be devoted to cell–cell communication, such as in the Drosophila wing imaginal disc where long, thin filopodial extensions (cytonemes) provide sites of contact and signaling between the disc anterior/posterior center and outlying cells (Ramirez-Weber and Kornberg, 2000). Neuronal growth cones use filopodia to probe their environment for guidance cues but also to migrate (Kater and Rehder, 1995; Tessier-Lavigne and Goodman, 1996). Filopodia and lamellipodia are regularly seen at the leading edge of migrating cells in culture, such as fibroblasts and keratinocytes, from which they reach out and adhere to the substrate, allowing the cell to pull itself forward.
In endothelial cells, filopodia may serve several of these functions. Our data indicate that in addition to tip cell filopodia sensing the VEGF gradient generated by retinal astrocytes, it is also probable that tip cells use the filopodia to attach to the astrocytes or astrocyte-derived matrix and to migrate. It is tempting to speculate that the latter may involve transient fibronectin deposition on astrocytes, which we have observed together with fibronectin-type integrin receptors localized at the tips of the endothelial filopodia (unpublished data)
The pleiotrophic actions of VEGF-A
Genetic ablation experiments of VEGF-A and VEGFR2 in mice (Shalaby et al., 1995; Carmeliet et al., 1996; Ferrara et al., 1996) revealed essential functions in the specification, differentiation, and assembly of angioblasts into primary vascular plexuses. VEGF also induces angiogenic sprouting (for review see Ferrara, 2000), vascular permeability (Senger et al., 1983), and recently proposed, the patterning of arteries (Lawson et al., 2002; Mukouyama et al., 2002; Stalmans et al., 2002). Thus, VEGF regulates several different processes critical for blood vessel formation and function; however, it is not clear how different cellular effects, like proliferation, migration, and survival, translate into higher order morphogenetic phenomena, such as vascular assembly and sprouting.
Our present results confirm that VEGF-A independently controls endothelial migration at the tip and proliferation in the stalk of the angiogenic sprout. Remarkably, these processes are functionally independent in that they affect distinct subpopulations of endothelial cells, and separable, since stalk cell proliferation can proceed in the absence of tip cell migration (this study), and vice versa (unpublished data). This is consistent with earlier studies, demonstrating separate migratory and proliferative activities during the process of angiogenesis (Ausprunk and Folkman, 1977; Sholley et al., 1984). As both functions are mediated by VEGFR2, the signals must be interpreted differently by the two endothelial subtypes. Moreover, the pattern of extracellular distribution of VEGF-A is interpreted differently; an uneven distribution in the form of steep soluble gradients or focal matrix depositions is a prerequisite for directional tip cell migration and filopodial extension. Proliferation apparently reflects the local concentration of VEGFR2 agonist.
Can the retinal angiogenic guidance paradigm be generalized?
Clearly, the tip-stalk paradigm is not restricted to the retina; tip cells with the described characteristics are found throughout the developing CNS (Ruhrberg et al., 2002; unpublished data) during angiogenesis in the aortic ring assay (this study) and in certain tumor models (unpublished data). The abnormal vessel profiles resulting from the manipulations of retinal VEGF gradients and levels are reminiscent of tumor angiogenesis and diabetic microangiopathy. This implicates abnormalities in the spatial regulation of VEGF/VEGFR signaling with the abnormal vessel phenotypes seen in these pathologies. We expect that our suggested model for VEGF-A–driven developmental angiogenesis will provide a helpful conceptual framework for the design and interpretation of future studies of pathological angiogenesis.
Materials and methods
Animals
Descriptive analysis and intraocular injections were performed on wild-type mice of various strains in order to rule out strain-dependent differences. The numbers of transgenic mice were as follow: VEGF120/120 (8), 120/wt (22); GFAP-PDGFAtg (15), littermate controls (15); PDGF-A −/− (3), +/− (6), +/+ (2); αA–crystallin VEGF120tg (12), littermate controls (14); αA–crystallin VEGF164tg (10), littermate controls (7); and αA–crystallin VEGF188tg (10), littermate controls (8).
Whole mount immunohistochemistry and in situ hybridization
Eyes were fixed in 4% PFA in PBS at 4°C overnight and washed in PBS. Retinas were dissected, permeabilized in PBS, 1% BSA, and 0.5% Triton X-100 at 4°C overnight, rinsed in PBS, washed twice in PBlec (PBS, pH 6.8, 1% Triton-X100, 0.1 mM CaCl2, 0.1 mM MgCl2, 0.1 mM MnCl2), and incubated in biotinylated isolectin B4 (Bandeiraea simplicifolia; L-2140; Sigma-Aldrich) 20 μg/ml in PBlec at 4°C overnight. After five washes in PBS, samples were incubated with streptavidin conjugates (Alexa 488, 568, or 633; Molecular Probes) diluted 1:100 in PBS, 0.5% BSA, and 0.25% Triton X-100 at 4°C for 6 h. TO-PRO 3 (1:1,000; Molecular Probes) served for nuclear staining. After washing and a brief postfixation in PFA, the retinas were either flat mounted using Mowiol/DABCO (Sigma-Aldrich) or processed for multiple labeling. The following antibodies were used: GFAP (1:75; Z 0334; Dako), VEGFR2 (1:50; 555307; BD PharMingen), VEGFR2 (1:50; AF644; R&D Systems), F4/80 (1:100; MCAP497; Serotec), fibronectin (1:200; A 0245; Dako), VE-cadherin (1:1, culture supernatant provided by Dietmar Vestweber, University of Muenster, Muenster, Germany), and Ki67 (1:200; NCL-Ki67p; Novo Castra). Alexa-488, 568, or 633 conjugated secondary antibodies (Molecular Probes). Rhodamine-phalloidin served for actin staining (1:40; Molecular Probes). VEGFR2 signal was amplified using the TSATM Fluorescein System (NEL701; NEN) according to instructions. Flat mounted retinas were analyzed by fluorescence microscopy using a Nikon E1000 microscope equipped with a digital camera (Nikon Coolpix 990) and by confocal laser scanning microscopy using a Leica LCS NT. Images were processed using Adobe Photoshop®.
For visualization of vascular lumina, FITC-conjugated Dextran (FD-2000S; Sigma-Aldrich) was warmed to 37°C and perfused through the heart of deeply anaesthetized mice (Avertin, intraperitoneally 10 μl/g body weight).
Mouse VEGF-A, PDGF-B, VEGFR1, and VEGFR2 cDNA fragments were used for whole mount in situ hybridization as described (Fruttiger, 2002). For double labeling, immunolabeling was performed after a 10-min postfixation in 4% PFA.
BrdU labeling was achieved by a 2-h BrdU pulse before fixation (100 μg Brd U/g body weight, intraperitoneally). For double labeling, isolectin labeling was followed by a 30 min 4% PFA fixation, three washes in PBS, a 1-h incubation in 6 M HCl and 0.1% Triton X-100, six washes in PBT, blocking, and anti-BrdU antibody (1:50, 347580; BD PharMingen) incubation.
Rat aortic ring assay
Serum-free aortic ring cultures were established according to the methodology of Bonanno et al. (2000) with modifications (unpublished data). Time-lapse microscopy was performed under low light fluorescence illumination using a Nikon Diaphot 200 inverted microscope and a Hamamatsu C4742–95 Orca1 CCD camera (Hamamatsu Photonic Systems). Aortic rings were maintained under normal culture conditions throughout the filming period. For vital immunostaining, cultures were incubated at 37°C for 30 min with a phycoerythrin-conjugated PECAM-1 antibody (clone TLD-1A2; BD PharMingen) diluted 1:100 in growth medium. After rinsing with growth medium a minimum of five times, the cultures were set up for filming, and images were captured every 10 or 15 min. Movies were created and analyzed using Kinetic AQM 2000 software (Kinetic Imaging).
For immunostaining, cultures were fixed in ice cold methanol for 10 min, rehydrated in a graded series of methanol/PBS. After a 10-min block in normal goat serum, PECAM-1 (phycoerythrin or biotin conjugate of the TLD-1A2 clone; 1:100 in PBS) and rabbit–anti–phospho-histone-H3 (1:200; Upstate Biotechnology) antibodies were added for 1 h at RT. After washing, labeling was detected using Alexa-488 streptavidin or goat anti–rabbit IgG (Molecular Probes). After rinsing with PBS, the areas of sprouting growth were cut out and mounted in SlowFade AntiFade (Molecular Probes). Imaging was performed on a Carl Zeiss MicroImaging Corp. LSM 510 confocal microscope, and quantification of sprout and lamellipodia lengths was performed using IPLab Spectrum software (Signal Analytics Corporation).
Intraocular injections
Pups were deeply anaesthetized by isofluran inhalation. Injections were performed using 10 μl gastight Hamilton syringes equipped with 33 gauge needles attached to a micromanipulator. Approximately 0.5 μl (1 μg/μl sterile filtered solution) was injected (variation due to reflux; delivery was checked by antibody staining). The following substances were used: Flt-1/Fc chimera (471-F1–100; R&D Systems), neutralizing VEGFR1 antibody (AF471; R&D Systems), neutralizing VEGFR2 (AF644; R&D Systems), recombinant mouse VEGF-A 164 (493-MV/CF; R&D Systems), recombinant mouse PlGF-2 (465-PL/CF; R&D Systems), VEGF-E NZ2, VEGF-E NZ7, and VEGF-C156S (see Protein production).
Binding to soluble VEGFR–IgG fusion proteins
100 ng of purified protein was incubated for 8 h at 4°C under gentle agitation with equimolar amounts of VEGFR–IgG fusion proteins (Achen et al., 1998) in PBS supplemented with 0.5% BSA and 0.05% Tween-20 and 1 μg/ml heparin. Receptor-bound growth factors were precipitated with protein A sepharose, subjected to reducing SDS-PAGE, and after blotting visualized using pentahistidine antibody (QIAGEN) and the ECL detection system (Amersham Biosciences).
Protein production
Recombinant proteins were produced using the Bac-to-Bac system (Invitrogen). Nucleotides 658–996 (VEGF-C; sequence data available from GenBank/EMBL/DDBJ under accession no. X94216), 235–801 (VEGF; sequence data available from GenBank/EMBL/DDBJ under accession no. M27281), 329–670 (VEGF-ENZ2; sequence data available from GenBank/EMBL/DDBJ under accession no. S67520), 387–755 (VEGF-ENZ7; sequence data available from GenBank/EMBL/DDBJ under accession no. S67522), and 112–1866 (human serum albumin; sequence data available from GenBank/EMBL/DDBJ under accession no. V00494) were cloned in-frame into a modified transfer vector (pFASTBAC1; Invitrogen) in between sequences coding for a melittin signal peptide and a hexahistidine tag. Human serum albumin was additionally hexahistidine tagged at the NH2 terminus of the mature protein in order to deploy the same stringent washing conditions in the purification as for the dimeric VEGFs. The VEGFR-3–specific mutation in VEGF-C (C156S) has been described previously (Joukov et al., 1998). Conditioned serum-free medium (Sf900II; Invitrogen) of High Five cells was harvested 72 h postinfection and dialyzed against 30 mM sodium phosphate, 0.4 M sodium chloride, pH 6. The pH was adjusted to 8.0, and Ni2+NTA Superflow resin (QIAGEN) was added. Samples were agitated over night at +4°C, and the resin was then collected and applied to chromatography columns. The columns were washed with 30 mM sodium phosphate, 400 mM sodium chloride, 0.6 M glycerol, 20 mM imidazole at pH 8.0, and bound proteins were eluted with an imidazole step gradient. The eluate was dialyzed against PBS or 0.1% TFA and sterilized using Millex-GV filters (Millipore). The proteins were checked on silver-stained reducing SDS-PAGE gels and quantitated using the BCA protein assay (Pierce Chemical Co.).
Online supplemental material
Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200302047/DC1, illustrates the time-lapse analysis of tip cell migration and lamellipodia extension after VEGF sequestration in the aortic ring assay model. Quantitation of migration and lamellipodia length is presented.
Supplemental Material
Acknowledgments
We thank Helen Hjelm, Monica Elmestam, M. Mitchell, C. Rutland, and S. Cooper for excellent help with mice, Carolina Mailhos for assistance in modification of the rat aortic ring model, Per Lindahl and Mats Hellström for valuable comments on the manuscript, and Fredrik Wolfhagen for help with the in situ hybridization.
The study was supported by the Novo Nordisk Foundation, the Swedish Cancer Foundation, and the IngaBritt and Arne Lundberg Foundation to C. Betsholtz. H. Gerhardt is supported by a European Molecular Biology Organization postdoctoral fellowship and by the Swedish Cancer Foundation.
The online version of this article includes supplemental material.
David Shima's present address is Eyetech Research Center, Eyetech Pharmaceuticals Inc., 42 Cummings Park, Woburn, MA 01801.
Footnotes
Abbreviations used in this paper: CNS, central nervous system; GFAP, glial fibrillary acidic protein; ILM, inner limiting membrane; P, postnatal day; PECAM, platelet–endothelial cell adhesion molecule; PlGF, placenta growth factor; VEGFR, vascular endothelial growth factor receptor.
References
- Achen, M.G., M. Jeltsch, E. Kukk, T. Makinen, A. Vitali, A.F. Wilks, K. Alitalo, and S.A. Stacker. 1998. Vascular endothelial growth factor D (VEGF-D) is a ligand for the tyrosine kinases VEGF receptor 2 (Flk1) and VEGF receptor 3 (Flt4). Proc. Natl. Acad. Sci. USA. 95:548–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ausprunk, D.H., and J. Folkman. 1977. Migration and proliferation of endothelial cells in preformed and newly formed blood vessels during tumor angiogenesis. Microvasc. Res. 14:53–65. [DOI] [PubMed] [Google Scholar]
- Bär, T., and J.R. Wolff. 1972. The formation of capillary basement membranes during internal vascularization of the rat's cerebral cortex. Z Zellforsch Mikrosk. Anat. 133:231–248. [DOI] [PubMed] [Google Scholar]
- Benjamin, L.E., D. Golijanin, A. Itin, D. Pode, and E. Keshet. 1999. Selective ablation of immature blood vessels in established human tumors follows vascular endothelial growth factor withdrawal. J. Clin. Invest. 103:159–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonanno, E., M. Iurlaro, J.A. Madri, and R.F. Nicosia. 2000. Type IV collagen modulates angiogenesis and neovessel survival in the rat aorta model. In Vitro Cell. Dev. Biol. Anim. 36:336–340. [DOI] [PubMed] [Google Scholar]
- Carmeliet, P. 2000. VEGF gene therapy: stimulating angiogenesis or angiomagenesis? Nat. Med. 6:1102–1103. [DOI] [PubMed] [Google Scholar]
- Carmeliet, P., and R.K. Jain. 2000. Angiogenesis in cancer and other diseases. Nature. 407:249–257. [DOI] [PubMed] [Google Scholar]
- Carmeliet, P., V. Ferreira, G. Breier, S. Pollefeyt, L. Kieckens, M. Gertsenstein, M. Fahrig, A. Vandenhoeck, K. Harpal, C. Eberhardt, et al. 1996. Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature. 380:435–439. [DOI] [PubMed] [Google Scholar]
- Dorrell, I., E. Aguilar, and M. Friedlander. 2002. Retinal vascular development is mediated by endothelial filopodia, a preexisting astrocytic template and specific R-cadherin adhesion. Invest. Ophthalmol. Vis. Sci. 43:3500–3510. [PubMed] [Google Scholar]
- Dumont, D.J., L. Jussila, J. Taipale, A. Lymboussaki, T. Mustonen, K. Pajusola, M. Breitman, and K. Alitalo. 1998. Cardiovascular failure in mouse embryos deficient in VEGF receptor-3. Science. 282:946–949. [DOI] [PubMed] [Google Scholar]
- Eriksson, U., and K. Alitalo. 1999. Structure, expression and receptor-binding properties of novel vascular endothelial growth factors. Curr. Top. Microbiol. Immunol. 237:41–57. [DOI] [PubMed] [Google Scholar]
- Ferrara, N. 1999. Vascular endothelial growth factor: molecular and biological aspects. Curr. Top. Microbiol. Immunol. 237:1–30. [DOI] [PubMed] [Google Scholar]
- Ferrara, N. 2000. VEGF: an update on biological and therapeutical aspects. Curr. Opin. Biotechnol. 11:617–624. [DOI] [PubMed] [Google Scholar]
- Ferrara, N., K. Carver-Moore, H. Chen, M. Dowd, L. Lu, S. O'Shea, L. Powell-Braxton, K.J. Hillan, and M.W. Moore. 1996. Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature. 380:439–442. [DOI] [PubMed] [Google Scholar]
- Fruttiger, M. 2002. Development of the mouse retinal vasculature: angiogenesis versus vasculogenesis. Invest. Ophthalmol. Vis. Sci. 43:522–527. [PubMed] [Google Scholar]
- Fruttiger, M., A.R. Calver, W.H. Kruger, H.S. Mudhar, D. Michalovich, N. Takakura, S. Nishikawa, and W.D. Richardson. 1996. PDGF mediates a neuron-astrocyte interaction in the developing retina. Neuron. 17:1117–1131. [DOI] [PubMed] [Google Scholar]
- Fruttiger, M., A.R. Calver, and W.D. Richardson. 2000. Platelet-derived growth factor is constitutively secreted from neuronal cell bodies but not from axons. Curr. Biol. 10:1283–1286. [DOI] [PubMed] [Google Scholar]
- Joukov, V., V. Kumar, T. Sorsa, E. Arighi, H. Weich, O. Saksela, and K. Alitalo. 1998. A recombinant mutant vascular endothelial growth factor-C that has lost vascular endothelial growth factor receptor-2 binding, activation and vascular permeability activities. J. Biol. Chem. 273:6599–6602. [DOI] [PubMed] [Google Scholar]
- Kater, S.B., and V. Rehder. 1995. The sensory-motor role of growth cone filopodia. Curr. Opin. Neurobiol. 5:68–74. [DOI] [PubMed] [Google Scholar]
- Lawson, N.D., A.M. Vogel, and B.M. Weinstein. 2002. Sonic hedgehog and vascular endothelial growth factor act upstream of the notch pathway during arterial endothelial differentiation. Dev. Cell. 3:127–136. [DOI] [PubMed] [Google Scholar]
- Lindahl, P., B.R. Johansson, P. Levéen, and C. Betsholtz. 1997. Pericyte loss and microaneurysm formation in PDGF-B-deficient mice. Science. 277:242–245. [DOI] [PubMed] [Google Scholar]
- Marin-Padilla, M. 1985. Early vascularization of the embryonic cerebral cortex: Golgi and electron microscopic studies. J. Comp. Neurol. 241:237–249. [DOI] [PubMed] [Google Scholar]
- Mukouyama, Y., D. Shin, S. Britsch, M. Taniguchi, and D.J. Anderson. 2002. Sensory nerves determine the pattern of arterial differentiation and blood vessel branching in the skin. Cell. 109:693–705. [DOI] [PubMed] [Google Scholar]
- Nicosia, R.F., and A. Ottinetti. 1990. Growth of microvessels in serum-free matrix culture of rat aorta. A quantitative assay of angiogenesis in vitro. Lab. Invest. 63:115–122. [PubMed] [Google Scholar]
- Ogawa, S., A. Oku, A. Sawano, S. Yamaguchi, Y. Yazaki, and M. Shibuya. 1998. A novel type of vascular endothelial growth factor: VEGF-E (NZ-7 VEGF) preferentially utilizes KDR/Flk-1 receptor and carries a potent mitotic activity without heparin-binding domain. J. Biol. Chem. 273:31273–31282. [DOI] [PubMed] [Google Scholar]
- Pierce, E.A., R.L. Avery, E.D. Foley, L.P. Aiello, and L.E. Smith. 1995. Vascular endothelial growth factor / vascular permeability factor expression in a mouse model of retinal neovascularization. Proc. Natl. Acad. Sci. USA. 92:905–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provis, J.M., J. Leech, C.M. Diaz, P.L. Penfold, J. Stone, and E. Keshet. 1997. Development of the human retinal vasculature: cellular relations and VEGF expression. Exp. Eye Res. 65:555–568. [DOI] [PubMed] [Google Scholar]
- Ramirez-Weber, F.A., and T.B. Kornberg. 2000. Signaling reaches to new dimensions in Drosophila imaginal discs. Cell. 103:189–192. [DOI] [PubMed] [Google Scholar]
- Ribeiro, C., A. Ebner, and M. Affolter. 2002. In vivo imaging reveals different cellular functions for FGF and Dpp signaling in tracheal branching morphogenesis. Dev. Cell. 2:677–683. [DOI] [PubMed] [Google Scholar]
- Risau, W., and I. Flamme. 1995. Vasculogenesis. Annu. Rev. Cell Dev. Biol. 11:73–91. [DOI] [PubMed] [Google Scholar]
- Ruhrberg, C., H. Gerhardt, M. Golding, R. Watson, S. Ioannidou, H. Fujisawa, C. Betsholtz, and D. Shima. 2002. Spatially restricted patterning cues provided by heparin-binding VEGF-A control blood vessel branching morphogenesis. Genes Dev. 16:2684–2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samakovlis, C., N. Hacohen, G. Manning, D.C. Sutherland, K. Guillemin, and M.A. Krasnow. 1996. Development of the Drosophila tracheal system occurs by a series of morphologically distinct but genetically coupled branching events. Development. 122:1395–1407. [DOI] [PubMed] [Google Scholar]
- Senger, D.R., S.J. Galli, A.M. Dvorak, C.A. Perruzzi, V.S. Harvey, and H.F. Dvorak. 1983. Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science. 219:983–985. [DOI] [PubMed] [Google Scholar]
- Shalaby, F., J. Rossant, T.P. Yamaguchi, M. Gertsenstein, X.-F. Wu, M.L. Breitman, and A.C. Schuh. 1995. Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature. 376:62–66. [DOI] [PubMed] [Google Scholar]
- Shima, D., and C. Mailhos. 2000. Vascular developmental biology: getting nervous. Curr. Opin. Genet. Dev. 10:536–542. [DOI] [PubMed] [Google Scholar]
- Sholley, M.M., G.P. Ferguson, H.R. Seibel, J.L. Montour, and J.D. Wilson. 1984. Mechanisms of neovascularization. Vascular sprouting can occur without proliferation of endothelial cells. Lab. Invest. 51:624–634. [PubMed] [Google Scholar]
- Soker, S., S. Takashima, H.Q. Miao, G. Neufeld, and M. Klagsbrun. 1998. Neuropilin-1 is expressed by endothelial and tumor cells as an isoform-specific receptor for vascular endothelial growth factor. Cell. 92:735–745. [DOI] [PubMed] [Google Scholar]
- Stalmans, I., N. Yin-Shan, R. Rohan, M. Fruttiger, A. Bouché, A. Yuce, H. Fijusawa, B. Hermans, M. Shani, S. Jansen, et al. 2002. Arteriolar and venolar patterning in retinas of mice selectively expressing VEGF isoforms. J. Clin. Invest. 109:327–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone, J., and Z. Dreher. 1987. Relationship between astrocytes, ganglion cells and vasculature of the retina. J. Comp. Neurol. 255:35–49. [DOI] [PubMed] [Google Scholar]
- Stone, J., A. Itin, T. Alon, J. Peer, H. Gnessin, T. Chan-Ling, and E. Keshet. 1995. Development of retinal vasculature is mediated by hypoxia-induced vascular endothelial growth factor (VEGF) expression by neuroglia. J. Neurosci. 15:4738–4747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tessier-Lavigne, M., and C.S. Goodman. 1996. The molecular biology of axon guidance. Science. 274:1123–1133. [DOI] [PubMed] [Google Scholar]
- Wise, L.M., T. Veikkola, A.A. Mercer, L.J. Savory, S.B. Fleming, C. Caesar, A. Vitali, T. Makinen, K. Alitalo, and S.A. Stacker. 1999. Vascular endothelial growth factor (VEGF)-like protein from orf virus NZ2 binds to VEGFR2 and neuropilin-1. Proc. Natl. Acad. Sci. USA. 96:3071–3076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood, W., and P. Martin. 2002. Structures in focus—filopodia. Int. J. Biochem. Cell Biol. 34:726–730. [DOI] [PubMed] [Google Scholar]
- Zelzer, E., and B.Z. Shilo. 2000. Cell fate choices in Drosophila trachela morphogenesis. Bioessays. 22:219–226. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}