

Abstract
In mammalian cells, the Golgi apparatus is disassembled at the onset of mitosis and reassembled at the end of mitosis. This disassembly–reassembly is generally believed to be essential for the equal partitioning of Golgi into two daughter cells. For Golgi disassembly, membrane fusion, which is mediated by NSF and p97, needs to be blocked. For the NSF pathway, the tethering of p115-GM130 is disrupted by the mitotic phosphorylation of GM130, resulting in the inhibition of NSF-mediated fusion. In contrast, the p97/p47 pathway does not require p115-GM130 tethering, and its mitotic inhibitory mechanism has been unclear. Now, we have found that p47, which mainly localizes to the nucleus during interphase, is phosphorylated on Serine-140 by Cdc2 at mitosis. The phosphorylated p47 does not bind to Golgi membranes. An in vitro assay shows that this phosphorylation is required for Golgi disassembly. Microinjection of p47(S140A), which is unable to be phosphorylated, allows the cell to keep Golgi stacks during mitosis and has no effect on the equal partitioning of Golgi into two daughter cells, suggesting that Golgi fragmentation-dispersion may not be obligatory for equal partitioning even in mammalian cells.
Keywords: Golgi; p47; p97; phosphorylation; mitosis
Introduction
The Golgi apparatus occupies a central position in the classical secretory pathway, where it receives the entire output of de novo synthesized proteins from the ER, and functions to distill, posttransitionally process, and sort cargo to their ultimate destinations (Mellman and Simons, 1992). For its inheritance, there are several strategies: de novo formation, fission, and disassembly–reassembly. Glick and coworkers examined the Golgi during mitosis in the budding yeast Pichia pastoris and found de novo formation of Golgi in a daughter cell (Bevis et al., 2002). In the protozoa Toxoplasma gondii, Warren and coworkers reported binary fission of Golgi for its inheritance (Pelletier et al., 2002). In this case, the Golgi splits in half before mitosis and segregates into the two daughter cells. In contrast, animal cells utilize the strategy of disassembly–reassembly (Warren, 1993). During mitosis, the Golgi apparatus is fragmented into thousands of vesicles and short tubules that are dispersed throughout the cytoplasm. Some or all of them might be absorbed into ER, which is still a matter of contention (Zaal et al., 1999; Jokitalo et al., 2001). At telophase, a Golgi apparatus is rapidly reassembled from the fragments within each daughter cell (Lucocq et al., 1989).
This disassembly–reassembly process is regulated by phosphorylation (Shorter and Warren, 2002), but there are continuing debates as to which kinases are responsible. Suggested kinases include Cdc2 kinase (Lowe et al., 1998; Kano et al., 2000), MAPK kinase (Acharya et al., 1998; Kano et al., 2000), and Polo-like kinase 1 (Sutterlin et al., 2001). Since the Golgi apparatus has a complicated structure, its disassembly is thought to occur in several steps. These kinases might be involved in these disassembly steps. Golgi disassembly–reassembly requires the blocking of membrane fusion at early mitosis and its unblocking at late mitosis (Warren, 1993). Experiments using an in vitro assay showed that Golgi reassembly requires at least two distinct membrane fusion pathways: the NSF pathway and the p97/p47 pathway (Rabouille et al., 1995c). NSF binds to SNAREs via α-SNAP and activates it for membrane fusion (Sollner et al., 1993). NSF also needs the assistance of p115-GM130 tethering (Nakamura et al., 1997). At early mitosis, GM130 is phosphorylated on Serine (Ser)*-25 by Cdc2 kinase. This phosphorylation disrupts the tethering of p115-GM130, resulting in the mitotic inhibition of the NSF pathway (Lowe et al., 1998). Similar to NSF, p97 binds to SNAREs via the cofactor p47 (Kondo et al., 1997) and dissociates the SNARE complex with the assistance of the valosin-containing protein (p97)/p47 complex–interacting protein (VCIP135) during membrane fusion (Uchiyama et al., 2002). It has also been suggested that the interaction between p47 and monoubiquitin might be important (Meyer et al., 2002). In contrast to NSF, p97 does not require p115-GM130 tethering (Rabouille et al., 1995c). This leads to the question: how is the p97 pathway regulated mitotically during the Golgi disassembly–assembly?
In this paper, we first observed the main localization of p47 in the nucleus, which suggests that the major population of p47 does not function in the maintenance of the Golgi apparatus during interphase. Next, we found that mitotic phosphorylation of p47 inhibits its binding to Golgi membranes. This phosphorylation was shown to be required for Golgi disassembly in vitro. Finally, we succeeded in maintaining Golgi stacks in mitotic mammalian cells by the microinjection of p47(S140A), which is unable to be phosphorylated. Using this model cell, we tested whether the fragmentation-dispersion of Golgi during mitosis is essential for its equal partitioning.
Results
p47 mainly localizes to the nucleus in an interphase cell
The subcellular distribution of p97 and p47 was determined in NRK cells by immunofluorescence microscopy using PFA fixation. Several groups have previously reported different staining patterns for p97: the nucleus-dominant pattern (Peters et al., 1990; Meyer et al., 2000) and the cytoplasm-dominant pattern (Madeo et al., 1998). Our results showed that p97 existed in both cytoplasm and nucleus (Fig. 1 A, left). Muller et al. (1999) reported that the amounts of p97 in the nucleus and cytoplasm varied widely in different tissues. Hence, these different staining patterns might depend on the cell lines used.
Figure 1.
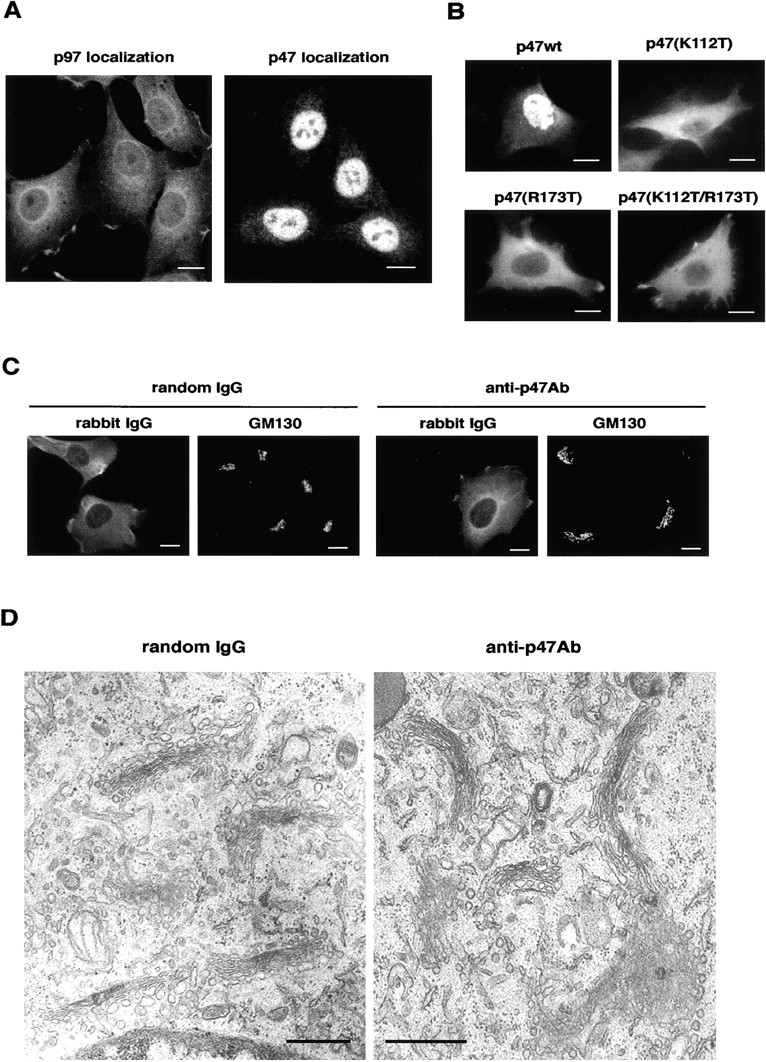
p47 is mainly localized to a nucleus at interphase. (A) The distribution of p47 and p97. NRK cells were fixed with PFA, permeabilized with Triton X-100, stained with polyclonal antibodies to p97 and p47, and observed by confocal microscopy. Bar, 10 μm. (B) HA-tagged p47 and its mutants were expressed in NRK cells, stained with antibodies to HA tag, and observed by conventional fluorescence microscopy. Bar, 10 μm. (C) NRK cells were microinjected in their cytoplasmic regions with either anti-p47 antibodies (8 g/l) or random IgG (8 g/l) and fixed after 2 h incubation. Injected antibodies and Golgi were stained by anti–rabbit antibodies and monoclonal anti-GM130 antibodies. Bar, 10 μm. (D) Representative EM images of Golgi in the cells microinjected with either anti-p47 antibodies (16 g/l) or random IgG (16 g/l). Bars, 0.5 μm.
In contrast to p97, p47 mainly localized to the nucleus with a small amount present in the cytoplasm (Fig. 1 A, right). This staining pattern was the same in HeLa and CHO cell lines (unpublished data). Using antibodies to p47 NH2- or COOH-terminal fragment also revealed nuclear-staining patterns (unpublished data). Because p47 forms a trimer of ∼150 kD (Kondo et al., 1997), it is difficult for it to be transported into the nucleus by simple diffusion. A sequence search for peptide motifs predicted two candidate NLSs in p47: (119)PPRKKSPN(116) and (172)RRRH(175) (Hicks and Raikhel, 1995). Hence, we induced a point mutation in each candidate sequence (K112T, 112K→T; R173T, 173R→T) and expressed the mutants in NRK cells (Fig. 1 B). The expressed p47 was detected by using antibodies to an HA tag in order to distinguish it from endogenous p47. p47wt was expressed in the nucleus (Fig. 1 B, top left), displaying a distribution similar to endogenous p47. When p47 was mutated in either or both of the two NLS motifs, mutated p47 was expressed mainly in the cytoplasm (Fig. 1 B). Together, p47 mainly localizes to the nucleus of interphase cells, and the two NLS motifs were found to be important for this nuclear localization. Since p47 is an essential cofactor of the p97 membrane fusion pathway (Kondo et al., 1997), its nuclear localization suggests a low activity of p97/p47-mediated membrane fusion in the cytoplasm of interphase cells.
Although the major population of p47 was localized to the nuclear region, a small amount of p47 was still observed in the cytoplasm (Fig. 1 A, right). The next question was whether the tiny amount of p47 in cytoplasm functions in Golgi membrane fusion in interphase cells. To address this, we microinjected anti-p47 antibodies into the cytoplasm of interphase cells and investigated Golgi morphology. When prophase cells were injected with the same antibody (8 g/l) and incubated for 1.5 h, Golgi reassembly at the end of mitosis was completely inhibited. Only clusters of vesicles and tubules, not stacks, were observed in daughter cells (Uchiyama et al., 2002). Since the amount of cytoplasmic p47 in interphase cells is much less than that in mitotic cells, this concentration (8 g/l) of the antibody must be sufficient to block the function of cytoplasmic p47. Fig. 1 C shows Golgi staining with anti-GM130 antibodies in the injected cells. Here, the Golgi apparatus was still located in perinuclear regions without dispersion (Fig. 1 C, second and fourth panels). Next, we investigated Golgi ultrastructure by EM. Interphase cells were injected with this antibody at 8 or 16 g/l and incubated for 2 h. As shown in Fig. 1 D (the injected antibody, 16 g/l), the Golgi apparatus still showed normal stacking structures without fragmentation (right). Thus, we could not find any obvious evidence of a Golgi maintenance function mediated by p97/p47. We concluded that the fusion activity mediated by p97/p47 is less critical during interphase than at the end of mitosis.
Once the cell enters mitosis, the nuclear envelope is broken down and p47 comes out from the nucleus, which enables p47 to function in Golgi membrane fusion. However, Golgi disassembly at early mitosis requires the inhibition of membrane fusion. This leads to the question: how is the p97/p47 pathway blocked during early mitosis?
p47 is mitotically phosphorylated
To investigate whether there was mitotic phosphorylation of p47, NRK cells were synchronized and incubated in medium containing [32P]orthophosphate. Mitotic cells were collected by mechanical shake-off and used for the preparation of 32P-labeled mitotic cytosol. Hoechst DNA staining showed that >95% of cells were mitotic (unpublished data). Nonsynchronized cells were used for the preparation of interphase cytosol. p47 was isolated from the cytosol by denatured immunoprecipitation (Fig. 2 A). Western blotting showed that the band of mitotic p47 was shifted upwards (Fig. 2 A, top), and autoradiography revealed that p47 was strongly phosphorylated at mitosis (Fig. 2 A, bottom).
Figure 2.

p47 is mitotically phosphorylated by Cdc2. (A) Mitotic phosphorylation of p47 in vivo. p47 was immunoprecipitated from either 32P-labeled interphase or mitotic cell lysates under a denatured condition. After fractionation by SDS-PAGE, total p47 amount was detected by Western blotting with anti-p47 antibodies (top), and 32P-labeled p47 was detected by autoradiography (bottom). (B) Effect of protein kinase inhibitors on p47 phosphorylation in mitotic cytosol. His-tagged p47 (40 μg/ml) was incubated for 1 h at 30°C in the presence of mitotic cytosol (10 mg protein/ml) and [γ-32P]ATP with the indicated protein kinase inhibitor as follows: 10 μM staurosporine, 1 mM olomoucine, 50 μM PD98059, 100 μM SB203580, 10 μM KT5720, and 500 nM calphostin C. After incubation, His-p47 was immunoprecipitated with anti-His antibodies under a denatured condition, and phospholabeled His-p47 was detected as in A. (C) Effect of Cdc2 depletion from mitotic cytosol on p47 phosphorylation. His-p47 (40 μg/ml) was incubated in the presence of [γ-32P]ATP with buffer, mitotic cytosol (8 mg protein/ml), Cdc2-depleted mitotic cytosol (8 mg protein/ml), or Cdc2-depleted mitotic cytosol supplemented with recombinant Cdc2 kinase complex. (D) Direct phosphorylation of p47 by Cdc2. His-p47 (40 μg/ml) was incubated in the presence of [γ-32P]ATP with buffer or recombinant Cdc2 complex (60 mU/ml). (E) Cdc2-phosphorylation site in p47. Either p47wt or p47 mutant (40 μg/ml) was incubated in the presence of [γ-32P]ATP with mitotic cytosol (10 mg protein/ml).
We tried to determine the responsible kinase. At first, we investigated the effect of several protein kinase inhibitors on the mitotic phosphorylation of p47 by incubating His-p47 with mitotic cytosol and [32P]ATP in the presence of the indicated protein kinase inhibitor (Fig. 2 B). Staurosporine (broad Ser/threonine (Thr) kinase inhibitor) and olomoucine (Cdc2 inhibitor) inhibited p47 phosphorylation (Fig. 2 B, fifth and sixth lanes). But, neither PD98059 (MEK inhibitor), SB203580 (p38 MAPK inhibitor), KT5720 (protein kinase A inhibitor), nor calphostin C (PKC inhibitor) had any effect on p47 phosphorylation. Hence, Cdc2 was thought to be a candidate kinase. This was supported by the previous report that p47 can be a substrate of Cdc2 in vitro (Mayr et al., 1999). To confirm this, we investigated whether Cdc2-depleted mitotic cytosol phosphorylated p47 or not (Fig. 2 C). p13-suc1 beads were used for the depletion of Cdc2 from cytosol, and Western blotting with anti-Cdc2 antibodies showed that >90% of Cdc2 was depleted (unpublished data). p47 phosphorylation was inhibited by Cdc2 depletion from mitotic cytosol (Fig. 2 C, lane 3), and this inhibition was rescued by supplementation with Cdc2 (Fig. 2 C, lane 4). In addition, the purified Cdc2 complex phosphorylated p47 in the absence of mitotic cytosol as shown in Fig. 2 D. Therefore, Cdc2 was confirmed as a responsible kinase.
The consensus motif for Cdc2 is Ser/Thr-Pro-X–Arg/Lys, with Pro at the +1 position being absolutely critical, and a basic residue at the +3 position preferred but not essential for kinase recognition (Holmes and Solomon, 1996). p47 has one Thr and three Ser residues with Pro at the +1 position: T57, S114, S140, and S272. His-p47 was mutated at each phosphorylation site and tested for phosphorylation. As presented in Fig. 2 E, all p47 mutants except p47(S140A) were phosphorylated by mitotic cytosol. Similar results were obtained by using purified Cdc2 instead of mitotic cytosol (unpublished data). Together, we concluded that p47 was mitotically phosphorylated on Ser-140 by Cdc2.
p47 forms a complex with p97 at mitosis
After determining the phosphorylation of p47 at mitosis, this led to the question of what effect the phosphorylation would have on the function of p47. p47 forms a tight complex with p97, which is essential for in vitro Golgi reassembly (Kondo et al., 1997). We investigated the effect of phosphorylation on the binding of p47 to p97. To test whether p97 binds phosphorylated p47, p97 and p47 were incubated in the presence of Cdc2 and [γ-32P]ATP (Fig. 3 A). p47 bound to p97 was coimmunoprecipitated with antibodies to the His tag on p97. Phosphorylated p47 was detected by autoradiography. Phosphorylated p47 was coimmunoprecipitated together with p97 (Fig. 3 A, lane 2), suggesting that phosphorylated p47 bound to p97. p97 is also reported to be phosphorylated at mitosis (Madeo et al., 1998), and therefore, we tested whether phosphorylated p97 bound to p47 (Fig. 3 B). Synchronized NRK cells were labeled with [32P]orthophosphate and used for the preparation of mitotic cytosol. p47 and its binding proteins were coimmunoprecipitated from the 32P-labeled mitotic cytosol using polyclonal anti-p47 antibodies and protein A beads. p47-binding proteins were eluted from the beads by 1 M KCl. p97 was isolated from the eluate by denatured immunoprecipitation using polyclonal anti-p97 antibodies, and phosphorylated p97 was detected by antoradiography. As shown in Fig. 3 B, lane 2, phosphorylated p97 bound to p47. We next compared p47 distributions in interphase and mitotic cell extracts. Extracts were size fractionated by gel filtration, and the localization of p97 and p47 were studied (Fig. 3 C). In both the interphase and mitotic extracts, p97 eluted at ∼670 kD, and p47 eluted at ∼670 and ∼200 kD. (Note that the bands of p47 were double in the mitotic extract.) Since p97-associated p47 is eluted at ∼670 kD (Kondo et al., 1997), these results indicate that p47 forms a complex with p97 in the mitotic extract as well as in the interphase extract. Thus, all the data from the binding experiments and gel filtration show that p47 phosphorylation has no effect on complex formation with p97.
Figure 3.

p47 forms a complex with p97 in mitotic cytosol. (A) Phosphorylated p47 binds to p97. His-tagged p97 (60 μg/ml), no tag p47 (30 μg/ml), or their combination was incubated with [γ-32P]ATP, and Cdc2 (80 mU/ml) at 30°C. After 1 h incubation, His-p97 and its binding proteins were immunoprecipitated with anti-His antibodies. Phosphorylated p47 bound to His-p97 was detected by autoradiography. (B) Mitotically phosphorylated p97 binds to p47. p47 and its binding proteins were isolated from in vivo 32P-labeled mitotic cytosol by native immunoprecipitation using anti-p47 antibodies and protein A beads. p47-binding proteins were eluted from the beads using 1 M KCl. p97 in the elution was isolated by denatured immunoprecipitation using polyclonal anti-p97 antibodies, and phosphorylated p97 was detected by autoradiography. (C) A p97/p47 complex is kept in mitotic cell extract. Synchronized and unsynchronized HeLa cells were completely homogenized until nuclear envelopes were broken and supercentrifuged to get mitotic and interphase cell extracts, respectively. Extracts were size fractionated by gel filtration, and the fractions were assayed by Western blotting with polyclonal antibodies to p97 and p47.
Phosphorylation of Ser-140 in p47 inhibits its binding to Golgi membranes
It has been reported that p47 mediates the binding of the p97/p47 complex to Golgi membranes (Rabouille et al., 1998). Therefore, we investigated the effect of the phosphorylation on the binding of p47 to Golgi membranes. Purified rat liver Golgi membranes were incubated with mitotic HeLa cytosol to obtain mitotic Golgi fragments. Golgi membranes were incubated with interphase cytosol as a control. Fig. 4 A shows the amounts of p97, p47, and syntaxin5 in both membranes by immunoblotting. Mitotic Golgi fragments had very little p97 and p47 compared with those in the membranes incubated with interphase cytosol (Fig. 4 A, top and middle), although the amounts of the membrane protein syntaxin5 were almost the same in both membranes (Fig. 4 A, bottom).
Figure 4.

Phosphorylated p47 does not bind to Golgi membranes. (A) The amounts of p97, p47, and syntaxin5 bound to Golgi membranes after incubation with either interphase or mitotic cytosol were determined by Western blotting using polyclonal antibodies to p97, p47, and syntaxin5 peptides, respectively. (B) Mitotic Golgi fragments can bind p47. Rat liver Golgi and mitotic Golgi fragments were salt washed after their phosphorylation conditions were “frozen” by the addition of staurosporin and microcystin-LR. These salt-washed membranes (30 μg) were incubated with p47 (1 μg) for 1 h on ice in the presence of staurosporin and maicrocystin-LR, and then bound p47 to the membranes were assayed by Western blotting using antibodies to the His tag on p47. Syntaxin5 and GM130 were detected with antibodies to syntaxin5 peptides and GM130. (C) p47 (1.5 μg) was incubated with Cdc2 (30 U) in the absence or presence of olomoucine (1 mM) for 1 h at 30°C, and then staurosporine and microcystin-LR were added. The solution was incubated with 1 M KCl-washed Golgi membranes (30 μg), and then the membranes were isolated from unbound p47 by centrifugation followed by Western blotting. Blots were probed by mAb to the His tag on p47 and polyclonal antibodies to syntaxin5 peptides and GM130. (D) p47(S140A) was used instead of p47wt in the experiments presented in F. Blots were probed by mAb to the His tag on p47(S140A) and polyclonal antibodies to syntaxin5 peptides and GM130. (E) Binding of p47 mutants to Golgi membranes. 1 M KCl-washed Golgi membranes (30 μg) were incubated with either p47wt or p47 mutant (0.5 or 2 μg). Blots were probed by mAb to the His tag on p47s and polyclonal antibodies to syntaxin5 peptides.
In Fig. 4 B, staurosporine and microcystin-LR were added to mitotic Golgi fragments to inactivate any remaining kinases and phosphatases. The resulting membranes were salt washed and incubated with p47 in the presence of staurosporine and microcystin-LR. The upward shifts of GM130 bands (Fig. 4 B, top, lanes 3 and 4) showed that the membranes remained “frozen” in a mitotic state. The frozen mitotic Golgi fragments could bind p47 (Fig. 4 B, middle, lane 4) as well as the control Golgi membranes (interphase Golgi membranes; Fig. 4 B, middle, lane 2). This indicates that the mitotic Golgi fragments still have the ability to bind p47. Accordingly, it is thought that the phosphorylation of p47 decreases its binding affinity to the membranes. To confirm this, we next performed several binding experiments using p47 and salt-washed Golgi membranes.
We first investigated whether phosphorylated p47 bound to Golgi membranes. p47 was phosphorylated by the incubation with Cdc2 and then its phosphorylation condition was frozen by the addition of staurosporine and microcystin-LR. The phosphorylated p47 was used for binding experiments with salt-washed Golgi membranes. As presented in Fig. 4 C, Cdc2-mediated p47 phosphorylation inhibited its binding to the membranes (Fig. 4 C, middle, lane 3). This inhibition was rescued by olomoucine, an inhibitor of Cdc2 (Fig. 4 C, middle, lane 4). These inhibition and rescue were also observed when a p97/p47 complex was added instead of p47 (unpublished data). Next, the mutated p47, p47(S140A), which cannot be phosphorylated in Ser-140, was used for the binding experiments (Fig. 4 D). p47(S140A) bound to Golgi membranes (Fig. 4 D, lane2), and Cdc2 had no effect on this binding (Fig. 4 D, lane 3). We finally tested another p47 mutant, p47(S140D), which mimics the phosphorylation of Ser-140, for the binding experiments (Fig. 4 E). Much less p47(S140D) bound to Golgi membranes (Fig. 4 E, top, lanes 4 and 5) when compared with p47wt (Fig. 4 E, top, lanes 2 and 3) and p47(S140A) (Fig. 4 E, top, lanes 6 and 7). In summary, our biochemical data showed that phosphorylation of Ser-140 in p47 inhibited its binding to Golgi membranes.
We also further investigated the localization of p47 in mitotic cells (Fig. 5). As shown in Fig. 1 A, when NRK cells were directly fixed using PFA, anti-p47 antibodies displayed very strong nuclear staining and weak cytoplasmic staining. However, when such cells were washed with 0.01% saponin-containing buffer to remove p47 that was not membrane-bound before fixation, perinuclear staining was prominent in addition to nuclear staining (Fig. 5 A, arrows). In interphase cells as shown in Fig. 5 A (the star-marked cells), the staining of p47 showed clear colocalization with the staining of GM130, a Golgi marker, at perinuclear regions. In early prometaphase cells (the top left cell in Fig. 5 A and its magnified images shown in Fig. 5 B), p47 staining did not colocalize with GM130 staining. These results strongly support the biochemical data that mitotically phosphorylated p47 does not bind to Golgi membranes. Fig. 5 C presents the localization of p47 in the cell at early telophase. Once the cell entered telophase, p47 staining again showed colocalization with GM130 (Fig. 5 C, arrowheads). It has been reported that GM130 is phosphorylated by Cdc2 at prophase and dephosphorylated at early telophase (Lowe et al., 2000). Similarly, it is likely that p47 was dephosphorylated at early telophase and rebound to Golgi membranes.
Figure 5.

p47 is dissociated from Golgi at prometaphase and rebinds to it at early telophase. NRK cells were washed with 0.01% saponin-containing buffer, fixed with PFA, stained with antibodies to p47 and GM130, and observed by confocal microscopy. Chromatin was stained with DAPI. (A) Interphase cells and an early prometaphase cell. In interphase cells (asterisk), p47 and GM130 showed colocalization in perinuclear regions (arrows). Bar, 10 μm. (B) Magnified images of the early prometaphase cell in A. p47 and GM130 were not colocalized. Bar, 2 μm. (C) An early telophase cell. Arrowheads show the colocalization of p47 and GM130. Bar, 5μm.
Phosphorylation of Ser-140 in p47 controls p97-mediated membrane fusion in vitro
We have established that p47 is mitotically phosphorylated at Ser-140 and that phosphorylated p47 is dissociated from Golgi membranes. Next, we wanted to ascertain whether this phosphorylation in p47 has any effect on Golgi disassembly and assembly in vitro.
If p47 is not phosphorylated at early mitosis, are Golgi membranes fragmented? We used mutated p47, p47(S140A), which cannot be phosphorylated on Ser-140. The early biochemical binding experiment had shown that p47(S140A) could bind to Golgi membranes in the presence of Cdc2 (Fig. 4 D). Purified Golgi membranes and mitotic cytosol were incubated with p97/p47wt or p97/p47(S140A) for the in vitro Golgi disassembly assay. Cisternal fragmentation was defined by the loss of cisternal membranes, with fragmentation induced by mitotic cytosol set to 100%. As shown in Fig. 6 A, the addition of p97/p47(S140A) inhibited cisternal fragmentation almost completely, whereas the addition of p97/p47wt had no effect. This indicates that the phosphorylation of Ser-140 in p47 is required for Golgi disassembly.
Figure 6.

The effects of p47 with a mutated phosphorylation site on Golgi disassembly/reassembly in vitro. (A) Golgi membranes were incubated with the indicated components at 37°C for 30 min: mitotic cytosol (10 mg protein/ml), p97/p47wt (60 μg p97/ml), and p97/p47(S140A) (60 μg p97/ml). The membranes were fixed and processed, and the percentage of membrane in cisternae was determined. Results are presented as the percentage of cisternal fragmentation ± SD (n = 4–5); 0% represents the buffer (41.0% in cisternal membranes), and 100% represents the mitotic cytosol (25.2% in cisternal membranes). (B) Mitotic Golgi membranes were incubated with the indicated components at 37°C for 60 min: p97/p47wt (60 μg p97/ml) and p97/p47(S140D) (60 μg p97/ml). Results are presented as the percentage of cisternal regrowth ± SD (n = 4): 0% represents the buffer (21.3% in cisternal membranes), and 100% represents p97/p47wt (44.7% in cisternal membranes).
We also tested the effect of p47(S140D), which mimics Ser-140 phosphorylation, on Golgi reassembly. Mitotic Golgi fragments were incubated with p97/p47wt or p97/p47(S140D) for the in vitro Golgi reassembly assay. Fig. 6 B shows that p97/p47(S140D) had a very small effect on the cisternal regrowth compared with p97/p47wt, suggesting that the dephosphorylation of Ser-140 in p47 is necessary for Golgi reassembly.
Phosphorylation of Ser-140 in p47 is required for Golgi fragmentation-dispersion during mitosis in living cells
The in vitro Golgi disassembly assay showed that the addition of p97/p47(S140A) to mitotic cytosol inhibited Golgi fragmentation (Fig. 6 A). We next investigated the effect of p47(S140A) in living cells. Either p97/p47wt or p97/p47(S140A) was microinjected into prophase (or early prometaphase) cells and fixed at several intervals. Fig. 7 A shows Golgi staining with anti-GM130 antibodies after injection. In the cells injected with p97/p47wt, the Golgi membranes dispersed all over the cell at prometaphase and metaphase, although there remained some small blobs, and they were reorganized to form Golgi clusters at telophase (Fig. 7 A, top). The cells injected with p97/p47(S140A) showed entirely different Golgi staining: the dispersion of Golgi membranes was mostly suppressed, and Golgi clusters showing bright staining were observed through all mitotic phases (Fig. 7 A, bottom). The Golgi clusters induced by p97/p47(S140A) were doubly stained with antibodies to GM130 and β1,4-galactosyltransferase (GalT), markers of cis- and trans-Golgi, respectively (Rabouille et al., 1995a). As shown in Fig. 7 B, some parts of the structures were stained by only one antibody, although others were stained by both. This suggests that these Golgi clusters were polar structures.
Figure 7.
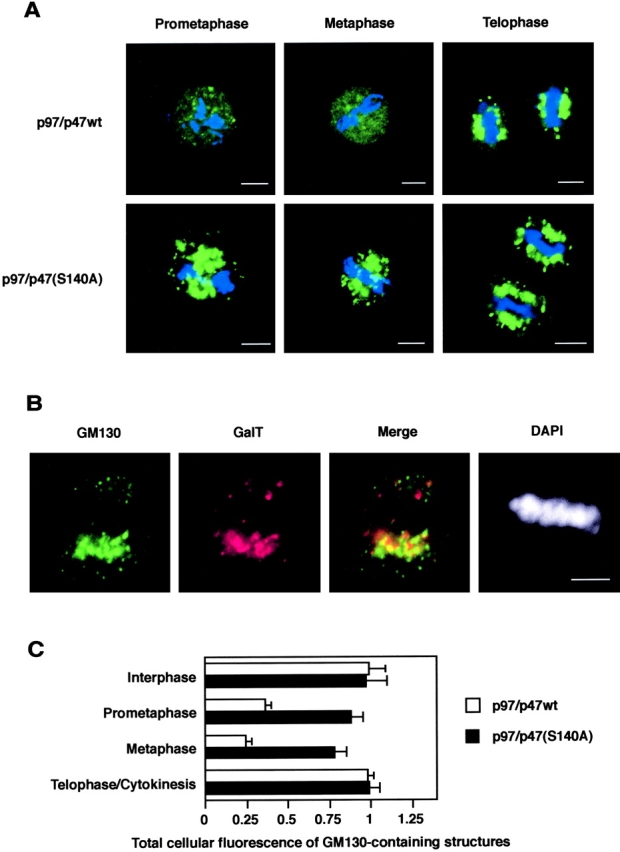
The microinjection of p47(S140A) prevents Golgi dispersion at mitosis in living cells. (A) The microinjection of p97/p47(S140A) into living cells. NRK cells at prophase (or early prometaphase) were injected with either p97/p47wt or p97/p47(S140A): p97 (13 g/l), p47wt (5 g/l), and p47(S140A) (5 g/l). After 0.5–1.5 h incubation, the cells were fixed with PFA and permeabilized with saponin. Golgi (green) and chromatin (blue) were stained by anti-GM130 antibodies and DAPI. The injected cells were identified by fluorescence of coinjected Cy3-BSA (unpublished data). Bar, 5μm. (B) The p97/p47(S140A)-injected cell was fixed by methanol for 4 min at −20°C and stained with polyclonal anti-GM130 antibodies, monoclonal anti-GalT antibodies, and DAPI. Confocal microscopy was used for the observation. Bar, 5 μm. (C) Quantitation of GM130-containing structures in the cells injected with either p97/p47wt or p97/p47(S140A). NRK cells were labeled with polyclonal antibodies to GM130 and imaged by confocal microscopy. Quantitation was performed as described in Materials and methods. The total cellular immunofluorescence of GM130-containing structures is expressed in arbitrary units as means ± SE (n = 9–11) and normalized to give a control (uninjected interphase cells) value of 1.
The Golgi apparatus generally vesiculates and disperses all over the cytoplasm from prometaphase to early telophase. We thus quantified the total cellular fluorescence of GM130-containing structures in the injected cells. As presented in Fig. 7 C, in the cells injected with p97/p47wt, the amount of GM130 in identifiable fragments fell to ∼24% at metaphase. In the p97/p47(S140A)-injected cells, the reduction was very small; it fell to only ∼80% at metaphase. Zaal et al. (1999) suggested that the absorption of Golgi vesicles to ER might be related to this reduction, although this is a matter of controversy (Jokitalo et al., 2001). Accordingly, our results suggest that p47(S140A) inhibited the vesiculation-dispersion of Golgi (and its absorption to ER, if this absorption exists).
The double staining images presented in Fig. 7 B suggest that the Golgi clusters induced by p97/p47(S140A) may be organized polar structures. Therefore, we investigated the ultrastructure of injected cells by EM. Fig. 8 A shows representative images of the injected cells. In the cells injected with p97/p47wt, Golgi stacks were almost lost and fragmented at late prometaphase, similar to uninjected cells (Fig. 8, left). In contrast, the cells injected with p97/p47(S140A) had highly organized Golgi stacks at the same mitotic phase: 80.1% of cisternal membranes were found in stacks (Fig. 8, right). The connections between stacks were lost, and the Golgi ribbon was broken into discrete stacks. Note that the nuclear envelope had been lost and chromatin was well condensed in this cell (Fig. 8, right, inset). Fig. 8 B presents the quantitative results. The injection of p97/p47(S140A) significantly increased the percentage of Golgi membranes in cisternal structures threefold and significantly decreased the percentage of Golgi membranes in vesicles by a third compared with the injection of p97/p47wt. The amount of tubules/tubular networks did not change. Neither Golgi stacks nor cisternae are generally observed in mitotic cells, whereas the injection of p97/p47wt also allowed a few cisternae to remain at early mitosis (Fig. 8 B). Some of the injected p47wt might not be phosphorylated, which enabled occasional cisternae to remain.
Figure 8.
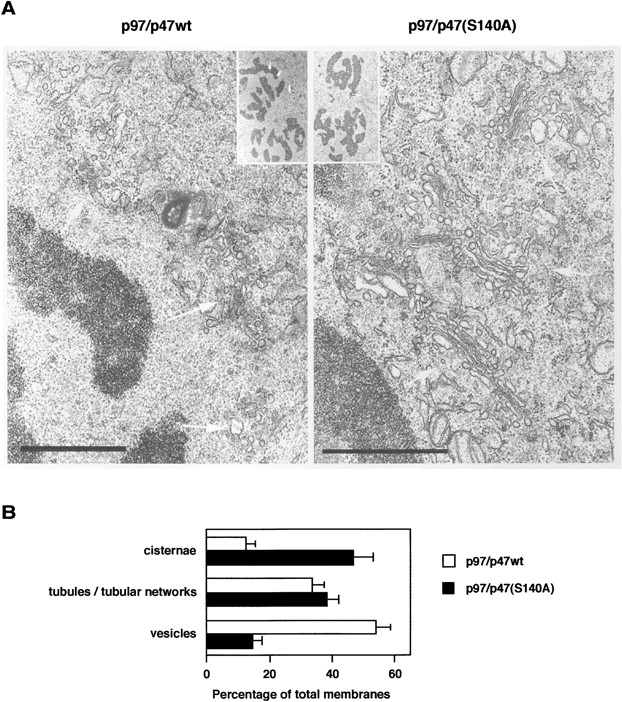
The microinjection of p47(S140A) preserves Golgi stacks in mitotic cells. (A) Representative EM images of Golgi at late prometaphase in the cells injected with either p97/p47wt or p97/p47(S140A) as described in Fig. 7 A. Insets show the whole cell images. White arrows show Golgi. Bars, 1 μm. (B) In the injected cells at prometaphase, Golgi membranes were classified and counted as described previously (Shorter and Warren, 1999; Seemann et al., 2000). Means ± SE (n = 9).
The fragmentation-dispersion of Golgi is generally believed to be necessary for its equal partitioning into two daughter animal cells (Warren, 1993). Now, we have succeeded in preventing Golgi fragmentation-dispersion at mitosis. This suggests that the equal partitioning of Golgi should not be achieved in the cells injected with p97/p47(S140A). Therefore, we tested the partitioning of Golgi in the injected cells. Because it is reported that GM130 remains in Golgi fragments at mitosis without being absorbed to ER (Seemann et al., 2002), the staining of GM130 was used to trace Golgi membranes. To estimate the accuracy of the partitioning, we calculated median deviations from equality. Uninjected cells were used to calculate the control value. Table I presents the results. There were no significant differences among the three groups. The cells injected with p97/p47(S140A) showed 3.60%, which was not bigger than those in the uninjected cells and p97/p47wt-injected cells. This indicates that the p97/p47(S140A)-injected cells achieved equal partitioning of Golgi at the same accuracy as the control cells.
Table I. Distribution of GM130 in daughter cells.
| Median deviation from equality | |||
|---|---|---|---|
|
|
No injection
|
p97/p47wt
|
p97/p47(S140A)
|
| Cytokinesis | |||
| 3.62% | 3.98% | 3.60% | |
| (n = 70) | (n = 67) | (n = 68) | |
NRK cells at prophase (or early prometaphase) were microinjected with either p97/p47wt or p97/p47(S140A) as in Fig. 7 A. The cells were fixed 90 min after injection and stained with anti-GM130 antibodies. Uninjected NRK cells were used as control. Total fluorescence on each side of the equatorial plate in each daughter cell pair at cytokinesis was quantified, and background fluorescence was subtracted. For each pair of values, the percentage of deviation from 50% was calculated, and the median deviation was determined from the number (n) of pairs evaluated.
Discussion
In animal cells, the Golgi apparatus is disassembled at the onset of mitosis and reassembled at the end of mitosis. Golgi disassembly requires the inhibition of membrane fusion, which is necessary for reassembly (Warren, 1993). Two ATPases are required for this membrane fusion: NSF and p97 (Rabouille et al., 1995b). The mitotic inhibition of the NSF pathway for Golgi disassembly is achieved by blocking the tethering of p115-GM130 (Nakamura et al., 1997), and its inhibition is rescued by dephosphorylation of GM130 at telophase (Lowe et al., 2000). In this paper, we have clarified the mechanism of the cell cycle–dependent regulation of the other membrane fusion pathway, the p97/p47 pathway. p47 is mainly localized to the nucleus during interphase and moves to the cytoplasm when cells enter mitosis. p47 is phosphorylated by Cdc2 at early mitosis, and this phosphorylation prevents the binding of the p97/p47 complex to Golgi membranes, resulting in the inhibition of p97-mediated membrane fusion. Thus, the fusion machinery itself is mitotically blocked in the case of the p97 pathway, in contrast to the NSF pathway; the function of NSF/SNAPs, the SNARE-priming activity, is intact through mitosis (Muller et al., 2002).
Phosphorylation of Ser-140 in p47 inhibited its binding to Golgi membranes (Fig. 4). The Ser-140 is in a highly unstructured region (Yuan. X., and S. Matthews, personal communications) which could become structured upon binding to SNAREs. The phosphorylation could alter the nature of the unstructured loop region, affecting the conformational changes. Also, the additional negative charges on phosphorylated Ser-140 could alter the local conformation of the structure, masking the interaction site with SNAREs. Our data show that p47 binds to Golgi membranes at interphase, comes off at prometaphase, and rebinds to the membranes at telophase (Fig. 5). These results indicate that p47 is phosphorylated at early mitosis and dephosphorylated at late mitosis, although we have no data on the responsible phosphatase at present. This is consistent with the results of the in vitro function assays which showed that Golgi disassembly requires the phosphorylation of p47 and that Golgi reassembly requires the dephosphorylation of p47 (Fig. 6). Therefore, we propose that the phosphorylation-dephosphorylation cycle of p47 at mitosis as well as its nuclear localization at interphase play key roles in Golgi disassembly–reassembly during the cell cycle. Fig. 9 shows a schematic model. In an interphase cell, p47 is mainly in the nucleus. Once the cell enters mitosis, its nuclear envelope is broken down, and p47 enters the cytoplasm. At the same time, p47 is phosphorylated by mitotically activated Cdc2 kinase. Since the phosphorylated p47 cannot bind to Golgi membranes, p97/p47-mediated membrane fusion is inhibited, resulting in the fragmentation of Golgi membranes. At late mitosis, p47 is dephosphorylated and binds to Golgi membranes. This allows p97/p47-mediated membrane fusion to reassemble the Golgi apparatus. When nuclear envelopes are formed in daughter cells, p47 moves to the nucleus, and the membrane fusion mediated by p97/p47 is suppressed. Hetzer et al. (2001) has reported that p97/p47 is also required for nuclear envelope formation. This implies that, when the nuclear envelope as well as the Golgi apparatus are reassembled and p97/p47-mediated fusion is not required anymore, membrane fusion will be suppressed by the nuclear envelope that p97/p47-mediated fusion assembled. This feedback control system seems to be very tight. Moreover, Ser-140 in p47 is conserved in Drosophila, Xenopus, mouse, and human but not in yeast. In yeast, the nuclear envelope is not broken down at mitosis, and hence, the phosphorylation-dephosphorylation of the yeast p47 homologue shp1p may not be necessary.
Figure 9.

A schematic model for the cell cycle- dependent control of the p97/p47 pathway. When a cell enters mitosis, p47 comes to the cytoplasm. However, p47 is phosphorylated by Cdc2 and its binding to Golgi membranes is inhibited, resulting in the Golgi fragmentation. At late mitosis, the p47 is dephosphorylated, and p97/p47 functions in membrane fusion for the Golgi reassembly. When a nuclear envelope is formed in a daughter cell, p47 is localized to the nucleus and the function of p97/p47 is suppressed. See Discussion for details.
Microinjection of p47(S140A), which cannot be phosphorylated, inhibits mitotic Golgi disassembly in living cells (Figs. 7 and 8). This is the first report showing that Golgi stacks can be successfully maintained during mitosis in mammalian cells. In p97/p47(S140A)-injected cells, the Golgi ribbon was broken into discrete stacks, but no further disassembly occurred (Fig. 8). This supports the general view that the disassembly occurs in two phases: first the ribbon is broken into stacks, and then each stack is converted into clusters of vesicles and tubules. p47(S140A) seems to have its effect on the second phase of disassembly. The function of p97/p47 is thought to be mediated by its membrane fusion activities, and there is no previous evidence concerning its function on Golgi stacking (Rabouille et al., 1995c; Kondo et al., 1997; Shorter and Warren, 1999). We observed no obvious effects of p97/p47(S140A) on the stacking in the in vitro assay (unpublished data). Therefore, it was a surprise that Golgi stacks remained in mitotic cells after injection of p97/p47(S140A). Nevertheless, this result is in good agreement with our previous observation in vivo that the injection of anti-p47 antibodies inhibited stacking formation as well as cisternal growth at the end of mitosis (Uchiyama et al., 2002). The discrepancy between in vitro and in vivo might be caused by lack of cytoskeletons in the in vitro system: cytoskeletal elements such as actin filaments were removed or depolymerized in the in vitro assay. The fragmentation of cisternae might be necessary for the effective destruction of Golgi stacks, because big cisternae are thought to prevent the access of mitotic modulators, such as kinases, to the stacking machinery between them. Fig. 8 B also shows the decrease of the amount of Golgi vesicles by the injection of p97/p47(S140A). In a previous paper, we showed that the in vivo inhibition of the p97/p47 pathway caused an accumulation of tubules, as opposed to vesicles, suggesting a function for p97/p47 in homotypic membrane fusion (Uchiyama et al., 2002). Accordingly, the vesicles might not be consumed by p97/p47(S140A)-mediated membrane fusion. The existence of big smooth cisternae at mitosis might result in the decrease of the regions for vesicle budding, and thus, it is most likely that budding of vesicles from Golgi was suppressed in p97/p47(S140A)-injected cells.
Normal prometaphase NRK cells had neither Golgi stacks nor cisternae, and vesiculated Golgi membranes were dispersed all over the cytoplasm. In contrast, the p97/p47(S140A)-injected prometaphase cell had Golgi cisternae and stacks (Fig. 8) and seemed to have very little absorption of Golgi vesicles into ER (Fig. 7 C). The injection of p97/p47(S140A) had no effect on cell cycle progression (e.g., chromatin segregation and cytokinesis were pursued normally; see Table S1, available at http://www.jcb.org/cgi/content/full/jcb.200303048/DC1). Hence, we used these p97/p47(S140A)-injected cells as model cells and tested whether Golgi fragmentation-dispersion is required for equal partitioning in mammalian cells. As a result, we found that the p97/p47(S140A)-injected cells achieved equal Golgi partitioning. Therefore, we propose that Golgi fragmentation-dispersion is not obligatory for its equal partitioning even in mammalian cells. Generally, organelles adopt one of two partitioning strategies: stochastic or ordered. Our result strongly suggests that the partitioning of the Golgi is controlled in a highly ordered way. A separate stack can be a minimum unit for partitioning even in mammalian cells. This is similar to the cases of algae and protozoa (Munro, 2002). Stochastic and ordered ways of partitioning need not be mutually exclusive and our results do not belittle the significance of mitotic Golgi fragmentation-dispersion. This might assist the ordered mechanism to increase the accuracy of partitioning. Sutterlin et al. (2002) also reported that Golgi fragmentation-dispersion is required for entry into mitosis, showing that this process is essential for mammalian cells. Taking our results into account, the fragmentation-dispersion of Golgi may have an important, presently unknown, significance besides partitioning.
Materials and methods
Proteins, constructs, antibodies, and reagents
His-tagged p97 and p47 were prepared as described previously (Kondo et al., 1997; Uchiyama et al., 2002). Endogenous p97 was purified from rat liver cytosol (Rabouille et al., 1995c; Kondo et al., 1997). Recombinant p47 without a tag was expressed in Escherichia coli and purified biochemically.
For the expression of HA-tagged p47 in cultured cells, p47 cDNA was subcloned to pCG-N-BL vector (a gift from Dr. Taketani, Kyoto Institute of Technology, Kyoto, Japan). Point mutations were directly introduced into the p47 cDNA in pQE30 or pCG-N-BL by PCR reactions, using the Quick-change mutagenesis kit (Stratagene). All clones were verified by DNA sequencing.
Polyclonal antibodies to p47 and syntaxin5 were prepared as described (Hui et al., 1997; Kondo et al., 1997). A rabbit anti-p97 polyclonal antibody was raised using His-p97(1–198) as an antigen. Monoclonal anti-GalT antibodies and polyclonal anti-GM130 antibodies were gifts from Dr. Suganuma (Miyazaki Medical College, Miyazaki, Japan) and Dr. Nakamura (Kanazawa University, Kanazawa, Japan), respectively. mAb to p97, His-tag, and GM130 were purchased from Progen, QIAGEN, and BD Transduction, respectively.
The following reagents were purchased from Calbiochem; staurosporine, olomoucine, PD98059, SB203580, KT5720, calphostin C, microcystin-LR. Cdc2 kinase (p34cdc2/cyclin B) was from New England Biolabs, Inc.
In vivo metabolic 32P labeling
For enrichment of mitotic NRK cells, aphidicolin (2.5 μg/ml) was added to the medium for 14 h. The cells were washed with fresh medium, released from the S phase block for 2 h, and labeled with [32P]orthophosphate (200 μCi/ml) for another 4 h at 37°C. Mitotic cells were flushed from the dish, washed with PBS, and extracted with buffer (20 mM Hepes, 0.1 M KCl, 5 mM EDTA, 2 mM EGTA, 40 mM β-glycerophosphate, 10 mM NaF, 1 mM sodium orthovanadate, 1 mM DTT, protease inhibitor cocktail, 0.5% Triton X-100, 10% glycerol, pH 7.4). The lysate was cleared by centrifugation and used for the native or denatured immunoprecipitation with anti-p47 antibodies.
In vitro phosphorylation
p47 or its mutant was incubated in buffer A (50 mM Tris, 50 mM KOAc, 10 mM MgOAc, 20 mM β-glycerophosphate, 0.2 mM DTT, 40 μM ATP, 30 μCi/μl [γ-32P]ATP, pH 7.4) with mitotic cytosol or purified kinase for 30 min at 30°C followed by denatured immunoprecipitation. The reactions were terminated by adding an equal volume of the buffer (100 mM Tris, 2 mM EDTA, 2% SDS, pH 7.4) and boiled for 4 min. After adding 20 vol of buffer (50 mM Tris, 0.15 M KCl, 0.5% Tween-20, 1 mM EDTA, 1 mM EGTA, 10 mM NaF, 40 mM β-glycerophosphate, 1 mM sodium orthovanadate, 1 mM DTT, pH 7.4), p47 or its mutant was immunoprecipitated with antibodies to a His tag and protein G beads followed by SDS-PAGE and autoradiography.
For the binding experiments of p47 to salt-washed Golgi membranes, 1 mM ATP was added to buffer A instead of 40 μM ATP and 30 μCi/μl [γ-32P]ATP. After incubation, the reactions were supplemented with microcystin-LR (10 μM) and staurosporine (25 μM) followed by the binding experiments using Golgi membranes.
Depletion of Cdc2 kinase was performed using p13-suc1 beads (Upstate Biotechnology) as described (Hirota et al., 2000).
Binding experiments and gel filtration
Golgi membranes were purified from rat liver (Hui et al., 1998). 1 M KCl-washed Golgi membranes and mitotic Golgi fragments were prepared as described previously (Uchiyama et al., 2002).
p47 or its mutant was incubated with salt-washed Golgi membranes in buffer (0.1 M KCl, 20 mM Tris, 1 mM MgCl2, 1 mM ATP, 0.4 g/l BSA, 0.2 M sucrose, pH 7.4) for 1 h on ice, and then the membranes were recovered by centrifugation.
Interphase and mitotic cytosol were prepared from HeLa cells (Rabouille et al., 1995b). The homogenizing buffer was supplemented with microcystin-LR (10 μM) and staurosporine (25 μM) in order to maintain their phosphorylation conditions. Cytosolic proteins were fractionated by a Superose 6 in the presence of microcystin-LR (2 μM) and staurosporine (5 μM). Molecular weight markers were thyroglobulin (1,340 and 670 k), apoferritin (440 k), and β-amylase (200 k).
In vitro Golgi disassembly and reassembly assays
The in vitro Golgi disassembly and reassembly assays were performed as reported previously (Lowe et al., 1998; Shorter and Warren, 1999).
Immunofluorescence microscopy
NRK cells grown on coverslips were fixed with 3% PFA/PBS for 20 min at room temperature and stained with antibodies after permeabilization, unless stated otherwise. 0.1% Triton X-100 and 0.1% saponin were used for the permeabilization of interphase and mitotic cells, respectively.
For the staining of mitotic cells with antibodies to p47 and GM130, cells were incubated in buffer (80 mM PIPES, 1 mM MgCl2, 5 mM EGTA, 0.01% saponin, pH 7) for 7 min at room temperature before the fixation with 3% PFA (Zerial et al., 1992). Saponin (0.01%) was used instead of Triton X-100 for the following permeabilization and labeling with antibodies.
The total cellular fluorescence of GM130-containing structures was calculated using a series of sections (1 μm in thickness) throughout a whole cell: 9–11 sections for an interphase cell and 17–19 sections for a mitotic cell. For the calculation, well-isolated cells were chosen. Background was subtracted and a threshold introduced to exclude diffuse cytoplasmic staining. All pixels above this threshold value were counted as GM130-containing structures in all sections and summed up. The resulting value was normalized to a uninjected interphase cells on the same coverslip.
Microinjection into living cells
The microinjection was performed as described previously (Uchiyama et al., 2002). Fluorescence of coinjected Cy3-BSA (∼1 μg/μl) was used as an injection marker to identify injected cells. For the injection into mitotic cells, the synchronized cells were used as described above. Prophase (or early prometaphase) cells were microinjected with p97/p47 samples 4 h after the release from aphidicolin block: a phase–contrast microscope was used for the identification of their phases. The injected cells were incubated for another 0.5–1.5 h and fixed.
For EM study, cells were grown on a coverslip on which a square area of ∼1 mm × ∼1 mm was outlined by a diamond pen (Uchiyama et al., 2002). The cells within the area were injected. In case of the injection into mitotic cells, all injections were done within 5–10 min. Uninjected cells within the area were completely removed by an injection needle using fluorescence of coinjected Cy3-BSA as a marker. After fixation and embedding into Epon, the cells within the area were ultrathin sectioned. Membrane profiles in the Golgi area were divided into three categories: cisternae, tubules, and vesicles. The relative proportion of each category of membranes was counted using an intersection method.
Online supplemental material
Table S1, showing the cell cycle progression after microinjection, is available at http://www.jcb.org/cgi/content/full/jcb.200303048/DC1.
Supplemental Material
Acknowledgments
We would like to thank M. Lowe for his kind and helpful advice, X. Yuan and S. Matthews for stimulating discussions; C. Rabouille for critically reading the manuscript; and K. Harasaki for his kind assistance in the preparation of the manuscript. Special thanks to P. Luzio and K. Maruyama for their kind support and encouragement.
E. Jokitalo is supported by Biocentrum Helsinki. This work was supported by a Wellcome Trust grant to H. Kondo.
The online version of this article includes supplemental material.
Footnotes
Abbreviations used in this paper: GalT, β1,4-galactosyltransferase; Ser, serine; Thr, threonine.
References
- Acharya, U., A. Mallabiabarrena, J.K. Acharya, and V. Malhotra. 1998. Signaling via mitogen-activated protein kinase kinase (MEK1) is required for Golgi fragmentation during mitosis. Cell. 92:183–192. [DOI] [PubMed] [Google Scholar]
- Bevis, B.J., A.T. Hammond, C.A. Reinke, and B.S. Glick. 2002. De novo formation of transitional ER sites and Golgi structures in Pichia pastoris. Nat. Cell Biol. 4:750–756. [DOI] [PubMed] [Google Scholar]
- Hetzer, M., H.H. Meyer, T.C. Walther, D. Bilbao-Cortes, G. Warren, and I.W. Mattaj. 2001. Distinct AAA-ATPase p97 complexes function in discrete steps of nuclear assembly. Nat. Cell Biol. 3:1086–1091. [DOI] [PubMed] [Google Scholar]
- Hicks, G.R., and N.V. Raikhel. 1995. Protein import into the nucleus: an integrated view. Annu. Rev. Cell Dev. Biol. 11:155–188. [DOI] [PubMed] [Google Scholar]
- Hirota, T., T. Morisaki, Y. Nishiyama, T. Marumoto, K. Tada, T. Hara, N. Masuko, M. Inagaki, K. Hatakeyama, and H. Saya. 2000. Zyxin, a regulator of actin filament assembly, targets the mitotic apparatus by interacting with h-warts/LATS1 tumor suppressor. J. Cell Biol. 149:1073–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes, J.K., and M.J. Solomon. 1996. A predictive scale for evaluating cyclin-dependent kinase substrates. A comparison of p34cdc2 and p33cdk2. J. Biol. Chem. 271:25240–25246. [DOI] [PubMed] [Google Scholar]
- Hui, N., N. Nakamura, B. Sonnichsen, D.T. Shima, T. Nilsson, and G. Warren. 1997. An isoform of the Golgi t-SNARE, syntaxin 5, with an endoplasmic reticulum retrieval signal. Mol. Biol. Cell. 8:1777–1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui, N., N. Nakamura, P. Slusarewicz, and G. Warren. 1998. Purification of rat liver golgi stacks. Cell Biology: A Laboratory Handbook. Vol. 2. J. Ceils, editor. Academic Press, Orlando, FL. 46–55.
- Jokitalo, E., N. Cabrera-Poch, G. Warren, and D.T. Shima. 2001. Golgi clusters and vesicles mediate mitotic inheritance independently of the endoplasmic reticulum. J. Cell Biol. 154:317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kano, F., K. Takenaka, A. Yamamoto, K. Nagayama, E. Nishida, and M. Murata. 2000. MEK and Cdc2 kinase are sequentially required for Golgi disassembly in MDCK cells by the mitotic Xenopus extracts. J. Cell Biol. 149:357–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo, H., C. Rabouille, R. Newman, T.P. Levine, D. Pappin, P. Freemont, and G. Warren. 1997. p47 is a cofactor for p97-mediated membrane fusion. Nature. 388:75–78. [DOI] [PubMed] [Google Scholar]
- Lowe, M., C. Rabouille, N. Nakamura, R. Watson, M. Jackman, E. Jamsa, D. Rahman, D.J. Pappin, and G. Warren. 1998. Cdc2 kinase directly phosphorylates the cis-Golgi matrix protein GM130 and is required for Golgi fragmentation in mitosis. Cell. 94:783–793. [DOI] [PubMed] [Google Scholar]
- Lowe, M., N.K. Gonatas, and G. Warren. 2000. The mitotic phosphorylation cycle of the cis-golgi matrix protein GM130. J. Cell Biol. 149:341–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucocq, J.M., E.G. Berger, and G. Warren. 1989. Mitotic Golgi fragments in HeLa cells and their role in the reassembly pathway. J. Cell Biol. 109:463–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madeo, F., J. Schlauer, H. Zischka, D. Mecke, and K.U. Frohlich. 1998. Tyrosine phosphorylation regulates cell cycle-dependent nuclear localization of Cdc48p. Mol. Biol. Cell. 9:131–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayr, P.S., V.J. Allan, and P.G. Woodman. 1999. Phosphorylation of p97(VCP) and p47 in vitro by p34cdc2 kinase. Eur. J. Cell Biol. 78:224–232. [DOI] [PubMed] [Google Scholar]
- Mellman, I., and K. Simons. 1992. The Golgi complex: in vitro veritas? Cell. 68:829–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer, H.H., J.G. Shorter, J. Seemann, D. Pappin, and G. Warren. 2000. A complex of mammalian ufd1 and npl4 links the AAA-ATPase, p97, to ubiquitin and nuclear transport pathways. EMBO J. 19:2181–2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer, H.H., Y. Wang, and G. Warren. 2002. Direct binding of ubiquitin conjugates by the mammalian p97 adaptor complexes, p47 and Ufd1-Npl4. EMBO J. 21:5645–5652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller, J.M., H.H. Meyer, C. Ruhrberg, G.W. Stamp, G. Warren, and D.T. Shima. 1999. The mouse p97 (CDC48) gene. Genomic structure, definition of transcriptional regulatory sequences, gene expression, and characterization of a pseudogene. J. Biol. Chem. 274:10154–10162. [DOI] [PubMed] [Google Scholar]
- Muller, J.M., J. Shorter, R. Newman, K. Deinhardt, Y. Sagiv, Z. Elazar, G. Warren, and D.T. Shima. 2002. Sequential SNARE disassembly and GATE-16–GOS-28 complex assembly mediated by distinct NSF activities drives Golgi membrane fusion. J. Cell Biol. 157:1161–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munro, S. 2002. More than one way to replicate the Golgi apparatus. Nat. Cell Biol. 4:E223–E224. [DOI] [PubMed] [Google Scholar]
- Nakamura, N., M. Lowe, T.P. Levine, C. Rabouille, and G. Warren. 1997. The vesicle docking protein p115 binds GM130, a cis-Golgi matrix protein, in a mitotically regulated manner. Cell. 89:445–455. [DOI] [PubMed] [Google Scholar]
- Pelletier, L., C.A. Stern, M. Pypaert, D. Sheff, H.M. Ngo, N. Roper, C.Y. He, K. Hu, D. Toomre, I. Coppens, et al. 2002. Golgi biogenesis in Toxoplasma gondii. Nature. 418:548–552. [DOI] [PubMed] [Google Scholar]
- Peters, J.M., M.J. Walsh, and W.W. Franke. 1990. An abundant and ubiquitous homo-oligomeric ring-shaped ATPase particle related to the putative vesicle fusion proteins Sec18p and NSF. EMBO J. 9:1757–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabouille, C., N. Hui, F. Hunte, R. Kieckbusch, E.G. Berger, G. Warren, and T. Nilsson. 1995. a. Mapping the distribution of Golgi enzymes involved in the construction of complex oligosaccharides. J. Cell Sci. 108:1617–1627. [DOI] [PubMed] [Google Scholar]
- Rabouille, C., T. Misteli, R. Watson, and G. Warren. 1995. b. Reassembly of Golgi stacks from mitotic Golgi fragments in a cell-free system. J. Cell Biol. 129:605–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabouille, C., T.P. Levine, J.M. Peters, and G. Warren. 1995. c. An NSF-like ATPase, p97, and NSF mediate cisternal regrowth from mitotic Golgi fragments. Cell. 82:905–914. [DOI] [PubMed] [Google Scholar]
- Rabouille, C., H. Kondo, R. Newman, N. Hui, P. Freemont, and G. Warren. 1998. Syntaxin 5 is a common component of the NSF- and p97-mediated reassembly pathways of Golgi cisternae from mitotic Golgi fragments in vitro. Cell. 92:603–610. [DOI] [PubMed] [Google Scholar]
- Seemann, J., E.J. Jokitalo, and G. Warren. 2000. The role of the tethering proteins p115 and GM130 in transport through the Golgi apparatus in vivo. Mol. Biol. Cell. 11:635–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seemann, J., M. Pypaert, T. Taguchi, J. Malsam, and G. Warren. 2002. Partitioning of the matrix fraction of the Golgi apparatus during mitosis in animal cells. Science. 295:848–851. [DOI] [PubMed] [Google Scholar]
- Shorter, J., and G. Warren. 1999. A role for the vesicle tethering protein, p115, in the post-mitotic stacking of reassembling Golgi cisternae in a cell-free system. J. Cell Biol. 146:57–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shorter, J., and G. Warren. 2002. Golgi architecture and inheritance. Annu. Rev. Cell Dev. Biol. 18:379–420. [DOI] [PubMed] [Google Scholar]
- Sollner, T., S.W. Whiteheart, M. Brunner, H. Erdjument-Bromage, S. Geromanos, P. Tempst, and J.E. Rothman. 1993. SNAP receptors implicated in vesicle targeting and fusion. Nature. 362:318–324. [DOI] [PubMed] [Google Scholar]
- Sutterlin, C., C.Y. Lin, Y. Feng, D.K. Ferris, R.L. Erikson, and V. Malhotra. 2001. Polo-like kinase is required for the fragmentation of pericentriolar Golgi stacks during mitosis. Proc. Natl. Acad. Sci. USA. 98:9128–9132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutterlin, C., P. Hsu, A. Mallabiabarrena, and V. Malhotra. 2002. Fragmentation and dispersal of the pericentriolar Golgi complex is required for entry into mitosis in mammalian cells. Cell. 109:359–369. [DOI] [PubMed] [Google Scholar]
- Uchiyama, K., E. Jokitalo, F. Kano, M. Murata, X. Zhang, B. Canas, R. Newman, C. Rabouille, D. Pappin, P. Freemont, and H. Kondo. 2002. VCIP135, a novel essential factor for p97/p47-mediated membrane fusion, is required for Golgi and ER assembly in vivo. J. Cell Biol. 159:855–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren, G. 1993. Membrane partitioning during cell division. Annu. Rev. Biochem. 62:323–348. [DOI] [PubMed] [Google Scholar]
- Zaal, K.J., C.L. Smith, R.S. Polishchuk, N. Altan, N.B. Cole, J. Ellenberg, K. Hirschberg, J.F. Presley, T.H. Roberts, E. Siggia, et al. 1999. Golgi membranes are absorbed into and reemerge from the ER during mitosis. Cell. 99:589–601. [DOI] [PubMed] [Google Scholar]
- Zerial, M., R. Parton, P. Chavrier, and F. Rainer. 1992. Localization of Rab family members in animal cells. Methods Enzymol. 219:398–408. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.