

Abstract
Downstream of kinase (Dok)–related protein (DokR, also known as p56dok/FRIP/Dok-R) is implicated in cytokine and immunoreceptor signaling in myeloid and T cells. Tyrosine phosphorylation induces DokR to bind the signal relay molecules, RasGTPase-activating protein (RasGAP) and Nck. Here, we have examined the function of DokR during hematopoietic development and the requirement for RasGAP and Nck binding sites in its biological function. Retroviral-mediated expression of DokR in bone marrow cells dramatically inhibited their capacity to form colonies in vitro in response to the cytokines macrophage colony–stimulating factor and stem cell factor, whereas responses to interleukin-3 and granulocyte macrophage colony–stimulating factor were only weakly affected. When introduced into lethally irradiated mice, hematopoietic cells expressing DokR showed a drastically reduced capacity to repopulate lymphoid tissues. Most notably, DokR dramatically reduced repopulation of the thymus, in part by reducing the number of T cell precursors seeding in the thymus, but equally, through inhibiting the transition of CD4−CD8− to CD4+CD8+ T cells. Consequently, the number of mature peripheral T cells was markedly reduced. In contrast, a minimal effect on B cell and myeloid lineage development was observed. Importantly, functional RasGAP and Nck binding sites were found to be essential for the biological effects of DokR in vitro and in vivo.
Keywords: signal transduction; growth inhibition; hematopoiesis; thymocyte; progenitor cell
Introduction
The downstream of kinase (Dok)* proteins are a recently described family of docking molecules that interact in a tyrosine phosphorylation–dependent manner with a range of intracellular signaling molecules (Tomlinson et al., 2000). Five Dok family molecules have been described: p62dok, otherwise known as Dok1 (Carpino et al., 1997; Yamanashi and Baltimore, 1997); p56dok/FRIP/Dok-related protein (DokR) (Di Cristofano et al., 1998; Jones and Dumont, 1998; Nelms et al., 1998; Lock et al., 1999); DOKL/Dok-3 (Cong et al., 1999; Lemay et al., 2000); Dok-4; and Dok-5 (Grimm et al., 2001). Dok proteins contain a pleckstrin homology domain, a phosphotyrosine binding domain, and a COOH-terminal region containing multiple tyrosine phosphorylation sites. These proteins are implicated in the regulation of multiple biological processes, including cell proliferation (Suzu et al., 2000; Yamanashi et al., 2000; Di Cristofano et al., 2001), transformation and tumorigenesis (Cong et al., 1999; Di Cristofano et al., 2001; Songyang et al., 2001), cell motility (Noguchi et al., 1999; Hosooka et al., 2001; Master et al., 2001), and differentiation (Grimm et al., 2001).
DokR is expressed in adult and embryonic hematopoietic tissues including thymus, lymph node, spleen, and fetal liver (Di Cristofano et al., 1998; Nelms et al., 1998; Lock et al., 1999; Lemay et al., 2000; Grimm et al., 2001). Several hematopoietic lineages, including T cells, macrophages, mast cells, and hematopoietic stem cells, express DokR (Nelms et al., 1998; Lemay et al., 2000; Phillips et al., 2000; Suzu et al., 2000), whereas B cells appear to lack detectable levels of DokR (Nelms et al., 1998; Lemay et al., 2000). Intriguingly, DokR mRNA and protein levels increase rapidly in NFS-60 myeloid leukemia cells and in primary macrophages stimulated with macrophage colony–stimulating factor (M-CSF), implying a role for DokR as a feedback regulatory molecule (Suzu et al., 2000).
Cytokines such as M-CSF (Suzu et al., 2000), stem cell factor (SCF) (Wisniewski et al., 1996), and interleukin (IL)-4 (Nelms et al., 1998) have been shown to rapidly induce the tyrosine phosphorylation of endogenous DokR in myeloid cells, whereas cross-linking antibodies to various immunoreceptors, such as CD2 (Nemorin and Duplay, 2000) and signaling lymphocyte activation molecule (SLAM) (Latour et al., 2001), promote DokR tyrosine phosphorylation in T cells. DokR is therefore likely to be a common component in several tyrosine kinase–mediated signaling cascades in these hematopoietic lineages.
Once tyrosine phosphorylated, DokR binds to RasGTPase-activating protein (RasGAP) and Nck (Di Cristofano et al., 1998; Jones and Dumont, 1998, 1999; Nelms et al., 1998; Lock et al., 1999). We and others have shown previously that these interactions are mediated by tyrosine-phosphorylated YxxP motifs located in the COOH terminus of DokR and by SH2 domains in RasGAP and Nck (Di Cristofano et al., 1998; Nelms et al., 1998; Jones and Dumont, 1999; Lock et al., 1999). Specifically, Y351 of DokR is responsible for interaction with Nck, whereas Y276 and Y304 in combination mediate binding to RasGAP. The RasGAP protein is implicated as a negative regulator of Ras, as it is capable of stimulating the intrinsic GTPase activity of Ras, thereby converting it into an inactive GDP-bound form (Boguski and McCormick, 1993). The Nck protein is an adaptor molecule containing three SH3 domains followed by an SH2 domain, and is implicated in cytoskeletal regulation (McCarty, 1998).
Recent evidence suggests that DokR participates in signaling pathways that suppress cell proliferation. When overexpressed, DokR inhibits the proliferation of NFS-60 myeloid leukemia cells stimulated with cytokines, whereas overexpression of antisense DokR constructs enhances the growth of J774 cells (Suzu et al., 2000). The DokR gene has been mapped to the hairless (hr) locus on chromosome 14 (Nelms et al., 1998). This locus also contains the gene for a putative transcription factor (Cachon-Gonzalez et al., 1994). Interestingly, DokR levels are reduced in several hematopoietic cell types from hairless mice (Nelms et al., 1998; Suzu et al., 2000). Analysis of peripheral T cells from these mice showed that they are hyperresponsive to cytokine and T cell receptor (TCR) stimulation (Nelms et al., 1998), while macrophage numbers in these mice are modestly elevated (Suzu et al., 2000). Although these studies support a role for DokR in the negative regulation of cellular proliferation, the requirement for DokR interactions with Nck and RasGAP in mediating these effects has not been investigated. Moreover, the role of DokR in vivo remains unclear, as the involvement of genes such as the hr gene in causing the phenotype of hairless mice is not known.
In the following study, we explore the physiological role of DokR in hematopoiesis. We demonstrate that constitutive expression of DokR in bone marrow cells differentially inhibits cytokine-dependent proliferation of progenitor cells in vitro and selectively inhibits early T cell development in reconstituted mice, suppressing both the seeding of the thymus and the progression of thymocytes from the CD4−CD8− to the CD4+CD8+ stage, resulting in a marked decrease in peripheral T cells. Furthermore, both RasGAP and Nck are implicated in mediating the biological activity of DokR, as no inhibitory activity in vitro or in vivo was observed with DokR mutants deficient in binding sites for either protein.
Results
DokR selectively inhibits cytokine-dependent colony formation by hematopoietic progenitor cells
To study the role of DokR in primary hematopoietic cells, cDNAs encoding FLAG-tagged DokR or the FLAG–DokR mutants, Y351F and Y276/304F, shown previously to lack functional binding sites for Nck and RasGAP, respectively (Jones and Dumont, 1998, 1999; Lock et al., 1999; Fig. 1 A), were inserted into the retroviral vector pMY-EGFP (Onai et al., 2000; Fig. 1 B). This bicistronic vector is capable of driving transcription in a wide range of hematopoietic lineages (Onai et al., 2000), and contains an internal ribosomal entry site (IRES) and the gene encoding green fluorescent protein (GFP), enabling cells expressing DokR (or mutant thereof) to be detected by GFP fluorescence. High-titre stocks of the control (empty) retrovirus and those encoding the different forms of FLAG–DokR were generated by transfection of Bosc23 virus–producing cells (Pear et al., 1993). Bone marrow cells from C57BL/6 mice treated with 5-fluorouracil (to enrich for immature hematopoietic cells) were serially infected with equivalent numbers of the different retroviruses. As shown in Fig. 1 C, flow cytometric analysis of the cells 7 d after infection revealed that all four retroviruses had a similar infection capability, as judged by the similar fractions of GFP+ cells generated. Notably, although infection efficiency varied between experiments, in any given experiment each retrovirus infected a comparable number of cells (unpublished data). Immunofluorescent staining of bone marrow cells showed that a significant proportion of infected (GFP+) as well as noninfected cells expressed c-Kit (Fig. 1 C), a marker found mainly on immature cells, but also on mast cells. Cytospin analysis of infected cells revealed that <0.5% of cells were mast cells, whereas the majority of cells were immature myeloid cells, including blasts (40–46%) and promyelocytes (27–35%). Less than 9% of cells had differentiated into neutrophils (2–5%), eosinophils (1–2%), or macrophages (1–2.5%) (unpublished data). We could detect no clear differences in the proportions of these cell types in bone marrow infected with the various retroviruses. Immunoprecipitation analysis with a FLAG antibody and immunoblot analysis with DokR-specific antiserum confirmed that the bone marrow cells expressed the wild-type and mutant FLAG–DokR proteins (Fig. 1 D).
Figure 1.
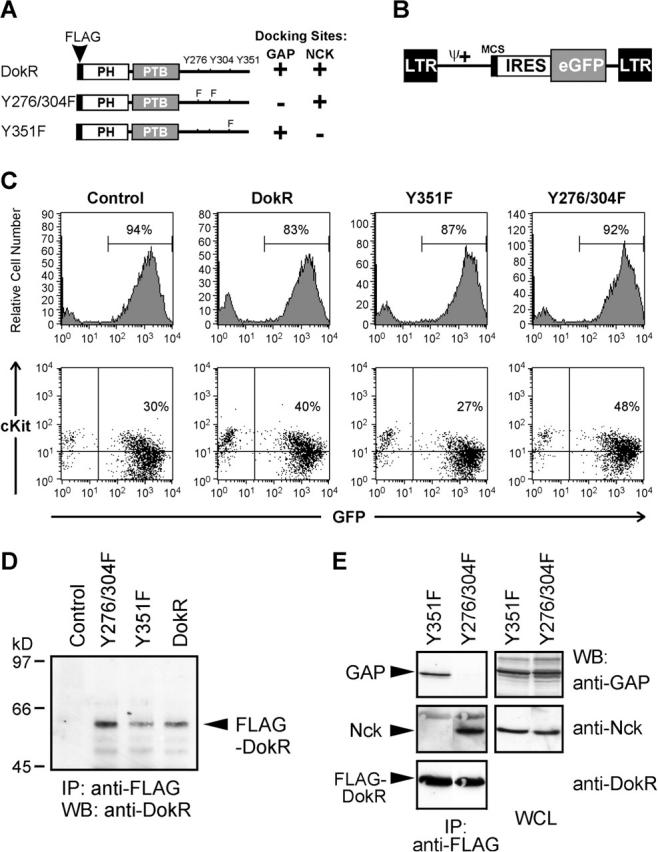
Retrovirus-mediated expression of FLAG–DokR proteins in murine bone marrow cells. (A) Schematic representation of FLAG–DokR and mutants thereof showing the location of tyrosine (Y) to phenylalanine (F) substitutions. The ability (+) or inability (−) of the DokR proteins to bind RasGAP and Nck is indicated (also see E). (B) Diagram of pMY-EGFP, the bicistronic retroviral vector used to express FLAG–DokR proteins. The viral long terminal repeats (LTR), extended packaging signal (ψ+), multiple cloning site (MCS), IRES, and enhanced GFP (eGFP) cassette are indicated. (C) Flow cytometric analysis of bone marrow cells 7 d after infection with the indicated retroviral constructs, showing the percentages of GFP+ (top) and GFP+/cKit+ cells (bottom). (D) Expression of retrovirally expressed FLAG–DokR proteins in bone marrow cells was examined by immunoprecipitation (IP) with an anti-FLAG antibody and Western blot (WB) analysis with an anti-DokR serum. (E) 293T cells were transfected with pMY-IRES–GFP retroviral vectors encoding Y351F or Y276/304F together with a pEF-BOS vector encoding Lyn. Cell lysates were analyzed 48 h after transfection by immunoprecipitation with anti-FLAG antibody and Western blotting with anti-GAP, anti-Nck, and anti-DokR sera. As a control, samples of whole cell lysate (WCL), containing 5% of the protein that was analyzed by IP, were probed with anti-GAP and anti-Nck.
We were interested in confirming the binding specificity of the retrovirus vector–encoded FLAG–DokR mutants. Retroviral constructs encoding Y351F and Y276/304F were therefore introduced into 293T cells with the Src kinase, Lyn, to induce DokR tyrosine phosphorylation. Immunoprecipitation analysis of the DokR mutants (Fig. 1 E) clearly demonstrated that the Y351F mutant binds selectively to RasGAP and, conversely, the Y276/304F mutant binds selectively to Nck, confirming our previous analysis of these mutants (Lock et al., 1999). Our results do not exclude the possibility that the Y351 and Y276/Y304 docking sites could also mediate binding to unknown proteins besides RasGAP and Nck. We also tried to demonstrate FLAG–DokR interactions in virus-infected bone marrow cells that were treated with pervanadate to enhance cellular tyrosine phosphorylation and SH2-dependent interactions (Harder et al., 2001). However, we were unable to detect interaction of wild-type or mutant FLAG–DokR proteins with RasGAP or Nck in these cells, presumably because we had only low numbers of cells for analysis, the endogenous levels of RasGAP and Nck were too low for detection by the antibodies used, and/or these interactions may normally only occur in a small fraction of the total cells. Consistent with the latter possibility, the biological effects of DokR that we observed in in vitro colony formation assays (Fig. 2) were apparent only in a minor fraction of cells.
Figure 2.

Retrovirus-mediated expression of FLAG–DokR but not DokR mutants in bone marrow cells differentially suppresses cytokine-dependent proliferation in vitro. Graphs show the numbers of GFP+ colonies formed by bone marrow cells infected with the indicated retroviral constructs and stimulated with the indicated cytokines. Numbers are the arithmetic means ± SEM of quadruplicate determinations in three independent experiments. *P < 0.05, **P < 0.01, and ***P < 0.001, compared with control (empty) retrovirus-infected cells (t test).
To assess the effects of retrovirus-mediated expression of DokR in primary hematopoietic progenitor cells, noninfected bone marrow cells or cells infected with the various retroviruses were subjected to in vitro colony formation assays in the presence of SCF, IL-3, granulocyte macrophage colony–stimulating factor (GM-CSF), or M-CSF. Cells infected with the control (empty) virus and noninfected cells formed equivalent numbers of colonies in each of the cytokines tested (unpublished data), indicating that the infection procedure per se did not affect colony forming ability. On the other hand, Fig. 2 shows that bone marrow cells infected with the DokR-encoding retrovirus formed fewer GFP+ colonies than control virus–infected cells in response to each cytokine, but the extent of this reduction varied dramatically. For instance, DokR virus–infected cells formed 35-fold fewer colonies than control virus–infected cells when cultured in the presence of M-CSF, whereas a fourfold reduction was observed in the presence of SCF and less than twofold reductions were observed with IL-3 and GM-CSF. In contrast, cells infected with viruses encoding the DokR mutants, Y351F and Y276/304F, formed similar or in some cases significantly greater colony numbers than control virus–infected cells (Fig. 2). These results imply that M-CSF and SCF signaling in hematopoietic progenitor cells is preferentially inhibited by enforced expression of DokR, and that tyrosines Y351 (which mediates binding to Nck) and Y276 and Y304 (which mediate RasGAP binding) are essential for its growth inhibitory activity.
Enforced expression of DokR in bone marrow cells suppresses reconstitution of lymphoid tissues in lethally irradiated mice
To explore the role of DokR in vivo, we compared the ability of bone marrow cells infected with the control, DokR, Y351F, or Y276/304F retroviruses to reconstitute hematopoiesis in lethally irradiated mice. For simplicity, reconstituted animals will be referred to as control, DokR, Y351F, or Y276/304F mice, respectively. 10 wk after reconstitution, the spleens, thymi, and bone marrow of reconstituted mice were collected and the cellularity of each tissue was determined. Table I shows that the total numbers of thymocytes and bone marrow cells (including GFP+ and GFP− cells) did not differ significantly between the different classes of reconstituted mice, whereas the Y351F and Y276/304F mice contained about twofold greater numbers of splenocytes than control mice. This increased cellularity appears to be related to reconstitution efficiency rather than to a direct effect of the DokR mutant proteins Y351F or Y276/304F, as the numbers of both GFP− as well as GFP+ splenocytes are increased (Table I; unpublished data).
Table I. DokR suppresses the capacity of bone marrow cells to repopulate lymphoid tissues in irradiated mice.
| Tissue | Group | n | Number of leukocytesa | GFP+ leukocytesb | GFPhi leukocytesc | Number of GFP+ leukocytesd | Number of GFPhi leukocytesd |
|---|---|---|---|---|---|---|---|
| ×106 | % | % | ×106 | ×106 | |||
| Spleen | |||||||
| Control | 18 | 126 ± 22 | 46 ± 5 | 28 ± 4 | 54 ± 11 | 35 ± 9 | |
| DokR | 17 | 106 ± 20 | 14 ± 4g | 3.5 ± 1.3g | 16 ± 6e | 4.2 ± 1.9f | |
| Y351F | 7 | 272 ± 30f | 35 ± 5 | 17 ± 4 | 93 ± 14 | 45 ± 9 | |
| Y276/304F | 9 | 212 ± 39e | 41 ± 7 | 21 ± 4 | 78 ± 20 | 46 ± 15 | |
| Thymus | |||||||
| Control | 18 | 104 ± 11 | 48 ± 9 | 45 ± 9 | 47 ± 10 | 43 ± 10 | |
| DokR | 17 | 75 ± 10 | 7 ± 3g | 1.2 ± 0.5g | 2.4 ± 1g | 0.4 ± 0.1g | |
| Y351F | 7 | 111 ± 28 | 40 ± 12 | 31 ± 13 | 36 ± 13 | 31 ± 14 | |
| Y276/304F | 9 | 74 ± 17 | 52 ± 14 | 44 ± 13 | 32 ± 11 | 29 ± 10 | |
| Bone marrow | |||||||
| Control | 16 | 34 ± 4 | 45 ± 8 | 33 ± 6 | 15 ± 3 | 10 ± 2 | |
| DokR | 16 | 34 ± 3 | 24 ± 7 | 17 ± 5 | 8 ± 3 | 6 ± 2 | |
| Y351F | 4 | 35 ± 2 | 37 ± 15 | 23 ± 9 | 12 ± 4 | 11 ± 3 | |
| Y276/304F | 6 | 41 ± 7 | 32 ± 11 | 22 ± 9 | 16 ± 9 | 11 ± 6 |
Splenocytes, thymocytes and bone marrow cells from the femurs of mice were harvested 10 wk after reconstitution with bone marrow cells infected with the indicated retroviral constructs. All values are the arithmetic means ± SEM of determinations from up to 18 animals per experimental group (see n values). P values were determined using an unpaired t test and are based on comparisons of the indicated data with corresponding control data.
Leukocyte numbers (GFP− plus GFP+ cells) were determined hemocytometrically after RBC lysis.
The percentage of GFP+ leukocytes was determined by flow cytometry.
GFPhi is defined as >103 log10 fluorescence units.
Numbers of GFP+ and GFPhi leukocytes were determined by multiplying the percentage of GFP+ or GFPhi cells by the total cell number.
P < 0.05.
P < 0.01.
P < 0.001.
Comparison of the proportion of leukocytes expressing GFP in control and DokR mice revealed that an average of 45% of bone marrow cells, 46% of splenocytes, and 48% of thymocytes were GFP+ in the control mice (Fig. 3 A; Table I), whereas only ∼24, 14, and 7% of the respective cell types were GFP+ in DokR mice (Table I; Fig. 3 A). The reduced percentage of GFP+ splenocytes and thymocytes, but not bone marrow cells, in DokR mice was highly statistically significant. In contrast, the percentages of these cells in Y351F and Y276/304F mice were not significantly different to those observed for the control mice (Fig. 3 A; Table I). Quantitation of the absolute numbers of GFP+ cells in reconstituted mice revealed that DokR mice contained about threefold fewer GFP+ splenocytes and nearly 20-fold fewer GFP+ thymocytes than control mice (Table I), whereas Y351F and Y276/304F mice contained slightly more GFP+ splenocytes and slightly fewer thymocytes than the control mice (Table I). Collectively, these results provide evidence that the capacity of hematopoietic precursor cells to repopulate the spleens and, even more dramatically, the thymi of irradiated mice was suppressed by enforced expression of DokR. These results also implicate the tyrosine phosphorylation sites that mediate binding of DokR to RasGAP and Nck as critical determinants of its biological activity.
Figure 3.
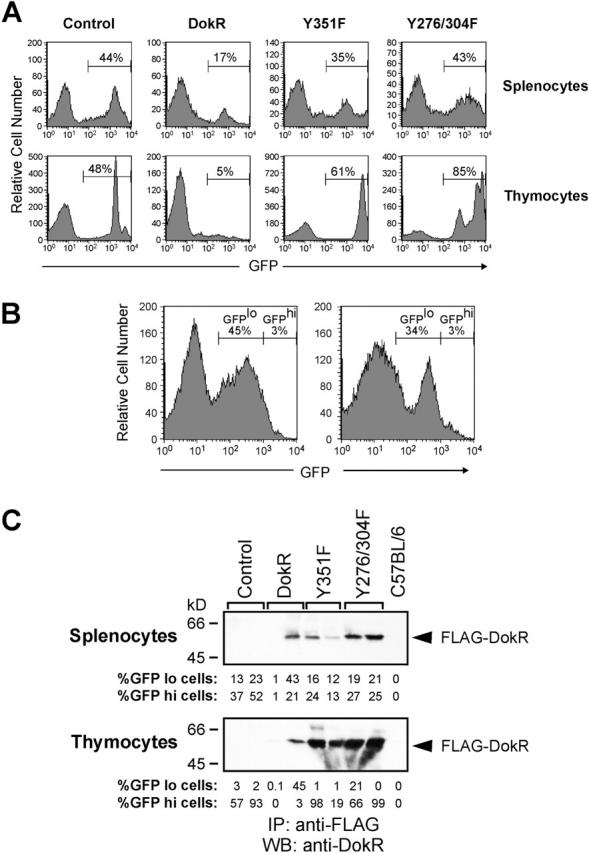
Retrovirus-mediated expression of FLAG–DokR but not DokR mutants inhibits the ability of bone marrow cells to repopulate lymphoid tissues in irradiated mice. (A) Flow cytometric analysis of splenocytes and thymocytes from reconstituted mice. Animals were reconstituted with bone marrow cells that had been infected in vitro with the indicated retroviral constructs. The histograms shown are representative of data obtained from 7 to 18 mice per reconstitution group. The percentage of GFP+ cells of each cell type is indicated (Table I). (B) Flow cytometric analysis of thymocytes from two mice reconstituted with bone marrow cells infected with the DokR- expressing retrovirus. These data are representative of a minority of DokR mice (3 out of 17) that contained >6% GFP+ cells. The percentages of GFPlo (defined arbitrarily as <103 log10 fluorescence units) and GFPhi cells (>103 log10 fluorescence units) are indicated. (C) Expression of retrovirus-encoded FLAG–DokR proteins in splenocytes and thymocytes of reconstituted mice was assessed by immunoprecipitation (IP) with anti-FLAG antibodies and Western blot (WB) analysis with an anti-DokR serum. The percentage of GFPlo and GFPhi cells (see legend in B) in each sample is indicated.
The thymi in a minority (<20%) of DokR mice contained a relatively high percentage (up to 48%) of GFP+ thymocytes but, strikingly, most of these cells expressed a relatively low level of GFP (GFPlo is henceforth arbitrarily defined as <103 log10 fluorescence units) (Fig. 3 B), whereas the vast majority of thymocytes from control, Y351F, or Y276/304F mice expressed GFP at high levels (GFPhi, >103 log10 fluorescence units) (Fig. 3 A; unpublished data). Comparison of the absolute numbers of GFPhi cells in reconstituted mice revealed that, on average, the thymi of DokR mice contained >110-fold fewer GFPhi cells than control mice (Table I), whereas Y351F and Y276/304F mice contained less than twofold fewer GFPhi thymocytes than control mice (Table I). Similarly, the spleens of DokR mice contained about eightfold fewer GFPhi cells than the control mice, whereas the number of GFPhi cells in the spleens of Y351F and Y276/304F mice was slightly greater than that in control mice (Table I). These results suggest that the ability of DokR to inhibit repopulation of the thymus and the spleen is concentration dependent.
To assess whether the levels of GFP measured by flow cytometry accurately represented the levels of FLAG–DokR proteins in cells, splenocytes and thymocytes from reconstituted mice were analyzed by immunoprecipitation with an anti-FLAG antibody and immunoblotting with an anti-DokR serum (Fig. 3 C). As expected, FLAG–DokR proteins were not detected in splenocytes or thymocytes from control mice or nonreconstituted C57BL/6 mice. However, wild-type and mutant forms of FLAG–DokR were detected in both cell types at a range of levels that correlated well with the percentage of GFPlo and GFPhi cells in the individual samples (Fig. 3 C). Notably, the levels of wild-type FLAG–DokR in thymocytes from DokR mice were much lower than those of DokR mutant proteins in Y351F and Y276/304F mice (Fig. 3 C; unpublished data), in agreement with our finding that thymi from DokR mice contained a significantly lower proportion of GFP+ cells than Y351F or Y276/304F mice (Table I). Importantly, thymocytes from DokR mice containing a relatively high percentage of GFPlo cells (e.g., Fig. 3 C, lane 4) were also found to contain very little FLAG–DokR protein, confirming that GFP levels closely reflected cellular FLAG–DokR protein levels.
Retrovirus-mediated expression of DokR suppresses seeding of the thymus and thymocyte maturation
The most dramatic effect of DokR was its ability to inhibit repopulation of the thymus (Table I). This could be due to reduced numbers of thymocyte precursors, inhibition of migration of thymocyte precursors to the thymus, and/or maturation of thymocyte precursors into the more abundant mature thymocyte populations. To examine more carefully the effect of DokR on thymic repopulation, thymocytes from reconstituted mice were stained with immunofluorescent antibodies to CD4 and CD8 and analyzed by flow cytometry. Although the number of total thymic GFP+ cells was dramatically reduced in DokR mice, we were nevertheless able to detect GFP+ thymocytes corresponding to each of the four major classes of thymic T cells, including CD4−CD8− double negative (DN) cells (including pro–T cells), CD4+CD8+ double positive (DP) cells (immature T cells), and CD4+ and CD8+ single positive (SP) cells (representing mature T cells) (Fig. 4 A). Interestingly however, the percentage of GFP+ cells of the DN phenotype was ∼4.5-fold higher in DokR mice compared with control mice and up to sevenfold higher than in DokR mutant mice (Fig. 4 A; unpublished data). By contrast, the percentage of GFP+ DP cells in DokR mice was consistently ∼20% lower than in control, Y351F, and Y276/304F mice (Fig. 4 A; unpublished data).
Figure 4.
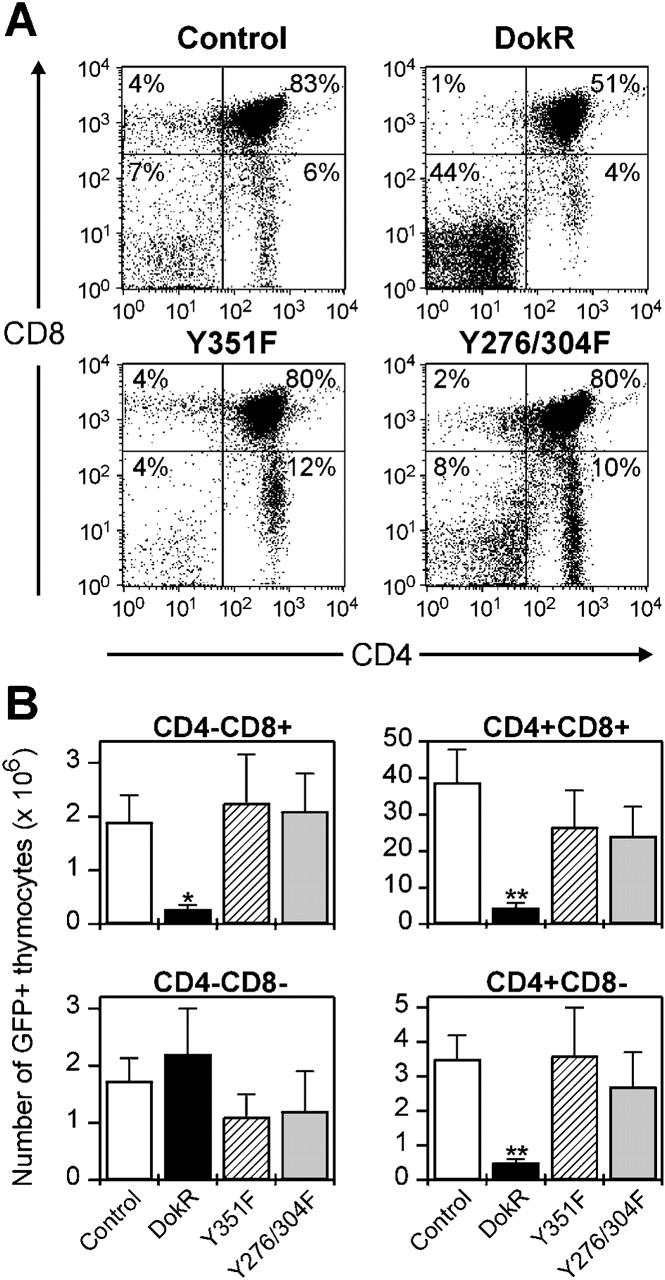
Constitutive expression of DokR suppresses CD4 − CD8 − to CD4 + CD8 + thymocyte progression. (A) Flow cytometric analysis showing the expression of CD4 and CD8 (detected by immunofluorescent staining with tricolor- and PE-conjugated antibodies, respectively) on GFP+ thymocytes from reconstituted mice. The percentage of GFP+ thymocytes of each thymic subset is indicated. Dot plots shown are representative of data corresponding to between 7 and 13 mice per reconstitution group. (B) Bar graphs showing the absolute numbers of GFP+ thymocytes of the indicated thymocyte subsets in groups of reconstituted mice. These figures were calculated by determining the proportion of GFP+ thymocytes of each subset by immunofluorescence staining and flow cytometry and multiplication by the total thymocyte number (Table I). The data represent the arithmetic means ± SEM of determinations from n = 7 to n = 13 mice per group of reconstituted mice. *P < 0.05 and **P < 0.01, compared with control retrovirus–infected cells (unpaired t test).
Because the total numbers of GFP+ thymocytes in DokR mice were dramatically reduced (Table I), we were interested in knowing what the effect of DokR was on the absolute numbers of GFP+ cells at each stage of development (Fig. 4 B). Also, although the fraction of GFP+ cells that were DN was significantly increased in DokR mice, these cells normally represent only a minor population and their relative proportion could easily be affected by changes in the frequencies of other thymocyte populations. Indeed, analysis of absolute numbers demonstrated that, on average, DokR mice had similar total numbers of GFP+ DN cells to the other classes of reconstituted mice (Fig. 4 B, see legend). By contrast, however, there were about eightfold fewer GFP+ DP thymocytes in DokR mice than the control mice. Because these cells normally account for ∼80% of total thymocytes, it is their reduced numbers in DokR mice that is primarily responsible for the marked reduction in total GFP+ thymocytes. In parallel with this, DokR mice also contained six- to sevenfold fewer mature GFP+ SP thymocytes compared with the control mice. In the Y351F and Y276/304F mice, the numbers of GFP+ thymocytes of each subtype were not significantly different from those in the control mice.
Because DN cells can include cells of non–T cell lineages, such as B cells, NK cells, dendritic cells, and myeloid cells, we wanted to exclude these non–T cells from our analysis. To do this, all cells expressing the markers Gr-1, Mac-1, B220, and MHC class II, as well as T cells expressing CD4 and CD8 were eliminated by cell sorting. The remaining DN T cell precursors were then examined by flow cytometry for expression of GFP. Fig. 5 shows that a high proportion (on average 20%) of the resulting DN T cell precursors derived from DokR mice were GFP+, confirming that DokR-expressing thymocyte precursors are generated in the bone marrow and are able to migrate to the thymus. Nevertheless, the proportion of GFP+ cells was approximately two- to threefold higher in the control mice, suggesting that DokR partially inhibits the generation of these cells from infected bone marrow cells or suppresses their ability to migrate into the thymus. However, because the total numbers of immature (DP) and mature (SP) thymocytes were more dramatically reduced (by up to eightfold; Fig. 4 B), DokR also clearly inhibits progression of DN T cells into more mature T cells. Collectively these results suggest that DokR inhibits repopulation of the thymus in two ways; first by inhibiting the seeding of the thymus with T cell precursors, either by inhibiting their formation or their migration to the thymus and, second, by inhibiting their maturation from DN T cell precursors into DP T cells. Furthermore, the activity of DokR is dependent on both its RasGAP and Nck binding sites.
Figure 5.

DokR partially suppresses seeding of the thymus by T cell precursors. Representative histograms showing flow cytometric analysis of thymic T cell precursors from control and DokR mice (n = 4 for each class of mice). The cells analyzed were prepared by sorting for cells that lacked surface expression of the following lineage markers: CD4, CD8, MHC class II, B220, Gr-1, and Mac-1 (see Materials and methods). The percentage of GFP+ cells is indicated.
DokR selectively inhibits T lymphoid development
Mature T lymphocytes leave the thymus and migrate to peripheral lymphoid tissues, such as the spleen and lymph nodes. To assess the effects of DokR on peripheral T cells, splenocytes were stained with antibodies to CD4 and CD8 and analyzed by flow cytometry. Consistent with the greatly reduced numbers of GFP+ SP T cells in the thymi of DokR mice (Fig. 4 B), we found that the frequency of GFP+ splenocytes expressing CD4 or CD8 in DokR mice (Fig. 6 A, bottom) was substantially lower than in the control or the DokR mutant mice. By contrast, the percentage of CD4+ or CD8+ cells derived from GFP− (noninfected) precursor cells was similar in DokR mice to that in control and DokR mutant mice (Fig. 6 A, top). In absolute terms, the spleens of DokR mice contained about eightfold fewer GFP+ T cells than control mice (unpublished data). An increase in the total numbers of GFP+ splenic T cells in the mutant mice was observed, suggesting a possible dominant negative effect, although the significance of this finding is unclear, as, overall, the total splenic cellularity (including GFP− cells) was also increased (Table I).
Figure 6.

Constitutive expression of DokR suppresses production of mature T cells but not mature B cells. (A) Flow cytometric analysis showing the percentage of GFP− (top) and GFP+ (bottom) splenocytes expressing CD4 and CD8. The results shown are representative of data obtained from 7 to 13 mice per experimental group. (B) Flow cytometric analysis showing the expression of IgM and B220 on GFP− (top) and GFP+ (bottom) splenocytes from reconstituted mice. The percentages of GFP− and GFP+ B lymphocytes (IgM+B220+ cells) are indicated. Note that because of compensation for the high levels of fluorescence emitted by the GFP+ (B220+IgM+) B cells, the DN (B220−IgM−) population (bottom left) was partially obscured. Dot plots shown are representative of data pertaining to 7–13 mice per experimental group.
The decreased percentage of GFP+ splenic T cells in the DokR mice was accompanied by a small corresponding increase in the percentage of GFP+ B cells (Fig. 6 B). Despite this, a small reduction (approximately twofold) in the total numbers of GFP+ splenic B cells in DokR mice was observed, but this was not found to be significant (unpublished data). No significant differences were observed in the total numbers of splenic B cells for the four groups of reconstituted mice (unpublished data). These results suggest that constitutive expression of DokR in bone marrow cells does not greatly affect B cell development in vivo.
Our in vitro colony assay data showing inhibition of M-CSF– and SCF-dependent colony formation by DokR (Fig. 2) suggested that DokR might affect myeloid lineage development in vivo. Surprisingly, however, DokR mice showed only a slight reduction (although not significant) in the percentage of GFP+ bone marrow cells (which contain a high proportion of myeloid cells) compared with control mice (Table I). Moreover, as shown in Fig. 7 A, analysis of GFP+ cells from reconstituted mice revealed that similar percentages of these cells were of a myeloid (Gr-1+ and Mac-1+ cells) or erythroid lineage (Ter-119+ cells) in control and DokR mice.
Figure 7.
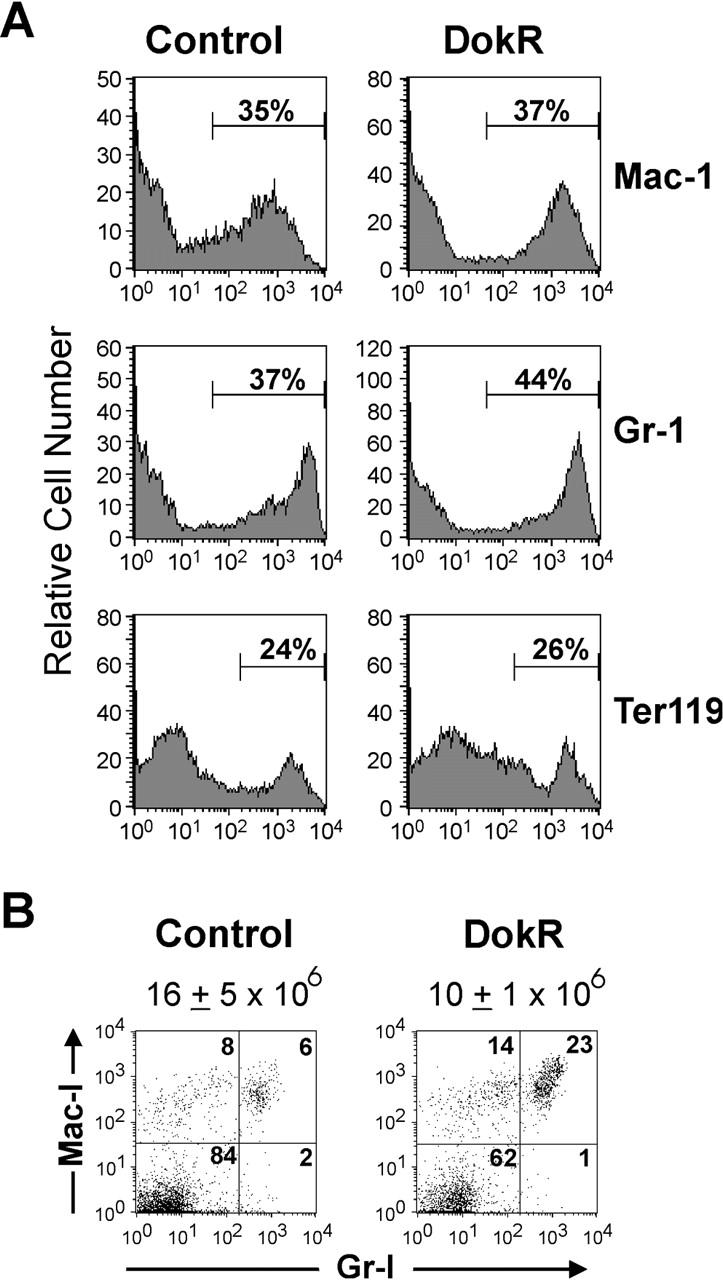
Phenotypic analysis of myeloid lineages in the bone marrow and blood of reconstituted mice. (A) Flow cytometric analysis of bone marrow leukocytes from reconstituted control and DokR mice. Histograms show the percentage of GFP+ cells that express the myeloid lineage cell surface markers Mac-1 and Gr-1 and the erythroid lineage marker Ter119. (B) Flow cytometric analysis of GFP+ blood leukocytes from control and DokR mice showing differential expression of Gr-1 and/or Mac-1. The numbers in each quadrant are percentages. The mean total leukocyte concentration ± SEM (per ml of blood) is indicated above the dot plots (control mice, n = 6; DokR mice, n = 5).
Although the bone marrow analysis suggested a minimal effect of DokR on myeloid lineages, we were interested in knowing whether these results were reflected in myeloid lineages from other hematopoietic compartments. We therefore analyzed leukocytes from the blood of control and DokR mice for expression of GFP, Gr-1, and Mac-1. Here, we again observed a small, but in this case significant (greater than twofold), reduction in the percentage of GFP+ cells in DokR mice versus control mice (33 ± 4% vs. 14 ± 2%, respectively; unpublished data). Analysis of the GFP+ cells shows that the percentages of Gr-1−/Mac-1+ (monocytes) and Gr-1+/Mac-1+ cells (neutrophils) were approximately two- to fourfold higher, respectively, in DokR mice versus the control mice (Fig. 7 B; unpublished data). However, when leukocyte numbers were taken into account (Fig. 7 B), we found that the absolute concentration of GFP+ monocytes in DokR mice was similar to that in control mice (1.3 ± 0.5 × 106/ml vs. 1.9 ± 0.4 × 106/ml, respectively) and the concentration of neutrophils was slightly higher (2 ± 106/ml vs. 1.6 ± 106/ml). Presumably, the reduced percentage of GFP+ cells not expressing Gr-1 and Mac-1 in DokR mice (Fig. 7 B) was due to a relative decrease in the representation of cells of other lineages (e.g., T lymphoid cells). These results suggest that constitutive expression of DokR does not significantly affect myeloid lineage development in vivo.
Expression of FLAG–DokR in T lymphoid, B lymphoid, and myeloid lineages
To examine the levels of FLAG–DokR protein in individual hematopoietic lineages and to be sure that GFP levels reflected accurately the FLAG–DokR expression levels, we pooled splenocytes from two control or two DokR mice and sorted them into populations containing Thy-1+, B220+, or Gr-1+/Mac-1+ plus Gr-1−/Mac-1+ populations (mature T cells, B cells, and myeloid cells, respectively). Flow cytometric analysis confirmed that all cell populations were of a high purity (85–97% pure; unpublished data). Cell lysates prepared from each lineage were then analyzed by Western blotting with anti-DokR antibodies. As shown in Fig. 8, endogenous DokR was just detectable in T cells and myeloid cells, but not in B cells from the control spleens, consistent with its reported distribution (Nelms et al., 1998; Lemay et al., 2000). Comparison of this expression pattern with that in the DokR spleens clearly demonstrates that FLAG–DokR was overexpressed in myeloid cells and B cells but, by contrast, was only slightly elevated compared with endogenous DokR in splenic T cells. The levels of FLAG–DokR protein closely matched the relative proportion of GFP+ cells in each sample (values indicated below immunoblot). By contrast, the levels of RasGAP and Nck in each sample were roughly equivalent. These results strongly support our contention, based on GFP expression data, that DokR selectively inhibits T cell development.
Figure 8.

Reduced expression of FLAG–DokR in T cells relative to B cells and myeloid cells. B cells, T cells, and myeloid cells were purified from pooled splenocytes from two control or two DokR mice by using cell sorting to isolate B220+, Thy-1+, or Gr-1+/Mac-1+ plus Gr-1−/Mac-1+ cells, respectively. The relative levels of expression of endogenous DokR and FLAG–DokR proteins in equivalent samples (5 × 104 cells) were determined by Western blot (WB) analysis of whole cell lysate (WCL) with an anti-DokR antibody. The blot was reprobed with anti-GAP and anti-Nck antibodies. The percentage of GFP+ cells in each sample is indicated.
Discussion
DokR is expressed in several hematopoietic tissues (Di Cristofano et al., 1998; Nelms et al., 1998; Lock et al., 1999; Lemay et al., 2000; Grimm et al., 2001) and implicated in cytokine and immunoreceptor signaling (Nelms et al., 1998; Nemorin and Duplay, 2000; Suzu et al., 2000; Latour et al., 2001), but its role in hematopoietic development is not known. Using bone marrow reconstitution to study DokR function in vivo, we found that DokR drastically inhibited the ability of bone marrow cells to repopulate the thymi and spleens of recipient mice. The efficiency with which cells reconstituted the thymus appeared to be inversely related to cellular DokR levels, suggesting that DokR inhibited thymic reconstitution in a dose-dependent manner. Phenotypic analysis of thymocytes and splenocytes derived from infected (GFP+) precursors using T lineage markers showed that DokR suppressed both thymocyte seeding and the progression of DN to DP thymocytes, leading to a marked deficit of mature SP T cells in the thymus and, consequently, in the spleen. In contrast to its inhibitory effects on T cell re-population, DokR had only a modest inhibitory effect on the ability of bone marrow cells to develop into mature (IgM+B220+) B cells and did not markedly affect the development of myeloid lineages, such as neutrophils (Gr-1+/Mac-1+ cells) or monocytes (Gr-1−/Mac-1+ cells) (see below).
DokR partially inhibited the seeding of T cell precursors in the thymus. This could be due to reduced expansion of bone marrow–derived T cell precursors, leading to fewer cells capable of repopulating the thymus. Alternatively, this could be a consequence of an impaired homing capacity of these precursors such that these cells develop normally but fail to migrate to the thymus. At present we cannot distinguish between these two possibilities. We also detected a marked inhibitory effect of DokR on T cell maturation within the thymus. This effect was largely confined to a specific developmental checkpoint: the transition of primitive CD4−CD8− DN thymocytes into immature CD4+CD8+ DP thymocytes (Fig. 4). Intriguingly, similar T cell maturation defects occur in several null mutant mouse strains. Mice lacking Lck (Molina et al., 1992), SLP-76 (Clements et al., 1998; Pivniouk et al., 1998), linker of activated T cells (LAT) (Zhang et al., 1999), Gads (Yoder et al., 2001), or components of the pre-TCR complex (von Boehmer and Fehling, 1997) show impaired thymocyte maturation beyond the DN stage. All of these proteins appear to participate in a common signaling pathway, initiated at the pre-TCR, and controlling CD4−CD8− to CD4+CD8+ thymocyte progression (von Boehmer and Fehling, 1997; Pivniouk and Geha, 2000; Tomlinson et al., 2000). The pre-TCR is thought to trigger the activation of cytoplasmic protein tyrosine kinases, Lck, Fyn, and Zap70, leading to the phosphorylation of LAT and SLP-76, among other proteins (Pivniouk and Geha, 2000; Tomlinson et al., 2000; Latour and Veillette, 2001). In turn, LAT and SLP-76 recruit several effector proteins that together coordinate the activation of the Ras pathway, mobilization of intracellular Ca2+ stores, and reorganization of the actin cytoskeleton (Pivniouk and Geha, 2000; Tomlinson et al., 2000). Our results showing reduced DN thymocyte progression indicate that DokR might negatively regulate this signaling cascade. In support of this hypothesis, Lck and Fyn are strongly implicated in signaling pathways that control DokR tyrosine phosphorylation and its association with RasGAP in T cell lines and primary thymocytes (Nemorin and Duplay, 2000; Latour et al., 2001). Moreover, we showed that the RasGAP binding site of DokR (Y276 and Y304) was essential for its inhibitory activity. Thus, it is plausible that the reduced progression of CD4−CD8− into CD4+CD8+ thymocytes we observed is mediated, at least in part, by RasGAP–DokR complexes that facilitate inhibition of Ras signaling. However, our results strongly argue that, in addition to recruitment of RasGAP, binding of Nck (via Y351) is also critical for the inhibitory function of DokR.
Nck is an adaptor protein that contains three SH3 domains that bind a number of signaling proteins involved in the regulation of actin cytoskeleton–dependent processes such as membrane ruffling and axon guidance (Rivero-Lezcano et al., 1995; Bokoch et al., 1996; Galisteo et al., 1996; Holland et al., 1997; Lu et al., 1997; Anton et al., 1998; McCarty, 1998). Interestingly, mutation of the Nck binding site in p62dok was found to suppress CHO cell migration in response to insulin, implying that p62dok-associated Nck also regulates cell motility (Noguchi et al., 1999). Very recently, interaction of DokR with Nck was shown to play a central role in mediating endothelial cell motility in response to angiopoietin-1 (Master et al., 2001). It is tempting to speculate that the migratory capacity of hematopoietic cells overexpressing DokR is also enhanced in an Nck-dependent manner and, consequently, cell growth might be indirectly affected, perhaps by altering exposure to growth stimulatory signals. Recent studies on a novel mammalian Nck isoform, Nckβ/Nck-2/Grb4 (Chen et al., 1998; Tu et al., 1998; Coutinho et al., 2000), revealed that this protein can suppress growth factor–stimulated DNA synthesis (Chen et al., 1998). It remains to be determined whether Nckβ/Nck-2/Grb4 associates with DokR in hematopoietic cells and contributes to its inhibitory activity. Mice deficient for both Nck isoforms have been generated recently, and although cells from these mice are resistant to actin cytoskeletal remodelling induced by the Tir protein of a pathogenic form of Escherichia coli, EPEC (Gruenheid et al., 2001), cell growth defects have not been reported. Irrespective of such a role, our results showing that the DokR Y276/304F mutant (which contains a functional Nck binding site) had little or no effect on T cell maturation or progenitor cell growth suggest that binding to Nck alone is insufficient for DokR to exert its effects. On this basis, we propose that DokR, Nck, and RasGAP might cooperate to negatively regulate cell growth.
In a previous study, DokR suppression of M-CSF– and SCF-dependent proliferation of NFS-60 leukemia cells was described (Suzu et al., 2000). We have extended these studies by demonstrating that DokR can also inhibit colony formation of primary hematopoietic cells, namely bone marrow–derived myeloid progenitor cells, in the presence of M-CSF and SCF. In addition, we have demonstrated a requirement for both Nck and RasGAP binding sites for this activity. Given the ability of DokR to suppress colony formation in the presence of cytokines such as M-CSF, we predicted that it might also negatively regulate the generation of myeloid lineages in vivo. However, our studies did not provide evidence for DokR in this role. Although we were surprised by these results, it is possible that other signaling pathways that cannot be inhibited by DokR have a greater role in the generation of these myeloid cell lineages in vivo. For example, IL-3– and GM-CSF–dependent colony formation in vitro was only mildly inhibited by expression of DokR and, therefore, their ability to regulate the generation of myeloid cells in vivo would probably also be unaffected.
As yet, DokR-deficient mice generated by gene targeting have not been reported. However, two groups have created p62dok−/− mice, and although studies on these animals have not revealed any effects on hematopoietic development, several hematopoietic cell types exhibit defects in negative regulation of proliferation in vitro (Yamanashi et al., 2000; Di Cristofano et al., 2001). For example, p62dok−/− B cells are hyperresponsive to cross-linking of the B cell antigen receptor and the inhibitory receptor FcγRIIB (Yamanashi et al., 2000). In addition, thymocytes costimulated with concanavalin A and IL-2 and bone marrow cells stimulated with SCF or IL-3 show enhanced responsiveness to these treatments compared with normal cells (Di Cristofano et al., 2001). Interestingly, loss of p62dok also enhanced the rate at which bone marrow expressing the Bcr-Abl oncoprotein can induce a lethal myeloid leukemia in reconstituted mice (Di Cristofano et al., 2001). At a molecular level, p62dok−/− cells show sustained activation of Ras- and mitogen-activated protein kinases in response to various stimuli (Yamanashi et al., 2000; Di Cristofano et al., 2001), presumably at least partly due to reduced RasGAP-mediated attenuation of Ras signaling. Given that p62dok and DokR are both capable of recruiting RasGAP and Nck and are expressed simultaneously in many hematopoietic cell types (except B cells, which lack DokR), it is possible that these proteins are functionally redundant. We cannot rule out the possibility that some of the effects we observed here would, under physiological conditions, be mediated by p62dok and are simply a consequence of overexpressing a Dok protein in cells. It is also likely that DokR and p62dok have unique functions. It is particularly interesting in this respect that although p62dok−/− bone marrow cells showed dramatically enhanced IL-3 signaling in vitro, overexpression of DokR in bone marrow cells had very little effect on IL-3–dependent colony formation in our hands. The question of functional redundancy will only be clearly resolved when mice deficient for both p62dok and DokR are generated and analyzed.
In summary, we have examined the role of DokR in regulating haematopoiesis. Our results provide compelling evidence for DokR-mediated negative regulation of T cell development and suggest that both RasGAP and Nck, through interaction with DokR at distinct sites, are critical for DokR function.
Materials and methods
Construction and characterization of retroviral constructs
Complementary DNAs encoding wild-type and mutant forms of FLAG–DokR (Lock et al., 1999) were inserted into the retroviral plasmid, pMY-EGFP (Onai et al., 2000). Bosc23 virus packaging cells (Pear et al., 1993) were transfected with pMY-DokR-EGFP constructs as previously described (Pear et al., 1993). Retrovirus-containing supernatants were harvested 48 h after transfection, adjusted to 10 mM Hepes, pH 7.0, and stored at −70°C. The infectious titres of frozen viral stocks were determined as previously described (Cong et al., 1999). Titres of between 1 and 2 × 106 infectious units/ml were routinely observed for each retroviral construct. To confirm the binding specificity of DokR mutants, 293T cells were cotransfected with pMY-DokR-EGFP constructs encoding Y351F or Y276/304F together with an expression vector encoding Lyn. Cell extracts were analyzed as described previously (Lock et al., 1999).
Retrovirus-mediated infection of bone marrow cells
Permission to use mice for this research was obtained from the animal ethics committee of the Ludwig Institute for Cancer Research and Department of Surgery, University of Melbourne. A spin infection procedure was used to infect bone marrow cells (van Parijs et al., 1999). In brief, bone marrow cells were harvested from the femurs of 8-wk-old female C57BL/6 mice 4 d after intravenous injection with 3.75 mg of 5-fluorouracil. RBCs were lysed in RBC lysis buffer (17 mM Tris-HCl, pH 7.65, 144 mM NH4Cl) for 5 min at 37°C. Washed leukocytes were resuspended at a density of 2 × 106 cells/ml in growth medium (DME medium supplemented with 15% FCS, 2 mM glutamine, 10% WEHI-3B cell-conditioned medium, 50 ng/ml murine SCF [a gift from N. Nicola, Walter and Eliza Hall Institute of Medical Research], 10 ng/ml human IL-3 [PeproTech], and 50 ng/ml murine IL-6 [a gift from R. Moritz, Ludwig Institute for Cancer Research, Parkville, Australia]) in 24-well plates (Nunc) and cultured for 3 h in 10% CO2 at 37°C. Growth medium was removed from the cells and replaced with retroviral supernatants containing equivalent numbers of infectious retroviruses (see above) and 4 μg/ml polybrene (Sigma-Aldrich). Plates were then centrifuged at 1,200 rpm at 37°C for 45 min (4K15 centrifuge; Sigma-Aldrich). Cells were replenished with growth medium, cultured overnight, and infected again 24, 48, and 72 h later.
Cytospin analysis of retrovirally infected bone marrow cells
Day 7 cultures of bone marrow cells infected in vitro with control, DokR, and DokR mutant retroviruses were deposited onto microscope slides at 800 rpm for 5 min at RT, stained with May-Grunwald Giemsa (Sigma-Aldrich), and mounted with DePeX (BDH). Cells were identified microscopically based on morphology, and the percentage of an individual cell type was calculated based on the analysis of 200 cells.
In vitro colony formation assays
Retrovirally infected bone marrow cells were washed in PBS containing 1% FCS and then cultured in 0.3% agar containing DME, 20% FCS, and the following cytokines: M-CSF (10 ng/ml); SCF (50 ng/ml); GM-CSF (10 ng/ml); or IL-3 (10 ng/ml). For M-CSF and SCF cultures, 5,000 cells were plated per 35-mm dish. For GM-CSF and IL-3 cultures, 2,000 and 1,000 cells were plated per dish, respectively. Colonies of >50 GFP+ cells were scored by fluorescence microscopy after 7 d. Assays were performed in quadruplicate three times.
Bone marrow reconstitution experiments
Female C57BL/6 mice were γ irradiated (two doses of 550 rads, 3 h apart) and injected intravenously with 2–5 × 105 retrovirally infected bone marrow cells. Mice were administered the antibiotic Baytril (Bayer) (0.17 mg/ml) in their drinking water and housed in microisolators. Mice were killed 10 wk after bone marrow reconstitution and the spleens, thymi, and bone marrow were dissected.
Preparation of hematopoietic cells from reconstituted mice
Bone marrow cells were flushed from the femurs of reconstituted mice in 5 ml of FACS buffer (PBS containing 1% FCS). Suspensions of splenocytes and thymocytes were prepared by gently teasing spleens and thymi respectively through a stainless steel mesh in 5 ml of FACS buffer. Blood was obtained by eye bleeding. Crude cell suspensions were depleted of RBCs by one or two incubations in RBC lysis buffer at RT or 37°C and then washed two times in FACS buffer. Numbers of viable leukocytes from spleen, thymus, and bone marrow were determined on the basis of eosin exclusion using a hemocytometer. For absolute blood leukocyte counts, 5 μl of heparinized blood was diluted in 20 ml PBS buffer containing 1% Zap-oglobin lytic reagent (Beckman Coulter) for lysis of RBCs. The cells were then counted using a particle count and size analyzer (counter model Z2; Beckman Coulter).
Flow cytometry
Flow cytometric analyses were performed using a FACScan® flow cytometer and CellQuest software (Becton Dickinson). Leukocytes were resuspended at a density of 20 × 106 cells/ml in FACS buffer. Cells were then either analyzed for GFP expression immediately, or, for two- and three-color analyses, stained with phycoerythrin (PE)-conjugated monoclonal antibodies (anti-CD8, anti-B220, anti–Gr-1, anti–Mac-1; BD PharMingen) and/or biotinylated monoclonal antibodies (anti-CD4, anti-IgM, anti-cKit, anti-Ter119) for 30 min at 4°C. For detection of biotinylated antibodies, cells were stained with streptavidin-PE (Caltag) or streptavidin-tricolor (Caltag) where appropriate for 30 min at 4°C. For one- and two-color analyses, cells were washed and resuspended in FACS buffer containing 2 μg/ml propidium iodide (PI) to exclude nonviable cells. For three-color analyses, 20 μg/ml PI was added to distinguish PI-positive from tricolor-positive cells, as both are in the FL-3 channel. Cell viability was generally >90%. Residual RBCs were excluded from all analyses on the basis of forward scatter. Antibodies were provided by A. Strasser (Walter and Eliza Hall Institute of Medical Research), M. Hibbs, and S. Basu (Ludwig Institute for Cancer Research) unless otherwise indicated.
Cell sorting
Cells were sorted using a MoFlo cell sorter (Cytomation, Inc.). For purification of splenic B cells, T cells, and myeloid cells, splenocytes from two control mice or two DokR mice were pooled, resuspended in 0. 5 ml of balanced salt solution (BSS) (149 mM NaCl, 3.7 mM KCl, 2.5 mM CaCl2, 1.2 mM MgSO4, 2.5 mM potassium phosphate, 14.8 mM Hepes, pH 7.2) containing 4% FCS (BSS/FCS) and divided into two equal fractions. Cells were then stained either with anti–B220-PE plus anti–Thy-1 biotin or with anti–Mac-1–PE plus anti–Gr-1 biotin. Cells were incubated for 30 min at 4°C, washed in BSS/FCS, collected by centrifugation, and incubated with streptavidin-tricolor (Caltag) for 30 min at 4°C. Cells were washed as above and then separated from cell clumps using a cell debris removal column. Cells were resuspended in BSS/FCS containing excess PI (20 μg/ml) to distinguish between tricolor and PI-positive populations. Cells were then sorted into B220+ (B lymphoid), Thy-1+ (T lymphoid), or Gr-1+/Mac-1+ plus Gr-1−/Mac-1+ (myeloid) populations. Only PI-negative cells were included in the sorted populations. For purification of primitive thymic T cells, thymocytes from individual control or DokR mice were resuspended in 0.25 ml BSS/FCS, stained with a cocktail of PE-coupled (anti-CD8, anti-B220, anti–Gr-1, anti–Mac-1) and biotinylated antibodies (anti-CD4, MHC class II) for 30 min at 4°C, washed, and then stained with streptavidin-PE. Cells were washed and resuspended in BSS/FCS containing PI as above, and cells that did not stain with PE or PI were collected by cell sorting.
Immunoprecipitation and immunoblot analysis
Splenocyte and thymocyte extracts were prepared by lysing cells in lysis buffer (50 mM Tris, pH 8.0, 1% Triton X-100, 150 mM NaCl) supplemented with a cocktail of protease inhibitors (complete; Roche) for 15 min at 4°C. Extracts of purified splenic T cells, B cells, and myeloid cells were prepared by washing cells once in ice cold TBS containing a twofold excess of protease inhibitors before extraction in lysis buffer as above. Cell lysates were clarified by centrifugation at 13,000 rpm for 15 min at 4°C. Splenocyte and thymocyte extracts containing ∼1 × 106 cells were analyzed by immunoprecipitation with anti-FLAG sepharose beads (Sigma-Aldrich), followed by SDS-PAGE and Western analysis using an anti-DokR serum (Lock et al., 1999). Lysates containing 5 × 104 splenic B cells, T cells, or myeloid cells were analyzed sequentially by Western blotting with anti-DokR, anti-GAP, and anti-Nck sera.
Statistical analyses
An unpaired t test was performed to compare the arithmetic means ± SEM of values relating to the control group of reconstituted mice with those pertaining to the DokR, Y351F, or Y276/304F mice.
Acknowledgments
We are grateful to Robert Moritz and Nicos Nicola for providing cytokines, Margaret Hibbs, Sunanda Basu, and Andreas Strasser for providing antibodies, Janna Taylor (University of Melbourne, Parkville, Australia) for preparing artwork, and Viki Lapartis for technical help with cell sorting.
This work was supported in part by a grant from the National Health and Medical Research Council, Australia.
Footnotes
Abbreviations used in this paper: DN, double negative; Dok, downstream of kinase; DokR, Dok-related protein; DP, double positive; GFP, green fluorescent protein; GM-CSF, granulocyte macrophage colony–stimulating factor; IL; interleukin; IRES, internal ribosomal entry site; LAT, linker of activated T cells; M-CSF, macrophage colony–stimulating factor; PE, phycoerythrin; PI, propidium iodide; RasGAP, RasGTPase-activating protein; SCF, stem cell factor; SP, single positive; TCR, T cell receptor.
References
- Anton, I.M., W. Lu, B.J. Mayer, N. Ramesh, and R.S. Geha. 1998. The Wiskott-Aldrich syndrome protein-interacting protein (WIP) binds to the adaptor protein Nck. J. Biol. Chem. 273:20992–20995. [DOI] [PubMed] [Google Scholar]
- Boguski, M.S., and F. McCormick. 1993. Proteins regulating Ras and its relatives. Nature. 366:643–654. [DOI] [PubMed] [Google Scholar]
- Bokoch, G.M., Y. Wang, B.P. Bohl, M.A. Sells, L.A. Quilliam, and U.G. Knaus. 1996. Interaction of the Nck adapter protein with p21-activated kinase (PAK1). J. Biol. Chem. 271:25746–25749. [DOI] [PubMed] [Google Scholar]
- Cachon-Gonzalez, M.B., S. Fenner, J.M. Coffin, C. Moran, S. Best, and J.P. Stoye. 1994. Structure and expression of the hairless gene of mice. Proc. Natl. Acad. Sci. USA. 91:7717–7721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpino, N., D. Wisniewski, A. Strife, D. Marshak, R. Kobayashi, B. Stillman, and B. Clarkson. 1997. p62(dok): a constitutively tyrosine-phosphorylated, GAP-associated protein in chronic myelogenous leukemia progenitor cells. Cell. 88:197–204. [DOI] [PubMed] [Google Scholar]
- Cong, F., B. Yuan, and S.P. Goff. 1999. Characterization of a novel member of the DOK family that binds and modulates Abl signaling. Mol. Cell. Biol. 19:8314–8325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, M., H. She, E.M. Davis, C.M. Spicer, L. Kim, R. Ren, M.M. Le Beau, and W. Li. 1998. Identification of Nck family genes, chromosomal localization, expression, and signaling specificity. J. Biol. Chem. 273:25171–25178. [DOI] [PubMed] [Google Scholar]
- Clements, J.L., B. Yang, S.E. Ross-Barta, S.L. Eliason, R.F. Hrstka, R.A. Williamson, and G.A. Koretzky. 1998. Requirement for the leukocyte-specific adapter protein SLP-76 for normal T cell development. Science. 281:416–419. [DOI] [PubMed] [Google Scholar]
- Coutinho, S., T. Jahn, M. Lewitzky, S. Feller, P. Hutzler, C. Peschel, and J. Duyster. 2000. Characterization of Grb4, an adapter protein interacting with Bcr-Abl. Blood. 96:618–624. [PubMed] [Google Scholar]
- Di Cristofano, A., N. Carpino, N. Dunant, G. Friedland, R. Kobayashi, A. Strife, D. Wisniewski, B. Clarkson, P.P. Pandolfi, and M.D. Resh. 1998. Molecular cloning and characterization of p56dok-2 defines a new family of RasGAP-binding proteins. J. Biol. Chem. 273:4827–4830. [DOI] [PubMed] [Google Scholar]
- Di Cristofano, A., M. Niki, M. Zhao, F.G. Karnell, B. Clarkson, W.S. Pear, L. Van Aelst, and P.P. Pandolfi. 2001. p62(dok), a negative regulator of Ras and mitogen-activated protein kinase (MAPK) activity, opposes leukemogenesis by p210(bcr-abl). J. Exp. Med. 194:275–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galisteo, M.L., J. Chernoff, Y.C. Su, E.Y. Skolnik, and J. Schlessinger. 1996. The adaptor protein Nck links receptor tyrosine kinases with the serine-threonine kinase Pak1. J. Biol. Chem. 271:20997–21000. [DOI] [PubMed] [Google Scholar]
- Grimm, J., M. Sachs, S. Britsch, S. Di Cesare, T. Schwarz-Romond, K. Alitalo, and W. Birchmeier. 2001. Novel p62dok family members, dok-4 and dok-5, are substrates of the c-Ret receptor tyrosine kinase and mediate neuronal differentiation. J. Cell Biol. 154:345–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruenheid, S., R. DeVinney, F. Bladt, D. Goosney, S. Gelkop, G.D. Gish, T. Pawson, and B.B. Finlay. 2001. Enteropathogenic E. coli Tir binds Nck to initiate actin pedestal formation in host cells. Nat. Cell Biol. 3:856–859. [DOI] [PubMed] [Google Scholar]
- Harder, K.W., L.M. Parsons, J. Armes, N. Evans, N. Kountouri, R. Clark, C. Quilici, D. Grail, G.S. Hodgson, A.R. Dunn, and M.L. Hibbs. 2001. Gain- and loss-of-function Lyn mutant mice define a critical inhibitory role for Lyn in the myeloid lineage. Immunity. 15:603–615. [DOI] [PubMed] [Google Scholar]
- Holland, S.J., N.W. Gale, G.D. Gish, R.A. Roth, Z. Songyang, L.C. Cantley, M. Henkemeyer, G.D. Yancopoulos, and T. Pawson. 1997. Juxtamembrane tyrosine residues couple the Eph family receptor EphB2/Nuk to specific SH2 domain proteins in neuronal cells. EMBO J. 16:3877–3888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosooka, T., T. Noguchi, H. Nagai, T. Horikawa, T. Matozaki, M. Ichihashi, and M. Kasuga. 2001. Inhibition of the motility and growth of B16F10 mouse melanoma cells by dominant negative mutants of Dok-1. Mol. Cell. Biol. 21:5437–5446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, N., and D.J. Dumont. 1998. The Tek/Tie2 receptor signals through a novel Dok-related docking protein, Dok-R. Oncogene. 17:1097–1108. [DOI] [PubMed] [Google Scholar]
- Jones, N., and D.J. Dumont. 1999. Recruitment of Dok-R to the EGF receptor through its PTB domain is required for attenuation of Erk MAP kinase activation. Curr. Biol. 9:1057–1060. [DOI] [PubMed] [Google Scholar]
- Lemay, S., D. Davidson, S. Latour, and A. Veillette. 2000. Dok-3, a novel adapter molecule involved in the negative regulation of immunoreceptor signaling. Mol. Cell. Biol. 20:2743–2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latour, S., and A. Veillette. 2001. Proximal protein tyrosine kinases in immunoreceptor signaling. Curr. Opin. Immunol. 13:299–306. [DOI] [PubMed] [Google Scholar]
- Latour, S., G. Gish, C.D. Helgason, R.K. Humphries, T. Pawson, and A. Veillette. 2001. Regulation of SLAM-mediated signal transduction by SAP, the X-linked lymphoproliferative gene product. Nat. Immunol. 2:681–690. [DOI] [PubMed] [Google Scholar]
- Lock, P., F. Casagranda, and A.R. Dunn. 1999. Independent SH2-binding sites mediate interaction of Dok-related protein with RasGTPase-activating protein and Nck. J. Biol. Chem. 274:22775–22784. [DOI] [PubMed] [Google Scholar]
- Lu, W., S. Katz, R. Gupta, and B.J. Mayer. 1997. Activation of Pak by membrane localization mediated by an SH3 domain from the adaptor protein Nck. Curr. Biol. 7:85–94. [DOI] [PubMed] [Google Scholar]
- Master, Z., N. Jones, J. Tran, J. Jones, R.S. Kerbel, and D.J. Dumont. 2001. Dok-R plays a pivotal role in angiopoietin-1-dependent cell migration through recruitment and activation of Pak. EMBO J. 20:5919–5928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarty, J.H. 1998. The Nck SH2/SH3 adaptor protein: a regulator of multiple intracellular signal transduction events. Bioessays. 20:913–921. [DOI] [PubMed] [Google Scholar]
- Molina, T.J., K. Kishihara, D.P. Siderovski, W. van Ewijk, A. Narendran, E. Timms, A. Wakeham, C.J. Paige, K.U. Hartmann, A. Veillette, et al. 1992. Profound block in thymocyte development in mice lacking p56lck. Nature. 357:161–164. [DOI] [PubMed] [Google Scholar]
- Nelms, K., A.L. Snow, J. Hu-Li, and W.E. Paul. 1998. FRIP, a hematopoietic cell-specific rasGAP-interacting protein phosphorylated in response to cytokine stimulation. Immunity. 9:13–24. [DOI] [PubMed] [Google Scholar]
- Nemorin, J.G., and P. Duplay. 2000. Evidence that Lck-mediated phosphorylation of p56dok and p62dok may play a role in CD2 signaling. J. Biol. Chem. 275:14590–14597. [DOI] [PubMed] [Google Scholar]
- Noguchi, T., T. Matozaki, K. Inagaki, M. Tsuda, K. Fukunaga, Y. Kitamura, T. Kitamura, K. Shii, Y. Yamanashi, and M. Kasuga. 1999. Tyrosine phosphorylation of p62(Dok) induced by cell adhesion and insulin: possible role in cell migration. EMBO J. 18:1748–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onai, N., Y. Zhang, H. Yoneyama, T. Kitamura, S. Ishikawa, and K. Matsushima. 2000. Impairment of lymphopoiesis and myelopoiesis in mice reconstituted with bone marrow-hematopoietic progenitor cells expressing SDF-1-intrakine. Blood. 96:2074–2080. [PubMed] [Google Scholar]
- Pear, W.S., G.P. Nolan, M.L. Scott, and D. Baltimore. 1993. Production of high-titer helper-free retroviruses by transient transfection. Proc. Natl. Acad. Sci. USA. 90:8392–8396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips, R.L., R.E. Ernst, B. Brunk, N. Ivanova, M.A. Mahan, J.K. Deanehan, K.A. Moore, G.C. Overton, and I.R. Lemischka. 2000. The genetic program of hematopoietic stem cells. Science. 288:1635–1640. [DOI] [PubMed] [Google Scholar]
- Pivniouk, V., E. Tsitsikov, P. Swinton, G. Rathbun, F.W. Alt, and R.S. Geha. 1998. Impaired viability and profound block in thymocyte development in mice lacking the adaptor protein SLP-76. Cell. 94:229–238. [DOI] [PubMed] [Google Scholar]
- Pivniouk, V.I., and R.S. Geha. 2000. The role of SLP-76 and LAT in lymphocyte development. Curr. Opin. Immunol. 12:173–178. [DOI] [PubMed] [Google Scholar]
- Rivero-Lezcano, O.M., A. Marcilla, J.H. Sameshima, and K.C. Robbins. 1995. Wiskott-Aldrich syndrome protein physically associates with Nck through Src homology 3 domains. Mol. Cell. Biol. 15:5725–5731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Songyang, Z., Y. Yamanashi, D. Liu, and D. Baltimore. 2001. Domain-dependent function of the rasGAP-binding protein p62Dok in cell signaling. J. Biol. Chem. 276:2459–2465. [DOI] [PubMed] [Google Scholar]
- Suzu, S., M. Tanaka-Douzono, K. Nomaguchi, M. Yamada, H. Hayasawa, F. Kimura, and K. Motoyoshi. 2000. p56(dok-2) as a cytokine-inducible inhibitor of cell proliferation and signal transduction. EMBO J. 19:5114–5122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomlinson, M.G., J. Lin, and A. Weiss. 2000. Lymphocytes with a complex: adapter proteins in antigen receptor signaling. Immunol. Today. 21:584–591. [DOI] [PubMed] [Google Scholar]
- Tu, Y., F. Li, and C. Wu. 1998. Nck-2, a novel Src homology2/3-containing adaptor protein that interacts with the LIM-only protein PINCH and components of growth factor receptor kinase-signaling pathways. Mol. Biol. Cell. 9:3367–3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Parijs, L., Y. Refaeli, J.D. Lord, B.H. Nelson, A.K. Abbas, and D. Baltimore. 1999. Uncoupling IL-2 signals that regulate T cell proliferation, survival, and Fas-mediated activation-induced cell death. Immunity. 11:281–288. [DOI] [PubMed] [Google Scholar]
- von Boehmer, H., and H.J. Fehling. 1997. Structure and function of the pre-T cell receptor. Annu. Rev. Immunol. 15:433–452. [DOI] [PubMed] [Google Scholar]
- Wisniewski, D., A. Strife, E. Berman, and B. Clarkson. 1996. c-kit ligand stimulates tyrosine phosphorylation of a similar pattern of phosphotyrosyl proteins in primary primitive normal hematopoietic progenitors that are constitutively phosphorylated in comparable primitive progenitors in chronic phase chronic myelogenous leukemia. Leukemia. 10:229–237. [PubMed] [Google Scholar]
- Yamanashi, Y., and D. Baltimore. 1997. Identification of the Abl- and rasGAP-associated 62 kDa protein as a docking protein, Dok. Cell. 88:205–211. [DOI] [PubMed] [Google Scholar]
- Yamanashi, Y., T. Tamura, T. Kanamori, H. Yamane, H. Nariuchi, T. Yamamoto, and D. Baltimore. 2000. Role of the rasGAP-associated docking protein p62(dok) in negative regulation of B cell receptor-mediated signaling. Genes Dev. 14:11–16. [PMC free article] [PubMed] [Google Scholar]
- Yoder, J., C. Pham, Y.M. Iizuka, O. Kanagawa, S.K. Liu, J. McGlade, and A.M. Cheng. 2001. Requirement for the SLP-76 adaptor GADS in T cell development. Science. 291:1987–1991. [DOI] [PubMed] [Google Scholar]
- Zhang, W., C.L. Sommers, D.N. Burshtyn, C.C. Stebbins, J.B. DeJarnette, R.P. Trible, A. Grinberg, H.C. Tsay, H.M. Jacobs, C.M. Kessler, et al. 1999. Essential role of LAT in T cell development. Immunity. 10:323–332. [DOI] [PubMed] [Google Scholar]