

Abstract
We define a novel mechanism by which integrins regulate growth factor expression and the survival of carcinoma cells. Specifically, we demonstrate that the α6β4 integrin enhances vascular endothelial growth factor (VEGF) translation in breast carcinoma cells. The mechanism involves the ability of this integrin to stimulate the phosphorylation and inactivation of 4E-binding protein (4E-BP1), a translational repressor that inhibits the function of eukaryotic translation initiation factor 4E (eIF-4E). The regulation of 4E-BP1 phosphorylation by α6β4 derives from the ability of this integrin to activate the PI-3K–Akt pathway and, consequently, the rapamycin-sensitive kinase mTOR that can phosphorylate 4E-BP1. Importantly, we show that this α6β4-dependent regulation of VEGF translation plays an important role in the survival of metastatic breast carcinoma cells by sustaining a VEGF autocrine signaling pathway that involves activation of PI-3K and Akt. These findings reveal that integrin-mediated activation of PI-3K–Akt is amplified by integrin-stimulated VEGF expression and they provide a mechanism that substantiates the reported role of α6β4 in carcinoma progression.
Keywords: integrin; VEGF; translation; carcinoma; eIF-4E
Introduction
An understanding of the mechanisms that sustain the survival of tumor cells in adverse physiological conditions is one of the most important problems in cancer biology. As argued recently, cancer progression is an evolutionary process that selects for cells that exhibit the capacity for survival in environmental conditions not present in normal tissue (Fearon, 1999; Hanahan and Weinberg, 2000). One survival strategy used by tumor cells is the secretion of proteins that elicit an angiogenic response, such as vascular permeability factor or vascular endothelial growth factor (VEGF).* VEGF appears to be an essential factor for the progression of many solid tumors (Shweiki et al., 1992; Brown et al., 1999; Dvorak et al., 1999). It is widely assumed that the function of VEGF produced by tumor and tumor stromal cells is to stimulate angiogenesis by acting in a paracrine fashion on vicinal endothelium (Hanahan and Folkman, 1996; Brown et al., 1999). Another mechanism for tumor cell survival is the establishment of autocrine signaling loops that act on tumor cells directly (Scotlandi et al., 1996; Tokunou et al., 2001; Wong et al., 2001). Although the significance of this mechanism has been overshadowed by angiogenesis, recent studies have substantiated the importance and necessity of such signaling loops for tumor survival (Scotlandi et al., 1996; Bachelder et al., 2001; Tokunou et al., 2001; Wong et al., 2001). Indeed, this mechanism probably contributes to the ability of cells to survive in hypoxic, poorly vascularized regions of tumors. In this direction, we described recently the existence of a VEGF autocrine signaling pathway in metastatic breast carcinoma cells that is essential for their survival (Bachelder et al., 2001).
An important issue that arises from the contribution of VEGF autocrine signaling to tumor survival is an understanding of the mechanisms that regulate VEGF expression. Such mechanisms are important not only for VEGF signaling in tumor cells, but also for angiogenesis as well. Clearly, hypoxia is a strong inducer of VEGF transcription and mRNA stability (von Marschall et al., 2001), but other factors are likely to be involved. Of note, our finding that the α6β4 integrin can promote the survival of breast carcinoma cells in stress conditions is intriguing (Bachelder et al., 1999b) and raised the novel possibility that a specific integrin, which has been implicated in cancer progression, could regulate VEGF expression. This possibility is substantiated by the finding reported here that the ability of the α6β4 integrin to promote survival is VEGF dependent.
The results described above prompted us to investigate the relationship between the α6β4 integrin and VEGF expression. We observed that the expression and signaling properties of this integrin have no impact on steady-state VEGF mRNA levels. Surprisingly, however, we detected a significant influence of α6β4 on VEGF translation and protein expression in these cells, an observation that reveals the ability of this integrin to regulate translation. The mechanism by which α6β4 regulates VEGF expression involves its ability to stimulate the phosphorylation of 4E-binding protein (4E-BP1). 4E-BP1 is phosphorylated by mammalian target of rapamycin (mTOR), a protein kinase whose catalytic domain is structurally related to that of phosphatidylinositol 3-kinase (PI-3K) (Dennis et al., 1999; Schmelzle and Hall, 2000). Phosphorylation of 4E-BP1 by mTOR disrupts its binding to eukaryotic translation initiation factor eIF-4E, which is present in rate-limiting amounts in most cells (De Benedetti and Harris, 1999; McKendrick et al., 1999). eIF-4E plays a critical role in the recruitment of the translational machinery to the 5′ end of mRNA, which is demarcated by an m7GpppN cap (where m is a methyl group and N is any nucleotide) (Raught and Gingras, 1999). The m7 cap is essential for the translation of most mRNAs including VEGF (De Benedetti and Harris, 1999; Raught and Gingras, 1999). Dissociation of 4E-BP1 from eIF-4E enables eIF-4E to initiate translation (Gingras et al., 1999, 2001b). The regulation of 4E-BP1 phosphorylation by α6β4 derives from the ability of this integrin to activate the PI-3K–Akt pathway and, consequently, mTOR. Our findings reveal a novel mechanism of tumor cell survival and they highlight the ability of a specific integrin to regulate protein translation by influencing eIF-4E activity.
Results
The ability of the α6β4 integrin to promote the survival of carcinoma cells is VEGF dependent
To examine the hypothesis that the ability of the α6β4 integrin to promote survival is VEGF dependent, we used MDA-MB-435 cells, which lack expression of this integrin. Stable expression of α6β4 in these cells enhances their ability to survive in stressful conditions (Bachelder et al., 1999b). Importantly, however, expression of α6β4 does not alter the expression of other integrin subunits in these cells and does not influence their adhesion to matrix (Shaw et al., 1997). As shown in Fig.1 A, a significant level of apoptosis was observed after 24 h of serum deprivation in the parental MDA-MB-435 cells and mock transfectants, as well as in transfectants that express α6β4 containing a cytoplasmic domain deletion of the β4 subunit that lacks the ability to signal (Shaw et al., 1997). Stable subclones that express the intact α6β4 integrin, however, were protected from apoptosis under these conditions. Based on these results and our previous finding that the survival of metastatic breast carcinoma cells is dependent on VEGF, we used a VEGF antisense oligonucleotide to reduce VEGF expression in the MDA-MB-435/β4 transfectants and assessed the impact of reducing VEGF expression on their survival (Fig. 1, B and C). The VEGF antisense oligonucleotide reduced VEGF protein expression significantly in the β4 transfectants (Fig. 1 C). As shown in Fig. 1 B, this reduction in VEGF expression abrogated the survival-enhancing effect of α6β4 under conditions of serum deprivation.
Figure 1.
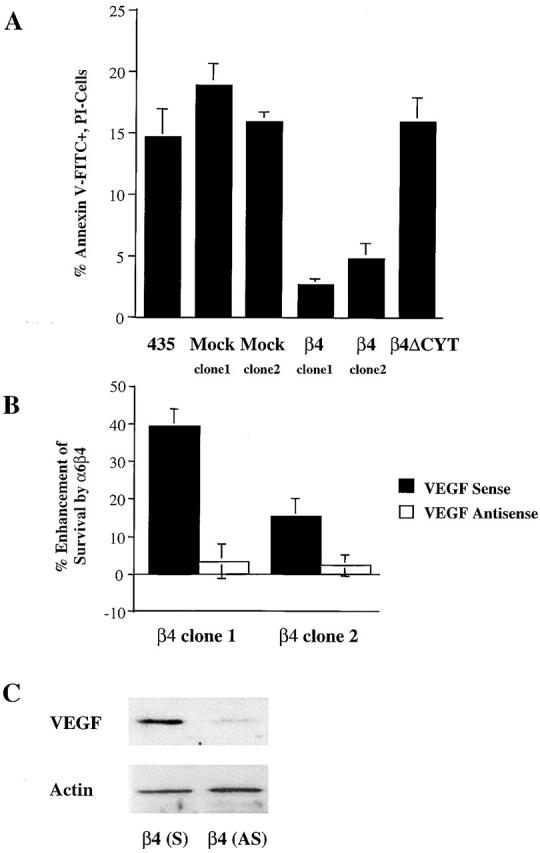
The α6β4-mediated survival of breast carcinoma cells is VEGF dependent. (A) Parental, mock (clone 1, 6D2; clone 2, 6D7), β4-ΔCYT–expressing (cytoplasmic tail deletion mutant), and β4 integrin–expressing (clone 1, 3A7; clone 2, 5B3) MDA-MB-435 subclones were maintained in low serum (0.5% FBS) medium for 24 h. To assess the level of apoptosis, these cells were stained with annexin V–FITC and propidium iodide (PI), and analyzed on a Becton Dickinson flow cytometer using CellQuest software. The percentage of annexin-positive, PI-negative cells (± SD) is indicated. Results were obtained from three independent experiments. Apoptosis was minimal in the presence of 10% FBS (unpublished data). (B) Mock-transfected clone 6D7 and β4 integrin– (clone 1, 3A7; clone 2, 5B3) expressing MDA-MB-435 subclones were transiently transfected with VEGF sense or antisense oligonucleotides and maintained in low serum (0.5% FBS) medium. After 24 h, the level of apoptosis in these cells was assessed as described above. The data are presented as the mean difference (± SD) in annexin positivity between mock-transfected and α6β4-expressing MDA-MB-435 cells. Similar results were observed in two separate experiments. (C) The relative amount of VEGF protein in extracts obtained from the MDA-MB-435/β4 cells transfected with either the VEGF sense (S) or antisense (AS) oligonucleotide was determined by immunoblotting using a polyclonal anti-VEGF immune serum.
The α6β4 integrin increases VEGF protein but not mRNA expression
Given that the survival effect of α6β4 expression is VEGF dependent, the novel possibility arose that VEGF expression could be regulated by this integrin. VEGF expression can be regulated at the level of both transcription and mRNA stability (Nabors et al., 2001; von Marschall et al., 2001), mechanisms that would alter the steady-state levels of VEGF mRNA. In addition, regulation can also occur at the level of VEGF translation (Kevil et al., 1996; Akiri et al., 1998; Stein et al., 1998). As shown in Fig. 2 A, quantitative analysis of VEGF mRNA levels in two clones of MDA-MB-435/mock and β4 transfectants using real-time PCR revealed no significant difference in the steady-state mRNA levels in these two populations. However, we detected a substantial increase in VEGF protein expression in the MDA-MB-435/β4 transfectants relative to either the parental cells, mock transfectants, or cells that express a cytoplasmic domain deletion of the β4 subunit (β4-ΔCYT) (Fig. 2 B). These results indicate that the α6β4 integrin regulates VEGF protein expression. It is also worth noting that the level of apoptosis observed in these populations in response to serum deprivation correlates inversely with their expression of VEGF (Fig. 1 A and Fig. 2 B).
Figure 2.
Expression of the α6β4 integrin increases VEGF protein but not steady-state mRNA. (A) The amount of VEGF mRNA in extracts obtained from mock- (clone 1, 6D2; clone 2, 6D7) and β4 integrin– (clone 1, 3A7; clone 2, 5B3) transfected MDA-MB-435 subclones was quantified by real-time PCR. The data are presented as the mean ratio of VEGF to β-actin mRNA (± SD) obtained from triplicate samples. (B) Parental (435), mock (clone 1, 6D2; clone 2, 6D7), β4-ΔCYT–expressing (clone 1E10), and β4 integrin–expressing (clone 1: 3A7, clone 2: 5B3) MDA-MB-435 subclones were cultured in low serum (0.5% FBS) medium for 24 h. Extracts of these cells containing equivalent amounts of protein were analyzed for their relative expression of VEGF and actin by immunoblotting. Similar results were observed in four independent experiments. (C) Mock (clone 6D7) and β4 integrin–expressing (clone 3A7) MDA-MB-435 subclones were maintained in low serum (0.5% FBS) medium for 24 h. These cells were detached with trypsin and incubated with integrin-specific antibodies (α6 integrin, 2B7; β4 integrin, A9; α5 integrin, Sam1) or IgG for 30 min in suspension and allowed to adhere on anti-IgG–coated plates for 60 min before lysis. In addition, cells were preincubated in cycloheximide (CHX) at a concentration of 10 μg/ml for 30 min and then incubated with either the α6 or β4 integrin antibodies in the presence of cycloheximide. Extracts of these cells containing equivalent amounts of protein were analyzed for their relative expression of VEGF and actin by immunoblotting. Similar results were observed in two independent experiments.
To substantiate the regulation of VEGF expression by α6β4, integrin-specific antibodies were used to cluster either α6β4 or α5β1 and the effects of integrin-mediated clustering on VEGF expression were assessed by immunoblotting. Of note, the MDA-MB-435/β4 transfectants express equivalent levels of α6β4 and α5β1 (unpublished data). An α6-specific antibody (mAb 2B7) was used to cluster the α6β1 integrin in the mock transfectants and the α6β4 integrin in the β4 transfectants, a β4-specific antibody (mAb A9) was used to cluster the α6β4 integrin in the β4 transfectants, and an α5-specific antibody (mAb Sam1) was used to cluster α5β1 in both the mock and β4 transfectants. A substantial induction of VEGF expression was observed upon α6β4 integrin clustering in the β4 transfectants but not in the mock transfectants, and no induction was seen in response to α5β1 clustering (Fig. 2 C). Importantly, the induction of VEGF expression that occurs in response to α6β4 clustering was inhibited by cycloheximide (Fig. 2 C). This result, together with the real-time PCR data (Fig. 2 A), indicates that α6β4 is influencing VEGF translation.
To obtain more definitive evidence that α6β4 is regulating VEGF translation, we performed polysome analysis of the VEGF message. mRNA isolated from the MDA-MB-435/mock and β4 transfectants was fractionated on a sucrose gradient (Fig. 3 A) and the relative amount of VEGF mRNA in each fraction was determined by real-time PCR (Fig. 3 B). As shown in Fig. 3 B, a striking difference in the distribution of VEGF mRNA was evident in the two populations of cells. In the MDA-MB-435/β4 transfectants, VEGF mRNA fractionated in the heavy polysomal region, whereas in the mock transfectants, the majority of VEGF mRNA was associated with light polysomal to ribosomal subunit fractions. This result indicates that the translation of VEGF in the MDA-MB-435/β4 transfectants is cap dependent.
Figure 3.
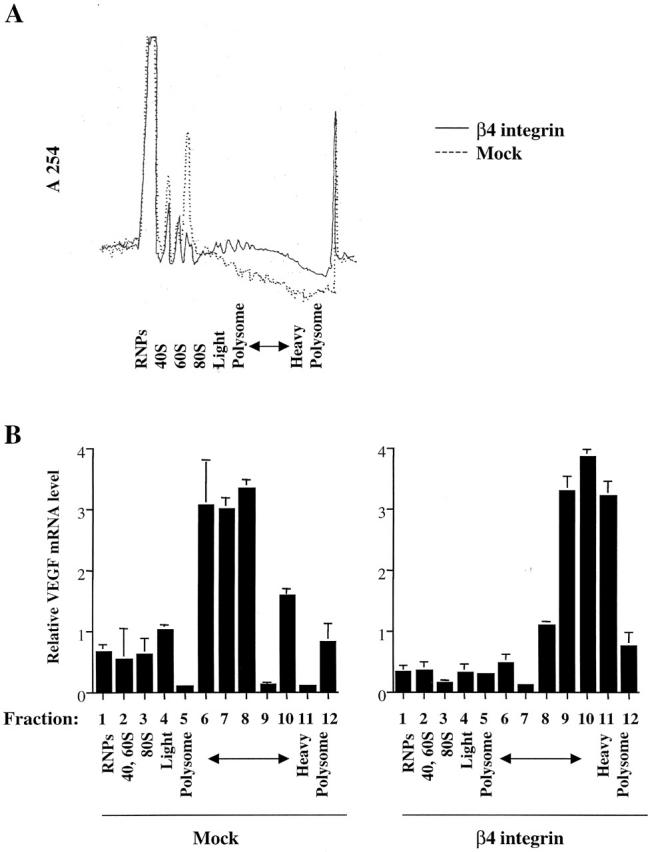
Polysome analysis of VEGF mRNA. (A) The distribution of RNA from MDA-MB-435/β4 and mock transfectants that had been fractionated on sucrose gradients as described in the Materials and methods was determined by measuring the A254 of each fraction. (B) The relative VEGF mRNA content of each sucrose gradient fraction was measured by real-time PCR as described in the Materials and methods. Fraction 1 contains unbound RNA present in the ribonucleoprotein fraction, fraction 2 contains 40S and 60S monosomes, fraction 3 contains 80S monosomes, fractions 4–7 contain light polysomes, and fractions 8–12 contain heavy polysomes. The data are presented as the mean ratio of VEGF to β-actin mRNA (± SD) obtained from triplicate samples. Similar results were obtained from three independent experiments.
Identification of an α6β4 integrin–mediated signaling pathway that regulates VEGF expression
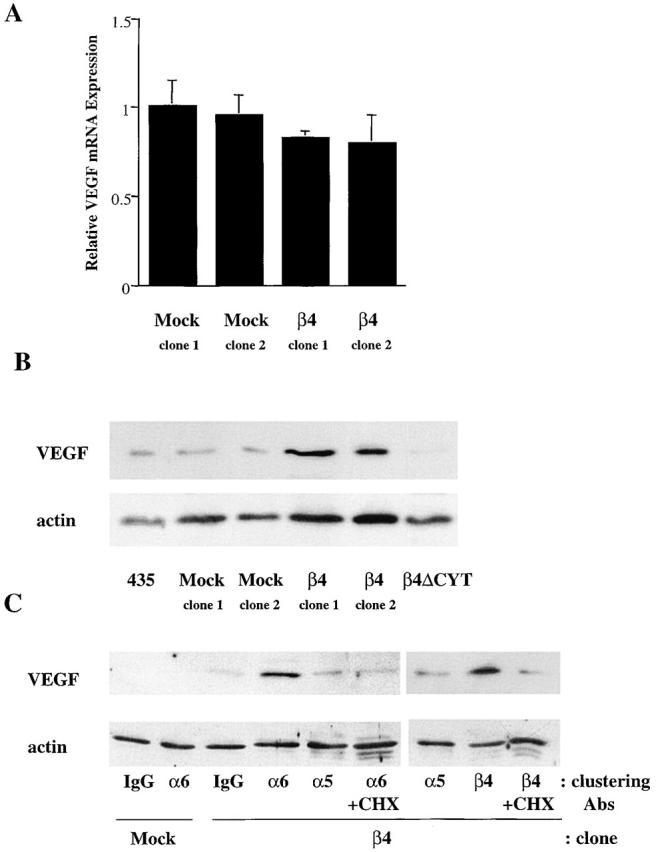
Our finding that α6β4 regulates the cap-dependent translation of VEGF prompted us to assess the ability of this integrin to stimulate the activity of the eIF-4E translation initiation factor. The α6β4 integrin is a potent activator of the PI-3K–Akt signaling pathway in MDA-MB-435 and other carcinoma cells (Shaw et al., 1997; Bachelder et al., 1999a; Gambaletta et al., 2000; Nguyen et al., 2000; Hintermann et al., 2001), and this pathway has been linked to the regulation of protein translation. Specifically, the serine/threonine kinase mTOR is activated by Akt-mediated phosphorylation events (Sekulic et al., 2000). Phosphorylation of 4E-BP1 by mTOR disrupts its binding to eIF-4E, enabling eIF-4E to initiate translation of VEGF and other molecules (De Benedetti and Harris, 1999). We hypothesized, based on this information, that α6β4 regulates 4E-BP1 phosphorylation and, as a consequence, VEGF expression. Initially, we assessed the steady-state phosphorylation levels of 4E-BP1 and S6 kinase (p70S6K), which are both downstream targets of mTOR, in cells that had been serum deprived for 24 h. Indeed, a marked increase in the level of phosphorylation of 4E-BP1 (on Ser65) and p70S6K (on Thr389) was evident in the MDA-MB-435/β4 transfectants relative to either the mock transfectants or the parental cells (Fig. 4 A). Phosphorylation of Ser65 of 4E-BP1 has been shown to be critical for dissociation of 4E-BP from eIF-4E (Gingras et al., 2001a). The reduced expression of 4E-BP1 in the β4 transfectants compared with the mock transfectants that is apparent in Fig. 4 A may reflect the possibility that the 4E-BP Ab does not recognize the hyperphosphorylated form of the protein as well as it recognizes the hypophosphorylated form.
Figure 4.
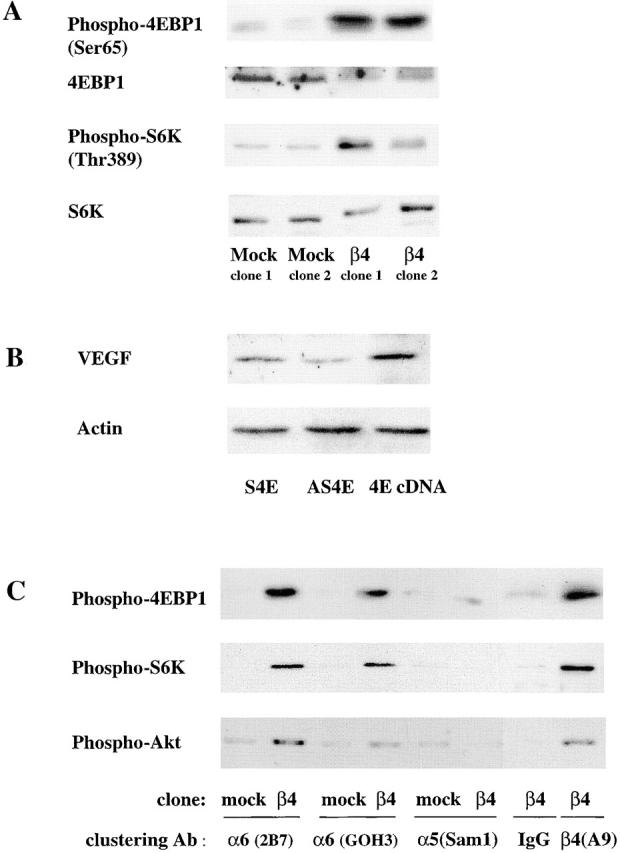
The α6β4 integrin stimulates the phosphorylation of Akt, 4E-BP1, and p70 S6K. (A) MDA-MB-435 parental cells, mock transfectants, and β4 transfectants were maintained in medium containing low serum (0.5% FBS) for 24 h. The phosphorylation status of 4E-BP1 on Ser 65 and S6K on Thr 389 was assessed in extracts from these cells using phosphospecific antibodies as described in the Materials and methods. In addition, the total amount of 4E-BP1 and p70S6K in these extracts was assessed by immunoblotting. (B) The MDA-MB-435/β4 cells were transiently transfected with either an eIF-4E sense (S) or antisense (AS) oligonucleotide, or a full-length eIF-4E cDNA (4E). Extracts of these cells containing equivalent amounts of protein were analyzed for their relative expression of VEGF and actin by immunoblotting. (C) MDA-MB-435 mock (clone 6D7) and β4 (clone 3A7) transfectants were maintained in low serum (0.5% FBS) medium for 24 h. These cells were detached with trypsin and incubated with integrin-specific antibodies (α6 integrin, 2B7; α6 integrin, GOH3; α5 integrin, Sam1; β4 integrin, A9) or IgG for 30 min as described in the legend to Fig. 2. The phosphorylation status of 4E-BP1 (Ser 65), S6K (Thr 389), and Akt (Ser 473) was assessed in extracts from these cells using phosphospecific antibodies. Similar results were observed in four independent experiments.
The involvement of eIF-4E in VEGF translation was confirmed by the expression of an antisense eIF-4E oligonucleotide in the MDA-MB-435/β4 transfectants. As shown in Fig. 4 B, expression of this oligonucleotide reduced the level of VEGF protein significantly. In contrast, expression of the full-length eIF-4E cDNA increased the VEGF protein level by approximately twofold. These results, together with the polysome analysis data (Fig. 3), indicate that α6β4 regulates VEGF expression by eIF-4E–mediated, cap-dependent translation.
To confirm the specificity of the α6β4 integrin in mTOR signaling, the effects of integrin-mediated clustering on 4E-BP1 phosphorylation were assessed. A substantial induction of Akt, 4E-BP1, and p70S6K phosphorylation was observed upon α6β4 integrin clustering in the β4 transfectants but not in the mock transfectants (Fig. 4 C). In contrast, clustering of the α5β1 integrin did not stimulate phosphorylation of these molecules in either the mock or β4 transfectants. Collectively, these data demonstrate the preferential ability of the α6β4 integrin to regulate the mTOR signaling pathway and, more importantly, the phosphorylation of 4E-BP1.
To establish that PI-3K and mTOR are required for 4E-BP1 phosphorylation and VEGF expression, we performed the antibody clustering experiments in the presence of the PI-3K–specific inhibitor LY294002 and the mTOR-specific inhibitor rapamycin (Fig. 5). As shown in Fig. 5 A, both of these inhibitors blocked the α6β4-mediated induction of 4E-BP1 phosphorylation and VEGF expression. Although rapamycin did not block Akt phosphorylation, LY294002 did inhibit its phosphorylation, confirming that Akt acts upstream of mTOR and downstream of PI-3K (Fig. 5 A). These inhibitors did not block the phosphorylation of ERK1 and ERK2 (Fig. 5 A).
Figure 5.
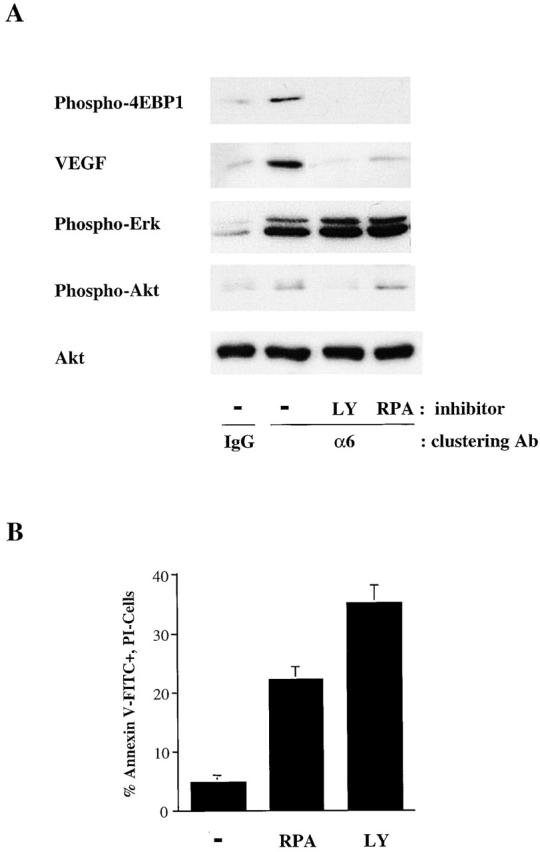
Stimulation of 4E-BP1 phosphorylation, VEGF expression, and survival by the α6β4 integrin requires PI-3K and mTOR. (A) MDA-MB-435 β4 transfectants (clone 3A7) were incubated with either DMSO (−), the PI-3K inhibitor LY 294002 (10 μM) (LY), or the mTOR-specific inhibitor rapamycin (50nM) (RPA) for 30 min and then incubated with either IgG or the α6 integrin antibody 2B7 as described in the legend to Fig. 2. Extracts of these cells were immunoblotted for phospho–4E-BP1 (Ser65), VEGF, phospho-Erk (recognizing phosphorylated isoforms of ERK1 and ERK2), phospho-Akt (Ser 473), and total Akt. Similar data were obtained in three experiments. (B) MDA-MB-435 β4 transfectants (clone 3A7) were maintained at low serum (0.5%) medium for 24 h in the presence of either rapamycin (50nM) (RPA), LY 294002 (10 μM) (LY), or DMSO (−). Apoptosis was assayed as described in the Materials and methods and is reported as the percentage of annexin V–FITC- positive, PI-negative cells. The data shown are mean values (± SD) of a representative experiment performed in triplicate.
Finally, we investigated the importance of the mTOR pathway in survival, using rapamycin and LY294002. As shown in Fig. 5 B, rapamycin treatment increased the apoptosis of the MDA-MB-435/β4 transfectants fivefold and LY294002 treatment increased their apoptosis eightfold. These results indicate that the PI-3K–mTOR pathway is critical for the survival of these cells.
Identification of a specific tyrosine residue in the β4 cytoplasmic domain required for α6β4 stimulation of 4E-BP1 phosphorylation and VEGF expression
Recently, a critical tyrosine residue (Y1494) was identified in the third fibronectin type III repeat of the β4 cytoplasmic domain, and this tyrosine was shown to be essential for activation of PI-3K by α6β4 (Shaw, 2001). To assess the importance of Y1494 in 4E-BP1 phosphorylation and VEGF expression, stable subclones of MDA-MB-435 cells were generated that expressed α6β4 containing a Y1494F mutation. As shown in Fig. 6 A, VEGF protein expression was barely detectable in these transfectants compared with the wild-type transfectants. Also, the steady-state level of 4E-BP1 phosphorylation was substantially lower in the Y1494F mutant transfectants than in the wild-type β4 transfectants. Interestingly, these mutant transfectants also exhibited an eightfold higher level of apoptosis than the wild-type β4 transfectants in response to serum deprivation (Fig. 6 B). The apoptosis of the mutant cells was reduced substantially by the addition of recombinant VEGF (Fig. 6 B), a result that substantiates the importance of VEGF in the survival of these cells. Together, these findings highlight the importance of the β4 cytoplasmic domain and PI-3K signaling in the regulation of VEGF expression and tumor cell survival.
Figure 6.
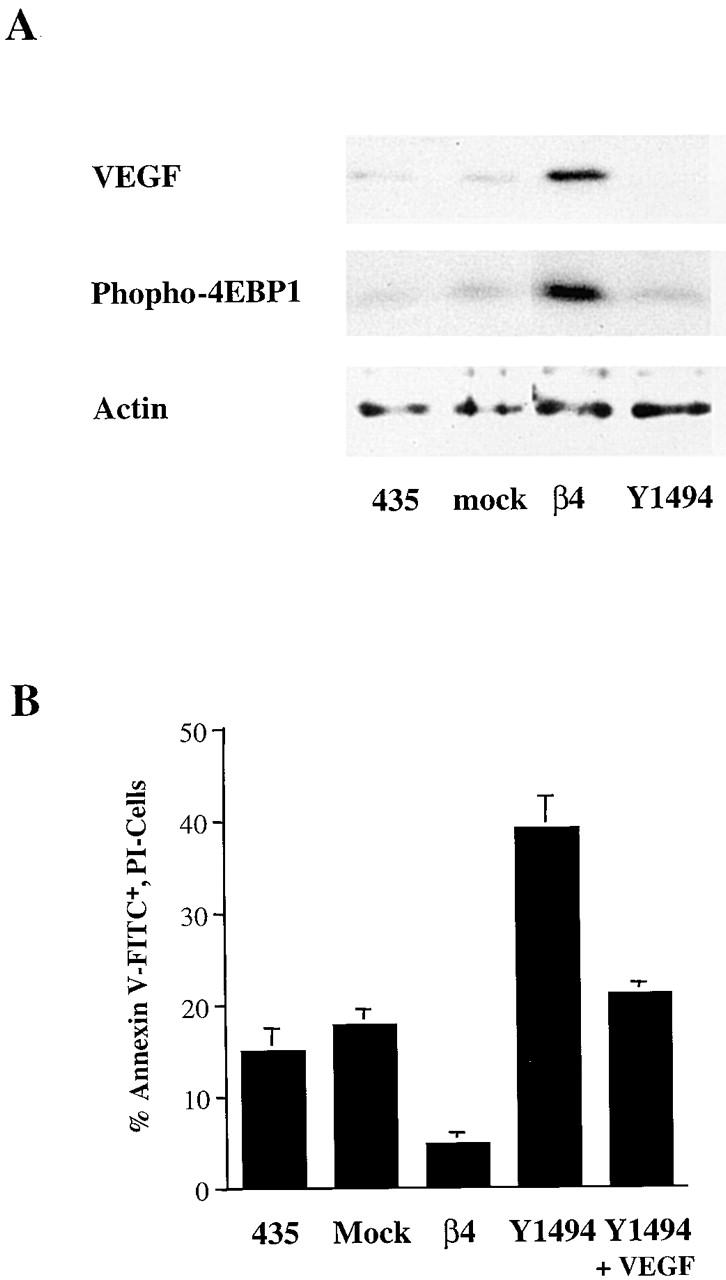
Y1494 in the β4 cytoplasmic domain is required for α6β4 stimulation of 4E-BP1 phosphorylation, VEGF expression, and survival. (A) MDA-MB-435 parental cells (435), mock transfectants (clone 6D7), wild-type β4 transfectants (clone 3A7), and Y1494F mutant transfectants (clone E1h) were maintained in low serum (0.5% FBS) for 24 h. Extracts from these cells were analyzed by immunoblotting to assess the relative expression of VEGF and 4E-BP1 phosphorylation. The relative amount of actin was also determined as a control for protein loading. Similar results were obtained in three experiments. (B) Aliquots of the same cell populations described in A were assayed for the level of apoptosis after a 24-h incubation in low serum (0.5% FBS) medium. Apoptosis was assayed as described in the Materials and methods and is reported as the percentage of annexin V–FITC-positive, PI-negative cells. The data shown are mean values (± SD) of three experiments performed in triplicate.
Expression of constitutively active Akt stimulates 4E-BP1 phosphorylation and VEGF expression in the absence of α6β4 signaling
The hypothesis that activation of Akt is a major determinant for the stimulation of 4E-BP1 phosphorylation and VEGF expression was assessed by expressing a constitutively active Akt construct in MDA-MB-435 cells that are deficient in α6β4 signaling. For this purpose, we used MDA-MB-435/mock transfectants that lack α6β4 expression and the MDA-MB-435/β4 Y1494F transfectants, described above, which are deficient in α6β4-mediated activation of PI-3K. These cells were infected with adenoviruses that encoded either a myristoylated Akt (Myr-Akt) construct or β-galactosidase as a control. As shown in Fig. 7, expression of Myr-Akt stimulated 4E-BP1 phosphorylation and VEGF expression substantially in both populations of transfectants in comparison to cells that expressed β-galactosidase. This result indicates the critical importance of Akt activation by α6β4 for stimulating VEGF expression.
Figure 7.

Expression of a constitutively active Akt construct mimics the effects of α6β4 integrin expression and signaling. MDA-MB-435 mock transfectants (clone 6D7) and Y1494F mutant transfectants (clone E1h) were infected with adenoviruses that expressed either β-galactosidase or Myr-Akt as described in the Materials and methods. Subsequently, the cells were incubated in low serum (0.5% FBS) medium for 24 h. Extracts of these cells were immunoblotted to assess the relative phosphorylation of Akt and 4E-BP1, as well as total expression of VEGF and actin.
α6β4 regulates 4E-BP1 phosphorylation, VEGF expression, and survival in carcinoma cells that express this integrin endogenously
Given that the data reported above are based on the exogenous expression of α6β4 in α6β4-deficient carcinoma cells, we sought to extend our findings to cells that express this integrin endogenously, a pattern that is typical of most carcinoma cells. For this purpose, we used MDA-MB-231 breast carcinoma cells because they express the α6β4 and α5β1 integrins (Plopper et al., 1998; Mukhopadhyay et al., 1999; Saad et al., 2000). Initially, we confirmed that the survival of these cells is dependent on their expression of VEGF. As shown in Fig. 8 A, expression of a VEGF antisense oligonucleotide in these cells (Bachelder et al., 2001) resulted in an approximate fourfold increase in annexin V staining upon serum starvation compared with either untreated cells or cells that expressed the sense oligonucleotide.
Figure 8.
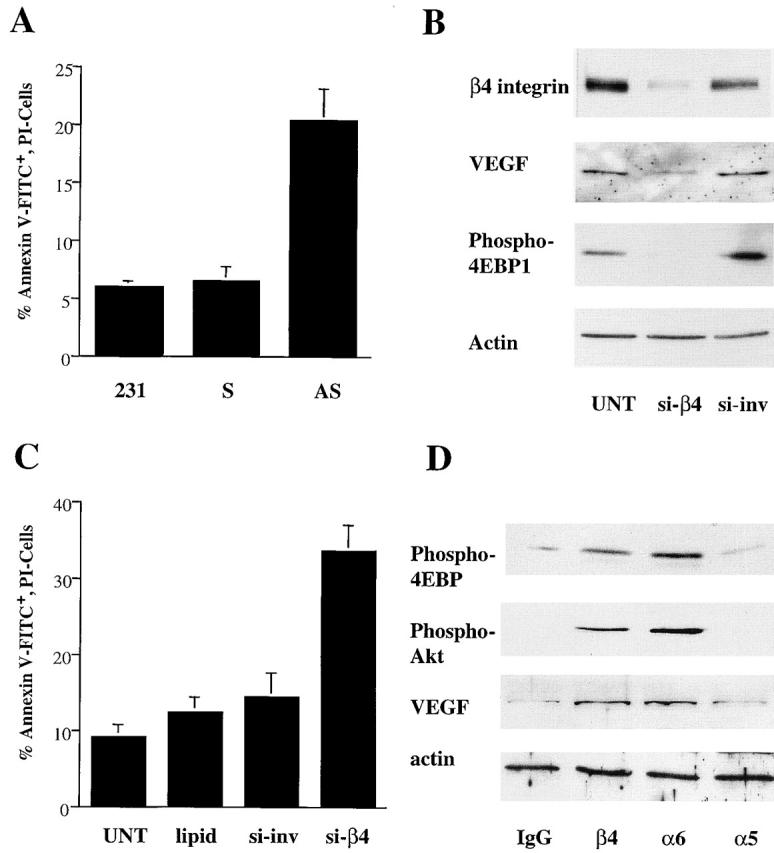
α6β4 regulates 4E-BP1 phosphorylation, VEGF expression, and survival in carcinoma cells that express this integrin endogenously. (A) Parental MDA-MB-231 cells and cells transfected with antisense or sense VEGF oligonucleotides were maintained in low serum (0.5% FBS) medium for 24 h. Apoptosis was assayed as described in the Materials and methods and is reported as the percentage of annexin V–FITC-positive, PI-negative cells. The data shown are mean values (± SD) of two separate experiments performed in triplicate. (B) MDA-MB-231 cells were left untreated (UNT) or were transfected with either an RNAi specific for the β4 integrin (si-β4) or the corresponding inverted sequence (si-inv). After 72 h, the cells were placed in medium containing low serum (0.5% FBS) for an additional 24 h and then extracted. Extracts of these cells were immunoblotted as described in the legend to Fig. 4 to assess expression of β4 integrin, VEGF, and actin, as well as the phosphorylation of 4E-BP1. Similar results were observed in three independent trials. (C) Apoptosis was assessed in the same populations of cells and is reported as the percentage of annexin V–FITC-positive, PI-negative cells. The data shown are mean values (± SD) of three independent experiments performed in triplicate. (D) MDA-MB-231 cells were maintained in low serum (0.5% FBS) medium for 24 h and harvested by trypsin treatment. The suspended cells were incubated with integrin-specific antibodies (β4 integrin, A9; α6 integrin, 2B7; α5 integrin, Sam1) or IgG for 30 min in suspension and allowed to adhere on anti-IgG–coated plates for 30 min. Extracts of these cells were immunoblotted with phosphospecific antibodies to assess the relative phosphorylation of Akt and 4E-BP1, as well as with antibodies specific for VEGF and actin. Similar results were obtained in five experiments.
To establish a role for α6β4 in regulating VEGF expression and survival rigorously, we used a small interfering RNA (RNAi) approach to inhibit β4 expression in MDA-231 cells. RNAis specific for the β4 subunit and the corresponding inverted sequence were designed and expressed in these cells by transfection. The cells were maintained in low serum (0.5%) for 24 h after transfection and then analyzed. As shown in Fig. 8 B, cells transfected with the RNAi specific for β4 exhibited a significant reduction in β4 expression in comparison with either untransfected cells or cells transfected with the inverted sequence. Importantly, the reduction in β4 expression by RNAi coincided with a marked reduction in 4E-BP1 phosphorylation and in the steady-state level of VEGF (Fig. 8 B), as well as an approximate threefold increase in annexin V staining (Fig. 8 C). These results link α6β4 expression directly to 4E-BP1 phosphorylation, VEGF expression, and survival in a carcinoma cell line that expresses endogenous α6β4.
Subsequently, we performed antibody clustering experiments to substantiate the regulation of VEGF expression by α6β4 (Fig. 8 D). Clustering of the α6β4 integrin with either an α6 integrin–specific antibody (mAb 2B7) or a β4 integrin–specific antibody (mAb A9) stimulated the phosphorylation of 4E-BP1 and Akt, and increased VEGF expression. In contrast, no induction of VEGF expression or stimulation of either 4E-BP1 or Akt phosphorylation was observed upon clustering with an α5 integrin–specific antibody (mAb Sam1) or IgG.
Discussion
This study establishes a novel mechanism by which integrins regulate growth factor expression. Specifically, our findings demonstrate the ability of a specific integrin (α6β4), which has been implicated in carcinoma progression (Mercurio and Rabinovitz, 2001), to stimulate the translation of VEGF and sustain a VEGF autocrine loop that is essential for survival. More specifically, we define a signaling pathway regulated by α6β4 that involves the preferential ability of this integrin to stimulate the phosphorylation of 4E-BP1 by activating the PI-3K–Akt pathway. As shown previously, this phosphorylation event dissociates 4E-BP1 from eIF-4E, enabling this key elongation factor to mediate the translation of VEGF and other functionally important molecules (De Benedetti and Harris, 1999; Gingras et al., 1999, 2001b; McKendrick et al., 1999). Moreover, the polysome analysis and antisense eIF-4E results we provide indicate that α6β4 stimulation of VEGF translation is cap dependent and probably doesn't involve the internal ribosome entry sites that are present in the VEGF mRNA (Huez et al., 1998; van der Velden and Thomas, 1999). Our data extend earlier reports on the involvement of eIF-4E, VEGF, and α6β4 in carcinoma progression by linking these molecules in a common signaling pathway that promotes tumor survival. Furthermore, they reveal a role for integrins in regulating growth factor expression by stimulating protein translation.
An important and novel aspect of our findings is that they add a new dimension to the understanding of how integrins promote cell survival. The widely accepted notion is that integrins, often in concert with growth factor receptors, activate specific signaling pathways that sustain survival (Taylor et al., 1999; Liu et al., 2000). We demonstrate here that the survival function of integrins may not only be mediated by the activation of a key survival kinase such as Akt and the consequent effects of Akt on apoptotic signaling (Datta et al., 1999) but also by the Akt-dependent translation and expression of growth factors, such as VEGF, that promote survival in an autocrine, and possibly paracrine, fashion. In other terms, our results reveal that VEGF is a novel target of Akt signaling by integrins that is important for survival and distinct from known survival factors that are downstream of Akt, such as Bad (Datta et al., 1999). Importantly, our recent observation that VEGF stimulates the PI-3K–Akt pathway in carcinoma cells (Bachelder et al., 2001), in conjunction with our finding that α6β4 signaling enhances VEGF expression, leads to the conclusion that integrin-mediated activation of PI-3K–Akt is amplified by integrin-stimulated VEGF expression. Moreover, we show that this amplification of PI-3K–Akt activity is important for carcinoma survival.
Although α6β4 activates PI-3K in carcinoma cells (Gambaletta et al., 2000; Nguyen et al., 2000; Hintermann et al., 2001; Trusolino et al., 2001), no attempt had been made to link this signaling event with downstream effectors that regulate protein translation, namely mTOR and 4E-BP1. One reason that this possibility had not been explored is because a role for α6β4 in regulating either protein translation or growth factor expression was not obvious. In fact, almost all of the functional studies on α6β4 in carcinoma cells have focused on its role in promoting migration and invasion, and on the mechanism by which α6β4-mediated signaling influences these processes (Mercurio, 1990; Shaw et al., 1997; Gambaletta et al., 2000; Trusolino et al., 2001). Our motivation to examine a possible connection between α6β4 and VEGF translation was based on our interest in understanding the mechanisms by which these molecules promote the survival of carcinoma cells. Indeed, our results establish a role for α6β4 in survival signaling by regulating VEGF translation, but the implications of these findings are more widespread. For example, recent studies that have argued that α6β4 is necessary for growth factor receptor (erbB2, c-met) activation of PI-3K (Gambaletta et al., 2000; Trusolino et al., 2001) raise the interesting possibility of an intimate functional association among specific growth factor receptors, α6β4, VEGF, and PI-3K, all of which have been implicated in tumor progression.
Surprisingly, few studies have examined the role of integrin signaling in regulating protein translation (e.g., Pabla et al., 1999). Indeed, there has been much more interest in defining the contribution of integrins to transcription. The ability of integrins to regulate translation, however, provides a mechanism for altering cell function rapidly, by increasing the expression of specific proteins. This possibility is exemplified by our finding that ligation of the α6β4 integrin resulted in a significant increase in VEGF protein within 60 min (Fig. 2 C). Given the fact that eIF-4E is rate limiting for the translation of proteins involved in growth control and other critical cell functions (De Benedetti and Harris, 1999), the hypothesis can be formulated that integrin-mediated regulation of translation contributes to the ability of cells to alter their behavior rapidly in response to changes in their microenvironment. This hypothesis is particularly relevant to our interest in the regulation of VEGF expression. Although much of the work in this area has focused on the ability of hypoxia to stimulate VEGF transcription and increase the stability of VEGF mRNA (von Marschall et al., 2001), it has become apparent that translational control is also important (Kevil et al., 1996; De Benedetti and Harris, 1999). Moreover, our recent finding that VEGF is essential for the survival of breast carcinoma cells in normoxia substantiates the functional importance of integrin-mediated regulation of VEGF expression (Bachelder et al., 2001).
The fact that our data implicate eIF-4E in tumor cell survival is of considerable interest because recent studies have revealed an important role for this elongation factor in cancer (DeFatta et al., 1999, 2000; Ernst-Stecken, 2000; Berkel et al., 2001). Overexpression of this factor in NIH3T3 cells, as well as other “normal” cells, stimulates division and can induce their transformation (Fukuchi-Shimogori et al., 1997). These findings are consistent with the reports that the expression of eIF-4E is elevated in solid tumors compared with normal tissue (De Benedetti and Harris, 1999). Moreover, hypoxia, a pathophysiological stress that provides a selective pressure for the survival of aggressive tumor cells, enhances eIF-4E expression (DeFatta et al., 1999). Together, these observations highlight an important role for translational control in human cancer. This role is substantiated by the fact that eIF-4E controls the translation of not only VEGF but also other molecules that influence tumor growth and survival such as c-Myc, cyclin D1, and FGF-2 (De Benedetti and Harris, 1999). From our perspective, we are intrigued by the reports that the α6β4 integrin is associated with the progression of many solid tumors, and its expression has been correlated with a poorer prognosis in patients with some of these tumors (Mercurio and Rabinovitz, 2001). Our finding that α6β4 can induce the translational function of eIF-4E by regulating the phosphorylation of 4E-BP1 provides one mechanism to account for the role of this integrin in cancer.
Materials and methods
Cells
MDA-MB-231 and MDA-MB-435 breast carcinoma cells were obtained from the Lombardi Breast Cancer Depository at Georgetown University. They were grown in low glucose DME containing 10% FBS, 1% penicillin–streptomycin, and 25 mM Hepes. For inhibitor experiments, cells were harvested by trypsinization and suspended cells were incubated with rapamycin (Calbiochem) at 100 nM or LY 294002 (Calbiochem) at 10 μM on ice for 30 min before they were plated at 37°C for the experiment.
The generation of MDA-MB-435 subclones expressing the α6β4 integrin has been described previously (Shaw et al., 1997). Tyrosine residue 1494 in the β4 subunit was mutated to a phenylalanine residue using the Quickchange site-directed mutagenesis kit (Stratagene), and stable subclones of MDA-MB-435 cells that expressed α6β4 containing this mutant β4 subunit were generated (Shaw, 2001).
For adenoviral infection, cells were grown in DME containing 10% FBS until they reached 50% confluency. At this point, the culture medium was changed to DME containing 0.5% FBS. Viral dilutions were prepared from purified viral stocks in DME containing 0.5% FBS and the cells were infected for 4 h. At the end of the infection period, the virus-containing medium was removed and the cells were washed once with PBS, and incubated for an additional 12 h in DME containing 10% FBS.
Apoptosis assays
To induce apoptosis, cells were incubated in DME containing 0.5% FBS for 24 h at 37°C. Subsequently, both adherent and nonadherent cells were harvested and their level of apoptosis was assessed using annexin V–FITC. In brief, cells were washed once with serum-containing medium, once with PBS, once with annexin V–FITC buffer (10 mM Hepes-NaOH, pH 7.4, 140 mM NaCl, 2.5 mM CaCl2), and then incubated for 15 min at room temperature with 5 μg/ml annexin V–FITC (Biosource International). After washing once with annexin V buffer, the samples were resuspended in the same buffer and analyzed by flow cytometry. Immediately before the analysis, 5 μg/ml propidium iodide (PI) (Biosource International) was added to distinguish apoptotic cells from necrotic cells.
Quantitative real-time PCR
Quantitative analysis of VEGF mRNA expression was performed by real-time PCR using an ABI Prism 7700 sequence detection system (PerkinElmer) and SYBR green master mix kit as described previously (Bachelder et al., 2001). Sequences of primers and probes were as follows: VEGF forward primer, 5′-GAAGTGGTGAAGTTCATGGATGTCTA-3′; VEGF reverse primer, 5′-TGGAAGATGTCCACCAGGGT-3′; VEGF probe, 5′-/TET/AGCGCAGCTACTGCCATCCAATCG/TAM/-3′; β-actin forward primer, 5′-TCACCATGGATGATGATATCGC-3′; β-actin reverse primer, 5′-AAGCCGGCCTTGCACAT-3′; and β-actin probe, 5′-/FAM/CGCTCGTCGTCGACAACGGCT/TAM/-3′.The data obtained are presented as the mean ratio of VEGF to β-actin mRNA (± SD) obtained from triplicate samples.
VEGF antisense oligonucleotide experiments
A VEGF antisense 2'-O-methyl phosphorothioate oligodeoxynucleotide (5′-CACCCAAGACAGCAGAA-3′) and a sense 2'-O-methyl phosphorothioate oligodeoxynucleotide (5′-CTTTCTGCTGTCTTGGGTG) (provided by Greg Robinson, Children's Hospital, Boston, MA) were used to transfect MDA-MB-435 β4 transfectants at a concentration of 0.3 μM in the presence of lipofectin reagent (2 μg/ml; GIBCO BRL). The experimental details for this approach have been described previously (Bachelder et al., 2001). In addition, the same protocol was used to express antisense and sense eIF-4E oligonucleotides, which were gifts from Arigo De Benedetti (Louisiana State University, Shreveport, LA) (DeFatta et al., 2000).
RNAi experiments
An RNAi specific for the β4 integrin subunit (GAGCUGCACGGAGUGUGUC) as well as the inverted sequence (CUGUGUGAGGCACGUCGAG) were designed and synthesized by Dharmacon, Inc. MDA-231 cells at 30% confluency were transfected with 300 pmoles of RNAi using TKO lipids (Mirus). Subsequently, the cells were maintained in complete medium for 72 h and in medium containing 0.5% FBS for an additional 24 h before analysis.
Polysome analysis
Cells (3 × 107) were maintained in medium containing low serum (0.5% FBS) for 24 h and then pretreated with 100 μg/ml cycloheximide (Calbiochem) for 15 min at 37°C before being harvested. After washing once with PBS containing 100 μg/ml cycloheximide, the cells were resuspended in 0.5 ml of a modified U+S buffer (Davies and Abe, 1995). This buffer was composed of 200 mM Tris-HCl (pH 8.8), 25 mM MgCl2, 5 mM EGTA (pH 8.0), 150 mM KCl, 10 μg/ml heparin, 5 mM DTT, 1% sodium deoxycholate, 2% polyoxyethylene 10-tridecy ester, 100 μg/ml cycloheximide, and 200 mM sucrose. Ribonuclease inhibitor (Amersham Biosciences) was added to a final concentration of 0.5 U/μl. Cells were homogenized with 20–25 strokes in a Kontes tissue homogenizer, followed by centrifugation for 5 min at 14,000 g. The supernatant was collected and frozen at −80°C until further use. Sucrose gradients (15–50%, wt/wt) were layered with 300 μl of cleared cell extract, which was then centrifuged at 160,000 g for 2 h. Fractions (0.75–0.375 ml) were withdrawn from the top of the gradient and monitored for absorbency at 254 nm using an ISCO syringe pump with UV-6 detector. Total RNA from the sucrose gradient fractions was extracted using Trizol LS (Life Technologies) according to the manufacturer's instructions. Quantitative real-time PCR was used to measure the VEGF mRNA level in each fraction as described above.
Integrin signaling experiments
Cells were harvested by trypsin treatment and washed twice with DME containing 25 mM Hepes and 0.1% BSA. After washing, the cells were resuspended in the same buffer at a concentration of 2 × 106 cells/ml and incubated for 30 min with integrin-specific antibodies (4 μg/ml) or with either mouse or rat IgG (4 μg/ml). The cells were washed once, resuspended in the same buffer, and added to plates that had been coated overnight with either the anti–mouse or rat IgG. After a 60-min incubation at 37°C, the cells that had attached to integrin-specific antibodies were washed twice with cold PBS and solubilized at 4°C for 10 min using RIPA buffer (20 mM Tris buffer, pH 7.4, containing 0.14 M NaCl, 1% NP-40, 10% glycerol, 1 mM sodium orthovanadate, 2 mM PMSF, 5 μg/ml aprotinin, pepstatin, and leupeptin). The IgG-treated cells were harvested by centrifugation and solubilized with RIPA buffer.
Protein analysis
Aliquots of cell extracts containing equivalent amounts of protein were solubilized using 5× sample buffer containing 100 mM DTT and then incubated at 100°C for 15 min. These extracts were resolved by SDS-PAGE and transferred to nitrocellulose filters. The filters were blocked for 1 h using a 50 mM Tris buffer, pH 7.5, containing 0.15 M NaCl, 0.05% Tween-20 (TBST), and 5% (wt/vol) Carnation dry milk. The filters were incubated overnight in the same buffer containing antibodies specific for p70S6K, 4EBP antibodies (Santa Cruz Biotechnology, Inc.), actin (ICN Biomedicals), and VEGF (clone 618, provided by Donald Senger, Beth Israel Deaconess Medical Center). After three, 10-min washes in TBST, the filters were incubated for 1 h in blocking buffer containing HRP-conjugated secondary antibodies. After three 10-min washes in TBST, proteins were detected by ECL (Pierce Chemical Co.).
For immunoblots involving phosphospecific antibodies, the filters were blocked for 1 h using a 10 mM Tris buffer, pH 7.5, containing 0.5 M NaCl, 0.1% Tween-20, and 2% (wt/vol) BSA. The filters were washed briefly and then incubated overnight at 4°C in the same blocking buffer containing antibodies specific for phospho-p70S6K (Thr-389; Cell Signaling Technology), phospho–4E-BP1 (Ser-65; Cell Signaling Technology), phospho-Erk (E10; Cell Signaling Technology), and phospho-Akt (Ser-473 clone 4E2; Cell Signaling Technology). After washing, the filters were incubated for 1 h in blocking buffer containing HRP-conjugated secondary antibody and the proteins were detected by ECL.
Acknowledgments
We thank Aisling S. Dugan and Melissa A. Wendt for expert technical assistance and Ken Walsh (Boston University Medical Center, Boston, MA) for help with the adenoviral experiments. We also thank Lee Gehrke and Trei Martin (Massachusetts Institute of Technology, Cambridge, MA) for assisting with the polysome analysis. Helpful discussions were had with Aimee Crago (Harvard Medical School) and Don Senger.
This work was supported by National Institutes of Health grants CA89209 and CA80789 (A.M. Mercurio), CA81697 (R.E. Bachelder), and CA81325 (L.M. Shaw) and US Army Medical Research grants BC001077 (J. Chung) and BC000697 (A.M. Mercurio).
Footnotes
Abbreviations used in this paper: 4E-BP1, 4E-binding protein; eIF-4E, eukaryotic initiation factor-4E; mTOR, mammalian target of rapamycin; Myr-Akt, myristoylated Akt; PI, propidium iodide; PI-3K, phosphatidylinositol 3-kinase; RNAi, small interfering RNA; VEGF, vascular endothelial growth factor.
References
- Akiri, G., D. Nahari, Y. Finkelstein, S.Y. Le, O. Elroy-Stein, and B.Z. Levi. 1998. Regulation of vascular endothelial growth factor (VEGF) expression is mediated by internal initiation of translation and alternative initiation of transcription. Oncogene. 17:227–236. [DOI] [PubMed] [Google Scholar]
- Bachelder, R.E., A. Marchetti, R. Falcioni, S. Soddu, and A.M. Mercurio. 1999. a. Activation of p53 function in carcinoma cells by the alpha6beta4 integrin. J. Biol. Chem. 274:20733–20737. [DOI] [PubMed] [Google Scholar]
- Bachelder, R.E., M.J. Ribick, A. Marchetti, R. Falcioni, S. Soddu, K.R. Davis, and A.M. Mercurio. 1999. b. p53 inhibits alpha 6 beta 4 integrin survival signaling by promoting the caspase 3–dependent cleavage of AKT/PKB. J. Cell Biol. 147:1063–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachelder, R.E., A. Crago, J. Chung, M.A. Wendt, L.M. Shaw, G. Robinson, and A.M. Mercurio. 2001. Vascular endothelial growth factor is an autocrine survival factor for neuropilin-expressing breast carcinoma cells. Cancer Res. 61:5736–5740. [PubMed] [Google Scholar]
- Berkel, H.J., E.A. Turbat-Herrera, R. Shi, and A. de Benedetti. 2001. Expression of the translation initiation factor eIF4E in the polyp-cancer sequence in the colon. Cancer Epidemiol. Biomarkers Prev. 10:663–666. [PubMed] [Google Scholar]
- Brown, L.F., A.J. Guidi, S.J. Schnitt, L. Van De Water, M.L. Iruela-Arispe, T.K. Yeo, K. Tognazzi, and H.F. Dvorak. 1999. Vascular stroma formation in carcinoma in situ, invasive carcinoma, and metastatic carcinoma of the breast. Clin. Cancer Res. 5:1041–1056. [PubMed] [Google Scholar]
- Datta, S.R., A. Brunet, and M.E. Greenberg. 1999. Cellular survival: a play in three Akts. Genes Dev. 13:2905–2927. [DOI] [PubMed] [Google Scholar]
- Davies, E., and S. Abe. 1995. Methods for isolation and analysis of polyribosomes. Methods Cell Biol. 50:209–222. [DOI] [PubMed] [Google Scholar]
- De Benedetti, A., and A.L. Harris. 1999. eIF4E expression in tumors: its possible role in progression of malignancies. Int. J. Biochem. Cell Biol. 31:59–72. [DOI] [PubMed] [Google Scholar]
- DeFatta, R.J., E.A. Turbat-Herrera, B.D. Li, W. Anderson, and A. De Benedetti. 1999. Elevated expression of eIF4E in confined early breast cancer lesions: possible role of hypoxia. Int. J. Cancer. 80:516–522. [DOI] [PubMed] [Google Scholar]
- DeFatta, R.J., C.A. Nathan, and A. De Benedetti. 2000. Antisense RNA to eIF4E suppresses oncogenic properties of a head and neck squamous cell carcinoma cell line. Laryngoscope. 110:928–933. [DOI] [PubMed] [Google Scholar]
- Dennis, P.B., S. Fumagalli, and G. Thomas. 1999. Target of rapamycin (TOR): balancing the opposing forces of protein synthesis and degradation. Curr. Opin. Genet. Dev. 9:49–54. [DOI] [PubMed] [Google Scholar]
- Dvorak, H.F., J.A. Nagy, D. Feng, L.F. Brown, and A.M. Dvorak. 1999. Vascular permeability factor/vascular endothelial growth factor and the significance of microvascular hyperpermeability in angiogenesis. Curr. Top. Microbiol. Immunol. 237:97–132. [DOI] [PubMed] [Google Scholar]
- Ernst-Stecken, A. 2000. The molecular marker eIF4E in the surgical margins of the resection preparations of head-neck tumors as a prognostic factor. Strahlenther. Onkol. 176:383–384. [PubMed] [Google Scholar]
- Fearon, E.R. 1999. Cancer progression. Curr. Biol. 9:R873–R875. [DOI] [PubMed] [Google Scholar]
- Fukuchi-Shimogori, T., I. Ishii, K. Kashiwagi, H. Mashiba, H. Ekimoto, and K. Igarashi. 1997. Malignant transformation by overproduction of translation initiation factor eIF4G. Cancer Res. 57:5041–5044. [PubMed] [Google Scholar]
- Gambaletta, D., A. Marchetti, L. Benedetti, A.M. Mercurio, A. Sacchi, and R. Falcioni. 2000. Cooperative signaling between alpha(6)beta(4) integrin and ErbB-2 receptor is required to promote phosphatidylinositol 3-kinase-dependent invasion. J. Biol. Chem. 275:10604–10610. [DOI] [PubMed] [Google Scholar]
- Gingras, A.C., S.P. Gygi, B. Raught, R.D. Polakiewicz, R.T. Abraham, M.F. Hoekstra, R. Aebersold, and N. Sonenberg. 1999. Regulation of 4E-BP1 phosphorylation: a novel two-step mechanism. Genes Dev. 13:1422–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingras, A.C., B. Raught, S.P. Gygi, A. Niedzwiecka, M. Miron, S.K. Burley, R.D. Polakiewicz, A. Wyslouch-Cieszynska, R. Aebersold, and N. Sonenberg. 2001. a. Hierarchical phosphorylation of the translation inhibitor 4E-BP1. Genes Dev. 15:2852–2864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingras, A.C., B. Raught, and N. Sonenberg. 2001. b. Regulation of translation initiation by FRAP/mTOR. Genes Dev. 15:807–826. [DOI] [PubMed] [Google Scholar]
- Hanahan, D., and J. Folkman. 1996. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell. 86:353–364. [DOI] [PubMed] [Google Scholar]
- Hanahan, D., and R.A. Weinberg. 2000. The hallmarks of cancer. Cell. 100:57–70. [DOI] [PubMed] [Google Scholar]
- Hintermann, E., M. Bilban, A. Sharabi, and V. Quaranta. 2001. Inhibitory role of α6β4-associated erbB-2 and phosphoinositide 3-kinase in keratinocyte haptotactic migration dependent on alpha3beta1 integrin. J. Cell Biol. 153:465–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huez, I., L. Creancier, S. Audigier, M.C. Gensac, A.C. Prats, and H. Prats. 1998. Two independent internal ribosome entry sites are involved in translation initiation of vascular endothelial growth factor mRNA. Mol. Cell. Biol. 18:6178–6190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kevil, C.G., A. De Benedetti, D.K. Payne, L.L. Coe, F.S. Laroux, and J.S. Alexander. 1996. Translational regulation of vascular permeability factor by eukaryotic initiation factor 4E: implications for tumor angiogenesis. Int. J. Cancer. 65:785–790. [DOI] [PubMed] [Google Scholar]
- Liu, W., S.A. Ahmad, N. Reinmuth, R.M. Shaheen, Y.D. Jung, F. Fan, and L.M. Ellis. 2000. Endothelial cell survival and apoptosis in the tumor vasculature. Apoptosis. 5:323–328. [DOI] [PubMed] [Google Scholar]
- McKendrick, L., V.M. Pain, and S.J. Morley. 1999. Translation initiation factor 4E. Int. J. Biochem. Cell Biol. 31:31–35. [DOI] [PubMed] [Google Scholar]
- Mercurio, A.M. 1990. Laminin: multiple forms, multiple receptors. Curr. Opin. Cell Biol. 2:845–849. [DOI] [PubMed] [Google Scholar]
- Mercurio, A.M., and I. Rabinovitz. 2001. Towards a mechanistic understanding of tumor invasion: lessons from the alpha6beta 4 integrin. Semin. Cancer Biol. 11:129–141. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay, R., R.L. Theriault, and J.E. Price. 1999. Increased levels of alpha6 integrins are associated with the metastatic phenotype of human breast cancer cells. Clin. Exp. Metastasis. 17:325–332. [DOI] [PubMed] [Google Scholar]
- Nabors, L.B., G.Y. Gillespie, L. Harkins, and P.H. King. 2001. HuR, a RNA stability factor, is expressed in malignant brain tumors and binds to adenine- and uridine-rich elements within the 3′ untranslated regions of cytokine and angiogenic factor mRNAs. Cancer Res. 61:2154–2161. [PubMed] [Google Scholar]
- Nguyen, B.P., S.G. Gil, and W.G. Carter. 2000. Deposition of laminin 5 by keratinocytes regulates integrin adhesion and signaling. J. Biol. Chem. 275:31896–31907. [DOI] [PubMed] [Google Scholar]
- Pabla, R., A.S. Weyrich, D.A. Dixon, P.F. Bray, T.M. McIntyre, S.M. Prescott, and G.A. Zimmerman. 1999. Integrin-dependent control of translation: engagement of integrin αIIbβ3 regulates synthesis of proteins in activated human platelets. J. Cell Biol. 144:175–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plopper, G.E., S.Z. Domanico, V. Cirulli, W.B. Kiosses, and V. Quaranta. 1998. Migration of breast epithelial cells on laminin-5: differential role of integrins in normal and transformed cell types. Breast Cancer Res. Treat. 51:57–69. [DOI] [PubMed] [Google Scholar]
- Raught, B., and A.C. Gingras. 1999. eIF4E activity is regulated at multiple levels. Int. J. Biochem. Cell Biol. 31:43–57. [DOI] [PubMed] [Google Scholar]
- Saad, S., L.J. Bendall, A. James, D.J. Gottlieb, and K.F. Bradstock. 2000. Induction of matrix metalloproteinases MMP-1 and MMP-2 by co-culture of breast cancer cells and bone marrow fibroblasts. Breast Cancer Res. Treat. 63:105–115. [DOI] [PubMed] [Google Scholar]
- Schmelzle, T., and M.N. Hall. 2000. TOR, a central controller of cell growth. Cell. 103:253–262. [DOI] [PubMed] [Google Scholar]
- Scotlandi, K., S. Benini, M. Sarti, M. Serra, P.L. Lollini, D. Maurici, P. Picci, M.C. Manara, and N. Baldini. 1996. Insulin-like growth factor I receptor-mediated circuit in Ewing's sarcoma/peripheral neuroectodermal tumor: a possible therapeutic target. Cancer Res. 56:4570–4574. [PubMed] [Google Scholar]
- Sekulic, A., C.C. Hudson, J.L. Homme, P. Yin, D.M. Otterness, L.M. Karnitz, and R.T. Abraham. 2000. A direct linkage between the phosphoinositide 3-kinase-AKT signaling pathway and the mammalian target of rapamycin in mitogen-stimulated and transformed cells. Cancer Res. 60:3504–3513. [PubMed] [Google Scholar]
- Shaw, L.M. 2001. Identification of insulin receptor substrate 1 (IRS-1) and IRS-2 as signaling intermediates in the α6β4 integrin-dependent activation of phosphoinositide 3-OH kinase and promotion of invasion. Mol. Cell. Biol. 21:5082–5093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw, L.M., I. Rabinovitz, H.H. Wang, A. Toker, and A.M. Mercurio. 1997. Activation of phosphoinositide 3-OH kinase by the alpha6beta4 integrin promotes carcinoma invasion. Cell. 91:949–960. [DOI] [PubMed] [Google Scholar]
- Shweiki, D., A. Itin, D. Soffer, and E. Keshet. 1992. Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature. 359:843–845. [DOI] [PubMed] [Google Scholar]
- Stein, I., A. Itin, P. Einat, R. Skaliter, Z. Grossman, and E. Keshet. 1998. Translation of vascular endothelial growth factor mRNA by internal ribosome entry: implications for translation under hypoxia. Mol. Cell. Biol. 18:3112–3119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor, S.T., J.A. Hickman, and C. Dive. 1999. Survival signals within the tumour microenvironment suppress drug-induced apoptosis: lessons learned from B lymphomas. Endocr. Relat. Cancer. 6:21–23. [DOI] [PubMed] [Google Scholar]
- Tokunou, M., T. Niki, K. Eguchi, S. Iba, H. Tsuda, T. Yamada, Y. Matsuno, H. Kondo, Y. Saitoh, H. Imamura, and S. Hirohashi. 2001. c-MET expression in myofibroblasts: role in autocrine activation and prognostic significance in lung adenocarcinoma. Am. J. Pathol. 158:1451–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trusolino, L., A. Bertotti, and P.M. Comoglio. 2001. A signaling adapter function for alpha6beta4 integrin in the control of HGF-dependent invasive growth. Cell. 107:643–654. [DOI] [PubMed] [Google Scholar]
- van der Velden, A.W., and A.A. Thomas. 1999. The role of the 5′ untranslated region of an mRNA in translation regulation during development. Int. J. Biochem. Cell Biol. 31:87–106. [DOI] [PubMed] [Google Scholar]
- von Marschall, Z., T. Cramer, M. Hocker, G. Finkenzeller, B. Wiedenmann, and S. Rosewicz. 2001. Dual mechanism of vascular endothelial growth factor upregulation by hypoxia in human hepatocellular carcinoma. Gut. 48:87–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong, A.S., S.L. Pelech, M.M. Woo, G. Yim, B. Rosen, T. Ehlen, P.C. Leung, and N. Auersperg. 2001. Coexpression of hepatocyte growth factor-Met: an early step in ovarian carcinogenesis? Oncogene. 20:1318–1328. [DOI] [PubMed] [Google Scholar]