

Abstract
We have identified a new pathway of ER-associated degradation in Saccharomyces cerevisiae that functions separately from the HRD/DER pathway comprised of Hrd1p, Hrd3p, Der1p, and Ubc7p. This pathway, termed Hrd1p independent-proteolysis (HIP), is capable of recognizing and degrading both lumenal (CPY* and PrA*), and integral membrane proteins (Sec61–2p) that misfold in the ER. CPY* overexpression likely saturates the HRD/DER pathway and activates the HIP pathway, so the slowed degradation kinetics of CPY* in a hrd1Δ strain is restored to a wild-type rate when CPY* is overexpressed. Substrates of HIP require vesicular trafficking between the ER and Golgi apparatus before degradation by the ubiquitin-proteasome system. Ubiquitination of HIP substrates does not involve the HRD/DER pathway ubiquitin ligase Hrd1p, but instead uses another ubiquitin ligase, Rsp5p. HIP is regulated by the unfolded protein response as Ire1p is necessary for the degradation of CPY* when overexpressed, but not when CPY* is expressed at normal levels. Both the HIP and HRD/DER pathways contribute to the degradation of CPY*, and only by eliminating both is CPY* degradation completely blocked.
Keywords: endoplasmic reticulum; ER-associated degradation; degradation; ubiquitin ligase; secretory pathway
Introduction
The highly conserved eukaryotic secretory pathway initiates at the ER, where proteins enter through the Sec61p-mediated translocon in an unfolded state (Deshaies et al., 1991). In the lumen of the ER, proteins undergo folding and maturation involving glycosylation, disulfide bond formation, and oligomerization. Before exiting the ER, proteins must first pass an ER quality control (ERQC)* system that ensures correct folding and processing has transpired (Brodsky and McCracken, 1999; Ellgaard et al., 1999). Failure to pass this quality control checkpoint results in the protein's degradation, preventing any toxic consequences that may result from an accumulation of aberrant proteins in the secretory pathway (Kopito, 1997; Plemper and Wolf, 1999). Quality control within the secretory pathway involves ER retention, ER-associated degradation (ERAD) and potential retrieval from downstream organelles (Ellgaard et al., 1999). ERQC uses molecular chaperones (Brodsky et al., 1999) and glycosylation events (Hebert et al., 1995; Jakob et al., 1998) to recognize misfolded or unassembled proteins. Such proteins are exported from the ER to the cytosol via the translocon and are degraded. The most well-characterized facet of ERAD occurs in yeast and utilizes the HRD/DER pathway (Wilhovsky et al., 2000) or HRD complex (Gardner et al., 2000). The HRD/DER pathway consists of Hrd1p/Der3p, Hrd3p, Der1p, and the ubiquitin-conjugating enzymes Ubc7p and Ubc1p, which together act to deliver misfolded proteins to the proteasome (Hampton et al., 1996; Knop et al., 1996; Bordallo et al., 1998; Friedlander et al., 2000). Hrd1p/Der3p, an ER-integral membrane protein, is the E3-ubiquitin ligase whose cytosolically positioned RING-H2 domain mediates the specific covalent attachment of ubiquitin to the target substrates (Bays et al., 2001; Deak and Wolf, 2001). Hrd3p regulates Hrd1p (Gardner et al., 2000), whereas the function of Der1p remains unknown (Knop et al., 1996).
ERQC contributes to the manifestation of a number of diseases by either depleting cells of essential proteins, or by the accumulation and aggregation of misfolded/mutant proteins it is incapable of degrading. Such substrates include the following: CFTR-ΔF508 (Ward et al., 1995), α1-antitrypsin inhibitor (Teckman and Perlmutter, 1996), unassembled T cell receptor subunits (Bonifacino et al., 1989), and apoB under conditions of limited lipid availability (Fisher et al., 1997). Pathogens are able to promote or conceal their infection by manipulating the ERQC system to degrade CD4 or MHC class I heavy chain molecules (Margottin et al., 1996; Wiertz et al., 1996), whereas bacterial toxins usurp ERQC to access cytosolic targets (Simpson et al., 1999). ERQC substrates characterized in yeast include CPY* and PrA*, which are mutant forms of the yeast vacuolar carboxypeptidase Y (CPY) and proteinase A (PrA), respectively, that are retained in the ER (Knop et al., 1996), and Sec61–2p, which is a mutant subunit of the translocon (Biederer et al., 1996). Finally, the ERQC-degradative machinery also acts in a regulatory role to moderate the half-life of correctly folded proteins such as HMG-CoA reductase in response to cellular signals so as to modulate the sterol-synthesizing mevalonate pathway (Hampton et al., 1996).
The ERQC is intimately associated with the unfolded protein response (UPR), a wide-ranging cellular response to transcriptionally up-regulate a number of distinct mechanisms to cope with the ER stress encountered when increased amounts of misfolded proteins accumulate in the ER (Friedlander et al., 2000). ER stress is thought to be sensed by Ire1p and the chaperone Kar2p, which transduce the UPR signal from the ER to the nucleus where in yeast almost 400 genes are transcriptionally up-regulated, including those encoding molecular chaperones, components of the HRD/DER pathway, and machinery responsible for vesicular transport between the ER and Golgi apparatus (Travers et al., 2000).
The HRD/DER pathway is able to target a number of ERQC substrates for proteasomal degradation, yet CPY* turnover, while impaired in HRD/DER-deficient strains, still proceeds at a substantial rate (Hill and Cooper, 2000). This slowed, but not abolished, degradation in the absence of Hrd1p is also the case for a number of other substrates (Wilhovsky et al., 2000). Furthermore, the inactivation of the HRD/DER pathway has no effect on the degradation of unassembled Vph1p (Hill and Cooper, 2000) and Fur4–430Np (Wilhovsky et al., 2000). This HRD/DER-independent means of degradation prompted the prediction of an additional degradative pathway (Hill and Cooper, 2000), or at a minimum an additional ubiquitin ligase (Wilhovsky et al., 2000).
Our interest in such an alternative degradative pathway resulted in the discovery of the requirement of ER-Golgi transport for efficient degradation of ERQC substrates (Caldwell et al., 2001). Inactivation of Sec12p (required for vesicle to exit the ER) or Sec18p (vesicle fusion with a post-ER compartment) significantly slowed the degradative rate of CPY* as did the absence of Erv29p (Caldwell et al., 2001). Erv29p is localized to the ER and Golgi apparatus, contains an ER-retrieval sequence, and behaves as a cargo loading/transport receptor that acts to transport a subset of lumenal proteins including CPY and PrA from the ER to the Golgi apparatus via COPII-coated vesicles (Belden and Barlowe, 2001; Caldwell et al., 2001). The absence of Erv29p severely retards the ER-Golgi transport of such ligands, yet the secretory pathway remains functional with invertase and alkaline phosphatase displaying normal transport kinetics (Caldwell et al., 2001). It is likely that CPY* also requires Erv29p to enter such transport vesicles and therefore, the absence of Erv29p severely impairs the transport of CPY* from the ER to the Golgi apparatus. This work investigates the role ER-Golgi trafficking plays in ERQC and resulted in the identification of an HRD/DER-independent ERQC mechanism capable of recognizing both lumenal and integral membrane substrates in the yeast Saccharomyces cerevisiae. This separate degradative mechanism requires ER-Golgi transport as evidenced by the requirement of Erv29p and Sec12p. We were able to directly observe this novel degradative mechanism by overexpressing CPY* because under these conditions the HRD/DER pathway appears saturated, causing CPY* degradation to be totally dependent on this novel mechanism. Substrates targeted for degradation through this mechanism are ubiquitinated, even in the absence of Hrd1p, by the ubiquitin ligase Rsp5p. Only by removing both ERQC pathways is CPY* degradation completely blocked. Our data call attention to the complexity of quality control in the ER and show that mechanisms in addition to the HRD/DER pathway are able to deliver misfolded proteins to the proteasome for degradation.
Results
CPY* degradation continues in HRD/DER-deficient cells and requires ER-Golgi transport
The ERQC machinery is responsible for the ERAD of misfolded proteins and unassembled subunits that arise in the ER. To date, much of the analyses have focused on the involvement of HRD/DER components in affecting the degradation of ERQC substrates. Although such components (Hrd1p/Der3p, Hrd3p, Der1p, Ubc7p, and Ubc1p) play a major role in the degradation of ERQC substrates such as CPY*, we have found that these substrates continue to be degraded at a significant rate in HRD/DER-deficient cells. Fig. 1 A shows the degradation kinetics of wild-type cells expressing CPY* compared with the congenic strain lacking the HRD1-encoded ubiquitin ligase. As expected, the disruption of HRD1 significantly slowed the degradation rate of CPY* compared with that in wild-type cells, but instead of completely blocking CPY* degradation, a substantial rate of CPY* degradation still persists. Disruption of HRD3, DER1, or UBC7 gave similar results to that shown for the hrd1Δ strain (unpublished data). Double disruptions of HRD/DER components (hrd1Δ der1Δ) did not further stabilize CPY* beyond that of the hrd1Δ disruption alone (Fig. 1 A), confirming that these components are in the same pathway.
Figure 1.
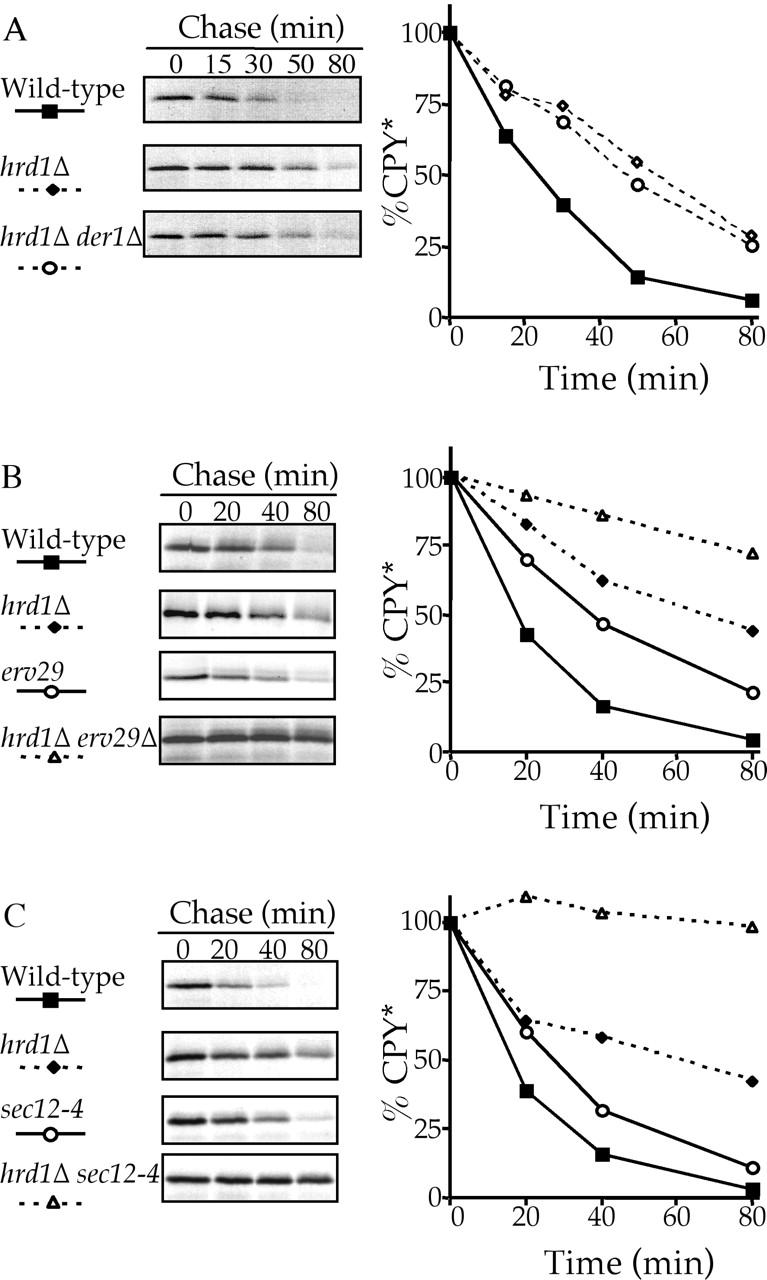
CPY* degradation is completely blocked upon inhibition of both the HRD/DER pathway and ER-Golgi transport. hrd1Δ (KHY171), hrd1Δ der1Δ (KHY237), erv29Δ (KHY270), erv29Δ hrd1Δ (KHY279), sec12–4 (KHY306), sec12–4 hrd1Δ (KHY308), and wild-type (KHY163) cells were radiolabeled, chased, and CPY* immunoprecipitated at various times. Samples were separated by SDS-PAGE, visualized by fluorography, and quantitated using a Phosphorimager as performed previously (Hill and Cooper, 2000). (A) HRD/DER pathway–deficient strains (hrd1Δ, hrd1Δ der1Δ) and wild-type cells expressing single copy levels of CPY*. (B) hrd1Δ, erv29Δ, hrd1Δ erv29Δ, or wild-type strains expressing single copy levels of CPY*. (C) hrd1Δ, sec12–4, hrd1Δ sec12–4, or wild-type strains expressing single copy levels of CPY* (34°C).
The finding that CPY* degradation continued in the absence of functional HRD/DER components raised the possibility that the ERQC system possesses an additional mechanism or pathway with which to achieve the degradation of ERQC substrates. Previously, we found that anterograde vesicular transport from the ER to the Golgi apparatus was required for the efficient degradation of the lumenal ERQC substrates CPY* and PrA* (Caldwell et al., 2001). The degradative rate of CPY* and PrA* was reduced when either all vesicular transport from the ER was blocked through the use of the temperature-sensitive sec12–4 allele or when exit from the ER of specific cargo proteins such as CPY* and PrA* was impaired by the disruption of ERV29 (Caldwell et al., 2001). Given the strong possibility of an additional degradative mechanism/pathway, we undertook to determine if this ER-Golgi transport step was a component of the HRD/DER pathway or of an independent pathway. Strains were constructed that were either lacking Hrd1p (hrd1Δ), blocked in ER-Golgi transport (erv29Δ or sec12–4), or both (hrd1Δ erv29Δ or hrd1Δ sec12–4). Fig. 1 B shows that hrd1Δ erv29Δ double mutant cells stabilize CPY* to a much greater extent than either single mutation alone. This additive effect demonstrates that the role of ER-Golgi transport in ERQC is independent of the HRD/DER components and therefore part of a distinct degradative pathway. The conclusion that the ERQC possesses an additional distinct pathway was further supported by the complete stabilization of CPY* in a hrd1Δ sec12–4 double mutant relative to either single mutant (Fig. 1 C).
CPY* overexpression results in HRD/DER-independent degradation
In a separate project investigating the association between CPY* and the translocon in HRD/DER-deficient strains, CPY* was overexpressed approximately eightfold. Surprisingly, we observed that CPY* was now degraded with wild-type kinetics in a hrd1Δ strain instead of the expected slowed degradative rate (Fig. 2 A). The wild-type degradative rate caused by overexpression of CPY* was also observed in both der1Δ and hrd3Δ cells (unpublished data). This HRD/DER-independent degradation was further investigated using the ERQC substrate PrA*. PrA* was overexpressed in both wild-type and hrd1Δ strains, and the effect on the rate of single copy CPY* degradation was examined. Fig. 2 B shows that the overexpression of PrA*–HA also resulted in CPY* being degraded with wild-type kinetics in a hrd1Δ strain. This suggests that this HRD/DER-independent degradative mechanism is not specific for CPY* and capable of recognizing and degrading a variety of lumenal ERQC substrates.
Figure 2.
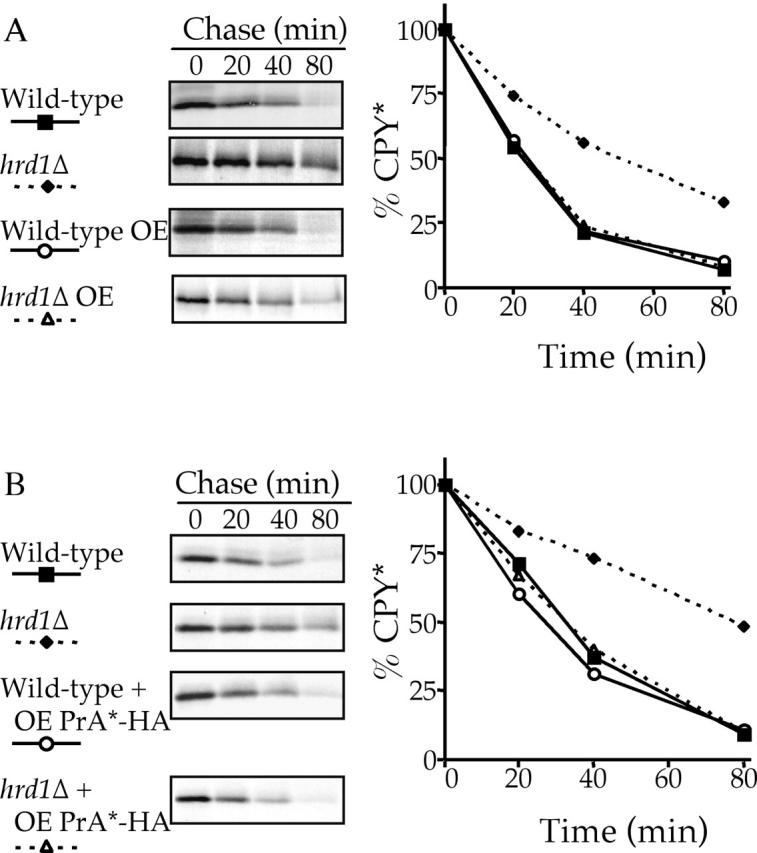
CPY* and PrA* can be degraded independently of the HRD/DER pathway. hrd1Δ and wild-type strains were radiolabeled, chased, and CPY* (A and B) immunoprecipitated at various times. Samples were analyzed as in Fig. 1. (A) Wild-type (KHY163) and hrd1Δ (KHY171) strains expressing single copy or overexpressed (OE) levels of CPY* (pAC453). (B) Wild-type (KHY163) and hrd1Δ (KHY171) strains expressing CPY* with and without overexpressed PrA*–HA (pAC540) present.
Our working model is that the ERQC uses at least two distinct mechanisms to identify and degrade misfolded proteins from the ER: (1) the HRD/DER pathway using the components Hrd1p/Der3p, Hrd3p, Der1p, and Ubc7p; and (2) an alternative mechanism distinct from the HRD/DER pathway that we term the Hrd1p-independent proteolysis (HIP) pathway. Upon overexpression of ERQC substrates, the capacity of the HRD/DER pathway is likely saturated, and the remaining CPY* is accommodated by the alternative HIP mechanism. The two lines of evidence for a HRD/DER-independent alternative pathway are as follows: (1) the continuing degradation of ERQC substrates in HRD/DER-deficient strains that requires ER-Golgi transport; and (2) overexpression of ERQC substrates accelerates/restores the degradative rate in HRD/DER-deficient strains. To confirm that a single mechanism is responsible for both of these observations, we formed several testable predictions that resulted from the integration of the ER-Golgi transport requirement and the HRD/DER pathway saturation model. First, in otherwise wild-type cells expressing single copy levels of CPY*, the absence of Erv29p should only moderately slow the degradative rate of CPY* as the HRD/DER pathway is functional. However, upon overexpression of CPY*, the HRD/DER components would be saturated and the vast majority of the CPY* would be diverted to the ER-Golgi transport–dependent pathway requiring Erv29p. Under these overexpression conditions, the disruption of ERV29, which impairs ER-Golgi transport, would now result in significantly enhanced stabilization of CPY*. Second, the high degree of CPY* stabilization in erv29Δ cells observed upon CPY* overexpression will be HRD/DER-independent, and therefore the disruption of HRD1 in such cells will have little additional stabilizing effect. Third, the stabilization of CPY* obtained by disabling both the HRD/DER components and ER-Golgi trafficking through a hrd1Δ erv29Δ double mutation should not be suppressed by overexpressing CPY*, as the alternative pathway has already been compromised by the disruption of ERV29. All three of these predictions were observed upon experimentation. Fig. 3 A shows that the degradation of single copy levels of CPY* is slowed somewhat in an erv29Δ strain, and that the extent of stabilization was greatly increased upon overexpression of CPY*. As predicted, the imposition of a hrd1Δ mutation into the erv29Δ strain overexpressing CPY* does not further stabilize CPY* degradation (Fig. 3 B). Finally, the very strong stabilization of CPY* observed in an erv29Δ hrd1Δ double mutant expressing single copy CPY* is not suppressed upon overexpression of CPY* (Fig. 3 C).
Figure 3.
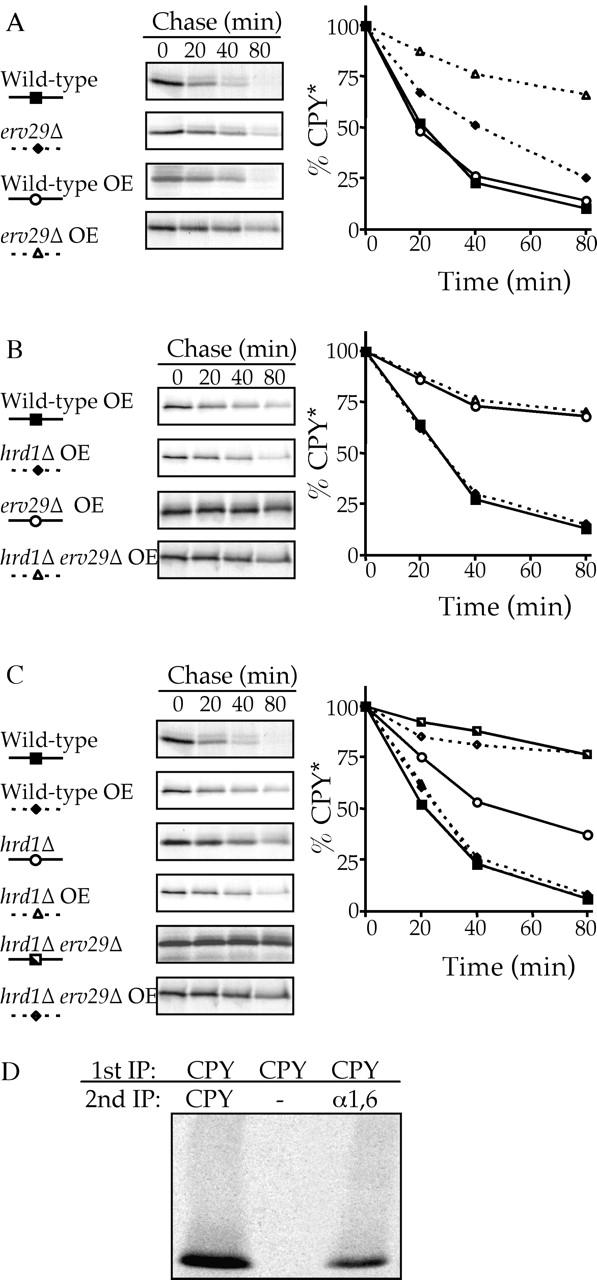
The inhibition of ER-Golgi transport ( erv29 Δ) but not the HRD/DER pathway (hrd1 Δ ) significantly slows the degradation of overexpressed (OE) CPY*. erv29Δ (KHY270), hrd1Δ (KHY171), hrd1Δ erv29Δ (KHY279), and wild-type (KHY163) cells were analyzed as in Fig. 1. (A) erv29Δ and wild-type strains expressing either single copy or overexpressed (OE) levels of CPY* (pAC453). (B) Wild-type, hrd1Δ, erv29Δ, and hrd1Δ erv29Δ strains overexpressing (OE) CPY*. (C) Wild-type, hrd1Δ, and hrd1Δ erv29Δ strains either expressing single copy or overexpressed levels of CPY*. (D) hrd1Δ (KHY171) cells overexpressing CPY* were radiolabeled and CPY* immunoprecipitated. The precipitates were reimmunoprecipitated with either CPY* or α1,6-mannose antibodies.
In summary, overexpression of CPY* and PrA* appears to saturate the capacity of the HRD/DER pathway and quantitatively shifts such substrates to the alternative pathway requiring delivery to the Golgi apparatus. To confirm that CPY* does itself transit to the Golgi apparatus, we performed sequential immunoprecipitations on radiolabeled hrd1Δ cells overexpressing CPY*, first with anti-CPY antibodies, and then with anti–α1,6-mannose antibodies. Fig. 3 D shows clearly that CPY* does indeed receive α1,6 mannose addition, which is indicative of delivery to the Golgi apparatus and confirms that this step is a requirement of HIP. Such a transport requirement for efficient degradation of overexpressed ERQC substrates might simply reflect that such substrates reach the Golgi apparatus, are subsequently secreted from the cell, and give the appearance of accelerated degradation or are alternatively sorted to the vacuole (lysosome equivalent) for degradation. Growth media was collected from radiolabeled wild-type and hrd1Δ cells overexpressing CPY* and any secreted CPY* was immunoprecipitated. Fig. 4 B shows that neither wild-type nor HRD/DER-deficient cells secrete detectable amounts of CPY*. Cells lacking the VPS10-encoded receptor responsible for sorting wild-type CPY to the vacuole were included as a control because such cells secrete large amounts of CPY (Cooper and Stevens, 1996). Additional evidence to eliminate the secretion possibility was provided by preventing secretory vesicles from fusing with the plasma membrane through the use of a temperature sensitive sec4–8 allele (Salminen and Novick, 1987), which had no stabilizing effect on overexpressed CPY* degradation (unpublished data). The potential role of the vacuole was investigated by disrupting PEP4 in a hrd1Δ strain and determining the degradation rate of CPY* expressed at both single copy and overexpressed levels. The proteolytic activity of the vacuole is controlled by the PEP4-encoded PrA that, when disrupted, results in the vacuole lacking proteolytic activity and consequential stabilization of substrates degraded there (Jones et al., 1982). Fig. 4 A shows that rendering the vacuole proteolytically inactive has no stabilizing effect on CPY* when expressed at either normal or elevated levels. This result also eliminates a role for autophagocytosis in HIP, as autophagocytosed substrates are degraded in the vacuole in a PEP4-dependent manner (Baba et al., 1997). We also investigated the degradation of CPY* in vps10Δ cells. Vps10p, the CPY sorting receptor, is also capable of sorting misfolded proteins from the trans-Golgi compartment to the vacuole (Hong et al., 1996). However, vps10Δ cells failed to stabilize overexpressed CPY* (unpublished data) and further indicated that the alternative degradation mechanism is not degrading substrates via delivery to the vacuole.
Figure 4.

Overexpressed (OE) CPY* is degraded independently of the vacuole and is not secreted from the cell. (A) hrd1Δ (KHY171) and hrd1Δ pep4Δ (KHY265) strains were analyzed as in Fig. 1. hrd1Δ PEP4 and hrd1Δ pep4Δ strains expressing single copy or overexpressed levels of CPY* (pAC519). (B) hrd1Δ (KHY171) and wild-type (KHY163) cells overexpressing (OE) CPY* and vps10Δ (AACY9) cells expressing CPY were radiolabeled, chased, and CPY* or CPY was immunoprecipitated from intracellular (I) and extracellular (E) fractions as described previously (Cooper and Stevens, 1996). Samples were visualized by fluorography.
The HIP pathway involves the proteasome and ubiquitination
Cells deficient in the HRD/DER pathway remain capable of degrading overexpressed intracellular CPY* independently of the vacuole, which suggests that the other major intracellular proteolytic activity provided by the 26S proteasome would be responsible. Strains containing pre1 pre2 mutations are deficient in 26S proteasome activity and have been previously characterized to impair CPY* turnover (Hiller et al., 1996). If overexpression of CPY* diverts it to the alternative pathway, then a strain deficient in this pathway should not be capable of accelerating/restoring the CPY* degradation upon CPY* overexpression. Fig. 5 shows wild-type and congenic pre1 pre2 cells expressing either single copy or overexpressed CPY*. As expected, the degradative rate of single copy levels of CPY* is slowed considerably in the proteasome-compromised strain, but overexpression of CPY* fails to accelerate the turnover of CPY* in the pre1 pre2 strain and demonstrates that the proteasome is responsible for degrading overexpressed CPY*.
Figure 5.

The proteasome is responsible for the degradation of overexpressed (OE) CPY*. pre1 pre2 (KHY293) and wild-type (KHY292) strains expressing single copy (pAC446) or overexpressed levels of CPY* (pAC519) were analyzed as in Fig. 1.
Both the HIP and HRD/DER pathways deliver ERQC substrates to the proteasome. The HRD/DER pathway achieves this by exporting CPY* through the translocon followed by Hrd1p-dependent ubiquitination (Plemper et al., 1997; Bays et al., 2001), but how is overexpressed CPY* delivered to the proteasome, and does it also involve ubiquitination? If HIP involves ubiquitination of overexpressed CPY*, then ubiquitinated forms of CPY* should be detectable in a hrd1Δ strain. This was tested by sequential immunoprecipitations of radiolabeled cells, first with anti-CPY antibodies followed by anti–ubiquitin-HA antibodies. Fig. 6 A (lanes 1–3) shows that CPY* in wild-type cells is heavily ubiquitinated, whereas ubiquitinated species of CPY* still existed in the hrd1Δ strain, albeit to a lesser extent. This HRD1-independent ubiquitin signal detected was specific to CPY*, as no ubiquitin signal was observed in a strain that lacked CPY* (prc1Δ).
Figure 6.
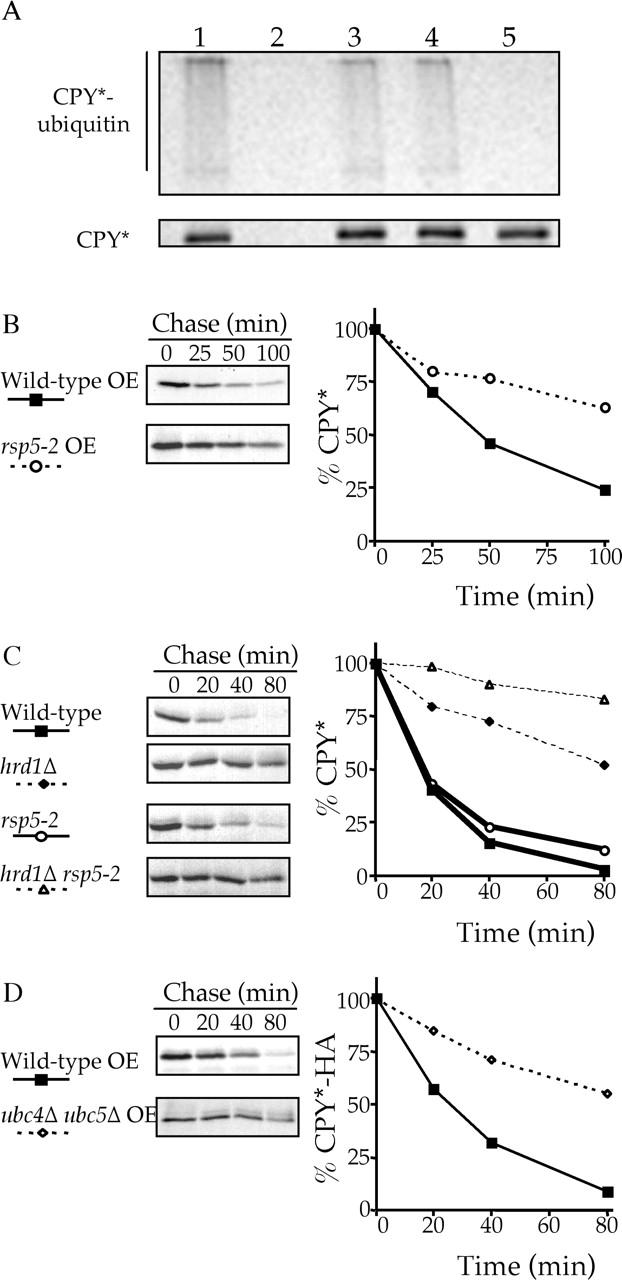
Rsp5p is the ubiquitin ligase of the HIP pathway and is responsible for the ubiquitination of CPY* in HRD/DER pathway–deficient cells. (A) Ubiquitination of CPY* in wild-type (lane 1), prc1Δ hrd1Δ (lane 2), hrd1Δ (lane 3), rsp5–2 (lane 4), and hrd1Δ rsp5–2 (lane 5) cells was assayed by radiolabeling at 37°C followed by sequential immunoprecipitations first with anti-CPY antibodies and then with anti–ubiquitin-HA antibodies. (B and C) rsp5–2 (KHY355), hrd1Δ (KHY171), hrd1Δ rsp5–2 (KHY359), and wild-type (KHY163) strains were radiolabeled, chased (at 37°C), and CPY* immunoprecipitated at various times. Samples were analyzed as in Fig. 1. (B) Wild-type and rsp5–2 cells overexpressing (OE) CPY* (pAC453). (C) Wild-type, hrd1Δ, rsp5–2, and hrd1Δ rsp5–2 expressing single copy levels of CPY*. (D) Wild-type (RH448) and ubc4Δ ubc5Δ (RH3097) overexpressing (OE) CPY*–HA were radiolabeled (at 30°C), chased, and CPY* immunoprecipitated.
Rsp5p is the ubiquitin ligase of the HIP pathway
The detection of ubiquitinated CPY* in a hrd1Δ strain requires that an additional E3 ubiquitin ligase participates in ERQC. We screened strains deficient in known or potential E3 enzymes, including the ER-localized DOA10 (Swanson et al., 2001), for impaired turnover rates of overexpressed CPY*. Ubiquitin ligases have either a RING-H2 motif or a HECT domain (Jackson et al., 2000), and using a PSI-BLAST homology search (Altschul et al., 1997), 31 proteins with significant homology to the RING-H2 domains of Hrd1p and Hrt1p were identified, whereas the 5 yeast proteins with HECT domains were previously known (Hochstrasser, 1996). The degradative rate of overexpressed CPY* was determined in the corresponding disruption or temperature-sensitive strains. Of the 36 potential ubiquitin ligases screened, only the essential gene RSP5 was necessary for the degradation of overexpressed CPY*. Fig. 6 B shows that degradation of overexpressed CPY* was significantly slowed in rsp5–2 cells at the restrictive temperature. If RSP5 represents the ubiquitin ligase of the alternative pathway, then inactivating both Rsp5p and Hrd1p should completely stabilize single copy CPY* as both the alternative and HRD/DER pathways would be incapacitated. This was indeed the case as single copy CPY* is almost completely stable in a hrd1Δ rsp5–2 strain (Fig. 6 C). Direct evidence that Rsp5p ubiquitinates ERQC substrates was that the ubiquitinated CPY* remaining in a hrd1Δ strain is not observed in the rsp5–2 hrd1Δ double mutant (Fig. 6 A, lanes 3–5).
To confirm that Rsp5p is a component of the HIP pathway instead of a potential third degradative pathway, we introduced the rsp5–2 mutation into a strain deficient for the HIP pathway (erv29Δ). The stabilization of CPY* in an erv29Δ rsp5–2 double mutant was found to be no greater than either of the single mutants, indicating that Rsp5p is indeed a component of the HIP pathway (unpublished data). Furthermore, overexpression of CPY* in a hrd1Δ rsp5–2 strain does not suppress stabilization of CPY* (unpublished data).
The RING-H2 ubiquitin ligase Hrd1p has been demonstrated to use the ubiquitin-conjugating enzymes Ubc7p and Ubc1p (Friedlander et al., 2000; Bays et al., 2001) in the HRD/DER pathway. In contrast to Hrd1p, Rsp5p is a HECT domain ubiquitin ligase, and previous work has indicated that Rsp5p uses the ubiquitin-conjugating enzymes Ubc4p and Ubc5p (Gitan and Eide, 2000). If Ubc4p and Ubc5p act in concert with Rsp5p to mediate the degradation of ERQC substrates by the HIP pathway, then the absence of both Ubc4p and Ubc5p should impair the degradation of that substrate. The degradative rate of overexpressed CPY* was examined in both wild-type and ubc4Δ ubc5Δ cells, where it was found that the absence of Ubc4p and Ubc5p significantly slowed the degradation of CPY* (Fig. 6 D).
The HIP pathway is also capable of degrading integral membrane ERQC substrates
The finding that the ERQC possesses an alternative degradative pathway capable of degrading lumenal ERQC substrates raised the issue of whether integral membrane ERQC proteins such as Sec61–2p might also be an alternative pathway substrate. The fact that Sec61–2p is not stabilized in a strain lacking Erv29p (Caldwell et al., 2001) does not exclude Sec61–2 from the alternative pathway because, Erv29p appears to act as a cargo receptor for a specific subset of ER lumenal proteins that would have no effect on integral membrane proteins. Sec61–2p seems likely to be a substrate of an alternative pathway as, analogous to CPY* in Fig. 1 A, the disabling of the HRD/DER pathway reduced, but did not abolish, the degradation of Sec61–2p (Fig. 7 A). Wild-type cells, hrd1Δ, rsp5–2, or hrd1Δ rsp5–2 double mutants expressing Sec61–2p were radiolabeled at the restrictive temperature and the degradative kinetics of Sec61–2p were examined (Fig. 7 A). Even when the HRD/DER pathway is functional, Rsp5p contributes to the degradation of Sec61–2p as shown by the retarded degradative kinetics in the rsp5–2 strain. Second, the inactivation of Rsp5p in cells lacking HRD/DER components (rsp5–2 hrd1Δ) further stabilized Sec61–2p. The findings that Sec61–2p is not fully stabilized in HRD/DER- deficient cells and that inactivation of the Rsp5p further stabilizes Sec61–2p suggest that integral membrane ERQC substrates can be degraded by the HIP pathway. The ability of the alternative pathway to degrade Sec61–2p was further demonstrated by overexpressing CPY* in hrd1Δ cells also expressing Sec61–2p with the result that the retarded Sec61–2p degradation kinetics seen in hrd1Δ cells was accelerated to those of a wild-type cell (Fig. 7 B).
Figure 7.
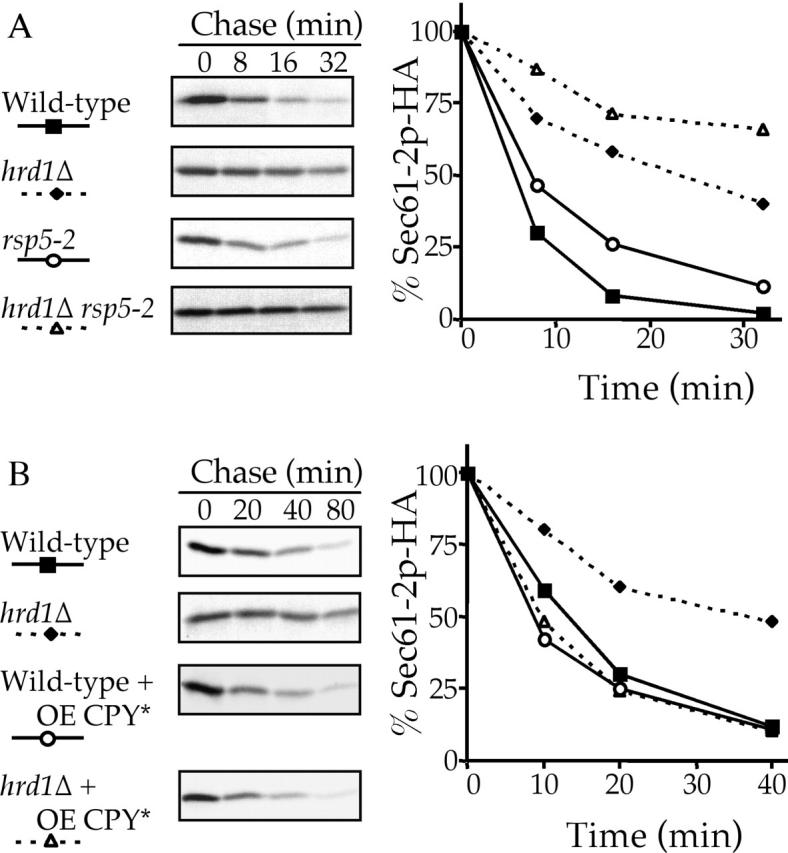
The integral membrane ERQC substrate Sec61–2p-HA can also be degraded by the HIP pathway. hrd1Δ (KHY171), rsp5–2 (KHY355), hrd1Δ rsp5–2 (KHY359), and wild-type (KHY163) cells were radiolabeled at 37°C (A) and 30°C (B), chased, Sec61–2p-HA immunoprecipitated at various times, and analyzed as in Fig. 1. (A) Wild-type, hrd1Δ, rsp5–2, and hrd1Δ rsp5–2 expressing Sec61–2p-HA (pAC460; Caldwell et al., 2001). (B) prc1Δ (KHY298) and prc1Δ hrd1Δ (KHY299) cells, in the absence of CPY*, expressing Sec61–2p-HA (pAC460) and wild-type (KHY163) and hrd1Δ (KHY171) in the presence of overexpressed (OE) CPY* expressing Sec61–2p-HA.
The UPR regulates the HIP pathway
Of the many genes transcriptionally up-regulated by the UPR during ER stress (Travers et al., 2000), half have no known function attributed to them (Fewell et al., 2001). It is conceivable that components of the HIP pathway, like those of the HRD/DER pathway (Friedlander et al., 2000), may also be up-regulated during times of stress and therefore would be under UPR transcriptional control. If so, then disabling the UPR pathway would diminish or inactivate the HIP pathway. To test this possibility, CPY* was expressed either at single copy or overexpressed levels in wild-type cells or cells disrupted for IRE1. The absence of Ire1p had little significant effect on the degradation of single copy CPY* (Fig. 8), which is consistent with that previously reported (Friedlander et al., 2000), presumably because in the absence of the UPR the capacity of the HRD/DER pathway is sufficient to cope with single copy levels of CPY*. Overexpression of CPY* in an ire1Δ strain significantly slowed the turnover kinetics of CPY* (Fig. 8), which demonstrates that the HIP pathway requires a functional UPR.
Figure 8.

The UPR is required for efficient degradation of overexpressed (OE) CPY*. ire1Δ (KHY280) and wild-type (KHY163) cells expressing single copy and overexpressed levels (pAC453) of CPY* were analyzed as in Fig. 1.
Discussion
ERQC substrates continue to be degraded in the absence of a functional HRD/DER pathway using a newly identified alternative mechanism that requires ER-Golgi vesicular transport of substrates and the ubiquitin ligase Rsp5p before proteasomal degradation. This alternative system is distinct from the HRD/DER pathway, but together the two comprise the ERQC degradative capacity because only by disabling both pathways is degradation completely blocked. We propose the acronym HIP for the alternative mechanism. Overexpression of ERQC substrates appears to saturate the capacity of the HRD/DER pathway, resulting in the degradation of ERQC substrates to be completely dependent on the HIP pathway.
The ubiquitin ligase Rsp5p is a component of the HIP pathway and together with Hrd1p is responsible for the ubiquitination of CPY*. Only by inactivating both Rsp5p and Hrd1p does CPY* fail to be ubiquitinated and remains completely stable. Rsp5p-dependent ubiquitination involves the ubiquitin-conjugating enzymes Ubc4p and Ubc5p (Gitan and Eide, 2000), and these enzymes were also required by the HIP pathway to degrade overexpressed CPY*. In addition to its role in ERQC, Rsp5p is responsible for mediating ubiquitin-dependent protein sorting and trafficking within the secretory pathway. The ubiquitination of plasma membrane proteins by Rsp5p results in their internalization and subsequent transport to the vacuole for degradation (Galan et al., 1996; Hicke and Riezman, 1996). Rsp5p, acting in concert with Bul1p and Bul2p, can also ubiquitinate proteins such as Gap1p to direct them from the trans-Golgi compartment to the vacuole for degradation (Helliwell et al., 2001). However, the role of Rsp5p in ERQC is distinct from that involving vacuolar sorting, as the HIP pathway is independent of vacuolar proteases and instead utilizes the proteasome. Furthermore, the disruption of both BUL1 and BUL2 has no effect on the degradation rate of CPY* (unpublished data).
Examination of the mechanisms used by mammalian cells to degrade ERQC substrates suggests extensive overlap with the HIP pathway described here. This similarity includes the observation of the following ERQC substrates in post-ER compartments (ER-Golgi intermediate compartment and/or Golgi subcompartments): unassembled MHC class I molecules (Hsu et al., 1991), misfolded G protein of vesicular stomatitis virus (Hammond and Helenius, 1994), mutant forms of sucrase-isomaltase and lysosomal α-glucosidase (Moolenaar et al., 1997), and precursors of human asialoglycoprotein receptor H2a (Kamhi-Nesher et al., 2001). The soluble secretory form of IgM molecules in differentiated B lymphocytes is degraded intracellularly in a manner completely dependent on transport to a post-ER compartment (Winitz et al., 1996), and truncated versions of CFTR reach the Golgi apparatus before degradation by the proteasome (Benharouga et al., 2001). Further evidence suggesting the involvement of post-ER compartment(s) is that some ERQC components are themselves found in post-ER locations: UDP-glucose:glycoprotein glucosyltransferase (UGGT), an important ERQC glycoprotein folding sensor, is found in post-ER vesicles (Zuber et al., 2001), whereas endo-α-mannosidase has been implicated in quality control and was localized to the cis/medial-Golgi apparatus (Zuber et al., 2000).
How far through the secretory pathway do ERQC substrates such as CPY* progress and what function would delivery of ERQC substrates to the Golgi apparatus serve? CPY* in either DER1-deficient cells (Knop et al., 1996) or those lacking an ER-localized proposed ERQC lectin (mnl1Δ/htm1Δ) (Jakob et al., 2001; Nakatsukasa et al., 2001) received α1,6-mannose addition but not α1,3-mannose addition, indicating that CPY* reached the cis-Golgi but not the trans-Golgi compartment (Brigance et al., 2000). Similarly, we have also found that CPY* is delivered to the cis-Golgi compartment when overexpressed in hrd1Δ cells as evidenced by the addition of α1,6-mannose. A recent report has shown that a heterologously expressed fusion ERQC substrate Kar2 hemagglutinin neuraminidase receives O-linked glycosylation indicative of it reaching the yeast cis/medial-Golgi apparatus, whereas both KHN and CPY* can be found in in vitro ER-derived COPII vesicles (Vashist et al., 2001). Vashist et al. (2001) concluded that all the KHN is delivered to the Golgi apparatus, and then is returned to the ER where it is exported to the cytosol and likely ubiquitinated by Hrd1p. However, we suggest another possibility wherein KHN (much like CPY*) is degraded via two distinct pathways (HIP and HRD/DER), in which a portion of KHN is retained in the ER and is exported and ubiquitinated by Hrd1p while the remaining KHN is transported to the Golgi apparatus and eventually ubiquitinated by Rsp5p.
After transportation to the Golgi apparatus, CPY* must then be exported from within the secretory pathway to the cytosol to be accessible to the cytosolically located Rsp5p. Therefore CPY* is likely returned from the Golgi apparatus to the ER where it is exported to the cytosol, presumably through the translocon, and subsequently ubiquitinated by Rsp5p before proteasomal degradation. A highly speculative alternative model involves the export of CPY* directly from the cis-Golgi compartment to the cytosol by an unidentified mechanism before ubiquitination by Rsp5p. These two possibilities are presented in the model shown in Fig. 9. Unfortunately, the broad subcellular distribution of Rsp5p provides no indication as to which model is correct (Gajewska et al., 2001). Rsp5p can presumably associate with both the Golgi apparatus to mediate Gap1p sorting (Helliwell et al., 2001) and the ER, as evidenced by its ubiquitination of the nuclear membrane/ER-restrained transcription factor Spt23p (Hoppe et al., 2000). Although ERQC substrates are likely returned to the ER from the Golgi apparatus, such a requirement has yet to be definitely demonstrated. The use of Golgi to ER retrograde trafficking mutants is problematic as those mutants tested also show defects in forward transport over the time course required for the degradation experiments (unpublished data). The rationale for delivering ERQC substrates to the Golgi apparatus may be either to (a) access a mechanism responsible for exporting it directly from the Golgi apparatus to the cytosol, or (b) to receive a Golgi apparatus-based modification that signals either its efficient translocation to the cytosol on its return to the ER or its efficient ubiquitination by Rsp5p on its retrotranslocation from the ER to the cytosol. We favor an alternative model in which ERQC substrates are not actively targeted to the Golgi apparatus, but instead are inadequately retained in the ER by the relevant components of the HRD/DER pathway and are transported to the Golgi apparatus. The insufficient ER retention may be because certain individual ERQC substrates interact poorly with the HRD/DER components or due to saturation of this pathway. Therefore the cis-Golgi apparatus may act as a quality control catchment system to capture ERQC substrates that have “escaped” the ER before returning them by retrograde transport to the ER.
Figure 9.

Summary: ERQC consists of two degradative mechanisms, the HIP and the HRD/DER pathway. Substrates destined for the HRD/DER pathway are recognized as being misfolded or unassembled, and are retained in the ER before being retrotranslocated through the translocon to the cytosol where the Hrd3p-regulated ubiquitin ligase (Hrd1p) attaches ubiquitin to the substrate and it is subsequently degraded by the 26S proteasome. The mechanism described here (HIP) eliminates misfolded proteins from the ER in a manner that requires vesicular transport from the ER to the Golgi apparatus involving Sec12p and the putative CPY* cargo receptor, Erv29p. Once in the cis-Golgi compartment, CPY* is modified by α1,6-mannose addition before ubiquitination by the ubiquitin ligase Rsp5p and subsequent degradation by the proteasome. Where the misfolded protein enters the cytosol and is exposed to Rsp5p is not clear, however, two locations seem possible: (1) it may enter the cytosol from the Golgi apparatus by an unknown mechanism, or (2) it may be transported back to the ER where it presumably would enter the cytosol through the translocon.
The existence of an alternative degradative system has previously been proposed by us (Hill and Cooper, 2000) and others (Friedlander et al., 2000) to explain either the continued degradation of ERQC substrates in HRD/DER-deficient cells or the surprising finding that cells deficient for the HRD/DER pathway display no growth defects even when expressing misfolded proteins in the ER. However, double mutants lacking both the UPR and HRD/DER pathways display significantly impaired growth phenotypes that are further exacerbated by ER stress (Friedlander et al., 2000; unpublished data). These observations are consistent with our findings that the HIP pathway is likely under UPR control such that the hrd1Δ ire1Δ mutant is deprived of both of its options in dealing with ER stress. Consistent with this model of UPR-regulated HIP is the identification of a number of genes under UPR transcriptional regulation that are involved in ER to Golgi vesicular transport and therefore likely necessary for the HIP pathway (Travers et al., 2000).
The importance of preventing both the aggregation of misfolded proteins in the ER and the transport of these proteins to the cell surface is underscored by the fact that the cell uses at least two distinct mechanisms to effect their degradation. However, this apparent redundancy raises the question of why one would observe any stabilization of ERQC substrates in HRD/DER-deficient cells while the HIP pathway is functional. It is possible that the two systems complement each other, with the HRD/DER pathway comprising a low capacity system for contending with the nominal loads of misfolded proteins accruing in the ER under optimal growth conditions. The HIP pathway might act as a high capacity mechanism that is up-regulated, potentially by the UPR, to accommodate increased levels of ERQC substrates.
In summary, we have identified an HRD/DER-independent degradation mechanism in which ER-Golgi trafficking and Rsp5p-dependent ubiquitination is required before degradation by the proteasome. Further work will identify the purpose of delivering ERQC substrates to the Golgi apparatus, where in the cell ERQC substrate ubiquitination by Rsp5p occurs, and how such substrates gain access to the Rsp5p in the cytosol.
Materials and methods
Plasmids, media, and strain construction
Media were prepared as described previously (Hill and Stevens, 1994). To introduce prc1Δ::KAN, ubc7Δ::KAN, ire1Δ::KAN, and hrd1Δ::KAN alleles into strains, oligonucleotides flanking the disrupted allele were used to amplify the allele from strains of the S. cerevisiae Genome Deletion Project (Research Genetics). The product was then transformed into a strain with subsequent selection on YEPD media containing 200 μg/ml G418. Disruptions were confirmed by PCR analysis on genomic DNA using oligonucleotides flanking each disrupted locus. WCG4a and WCGL-11/21a (provided by Dieter Wolf, Universitaet Stuttgart, Stuttgart, Germany) were transformed with the prc1Δ::KAN allele to make strains KHY292 and KHY293; ire1Δ::KAN was transformed into KHY163 to create KHY280, ubc7Δ::KAN was transformed into KHY163 to make KHY313; and hrd1Δ::KAN was transformed into KHY355 to create KHY359.
KHY163 was constructed as described previously (Caldwell et al., 2001). KHY171 was created by repairing the vma22Δ locus of KHY140 (Hill and Cooper, 2000). The prc1Δ::KAN allele was amplified by PCR and inserted into pTOPO 2.1 (Invitrogen) to create pAC550. A 0.9-kb ClaI-SalI fragment from pAC550 was replaced with the HIS3-containing ClaI-SalI fragment from pJJ217 to create pAC556. The prc1Δ::HIS3 allele from pAC556 was amplified by and transformed into KHY163 and KHY171, creating strains KHY298 and KHY299, respectively. The disruption was confirmed by PCR and Western blot (Caldwell et al., 2001). The pep4Δ::TRP1 allele from SacI-XhoI–digested pLS1–10 (provided by Dr. Steven Nothwehr, University of Missouri, Columbia, MO) was introduced into KHY171 to create KHY265. The ubc6Δ::HIS3 allele was amplified by from RH3140 (provided by Dr. Linda Hicke, Northwestern University, Evanston, IL), and transformed into KHY313, creating KHY333.
The hrd1Δ::TRP1 allele was constructed by inserting a TRP1-containing BglII-NsiI fragment from pJJ248 into BglII-NsiI–digested pAC370 (Hill and Cooper, 2000) creating pAC401. The hrd1Δ::TRP1 allele from DraI-digested pAC401 was introduced into KHY127 (Hill and Cooper, 2000) and KHY270 (Caldwell et al., 2001) to create strains KHY158 and KHY279, respectively. KHY237 was created by repairing the vma22Δ locus in KHY158.
To introduce the sec12–4 allele and create KHY306 and KHY308, SalI-digested pAC559, was transformed into KHY163 and KHY171, and Ura+ prototrophs were placed on 5-FOA. Ura− colonies were screened by failure to grow at 38°C. pAC559 was created by ligating a sec12–4 containing 3.4-kb XhoI-XbaI from pSHf2–1 (provided by Dr. Akihiko Nakano, RIKEN, Wako, Saitama, Japan) into XhoI-SpeI-digested pRS306. The rsp5–2 allele was introduced by transforming KHY163 with StuI-digested pKM017 (provided by Dr. Stefan Jentsch, Max Plank Institute of Biochemistry, Martinsried, Germany). RSP5 was then deleted by transforming the rsp5Δ::HIS3-containing fragment from SacI-SphI–digested pKM017.
Strains deleted for potential ubiquitin ligase genes were transformed with pAC578, radiolabeled, and CPY–HA was immunoprecipitated at various times (Hill and Cooper, 2000). Potential RING-H2–containing proteins were as follows: YFL010C, YMR075C-A, YKR073C, YKL034W, YDR128W, YOL138C, YCR066W, YHL010C, YOL054W, YLR247C, YLR024C, YGR184C, YHR115C, YDR265W, YDR266C, YOR191W, YOR027W, YLR427W, YMR247C, YDR143C, YLR032W, YPR093C, YNL116W, YBR114W, YLR148W, YER068W, YIL030C (DOA10/SSM4), YER116C, YDL074C, YBR062C, and YDL013W; and strains deleted/temperature sensitive allele for HECT domain–containing proteins are YJR036C, YDR457W, YKL010C, YGL141W, and YER125W.
pAC453 and pAC519 are 2-μm plasmids and pAC446 is a CEN plasmid in which a prc1–1-containing 2.8-kb SacI-SalI fragment from pAC356 (Hill and Cooper, 2000) was ligated into the SacI-SalI sites of pTV3,YEp352, and pRS315, respectively. pAC578 was created by ligating a CPY–3XHA–containing 4.3-kb SacI-XhoI fragment from pBG15 (provided by Dr. Scott Moye-Rowley, University of Iowa, Iowa City, IO) into SacI-SalI–digested YEp352. pAC540 is a 2-μm plasmid in which the 1.9-kb SacI-BglII fragment containing PrA–3XHA from pAC535 (Caldwell et al., 2001) was ligated in SacI-BamHI–digested YEp352.
Radiolabeling, immunoprecipitation, and antibodies
Radiolabeling and immunoprecipitation were performed as described previously (Hill and Cooper, 2000). Samples were resolved by SDS-PAGE, the gels were fixed, dried, and exposed either to a phosphor cassette (Molecular Dynamics) or to X-ray film. Quantification of gels was performed as described previously (Hill and Cooper, 2000).
Ubiquitin experiment
Yeast strains overexpressing HA-tagged ubiquitin from the CUP1 promoter (YEp112, provided by Dr. Mark Hochstrasser, Yale University, New Haven, CT) were grown overnight in 0.2% oleic acid minimal media containing 150 μM CuSO4. Cells were radiolabeled, spheroplasted, and lysed in buffer containing 10 mM NEM. Immunoprecipitation of CPY* was as described previously (Hill and Cooper, 2000), except with subsaturating amounts of antibody. A sequential immunoprecipitation was performed with HA antibodies. Samples were loaded on a 10% SDS-PAGE.
Acknowledgments
We thank Kathryn Hill, Douglas Crawford, and Tom Menees for critical reading of this manuscript. We thank Linda Hicke, Mark Hochstrasser, Stefan Jentsch, Akihiko Nakano, Steven Nothwehr, Rob Piper, Scott Moye-Rowley, and Dieter Wolf for strains, plasmids, and advice.
This work was supported by the National Institutes of Health grant GM55848.
Footnotes
Abbreviations used in this paper: CPY, carboxypeptidase Y; ERAD, ER-associated degradation; ERQC, ER quality control; HIP, Hrd1p-independent proteolysis; PrA, proteinase A; UPR, unfolded protein response.
References
- Altschul, S.F., T.L. Madden, A.A. Schaffer, J. Zhang, Z. Zhang, W. Miller, and D.J. Lipman. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba, M., M. Osumi, S.V. Scott, D.J. Klionsky, and Y. Ohsumi. 1997. Two distinct pathways for targeting proteins from the cytoplasm to the vacuole/lysosome. J. Cell Biol. 139:1687–1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bays, N.W., R.G. Gardner, L.P. Seelig, C.A. Joazeiro, and R.Y. Hampton. 2001. Hrd1p/Der3p is a membrane-anchored ubiquitin ligase required for ER-associated degradation. Nat. Cell Biol. 3:24–29. [DOI] [PubMed] [Google Scholar]
- Belden, W.J., and C. Barlowe. 2001. Role of Erv29p in collecting soluble secretory proteins into ER-derived transport vesicles. Science. 294:1528–1531. [DOI] [PubMed] [Google Scholar]
- Benharouga, M., M. Haardt, N. Kartner, and G.L. Lukacs. 2001. COOH-terminal truncations promote proteasome-dependent degradation of mature cystic fibrosis transmembrane conductance regulator from post-Golgi compartments. J. Cell Biol. 153:957–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biederer, T., C. Volkwein, and T. Sommer. 1996. Degradation of subunits of the Sec61p complex, an integral component of the ER membrane, by the ubiquitin-proteasome pathway. EMBO J. 15:2069–2076. [PMC free article] [PubMed] [Google Scholar]
- Bonifacino, J.S., C.K. Suzuki, J. Lippincott-Schwartz, A.M. Weissman, and R.D. Klausner. 1989. Pre-Golgi degradation of newly synthesized T-cell antigen receptor chains: intrinsic sensitivity and the role of subunit assembly. J. Cell Biol. 109:73–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordallo, J., R.K. Plemper, A. Finger, and D.H. Wolf. 1998. Der3p/Hrd1p is required for endoplasmic reticulum-associated degradation of misfolded lumenal and integral membrane proteins. Mol. Biol. Cell. 9:209–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brigance, W.T., C. Barlowe, and T.R. Graham. 2000. Organization of the yeast Golgi complex into at least four functionally distinct compartments. Mol. Biol. Cell. 11:171–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodsky, J.L., and A.A. McCracken. 1999. ER protein quality control and proteasome-mediated protein degradation. Semin. Cell Dev. Biol. 10:507–513. [DOI] [PubMed] [Google Scholar]
- Brodsky, J.L., E.D. Werner, M.E. Dubas, J.L. Goeckeler, K.B. Kruse, and A.A. McCracken. 1999. The requirement for molecular chaperones during endoplasmic reticulum-associated protein degradation demonstrates that protein export and import are mechanistically distinct. J. Biol. Chem. 274:3453–3460. [DOI] [PubMed] [Google Scholar]
- Caldwell, S.R., K.J. Hill, and A.A. Cooper. 2001. Degradation of endoplasmic reticulum (ER) quality control substrates requires transport between the ER and Golgi. J. Biol. Chem. 276:23296–23303. [DOI] [PubMed] [Google Scholar]
- Cooper, A.A., and T.H. Stevens. 1996. Vps10p cycles between the late-Golgi and prevacuolar compartments in its function as the sorting receptor for multiple yeast vacuolar hydrolases. J. Cell Biol. 133:529–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deak, P.M., and D.H. Wolf. 2001. Membrane topology and function of Der3/Hrd1p as a ubiquitin-protein ligase (E3) involved in endoplasmic reticulum degradation. J. Biol. Chem. 276:10663–10669. [DOI] [PubMed] [Google Scholar]
- Deshaies, R.J., S.L. Sanders, D.A. Feldheim, and R. Schekman. 1991. Assembly of yeast Sec proteins involved in translocation into the endoplasmic reticulum into a membrane-bound multisubunit complex. Nature. 349:806–808. [DOI] [PubMed] [Google Scholar]
- Ellgaard, L., M. Molinari, and A. Helenius. 1999. Setting the standards: quality control in the secretory pathway. Science. 286:1882–1888. [DOI] [PubMed] [Google Scholar]
- Fewell, S.W., K.J. Travers, J.S. Weissman, and J.L. Brodsky. 2001. The action of molecular chaperones in the early secretory pathway. Annu. Rev. Genet. 35:149–191. [DOI] [PubMed] [Google Scholar]
- Fisher, E.A., M. Zhou, D.M. Mitchell, X. Wu, S. Omura, H. Wang, A.L. Goldberg, and H.N. Ginsberg. 1997. The degradation of apolipoprotein B100 is mediated by the ubiquitin-proteasome pathway and involves heat shock protein 70. J. Biol. Chem. 272:20427–20434. [DOI] [PubMed] [Google Scholar]
- Friedlander, R., E. Jarosch, J. Urban, C. Volkwein, and T. Sommer. 2000. A regulatory link between ER-associated protein degradation and the unfolded-protein response. Nat. Cell Biol. 2:379–384. [DOI] [PubMed] [Google Scholar]
- Gajewska, B., J. Kaminska, A. Jesionowska, N.C. Martin, A.K. Hopper, and T. Zoladek. 2001. WW domains of Rsp5p define different functions: determination of roles in fluid phase and uracil permease endocytosis in Saccharomyces cerevisiae. Genetics. 157:91–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galan, J.M., V. Moreau, B. Andre, C. Volland, and R. Haguenauer-Tsapis. 1996. Ubiquitination mediated by the Npi1p/Rsp5p ubiquitin-protein ligase is required for endocytosis of the yeast uracil permease. J. Biol. Chem. 271:10946–10952. [DOI] [PubMed] [Google Scholar]
- Gardner, R.G., G.M. Swarbrick, N.W. Bays, S.R. Cronin, S. Wilhovsky, L. Seelig, C. Kim, and R.Y. Hampton. 2000. Endoplasmic reticulum degradation requires lumen to cytosol signaling. Transmembrane control of Hrd1p by Hrd3p. J. Cell Biol. 151:69–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitan, R.S., and D.J. Eide. 2000. Zinc-regulated ubiquitin conjugation signals endocytosis of the yeast ZRT1 zinc transporter. Biochem. J. 346:329–336. [PMC free article] [PubMed] [Google Scholar]
- Hammond, C., and A. Helenius. 1994. Quality control in the secretory pathway: retention of a misfolded viral membrane glycoprotein involves cycling between the ER, intermediate compartment, and Golgi apparatus. J. Cell Biol. 126:41–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton, R.Y., R.G. Gardner, and J. Rine. 1996. Role of 26S proteasome and HRD genes in the degradation of 3-hydroxy-3-methylglutaryl-CoA reductase, an integral endoplasmic reticulum membrane protein. Mol. Biol.Cell. 7:2029–2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert, D.N., B. Foellmer, and A. Helenius. 1995. Glucose trimming and reglucosylation determine glycoprotein association with calnexin in the endoplasmic reticulum. Cell. 81:425–433. [DOI] [PubMed] [Google Scholar]
- Helliwell, S.B., S. Losko, and C.A. Kaiser. 2001. Components of a ubiquitin ligase complex specify polyubiquitination and intracellular trafficking of the general amino acid permease. J. Cell Biol. 153:649–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicke, L., and H. Riezman. 1996. Ubiquitination of a yeast plasma membrane receptor signals its ligand-stimulated endocytosis. Cell. 84:277–287. [DOI] [PubMed] [Google Scholar]
- Hill, K., and A.A. Cooper. 2000. Degradation of unassembled Vph1p reveals novel aspects of the yeast ER quality control system. EMBO J. 19:550–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill, K.J., and T.H. Stevens. 1994. Vma21p is a yeast membrane protein retained in the endoplasmic reticulum by a di-lysine motif and is required for the assembly of the vacuolar H(+)-ATPase complex. Mol. Biol. Cell. 5:1039–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiller, M.M., A. Finger, M. Schweiger, and D.H. Wolf. 1996. ER degradation of a misfolded luminal protein by the cytosolic ubiquitin-proteasome pathway. Science. 273:1725–1728. [DOI] [PubMed] [Google Scholar]
- Hochstrasser, M. 1996. Ubiquitin-dependent protein degradation. Annu. Rev. Genet. 30:405–439. [DOI] [PubMed] [Google Scholar]
- Hong, E., A.R. Davidson, and C.A. Kaiser. 1996. A pathway for targeting soluble misfolded proteins to the yeast vacuole. J. Cell Biol. 135:623–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppe, T., K. Matuschewski, M. Rape, S. Schlenker, H.D. Ulrich, and S. Jentsch. 2000. Activation of a membrane-bound transcription factor by regulated ubiquitin/proteasome-dependent processing. Cell. 102:577–586. [DOI] [PubMed] [Google Scholar]
- Hsu, V.W., L.C. Yuan, J.G. Nuchtern, J. Lippincott-Schwartz, G.J. Hammerling, and R.D. Klausner. 1991. A recycling pathway between the endoplasmic reticulum and the Golgi apparatus for retention of unassembled MHC class I molecules. Nature. 352:441–444. [DOI] [PubMed] [Google Scholar]
- Jackson, P.K., A.G. Eldridge, E. Freed, L. Furstenthal, J.Y. Hsu, B.K. Kaiser, and J.D. Reimann. 2000. The lore of the RINGs: substrate recognition and catalysis by ubiquitin ligases. Trends Cell Biol. 10:429–439. [DOI] [PubMed] [Google Scholar]
- Jakob, C.A., P. Burda, J. Roth, and M. Aebi. 1998. Degradation of misfolded endoplasmic reticulum glycoproteins in Saccharomyces cerevisiae is determined by a specific oligosaccharide structure. J. Cell Biol. 142:1223–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakob, C.A., D. Bodmer, U. Spirig, P. Battig, A. Marcil, D. Dignard, J.J. Bergeron, D.Y. Thomas, and M. Aebi. 2001. Htm1p, a mannosidase-like protein, is involved in glycoprotein degradation in yeast. EMBO Rep. 2:423–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, E.W., G.S. Zubenko, and R.R. Parker. 1982. PEP4 gene function is required for expression of several vacuolar hydrolases in Saccharomyces cerevisiae. Genetics. 102:665–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamhi-Nesher, S., M. Shenkman, S. Tolchinsky, S.V. Fromm, R. Ehrlich, and G.Z. Lederkremer. 2001. A novel quality control compartment derived from the endoplasmic reticulum. Mol. Biol. Cell. 12:1711–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knop, M., A. Finger, T. Braun, K. Hellmuth, and D.H. Wolf. 1996. Der1, a novel protein specifically required for endoplasmic reticulum degradation in yeast. EMBO J. 15:753–763. [PMC free article] [PubMed] [Google Scholar]
- Kopito, R.R. 1997. ER quality control: the cytoplasmic connection. Cell. 88:427–430. [DOI] [PubMed] [Google Scholar]
- Margottin, F., S. Benichou, H. Durand, V. Richard, L.X. Liu, E. Gomas, and R. Benarous. 1996. Interaction between the cytoplasmic domains of HIV-1 Vpu and CD4: role of Vpu residues involved in CD4 interaction and in vitro CD4 degradation. Virology. 223:381–386. [DOI] [PubMed] [Google Scholar]
- Moolenaar, C.E., J. Ouwendijk, M. Wittpoth, H.A. Wisselaar, H.P. Hauri, L.A. Ginsel, H.Y. Naim, and J.A. Fransen. 1997. A mutation in a highly conserved region in brush-border sucrase-isomaltase and lysosomal alpha-glucosidase results in Golgi retention. J. Cell Sci. 110:557–567. [DOI] [PubMed] [Google Scholar]
- Nakatsukasa, K., S. Nishikawa, N. Hosokawa, K. Nagata, and T. Endo. 2001. Mnl1p, an alpha -mannosidase-like protein in yeast Saccharomyces cerevisiae, is required for endoplasmic reticulum-associated degradation of glycoproteins. J. Biol. Chem. 276:8635–8638. [DOI] [PubMed] [Google Scholar]
- Plemper, R.K., and D.H. Wolf. 1999. Retrograde protein translocation: ERADication of secretory proteins in health and disease. Trends Biochem. Sci. 24:266–270. [DOI] [PubMed] [Google Scholar]
- Plemper, R.K., S. Bohmler, J. Bordallo, T. Sommer, and D.H. Wolf. 1997. Mutant analysis links the translocon and BiP to retrograde protein transport for ER degradation. Nature. 388:891–895. [DOI] [PubMed] [Google Scholar]
- Salminen, A., and P.J. Novick. 1987. A ras-like protein is required for a post-Golgi event in yeast secretion. Cell. 49:527–538. [DOI] [PubMed] [Google Scholar]
- Simpson, J.C., L.M. Roberts, K. Romisch, J. Davey, D.H. Wolf, and J.M. Lord. 1999. Ricin A chain utilises the endoplasmic reticulum-associated protein degradation pathway to enter the cytosol of yeast. FEBS Lett. 459:80–84. [DOI] [PubMed] [Google Scholar]
- Swanson, R., M. Locher, and M. Hochstrasser. 2001. A conserved ubiquitin ligase of the nuclear envelope/endoplasmic reticulum that functions in both ER-associated and Matalpha2 repressor degradation. Genes Dev. 15:2660–2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teckman, J.H., and D.H. Perlmutter. 1996. The endoplasmic reticulum degradation pathway for mutant secretory proteins alpha1-antitrypsin Z and S is distinct from that for an unassembled membrane protein. J. Biol. Chem. 271:13215–13220. [DOI] [PubMed] [Google Scholar]
- Travers, K.J., C.K. Patil, L. Wodicka, D.J. Lockhart, J.S. Weissman, and P. Walter. 2000. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell. 101:249–258. [DOI] [PubMed] [Google Scholar]
- Vashist, S., W. Kim, W.J. Belden, E.D. Spear, C. Barlowe, and D.T. Ng. 2001. Distinct retrieval and retention mechanisms are required for the quality control of endoplasmic reticulum protein folding. J. Cell Biol. 155:355–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward, C.L., S. Omura, and R.R. Kopito. 1995. Degradation of CFTR by the ubiquitin-proteasome pathway. Cell. 83:121–127. [DOI] [PubMed] [Google Scholar]
- Wiertz, E.J., T.R. Jones, L. Sun, M. Bogyo, H.J. Geuze, and H.L. Ploegh. 1996. The human cytomegalovirus US11 gene product dislocates MHC class I heavy chains from the endoplasmic reticulum to the cytosol. Cell. 84:769–779. [DOI] [PubMed] [Google Scholar]
- Wilhovsky, S., R. Gardner, and R. Hampton. 2000. HRD gene dependence of endoplasmic reticulum-associated degradation. Mol. Biol. Cell. 11:1697–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winitz, D., I. Shachar, Y. Elkabetz, R. Amitay, M. Samuelov, and S. Bar-Nun. 1996. Degradation of distinct assembly forms of immunoglobulin M occurs in multiple sites in permeabilized B cells. J. Biol. Chem. 271:27645–27651. [DOI] [PubMed] [Google Scholar]
- Zuber, C., M.J. Spiro, B. Guhl, R.G. Spiro, and J. Roth. 2000. Golgi apparatus immunolocalization of endomannosidase suggests post-endoplasmic reticulum glucose trimming: implications for quality control. Mol. Biol. Cell. 11:4227–4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuber, C., J.Y. Fan, B. Guhl, A. Parodi, J.H. Fessler, C. Parker, and J. Roth. 2001. Immunolocalization of UDP-glucose:glycoprotein glucosyltransferase indicates involvement of pre-Golgi intermediates in protein quality control. Proc. Natl. Acad. Sci. USA. 98:10710–10715. [DOI] [PMC free article] [PubMed] [Google Scholar]