

Abstract
The toxic effect of cholera toxin (CT) on target cells is caused by its A1 chain. This polypeptide is released from the holotoxin and unfolded in the lumen of the ER by the action of protein disulfide isomerase (PDI), before being retrotranslocated into the cytosol. The polypeptide is initially unfolded by binding to the reduced form of PDI. We show that upon oxidation of the COOH-terminal disulfide bond in PDI by the enzyme Ero1, the A1 chain is released. Both yeast Ero1 and the mammalian Ero1α isoform are active in this reaction. Ero1 has a preference for the PDI–toxin complex. We further show that the complex is transferred to a protein at the lumenal side of the ER membrane, where the unfolded toxin is released from PDI by the action of Ero1. Taken together, our results identify Ero1 as the enzyme mediating the release of unfolded CT from PDI and characterize an additional step in retrotranslocation of the toxin.
Keywords: PDI; Ero1; cholera toxin; retrotranslocation; oxidation
Introduction
The bacterium Vibrio cholerae produces cholera toxin (CT)*which affects mammalian intestinal epithelial cells by causing salt and water secretion resulting in diarrhea (Sears and Kaper, 1996). CT consists of a ring of five B subunits and a single A subunit. The A subunit is cleaved into the A1 catalytic domain and the A2 domain upon secretion of the toxin by V. cholerae. The A1 and A2 fragments are connected by a disulfide bridge and by noncovalent interactions. The holotoxin is endocytosed and traffics in a retrograde manner along the secretory pathway to the lumen of the ER (Lencer et al., 1999). In this compartment, the disulfide bridge of the A subunit is reduced, and the A1 chain is released from the rest of the toxin and translocated to the cytosol. Based on coimmunoprecipitation and ribosome inhibition experiments, it seems that the A1 peptide is translocated through the Sec61p channel (Schmitz et al., 2000), the same channel used to translocate secretory, lumenal, and membrane proteins from the cytosol to the ER lumen (for review see Matlack et al., 1998). Although the signal transduction pathway induced by the A1 chain in the cytosol is well characterized, much less is known about the mechanism by which the A1 peptide is transported from the ER lumen to the cytosol, a process termed retrotranslocation (for review see Tsai et al., 2002).
Unfolding of the A1 peptide likely represents the first step in retrotranslocation of the toxin. Using a biochemical fractionation approach that made no assumptions about the nature of this unfolding activity, we previously identified the ER oxido-reductase protein disulfide isomerase (PDI) as the major activity that disassembles the toxin and unfolds the A1 chain (Tsai et al., 2001). More detailed analysis demonstrated that PDI acts as a redox-dependent chaperone; in its reduced state, PDI binds and unfolds the toxin, whereas in its oxidized state, PDI releases it. Release of the A1 chain from PDI upon oxidation must occur prior to its retrotranslocation across the ER membrane. When oxidation is induced with oxidized glutathione (GSSG), an unphysiologically high concentration was required to induce release (Tsai et al., 2001). Therefore, we hypothesized that this process must normally be catalyzed by an enzyme, i.e., an oxidase of PDI.
Here we identify the enzyme responsible for the release reaction, provide a mechanism for the release, and describe an additional step in retrotranslocation of the toxin. Our data demonstrate that the ER oxidase Ero1 is responsible for inducing release of the toxin from reduced PDI through oxidation of the COOH-terminal disulfide bond in PDI. Furthermore, we show that the complex of PDI and unfolded toxin is targeted to a protein on the lumenal side of the ER membrane. Subsequently, the toxin is released from PDI by the action of Ero1, presumably committing the toxin to retrotranslocation across the ER membrane.
Results and discussion
An ER activity induces toxin release from PDI
To identify an activity that caused the unfolding of purified A subunit of CT, we have previously used an ER extract from dog pancreatic microsomes that was obtained by the addition of a low concentration of detergent, and contained lumenal and some membrane proteins. The ER extract rendered the A and A1 peptides sensitive to trypsin digestion under reducing conditions (1 mM reduced glutathione, GSH; Fig. 1 A, lane 6; Tsai et al., 2001). BSA did not show this effect (Fig. 1 A, lane 2), indicating that a protein in the ER extract induces unfolding of the toxin. The protein was subsequently identified as PDI; indeed, incubation of purified PDI with toxin under reducing conditions similarly caused the A1 chain to be sensitive to trypsin digestion (Fig. 1 A, compare lane 4 with lane 2; Tsai et al., 2001).
Figure 1.
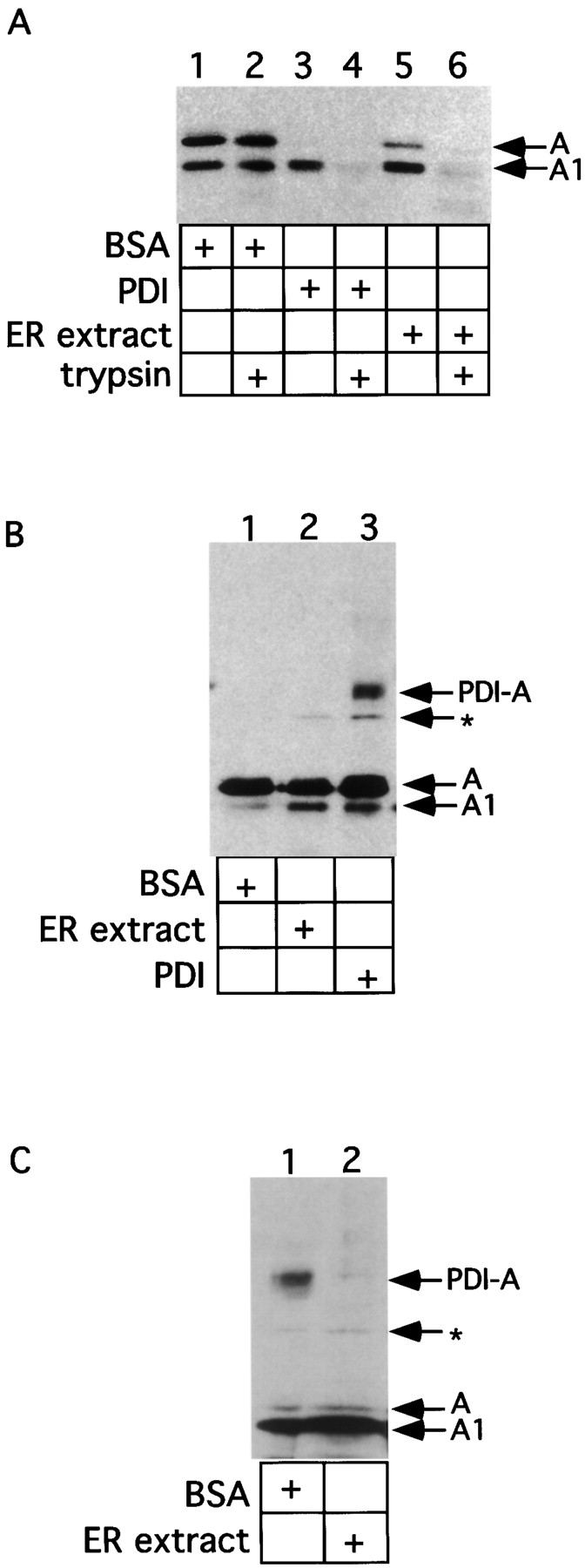
An ER activity induces release of the A1 chain from PDI. (A) Isolated A subunit (70 nM) was incubated with either BSA (3 μM), purified mammalian PDI (3 μM), or ER extract (3 mg/ml) in GSH (1 mM). Where indicated, trypsin (100 μg/ml) was added. Samples were analyzed by nonreducing SDS-PAGE and immunoblotting with a CT antibody. The positions of the proteolytically cleaved A subunit (containing A1 and A2 chains linked by a disulfide bond) and of the A1 chain are indicated. (B) Isolated A subunit (70 nM) was incubated with either BSA (3 μM), ER extract (3 mg/ml) or purified PDI (3 μM) in GSH (1 mM). A carbodiimide crosslinker (EDAC) was then added. Samples were analyzed as in (A). PDI-A represents a crosslinked product between PDI and the A subunit. * indicates an unidentified band. (C) Isolated A subunit (70 nM) was first incubated with purified PDI (3 μM) in GSH (1 mM), followed by addition of either BSA (3 μM) or ER extract (3 mg/ml). Samples were analyzed as in (B).
To probe the interaction between the toxin A chain and PDI, we developed a crosslinking assay (Tsai et al., 2001). Briefly, purified PDI or ER extract were incubated with the A subunit of CT under reducing conditions, followed by addition of a carbodiimide to induce crosslinks between carboxyl and amino groups of the two proteins. This complex is visualized as a higher molecular mass band in SDS-PAGE after immunoblotting with an antibody that recognizes the A subunit of CT. It should be noted that in both the trypsin digestion and crosslinking assays, PDI is in an ∼40-fold molar excess over the toxin.
Using this assay, we found that a PDI–A chain complex can be seen with purified PDI, but not with ER extract or BSA (Fig. 1 B, compare lane 3 with lanes 2 and 1; Tsai et al., 2001). Together with the unfolding assay, these results suggest that the toxin must be undergoing cycles of binding and release from PDI when incubated with ER extract, thereby preventing the formation of a stable PDI–toxin complex that can be captured by the crosslinker.
To directly test the possibility that a release activity exists in the ER extract, we first generated the PDI–A chain complex by incubating PDI and purified toxin together under reducing conditions. BSA or ER extract were then added to the preformed complex to induce toxin release, and the dissociation of the complex was probed with a bifunctional carbodiimide crosslinker. Indeed, addition of ER extract, but not BSA, resulted in loss of the crosslinked product (Fig. 1 C, compare lane 2 with lane 1). This result confirms the presence of an activity in the ER extract that induces the release of the A1 chain from PDI. Because addition of high concentrations of the oxidant GSSG can also induce toxin release (Tsai et al., 2001), the release activity is likely to be an oxidase of PDI.
Ero1 induces the release of unfolded toxin from reduced PDI
Using two different genetic screens in Saccharomyces cerevisiae, Ero1 was identified as an enzyme that functions as a PDI oxidase in the ER lumen (Frand and Kaiser, 1998, 1999; Pollard et al., 1998). Ero1 contains the cofactor FAD and may use molecular oxygen to reoxidize itself (Tu et al., 2000). We asked whether the mammalian homologue of Ero1 may be responsible for the release activity in the ER extract. In mammals, two different Ero1 isoforms have been identified, Ero1α (Cabibbo et al., 2000) and Ero1β (Pagani et al., 2000). To test whether both isoforms are present in dog microsomes, three different mammalian Ero1 antibodies were used, one that recognizes both mammalian Ero1 isoforms (α/β, antibody 194), one that only recognizes the Ero1α isoform, and one that only recognizes the Ero1β isoform. The Ero1α/β antibody recognized three distinct bands in the microsomes, with the top band corresponding to Ero1α, the middle band corresponding to Ero1β, and the bottom band corresponding possibly to a novel Ero1 isoform or a degradation product (Fig. 2 A, lane 1). The slower mobility of Ero1α compared to Ero1β is consistent with previous results (Pagani et al., 2000). Treatment of microsomes with endoglycosidase H (Endo H) increased the mobility of all three bands, confirming previous findings that Ero1α and Ero1β are glycoproteins (Fig. 2 A, lane 2; Pagani et al., 2000). The Ero1α- and Ero1β-specific antibodies each recognized only one Endo H–sensitive band in the microsomes (Fig. 2 A, lanes 3 and 5, respectively), which correspond to the positions of the top and middle bands identified using the Ero1α/β antibody, respectively. An additional Endo H–resistant band was observed using the Ero1α-specific antibody (Fig. 2 A, lane 4), which may represent an uncharacterized glycosylated form of Ero1α.
Figure 2.

Ero1 induces the release of the A1 peptide from reduced PDI. (A) Protein from five equivalents of dog pancreatic microsomes were treated with or without endo H and analyzed by SDS-PAGE followed by immunoblotting with antibodies to Ero1α/β (lanes 1 and 2), Ero1α (lanes 3 and 4), or Ero1β (lanes 5 and 6). Purified mammalian Ero1α (1 μg), analyzed in the Coomassie gel (lane 7), was tested with antibodies to Ero1α/β, Ero1α, or Ero1β (lanes 8–10). x represents an uncharacterized glycosylated form of Ero1α. (B) ER extract or Ero1- depleted extract (depleted by incubation with either antibody 193 or 194) were analyzed by immunoblotting with antibodies to Ero1α/β (antibody 194, lanes 1–3), Ero1α (lanes 4–6), or Ero1β (lanes 7–9). Lanes 10–14 show the results of release assays. Isolated A subunit (70 nM) and PDI (3 μM) were incubated in GSH (1 mM) followed by addition of BSA (2 mg/ml), ER extract (2 mg/ml), Ero1-depleted extract (depleted by antibody 193 or 194), or GSSG (30 mM; lanes 10–14). The samples were analyzed as in Fig. 1 B. (C) Purified mammalian Ero1α (0.3 μM or 3 μM) was added to the PDI–toxin complex and analyzed as in Fig. 1 B (lanes 1–3). Purified yeast Ero1 (Coomassie gel, lane 4) was added (0.3 μM or 1.5 μM) to the toxin-PDI complex and the samples were analyzed as in Fig. 1 B (lanes 5–7).
To test the specificity of the antibodies, we expressed and purified mammalian Ero1α from yeast cells (Fig. 2 A, lane 7). Both the Ero1α/β and Ero1α-specific antibodies recognized the purified Ero1α protein, whereas the Ero1β-specific antibody did not (Fig. 2 A, lanes 8–10). We were unable to purify the mammalian Ero1β protein from yeast.
We next used the antibodies to directly test whether the release activity observed in the crude ER extract can be attributed to Ero1. Both isoforms of Ero1 were present in the extract obtained with low detergent concentrations. To deplete Ero1α and Ero1β we used two different antibodies directed against both proteins (antibodies 193 and 194; Fig. 2 B, compare lanes 2 and 3 with lane 1; lanes 5 and 6 with lane 4; and lanes 8 and 9 with lane 7). Although addition of ER extract to the preformed PDI-toxin complex induced loss of the crosslinked product, addition of either of the two Ero1-depleted extracts did not (Fig. 2 B, compare lane 11 with lanes 12 and 13). In fact, with one antibody the crosslinked band was more intense than in the control, suggesting that in the absence of Ero1, the extract is more reducing than 1 mM GSH, perhaps due to the presence of a PDI-reductase. These results demonstrate that Ero1 is required for the release activity. As previously reported, addition of a high concentration of GSSG (30 mM) also resulted in the loss of the crosslinked product (Fig. 2 B, compare lane 14 with lane 10; Tsai et al., 2001).
We next asked whether Ero1 is sufficient for stimulating toxin release. Addition of purified Ero1α, either in substoichiometric (1:10) or stoichiometric (1:1) ratio to PDI, to the preformed PDI-toxin complex resulted in the disappearance of the crosslinked product (Fig. 2 C, compare lanes 2 and 3 with lane 1). The same result was obtained upon addition of purified yeast Ero1 (Fig. 2 C, lane 4), either in substoichiometric (1:10) or near-stoichiometric (1:2) ratio to PDI (Fig. 2 C, compare lanes 6 and 7 with lane 5). We conclude that Ero1 is both necessary and sufficient to induce release of the toxin from PDI.
We also separated the ER extract on an ion exchange column (Q-Sepharose) and tested each fraction for its content of Ero1 and its activity to release the toxin from reduced PDI. Two pools of Ero1 were found using the Ero1α/β antibody, one pool corresponding to fractions 5–9 (Fig. 3 A, top, lanes 1–5), identified as Ero1α (Fig. 3 A, middle, lanes 1–5), and another corresponding to fractions 17–21 (Fig. 3 A, top, lanes 13–17), identified as Ero1β (Fig. 3 A, bottom, lanes 13–17). Only fractions 5–9 were active in the release assay (Fig. 3 B, compare lanes 2–6 with lanes 14–18). These data confirm that Ero1α is able to induce toxin release from PDI and raise the possibility that Ero1β may be inactive in this assay. Interestingly, based on the theoretical pI values of the two isoforms, Ero1β is expected to elute before Ero1α, in contrast to the observation. It seems therefore likely that interactions with other proteins alter the properties of the isoforms. A novel ER protein, called Erp44, interacts with both Ero1 isoforms and is therefore unlikely to be responsible for the anomalous behavior of the two proteins (Anelli et al., 2002).
Figure 3.

Release activity is present in Ero1α-containing fractions. (A) The protein in an ER extract was bound to a Q-Sepharose column and eluted with a continuous salt gradient. Fractions were analyzed for its Ero1 content by SDS-PAGE and immunoblotting with antibodies that either recognize both Ero1 isoforms (top), Ero1α only (middle), or Ero1β only (bottom). (B) The fractions were tested for their ability to induce toxin release, as described in Fig. 1 B.
Ero1 oxidizes the COOH-terminal disulfide bond in PDI and acts preferentially on the PDI–toxin complex
To demonstrate that the mechanism of Ero1-mediated toxin release is due to oxidation of PDI, we designed an assay that allows us to examine PDI's oxidation state. Maleimide-PEG5000 is a chemical of ∼5,000 daltons that allows the modification of thiol groups in cysteines, and thereby significantly increases the size of the protein. The mobility shift is analyzed by SDS-PAGE followed by immunoblotting with an antibody directed against the modified protein. When PDI was fully reduced by 100 mM DTT and treated with maleimide-PEG5000, a dramatic upward mobility shift of PDI was seen (Fig. 4 A, compare lane 1 with lane 6). Under this condition, all six cysteines in PDI are expected to be in the reduced state and should therefore be modified. When PDI was incubated in the presence of a high concentration of the GSSG (100 mM), a much smaller upward mobility shift was observed (Fig. 4 A, compare lane 5 with lane 6). Under these conditions, only the two noncatalytic cysteines can be modified, whereas the cysteines within the two thioredoxin domains of PDI, each characterized by a CxxC motif, are expected to form disulfide bonds.
Figure 4.

Ero1 oxidizes the COOH-terminal disulfide bond in PDI and acts preferentially on the toxin–PDI complex. (A) Isolated A subunit (70 nM) and purified mammalian PDI (3 μM) were incubated in the presence of DTT (100 mM), GSSG (100 mM), or GSH (1 mM). Where indicated, purified mammalian Ero1α (0.3 μM or 3 μM) was added. 1/10 of the sample was TCA precipitated, washed with acetone, and treated with maleimide-PEG5000. The samples were subsequently analyzed by nonreducing SDS-PAGE followed by immunoblotting with an antibody against PDI. Arrows point to the number of cysteines in PDI modified with maleimide-PEG5000 (-SH-MP). (B) As in A except that purified yeast Ero1(0.3 μM or 1.5 μM) was used. (C) As in B except that 30 μM yeast Ero1 was added. (D) As in C except that 3 μM yeast mutant PDICxxC-AxxA or PDIAxxA-CxxC were used. (E) As in B except 70 nM or 3 μM of isolated A subunit were incubated with PDI (3 μM).
Incubation of PDI with GSH (1 mM) followed by maleimide-PEG5000 treatment resulted in an upward mobility shift similar to the mobility shift induced with DTT treatment (Fig. 4 A, compare lane 2 with lane 1), although some degree of smearing was consistently observed. Importantly, addition of stoichiometric amounts of mammalian Ero1α produced an upward mobility shift of PDI that was intermediate to the shifts seen when two or six cysteines in PDI were modified (Fig. 4 A, compare lane 4 with lane 2). Similar results were observed using near-stoichiometric amounts of yeast Ero1 (Fig. 4 B, compare lane 5 with lane 3). The simplest interpretation of this data is that Ero1 oxidizes only one of the two disulfide bonds in PDI, leaving four cysteines available for modification. The ability of Ero1 to oxidize PDI in the presence of 1 mM GSH is consistent with previous findings that yeast Ero1 can oxidize PDI even in 3 mM GSH (Tu et al., 2000).
To further establish that Ero1 can oxidize only one of PDI's two disulfide bonds, a tenfold excess of yeast Ero1 was added to reduced PDI. Again, only one of PDI's two disulfide bonds was oxidized (Fig. 4 C, compare lane 2 with lane 1). To determine whether Ero1 oxidizes the disulfide bond in the NH2- or COOH-terminal thioredoxin domain of PDI, we used purified mutants of yeast PDI in which the CxxC motif in either the NH2- or COOH-terminal domain is mutated to AxxA (PDIAxxA-CxxC and PDICxxC-AxxA,, respectively; Fig. 4 D, lanes 1 and 2). Using a tenfold excess of yeast Ero1 over PDI, we found that only the PDIAxxA-CxxC mutant was oxidized by Ero1 (Fig. 4 D, compare lane 6 with lane 8). Thus, Ero1 oxidizes only the disulfide bond in the carboxy-terminal thioredoxin domain.
Interestingly, addition of substoichiometric amounts of Ero1α or yeast Ero1 did not alter the mobility shift of PDI (Fig. 4, A, compare lane 3 with lane 2, B, compare lane 4 with lane 3), indicating that this amount of Ero1 is insufficient to oxidize the bulk of PDI. However, under the same conditions, both Ero1α and yeast Ero1 were able to fully induce release of toxin from PDI (Fig. 2 C, compare lane 2 with lane 1; compare lane 6 with lane 5). One explanation is that Ero1 preferentially binds to and oxidizes the PDI–toxin complex. Because the concentration of the toxin is much lower than that of PDI, oxidation of complexed PDI would not be visible in the modification assay. Indeed, when we added a stoichiometric concentration of toxin with respect to PDI, a mobility shift of PDI could be observed even at low concentrations of Ero1 (Fig. 4 E, compare lane 6 with lane 2). The band that runs between the ones corresponding to four and six free thiol groups may perhaps be caused by the formation of a mixed disulfide bond with either the toxin or GSH. Taken together, these data support the idea that Ero1 preferentially acts on the PDI-toxin complex.
PDI-unfolded toxin is transferred to the ER membrane
During retrotranslocation of the A1 chain, the unfolded A1 peptide must be targeted to the ER membrane and ultimately engage the translocation channel in order to be transported to the cytosol. We designed an assay to examine transfer of the unfolded A1 chain to the ER membrane, a process that likely mimics an intermediate step in retrotranslocation of the toxin. Purified A subunit of the toxin was initially incubated with ER extract under reducing conditions to stimulate toxin unfolding. Then proteoliposomes were added which were generated by dissolving microsomes in detergent followed by removal of the detergent with hydrophobic beads. These proteoliposomes contain essentially all ER membrane proteins with their orientations randomized. This should allow the PDI–toxin complex to interact with lumenal domains of membrane proteins. To assay for binding of the PDI–toxin complex, the proteoliposomes were sedimented and the pellet and supernatant fractions analyzed by immunoblotting with toxin antibodies.
Although addition of ER extract or proteoliposomes alone did not induce transfer of the A1 peptide to the pellet fraction, addition of ER extract followed by proteoliposomes caused a fraction of the A1 peptide (30%) to be transferred to the pellet (Fig. 5 A, compare lanes 1 and 3 with lane 5). Neither liposomes lacking proteins nor intact microsomes (which do not have the lumenal domains of membrane proteins exposed) stimulated toxin transfer (Fig. 5 A, compare lanes 7 and 9 with lane 11), indicating that the unfolded A1 chain does not bind to lipids or irrelevant proteins. This is supported by the fact that proteoliposomes generated from a proteinase K-treated detergent extract did not show binding activity (Fig. 5 A, compare lanes 15 and 13). Thus, it appears that the unfolded A1 peptide specifically binds to a protein at the lumenal side of the ER membrane. An unfolded state of the A1 peptide is insufficient to achieve binding to the ER membrane as demonstrated with A chains that were chemically unfolded with urea. The urea-treated A and A1 peptides were as sensitive to trypsin treatment as the A1 peptide unfolded with an ER extract (Fig. 5 B, compare lanes 6 and 4 with lane 2), but they did not bind to proteoliposomes (Fig. 5 B, compare lane 7 with lane 9). Thus, it appears that the formation of a PDI–toxin complex is necessary for transfer to the ER membrane, perhaps because PDI specifically interacts with a membrane protein. To further test this point, we asked whether PDI is required for the transfer reaction by using ER extract immunodepleted of PDI (Fig. 5 C, compare lane 3 with lane 2). Indeed, the PDI-depleted extract did not support toxin transfer to the membrane (Fig. 5 C, compare lane 6 with lane 4). Furthermore, purified PDI was competent in inducing transfer of the A1 chain to proteoliposomes (Fig. 5 C, compare lane 10 with lane 8). Therefore, PDI is both necessary and sufficient for the transfer of unfolded A1 peptide to the ER membrane. These experiments also show that Ero1 in the ER lumen is not required. About 10% of total Ero1 is found in the proteoliposomes, but this population also does not seem to be required for the transfer of the toxin to the ER membrane, because proteoliposomes generated from microsomes of the yeast ero1-1 mutant bound the PDI–toxin complex with unreduced efficiency (unpublished data). However, the mutant proteoliposomes were inactive in releasing the toxin from PDI, in contrast to wild-type vesicles (Fig. 5 D, compare lane 4 with lanes 2 and 3). Thus, these results indicate that the complex of toxin and PDI is first transferred to the ER membrane and then dissociated by the action of Ero1.
Figure 5.

PDI-unfolded A1 peptide is transferred to the ER membrane. (A) Isolated A subunit (70 nM) was incubated in the presence of GSH (1 mM) with ER extract, proteoliposomes, or ER extract followed by proteoliposomes (lanes 1–6). After sedimentation, the proteoliposome-bound toxin in the pellet fraction and the unbound fraction in the supernatant were analyzed by nonreducing SDS-PAGE followed by immunoblotting. Controls were performed with either native microsomes (lanes 9 and 10), liposomes lacking proteins (lanes 7 and 8), or proteoliposomes generated from a proteinase K-treated detergent extract (lanes 15 and 16). (B) Isolated A subunit (70 nM) was incubated with either BSA, ER extract, or 6M urea (lanes 1–6). Where indicated, trypsin (100 μg/ml) was added. Samples were analyzed as in A. In lanes 7–10, isolated A subunit (70 nM) was incubated with either ER extract or 6M urea (followed by addition of proteoliposomes. Samples were analyzed as in A. (C) In lanes 1–3, ER extract or PDI-depleted extract were analyzed by immunoblotting with PDI antibodies. In lanes 4–7, isolated A subunit (70 nM) was incubated with these extracts, followed by the addition of proteoliposomes and the samples were separated into pellet and supernatant fractions. In lanes 8–11, isolated A subunit (70 nM) was incubated with purified PDI or BSA followed by addition of proteoliposomes. (D) As in Fig. 1 C, except BSA, wild-type mammalian proteoliposomes, wild-type yeast proteoliposomes, or yeast mutant ero1–1 proteoliposomes were added to the preformed PDI–toxin complex.
Role of PDI-Ero1 unfolding in retrotranslocation of CT
Based on our findings, we propose the following model for retrotranslocation of CT. Upon reaching the ER lumen, the A1 chain is released from the rest of the toxin and unfolded by PDI. The reduced form of PDI binds to the A1 chain and unfolds it. We assume that the PDI-A1 complex is then targeted to the ER membrane because the A1 chain alone, even when unfolded, does not have a high affinity for the ER membrane. Because PDI, but not Ero1, is required for membrane targeting of the toxin, it appears that the PDI–toxin complex binds to a protein at the lumenal side of the ER membrane via PDI. Our results suggest that the next step is oxidation of PDI by Ero1 and release of the toxin from PDI at the membrane. Because the A1 chain refolds when it is released from PDI into buffer, we assume that the unfolded toxin is transferred directly into the translocation channel. This would be possible if either the PDI-interacting protein at the ER membrane or Ero1 were associated with the channel. In this connection it will be interesting to identify the proteins that appear to be associated with the two isoforms of Ero1 in the ER extract.
Although we have seen release of the toxin from PDI with both Ero1 in the ER detergent extract and with Ero1 present in reconstituted proteoliposomes, in vivo the active species is likely to be membrane-bound Ero1. Because Ero1 lacks a transmembrane domain and a K/HDEL sequence at its COOH terminus, it may be kept in the ER by an association with a membrane protein. Its appearance in the ER extract may be caused by disruption of this interaction by even low concentrations of detergent. Our results show that the α-isoform of Ero1 is capable of releasing the A1 chain from PDI, whereas fractions containing Ero1β were inactive. This raises the possibility that Ero1β may serve a different function in the cell than Ero1α, perhaps in the refolding of misfolded ER proteins, but we cannot exclude the possibility that Ero1β was inactivated during sample preparation.
Our data show that Ero1 only oxidizes the COOH-terminal disulfide bond in PDI to cause toxin release. This finding is consistent with previous results showing that the COOH-terminal thioredoxin domain plays a more critical role than its NH2-terminal counterpart during the in vitro refolding of carboxypeptidase Y (CPY), and of bovine pancreatic trypsin inhibitor (Westphal et al., 1999). In addition, the NH2-terminal thioredoxin domain does not appear to play a role in the PDI-mediated retrotranslocation of misfolded prepro-α factor (Gillece et al., 1999). However, in vivo the NH2-terminal thioredoxin domain was found to play a more important role than the COOH-terminal domain in the folding of CPY (Holst et al., 1997). The reasons for the differences between the in vitro and in vivo results remain to be clarified. Our results also indicate that Ero1 is more efficient in oxidizing toxin-associated PDI compared to free PDI. Although the molecular mechanism for this preference is unclear, it could prevent futile redox cycles of PDI in the absence of substrate.
Whether the PDI-Ero1–driven unfolding mechanism is used to translocate other substrates, such as misfolded ER proteins, remains to be explored. However, PDI and PDI-related proteins have been implicated in the retrotranslocation of misfolded ER substrates (Gillece et al., 1999; Wang and Chang, 1999), suggesting that the proposed pathway may be general.
Materials and methods
Antibodies
The A subunit of CT was purchased from Calbiochem and mammalian PDI from Takara. Antibodies against different proteins were kind gifts from the following sources: mammalian PDI from H. Ploegh (Harvard Medical School), Ero1α and β from H. Ploegh and C. Kaiser (Massachusetts Institute of Technology, Cambridge, MA), Ero1α from A. Benham (University of Durham, UK), A subunit of CT from W. Lencer (Harvard Medical School), and yeast PDI from T. Stevens (University of Orgeon, Eugene, OR). Antibodies against mammalian Ero1β were generated using a peptide containing the sequence SIKDCHVEPC with the NH2 terminus acetylated.
Purification of yeast PDI mutants
Plasmids expressing mutant yeast PDICxxC-AxxA and PDIAxxA-CxxC were gifts from J. Weissman (University of California, San Francisco, CA). Purification of the proteins was performed as described in Tu et al. (2000).
Purification of yeast Ero1 and mammalian Ero1α
The yeast strain overexpressing yeast Ero1-Myc-His6 (AFY383) was a gift from C. Kaiser. The yeast strain overexpressing the mammalian Ero1α-Myc-His6 protein was constructed as follows. The gene encoding Ero1α was excised from pcDNA3.1-Ero1α-Myc-His6, a gift from R. Sitia (Universita Vita-Salute San Raffaele, Milan, Italy) by digesting with XbaI and PmeI. The fragment was ligated between the XbaI and SmaI sites into a yeast overexpression vector (p416-GALL). Yeast cells containing this construct (p416-Ero1α) were grown overnight and then cultured in YEP Raf/Gal. The cells were harvested and homogenized in a french press, followed by centrifugation at 5000g to remove unbroken cells. The supernatant was centrifuged at 13,000 g for 30 min to collect the membrane fraction which was lysed in a buffer containing 1% Triton X-100, 5% glycerol, 50 mM Tris-HCl, pH 7.4, and protease inhibitors. The soluble material was collected by centrifugation at 100,000 g for 40 min. Affinity purification of the proteins was performed by binding to a Ni-NTA column. After washing with 10 mM imidazole, proteins were eluted with 500 mM imidazole in a buffer containing 0.1% Triton X-100, 5% glycerol, 50 mM Tris-HCl, pH 7.4 and protease inhibitors. The eluted sample was dialyzed and bound to a 1 ml Hi-Trap Q-Sepharose column (Amersham Biosciences). Bound material was eluted with a linear 0–1-M potassium acetate gradient. Fractions of 0.5 ml were collected and Ero1 content analyzed by SDS-PAGE and Coomassie staining.
Preparation of ER extract
An ER extract was prepared by adding 0.2% digitonin to a suspension of 2.2 equivalents/μl canine microsomes followed by centrifugation. The supernatant fraction represents the ER extract.
Immunodepletion of mammalian Ero1 and PDI
100 μl of ER extract derived from dog microsomes were incubated with 10 μl PDI antibodies or 10 μl Ero1α/β antibodies overnight at 4°C, followed by addition of 20 μl protein A Sepharose beads. Mock depletion was performed without antibodies.
Thiol modification with maleimide-PEG5000
Isolated A subunit (70 nM) was incubated with 3 μM PDI in the presence of DTT (100 mM), GSSG (100 mM), or GSH (1 mM) for 30 min at 30°C. To samples incubated in GSH (1 mM), Ero1 was added and incubated for 20 min at 30°C. 1/10 of the samples was precipitated with trichloroacetic acid, washed with acetone, and resuspended in a buffer containing 2% SDS, 50 mM Tris (pH 7.4), and 5 mM maleimide-PEG5000 (Shearwater). After incubation for 60 min at 30°C, the samples were analyzed by nonreducing SDS-PAGE and immunoblotted with an antibody directed against PDI.
Toxin transfer assay
Isolated A subunit (70 nM) was incubated with either ER extract, PDI-depleted extract, or 3 μM PDI for 20 min at 30°C. 10 equivalents of proteoliposomes (prepared as described in Gorlich and Rapoport [1993]) were added for 20 min at 30°C. Samples were sedimented for 20 min at 40,000 rpm in a tabletop ultracentrifuge using a TLA 100.4 rotor. The supernatant and pellet fractions were analyzed in nonreducing SDS-PAGE followed by immunoblotting. For preparation of protease-treated proteoliposomes, proteinase K-agarose beads were first incubated with solubilized microsomes for 30 min at room temperature, followed by removal of the protease by centrifugation. Hydrophobic SM2 beads were subsequently added to the solubilized microsomes to remove the detergent and allow formation of vesicles. Trypsin digestion assay, crosslinking assay, and fractionation of ER extract performed as described in Tsai et al. (2001).
Acknowledgments
We would like to thank H. Ploegh, C. Kaiser, A. Benham, W. Lencer, J. Weissman, T. Stevens, and R. Sitia for materials. We thank Y. Ye, C. Kaiser, and G. Guidotti for critical review of the manuscript.
B. Tsai is supported by a fellowship from the Damon Runyon Cancer Research Fund (DRG1579). T.A. Rapoport is a Howard Hughes Medical Institute Investigator.
Footnotes
Abbreviations used in this paper: CPY, carboxypeptidase Y; CT, cholera toxin; Endo H, endoglycosidase H; PDI, protein disulfide isomerase.
References
- Anelli, T., M. Alessio, A. Mezghrani, T. Simmen, F. Talamo, A. Bachi, and R. Sitia. 2002. ERp44, a novel endoplasmic reticulum folding assistant of the thioredoxin family. EMBO J. 21:835–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabibbo, A., M. Pagani, M. Fabbri, M. Rocchi, M.R. Farmery, N.J. Bulleid, and R. Sitia. 2000. ERO1-L, a human protein that favors disulfide bond formation in the endoplasmic reticulum. J. Biol. Chem. 275:4827–4833. [DOI] [PubMed] [Google Scholar]
- Frand, A.R., and C.A. Kaiser. 1998. The ERO1 gene of yeast is required for oxidation of protein dithiols in the endoplasmic reticulum. Mol. Cell. 1:161–170. [DOI] [PubMed] [Google Scholar]
- Frand, A.R., and C.A. Kaiser. 1999. Ero1p oxidizes protein disulfide isomerase in a pathway for disulfide bond formation in the endoplasmic reticulum. Mol. Cell. 4:469–477. [DOI] [PubMed] [Google Scholar]
- Gillece, P., J.M. Luz, W.J. Lennarz, F.J. de La Cruz, and K. Romisch. 1999. Export of a cysteine-free misfolded secretory protein from the endoplasmic reticulum for degradation requires interaction with protein disulfide isomerase. J. Cell Biol. 147:1443–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorlich, D., and T.A. Rapoport. 1993. Protein translocation into proteoliposomes reconstituted from purified components of the endoplasmic reticulum membrane. Cell. 75:615–630. [DOI] [PubMed] [Google Scholar]
- Holst, B., C. Tachibana, and J.R. Winther. 1997. Active site mutations in yeast protein disulfide isomerase cause dithiothreitol sensitivity and a reduced rate of protein folding in the endoplasmic reticulum. J. Cell Biol. 138:1229–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lencer, W.I., T.R. Hirst, and R.K. Holmes. 1999. Membrane traffic and the cellular uptake of cholera toxin. Biochim. Biophys. Acta. 1450:177–190. [DOI] [PubMed] [Google Scholar]
- Matlack, K.E., W. Mothes, and T.A. Rapoport. 1998. Protein translocation: tunnel vision. Cell. 92:381–390. [DOI] [PubMed] [Google Scholar]
- Pagani, M., M. Fabbri, C. Benedetti, A. Fassio, S. Pilati, N.J. Bulleid, A. Cabibbo, and R. Sitia. 2000. Endoplasmic reticulum oxidoreductin 1-lbeta (ERO1-Lbeta), a human gene induced in the course of the unfolded protein response. J. Biol. Chem. 275:23685–23692. [DOI] [PubMed] [Google Scholar]
- Pollard, M.G., K.J. Travers, and J.S. Weissman. 1998. Ero1p: a novel and ubiquitous protein with an essential role in oxidative protein folding in the endoplasmic reticulum. Mol. Cell. 1:171–182. [DOI] [PubMed] [Google Scholar]
- Schmitz, A., H. Herrgen, A. Winkeler, and V. Herzog. 2000. Cholera toxin is exported from microsomes by the Sec61p complex. J. Cell Biol. 148:1203–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sears, C.L., and J.B. Kaper. 1996. Enteric bacterial toxins: mechanisms of action and linkage to intestinal secretion. Microbiol. Rev. 60:167–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai, B., C. Rodighiero, W.I. Lencer, and T.A. Rapoport. 2001. Protein disulfide isomerase acts as a redox-dependent chaperone to unfold cholera toxin. Cell. 104:937–948. [DOI] [PubMed] [Google Scholar]
- Tsai, B., Y. Ye, and T.A. Rapoport. 2002. Retro-translocation of proteins from the endoplasmic reticulum into the cytosol. Nat. Rev. Mol. Cell Biol. 3:246–255. [DOI] [PubMed] [Google Scholar]
- Tu, B.P., S.C. Ho-Schleyer, K.J. Travers, and J.S. Weissman. 2000. Biochemical basis of oxidative protein folding in the endoplasmic reticulum. Science. 290:1571–1574. [DOI] [PubMed] [Google Scholar]
- Wang, Q., and A. Chang. 1999. Eps1, a novel PDI-related protein involved in ER quality control in yeast. EMBO J. 18:5972–5982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westphal, V., N.J. Darby, and J.R. Winther. 1999. Functional properties of the two redox-active sites in yeast protein disulphide isomerase in vitro and in vivo. J. Mol. Biol. 286:1229–1239. [DOI] [PubMed] [Google Scholar]