

Abstract
To track the behavior of human immunodeficiency virus (HIV)-1 in the cytoplasm of infected cells, we have tagged virions by incorporation of HIV Vpr fused to the GFP. Observation of the GFP-labeled particles in living cells revealed that they moved in curvilinear paths in the cytoplasm and accumulated in the perinuclear region, often near the microtubule-organizing center. Further studies show that HIV uses cytoplasmic dynein and the microtubule network to migrate toward the nucleus. By combining GFP fused to the NH2 terminus of HIV-1 Vpr tagging with other labeling techniques, it was possible to determine the state of progression of individual particles through the viral life cycle. Correlation of immunofluorescent and electron micrographs allowed high resolution imaging of microtubule-associated structures that are proposed to be reverse transcription complexes. Based on these observations, we propose that HIV uses dynein and the microtubule network to facilitate the delivery of the viral genome to the nucleus of the cell during early postentry steps of the HIV life cycle.
Keywords: HIV-1; reverse transcription complex; dynein; fluorescent microscopy; electron microscopy
Introduction
After CD4 and coreceptor-dependent fusion of human immunodeficiency virus (HIV)* with the cellular membrane, several events must occur before the HIV genomic DNA can be inserted into the genome of the cell. After entry into the cytoplasm, the infectious particle must lose some of its virion proteins, acquire some cellular factors, convert its RNA genome into the DNA preintegration form, and ultimately integrate into the genome of the infected cell (for review see Cullen, 2001). All of these events have been biochemically defined, but a clear understanding of these processes, how and when they occur, has been hampered by an inability to directly observe the events in infected cells.
HIV infects nondividing cells by delivering its genome into the nucleus through nuclear pores. To reach the pores in the nuclear envelope, the genome must travel from the plasma membrane through the cytoplasm. The integration competent particle, called the preintegration complex, has been estimated to be at least 50 nm in diameter (Miller et al., 1997). Because of the high viscosity of the cytoplasm, movement of these particles by diffusion is likely to be very limited (for review see Luby-Phelps, 2000). Some viruses overcome this obstacle by hijacking cytoplasmic motors to utilize the cellular cytoskeleton as a roadway. For instance, herpes simplex virus (HSV)-1 (Sodeik et al., 1997) and adenovirus (Ad; Suomalainen et al., 1999) are thought to use dynein motors to travel along the microtubule network for intracellular transport. To characterize the intracellular trafficking of HIV, we have developed several fluorescence-based methods that allow the detection and characterization of individual intracellular complexes of viral origin. Importantly, several of these labeling methods allow the visualization of individual virions in living cells. Observation of intracellular HIV in living cells revealed that HIV moves in the cytoplasm in curvilinear paths. Further analysis revealed that intracellular HIV is associated with microtubules and uses cytoplasmic dynein to move toward the nucleus of the cell. These studies suggest that the intracellular trafficking of the HIV genome is a highly ordered process using cellular motor pathways.
Results
GFP–Vpr is incorporated into intact infectious HIV-1 virions
To track viral-derived complexes in the cytoplasm of infected cells, we wanted to stably incorporate the GFP into the HIV virion core by fusion to the HIV accessory protein Vpr. Vpr is incorporated into the virion through interaction with the gag p6 region during viral assembly (Paxton et al., 1993). On average, 100–200 Vpr molecules are incorporated into a single virion produced during normal HIV replication, and much greater amounts can be incorporated when Vpr is overexpressed in trans (Muller et al., 2000; Singh et al., 2001). Importantly, Vpr remains associated with the DNA form of the viral genome in the cytoplasm (Fassati and Goff, 2001).
We generated labeled replication competent virus stocks by cotransfection of HIV proviral DNA with a plasmid that expresses GFP fused to the NH2 terminus of HIV-1 Vpr (GFP–Vpr). After target cells were exposed to the virus-containing supernatants, point sources of GFP fluorescence were found associated with the infected cells, presumably marking the presence of the GFP–Vpr-tagged intracellular particles (Fig. 1). Adherent human cells expressing CD4 were used in these studies because their flat morphology provides an extended cytoplasm, allowing greater resolution of cellular compartments. Early in infection, the GFP signal appeared as punctate signals spread throughout the cell (Fig. 1 A), and when the target cells were washed of free virus and allowed to incubate further, a significant proportion of the signal accumulated in the perinuclear region, often at the microtubule-organizing center (MTOC), in as little as 2 h (Fig. 1 B).
Figure 1.

Infection with EGFP–Vpr-labeled HIV results in perinuclear accumulation of point sources of Gfp fluorescence. Supernatant of 293T cells transfected with pLAI proviral and GFP–Vpr expression plasmids was collected, filtered through a 0.4-μm membrane, and transferred onto coverslips seeded with CD4-expressing target cells for 30 min at 37°C. Cells were rinsed and fixed in formaldehyde (A) or incubated for an additional 2 h (B) and fixed, then immunostained with an antitubulin antibody (red). GFP fluorescence is shown in green. Bars, 10 μm.
Indirect immunofluorescent staining of the GFP–Vpr-tagged HIV bound to glass coverslips confirmed that virtually all of the observed GFP fluorescence was associated with the viral matrix (p17MA) (Fig. 2 A) and capsid (p24CA) (Fig. 2 B) proteins. Typically 50–80% of the particles identified by HIV gag protein were strongly labeled with GFP–Vpr. 70% are labeled with GFP in the experiment shown as demonstrated by costaining for either p24CA or p17MA. The colocalization of HIV gag proteins with GFP fluorescence suggests that we can detect more than half of the virions by GFP fluorescence in these experiments.
Figure 2.

EGFP–Vpr colocalizes with viral gag proteins and cell-derived membranes. (A and B) GFP–Vpr colocalizes with HIV gag proteins. Supernatant from cells transfected with GFP–Vpr and pLAI proviral constructs was exposed to clean glass coverslips in the presence of 10 μg/ml Polybrene. Coverslips were rinsed, fixed, and immunostained with mAbs specific to p17MA (A) and p24CA (B). Top panels are the merged images so that overlapping red and green signals appear yellow. Bottom panels show the individual fluorescent images. White arrows denote double labeled particle; the gray arrow shows a single labeled (GFP-negative) particle. (C) GFP–Vpr colocalizes with cellular membranes. GFP–Vpr-labeled virus was prepared as in A with the addition of DiD (10 μM; Molecular Probes) to the culture medium overnight. Free DiD was washed from the culture, and supernatant was collected 4 h later, bound to a coverslip as above, and fixed with formaldehyde. Bottom panels are separated color images. (D) GFP–Vpr is incorporated into intact HIV virions. Supernatant was collected after transfection of 293T cells with GFP–Vpr, pLAI ∂env provirus, and VSV-G constructs. Virions were pelleted by centrifugation through a 20% sucrose cushion and applied to an Optiprep density gradient. Fractions were collected over the entire gradient, precipitated with 10% TCA, and subjected to SDS-PAGE and Western blotting with antibodies specific to p24CA (top) or GFP (bottom). A whole cell lysate of cells transfected with GFP was run as a control (lane C). Bars, 1 μm.
To determine whether the GFP-tagged HIV particles contained cell-derived membranes, we exposed the transfected virus-producing cells to the lipophilic membrane dye DiD (DiIC18; Molecular Probes) before collecting the supernatants. As expected, essentially all of the GFP fluorescence colocalized with the membrane dye (Fig. 2 C). As seen with gag-specific immunostaining, several GFP-low or -negative, dye-positive spots were observed. This suggested the presence either of viral particles that did not incorporate enough of the GFP–Vpr fusion to be easily detected or possibly of nonviral, lipid-containing cellular debris. We next established that the GFP–Vpr fusion migrated with intact virions on a sedimentation gradient. For this experiment, HIV lacking the native envelope, and therefore unable to replicate, was used to avoid biosafety concerns related to concentrating HIV. When GFP–Vpr-labeled viral supernatants were loaded onto the gradient, the GFP–Vpr fusion copurified with HIV p24CA, indicating specific incorporation into viral particles (Fig. 2 D). The GFP–Vpr fusion was absent from the bottom fraction where membrane debris and intracellular vesicles migrate. Importantly, the fusion protein remained largely intact, insuring that the fluorescent tag should remain attached to the complex containing the HIV genome. Together, these results indicate that the GFP–Vpr fusion protein allows the detection of individual HIV virions by fluorescent microscopy.
HIV uses the cellular cytoskeleton to move inside of cells
HIV particle behavior in living cells was analyzed next. Target cells expressing CD4 and coreceptor were infected with GFP–Vpr-labeled HIV for 30 min, and the unbound virus was washed away (Fig. 3). In this experiment, the M-trophic HIV strain YU-2 and target cells expressing the appropriate CCR5 coreceptor were used. The red stain identifies mitochondria to visualize general cellular structure. Virus location was monitored through a z-series of images taken every 5 min. The paths of movement of several particles during the 95 min of observation is shown in Fig. 3 A. Individual frames for the movement of a single particle is shown in Fig. 3 B. The particle moves in a curvilinear path toward the region of the nucleus (videos 1 and 2 available at http://www.jcb.org/cgi/content/full/jcb.200203150/DC1). To quantify the migration of HIV within cells, we determined the distances of individual particles from the nearest edge of the cell and from the nuclear membrane. We reasoned that a fractional representation of the distances (in this case the distance to the cell edge over the sum of the distances) should give an accurate measure of relative nuclear migration, so that particles at the periphery have positional values near 0 and particles close to the nucleus have values near 1 (Fig. 3 B). This allows the movement toward the nucleus to be quantified even though the particles have different distances to travel because the virus can attach anywhere on the exposed surface of the cell and each cell has a distinct morphology, which can change during the course of the time-lapse experiment. To further define particle movement, Ghost cells were used. Ghost cells contain a GFP reporter for HIV infection and express CD4 and CXCR4 coreceptor. Background levels of GFP expression allow the target cells to be simultaneously observed with the GFP-labeled HIV. Ghost cells were infected for 20 min at a low multiplicity of infection (MOI) with GFP–Vpr-labeled HIV and then imaged as a z-series every 5 min after washing to determine particle motility over time. The graphical representations of the migration of five typical particles (Fig. 3 C) shows both inward and outward motion over time, but in four of the five examples shown, significant nuclear migration occurred over the course of the experiment, and the particles tended to remain near the nucleus once they arrived there. Of 19 particles from 13 different cells examined in this way, 14 (74%) particles were in the perinuclear region at the end of the 75 min time course (unpublished data).
Figure 3.
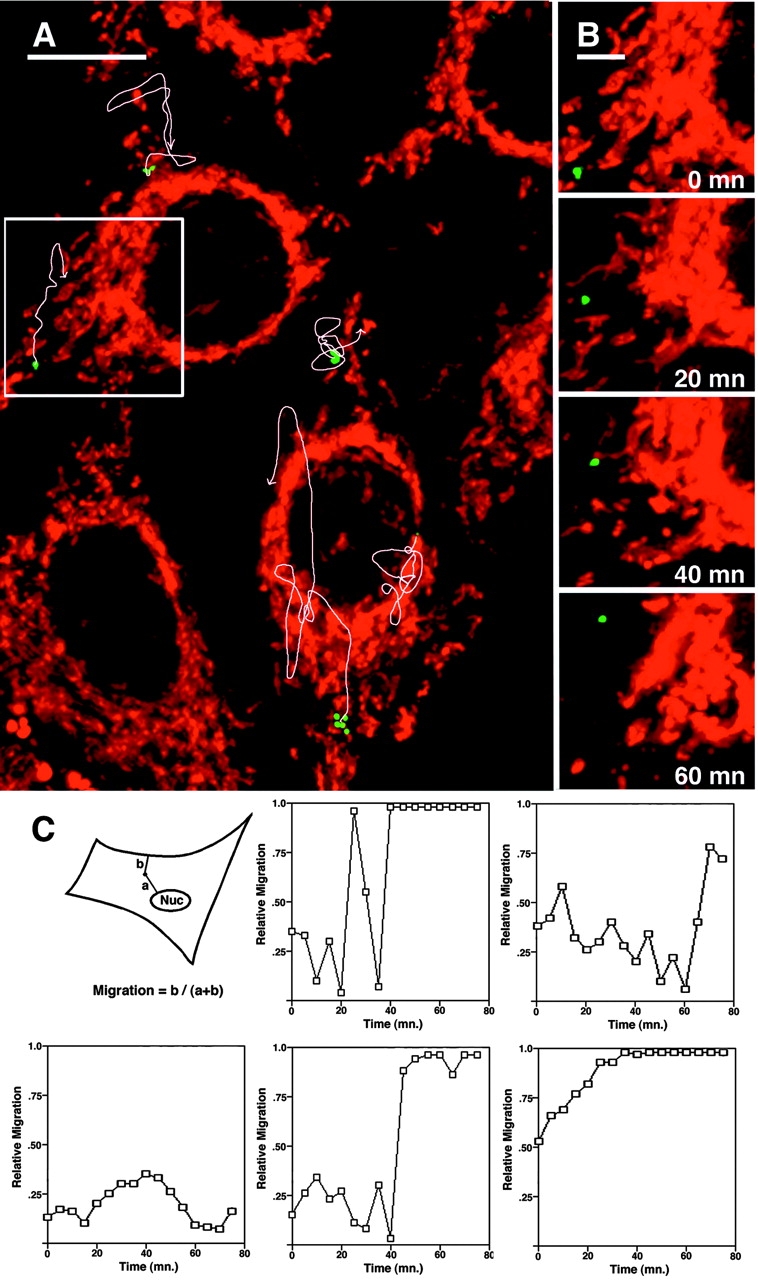
Movement of GFP–Vpr-tagged HIV particles in living cells. (A) HeLa/CD4/CCR5 cells were infected with GFP–Vpr-labeled HIV (Bru3 env, MOI <1) (green) for 30 mn at 23°C, stained with MitoTracker (red), and mounted in a live cell chamber for observation at 37°C. 14 0.5-μm optical z-sections were acquired every 5 mn for 95 min and rendered in single three-dimensional volume views. Oval areas without mitochondrial signal are the nuclei of the cells. Individual HIV particles appear as several spots in some cases due to short rapid movement during acquisition of the z-series. The image is of the first time point, and white arrows denote the trajectories of four different particles over time. (B) Four time points depict the progression of the particle in the inset box. (C) Quantification of GFP–Vpr-tagged HIV movement. HeLa/CD4 cells were infected for 20 min with GFP–Vpr-labeled HIV, washed, and imaged every 5 min. Particle position was measured at each time point, and relative nuclear migration is expressed as the fractional distance from the cell periphery to the nucleus (see diagram) so that low values represent peripheral localization and high values represent perinuclear localization. The five graphs track the movement of representative particles. Bars: (A) 10 μm; (B) 4 μm. See also videos 1 and 2 available at http://www.jcb.org/cgi/content/full/jcb.200203150/DC1.
The nuclear-directed motion of the GFP–Vpr-labeled particles suggested regulated movement through the cytoplasm. Such movement is usually a consequence of interactions with cytoskeletal elements (Goosney et al., 1999; Sodeik, 2000). To determine if HIV interacts with cytoskeletal elements, we fixed target cells at different time points after infection and stained for either actin filaments or microtubules. This analysis demonstrated a significant association of the particles with microtubules but not with actin filaments. In a typical experiment, >90% of the intracellular particles are within 0.5 μm of a microtubule immediately after infection, whereas only a few percent appear to coincide with filamentous actin (unpublished data). The majority of particles that are not associated with microtubules are in the peripheral regions of the cytoplasm. To extend these observations, we determined the effect of agents that specifically disrupt either the actin or microtubule networks. Target cells were infected with GFP–Vpr-labeled HIV for 1 h and then washed and observed at 37°C. Interestingly, after treatment with either the microtubule inhibitor nocodazole or the F-actin inhibitor latrunculin B movement of the GFP-labeled particles was still observed. However, treatment with both agents led to a cessation of movement. Washing out either drug led to the rapid restoration of particle movement, suggesting that HIV movement inside the cell depends on both the actin and microtubule networks. Treatments that retained or restored microtubule structure resulted in long range particle movement; in contrast, actin structure supported short range, more erratic motility (unpublished data).
To address the possibility that HIV might be tracking along microtubules, we performed time-lapse experiments with GFP–Vpr-tagged HIV-infected cells containing microinjected, fluorescently labeled tubulin (Fig. 4; see videos 1 and 2). For this study, virus with DiD-labeled membranes was used to allow the identification of particles, which had lost their membranes as would be expected after functional entry into the cytoplasm. The labeled HIV was spinoculated onto the target cells to increase the number of bound virions (O'Doherty et al., 2000), and the cells were washed and observed by fluorescent microscopy. Images were captured every minute for a 14-min period. Several DiD-negative particles are observed to migrate during the time course. Although the microtubule network is dense because of the injected tubulin, the tracking of DiD-negative particles along microtubules is apparent. Two examples of movement along microtubules are shown in Fig. 4 B. The particle designated by the white arrow is likely the same throughout the panel; however, the particle moved out of the plane of imaging for a single frame so that it is possible that the movement of two different DiD-negative particles is shown (see videos 1 and 2). In addition, the movement of DiD-negative particles not associated with microtubules is also observed (Fig. 4 B, yellow arrow).
Figure 4.

Microtubule-dependent and -independent movement of HIV particles in living cells. Hos/CD4 cells were microinjected with rhodamine-tubulin (Cytoskeleton) and incubated for 1 h at 37°C to label microtubules (blue), spinfected with GFP–Vpr (green), and DiD (red)-labeled HIV Bru3 for 1 h at 1,200 g, 23°C. The cells were washed and placed in medium supplemented with 50 mM Hepes and 0.1 μM taxol to maintain microtubule structure. Images were collected every minute in the three color spectra at 37°C. (A) Cell at the beginning time point. Nucleus is to the lower left, out of frame. (B) Two time segments depicting microtubule- dependent (white arrow) and -independent (colored arrow) movement of DiD-negative particles. The particle on microtubules appears to be the same in both time segments, although assignment is ambiguous in one intermediate frame so that this could represent two independent particles moving on microtubules. See also videos 1 and 2 available at http://www.jcb.org/cgi/content/full/jcb.200203150/DC1. Bars: (A) 10 μm; (B) 5 μm.
To assess a potential role for microtubules in the intracellular movement of GFP-labeled particles, we disrupted the function of cytoplasmic dynein. For this analysis, we microinjected target cells with a monoclonal antibody to the 74-kD intermediate chain of the dyenin motor complex (IC74). It has been reported that injection with this antibody can disrupt the microtubule-dependent minus end–directed movement of Ad (Leopold et al., 2000). Target cells were injected with anti-IC74 mixed with rhodamine-dextran as an injection marker and infected with GFP–Vpr-tagged HIV containing labeled membranes. Again we focused on DiD-negative particles, which have lost their membrane coats and entered the cytoplasm. In uninjected cells, most of the DiD-negative particles accumulated near the nucleus. In contrast, they accumulated in the periphery of the injected cells (Fig. 5, A–C). Quantification of the nuclear progression of the DiD-negative particles demonstrates that in the injected cells the average DiD-negative particle made significantly less progress toward the nucleus than in uninjected cells (Fig. 5 D, relative migration = 0.29 ± 0.03 versus 0.58 ± 0.04 in uninjected cells).
Figure 5.

Inhibition of dynein motor activity results in peripheral localization of HIV particles. (A) Cells were microinjected with antidynein IC74 mAb (1.5 mg/ml) and rhodamine-dextran marker (cell outlined in blue; white outlines are uninjected cells), infected for 1 h with DiD (red)-labeled GFP–Vpr-labeled HIV (green) at 37°C, and then fixed. (B and C) Magnification of boxed regions. Arrows show GFP-positive, DiD-negative particles in the perinuclear region of the uninjected cell (B) and peripheral region of the injected cell (C). (D) Quantification of DiD-negative particle progression toward the nucleus. Relative migration is calculated as described in Fig. 3. The graph represents the majority of particles in uninjected (none) and injected (IC74) cells from A. Bars, 10 μm.
Identification of cytoplasmic reverse transcription complexes
To detect reverse transcription associated with the internalized GFP-labeled particles, we injected a fluorescently labeled deoxynucleotide (Alexa-594–dUTP) into cells before infection. Coverslips carrying microinjected CD4-expressing target cells were infected with GFP–Vpr-labeled HIV for 2 h, washed, and incubated for an additional 2 h. This extended period was found to result in optimal incorporation of the fluorescent nucleotides into the cytoplasmic particles. The cells were then fixed and immunostained for microtubules (Fig. 6). The nucleus is brightly labeled with fluorescent dUTP as a consequence of incorporation of the microinjected nucleotide during cellular DNA replication. Two GFP-labeled particles that have incorporated fluorescent dUTP are readily apparent in the area of the MTOC (Fig. 6 B). The labeling is only seen under conditions where productive infection can take place. For example, Alexa–dUTP incorporation is only observed when the appropriate coreceptor is present as expected because proper receptor/coreceptor-mediated fusion and entry is required for the initiation of reverse transcription (unpublished data). Because the double labeled complexes have started the process of reverse transcription, we will refer to them as reverse transcription complexes (RTCs). Initial studies suggested that the RTCs are associated with microtubules. Detergent extraction of the cells before fixation resulted in significant loss of the RTCs along with the microtubule network, but if taxol was added to stabilize microtubules, RTCs were efficiently retained, suggesting a stable, specific interaction with microtubules (unpublished data).
Figure 6.
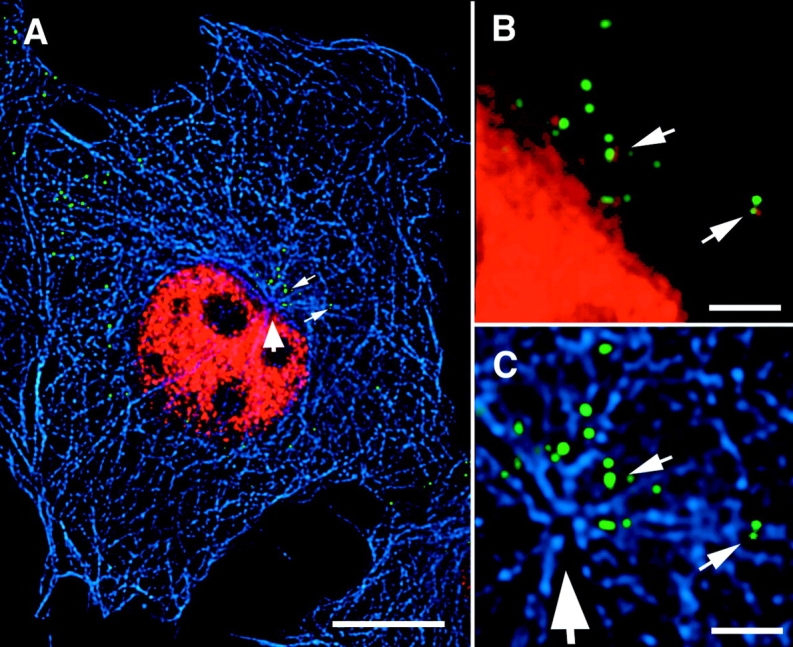
Deoxynucleotide labeling of cytoplasmic reverse transcription complexes. (A) Target cells expressing CD4 were injected with Alexa–dUTP (red) (1mM in PBS; Molecular Probes), infected with GFP–Vpr-labeled HIV (green) (MOI ∼10) for 2 h at 37°C, washed and incubated 2 h more before fixation. Microtubules (blue) were stained with antitubulin antibody. Large arrow denotes the MTOC, and small arrows show viral particles, which have incorporated the Alexa–dUTP. (B and C) Magnification of the MTOC region. Note the overlap of green (GFP) and red (dUTP) signals in the two particles, resulting in yellow color. Bars: (A) 10 μm; (B and C) 2 μm.
To further explore the association of RTCs with microtubules, we injected cells with fluorescent dUTP along with the dynein inhibitor antibody (Fig. 7). RTCs in cells injected with the anti-IC74 antibody exhibited reduced relative nuclear progression (0.23 ± 0.02) compared with RTCs in cells injected with a control antibody (0.57 ± 0.03; Fig. 7 A). In addition, approximately threefold more cytoplasmic RTCs could be identified in anti-IC74–injected cells (Fig. 7 B), presumably because in control injected cells the dynein motility resulted in nuclear accumulation and entry, in which case the nuclear fluorescent signal obscured the RTCs. These results reveal that HIV RTCs use dynein motors to move along microtubules to gain access to the nucleus.
Figure 7.

Inhibition of dynein motor activity results in reduced nuclear migration of HIV RTCs. (A and B) Hos/CD4/CCR5 cells were coinjected with Alexa–dUTP (0.5 mM) mixed with an isotype-matched IgG mAb (Control) or antidynein mAb (IC74), and infected with GFP–Vpr-labeled HIVYu (MOI ∼10) for 2 h at 37°C; medium was refreshed, and cells were incubated an additional 2 h. Cytoplasmic RTCs were identified, and their nuclear progression was quantified as in the legend to Fig. 3. Columns are average values ± SEM of four independent experiments. n, the total number of RTCs quantified. (B) The average number of RTCs/cell from the experiments described in A. Columns are the average number ± SEM. n, the number of cells evaluated.
We next determined whether HIV matrix and/or capsid proteins could be detected in the RTC. It has been demonstrated previously that HIV p17MA is detectable in the preintegration complex (PIC) but p24CA is not (Miller et al., 1997). Staining for p17MA revealed that 95% (19 out of 20) of the RTCs contained matrix. Surprisingly, we found that many of the RTCs also contained significant amounts of p24CA (Fig. 8 C). Quantification of the fluorescent signal in four independent experiments showed that 67 ± 6% (n = 95) of the RTCs contained amounts of p24CA similar to the amount of p24CA found in extracellular particles, suggesting that the capsid in these particles was largely intact. On the other hand, the remaining RTCs contained no detectable p24CA (Fig. 8 B). These results suggest that the capsid structure remains intact during the initiation of reverse transcription in the cytoplasm. Interestingly, both the p24CA-positive and -negative particles appear to associate with microtubules (Fig. 8, D and E), suggesting that the association occurs before loss of the capsid structure.
Figure 8.
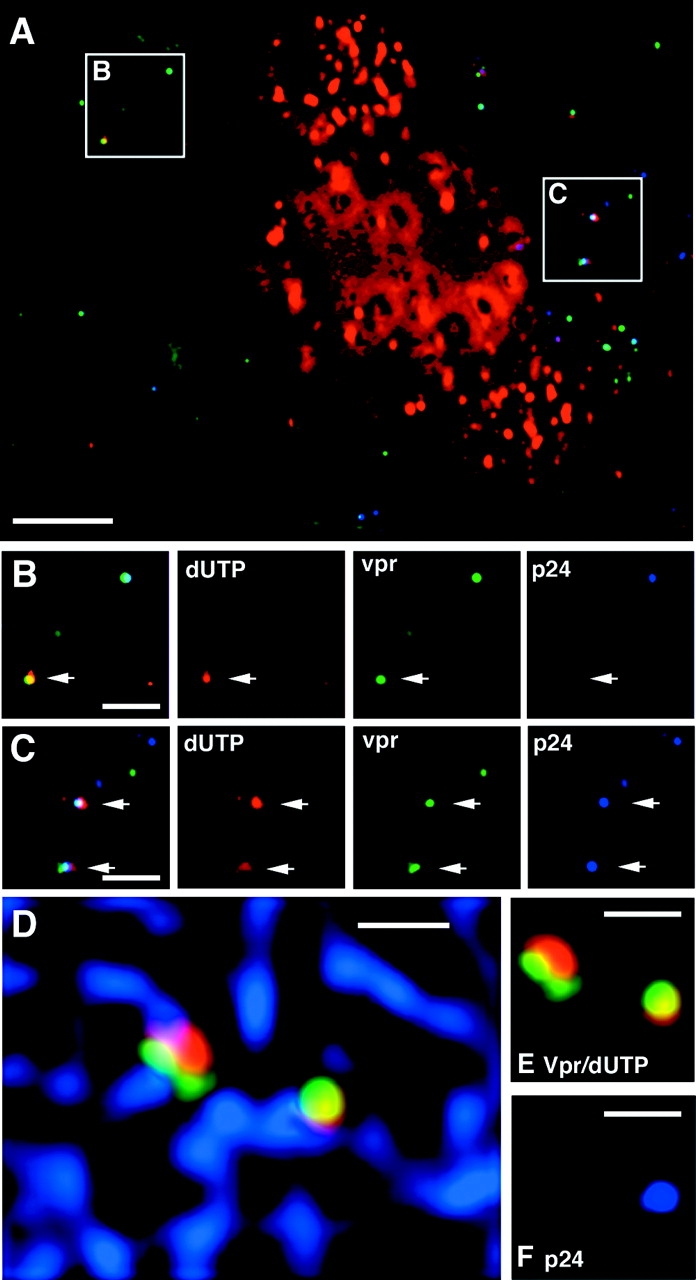
Some HIV reverse transcription complexes contain p24CA protein. Hos cells that express CD4 and CCR5 were injected with Alexa–dUTP (red) and infected with GFP–Vpr-labeled YU strain of HIV (green) (MOI ∼10) for 4 h, fixed, and stained with p24CA antibody. (A) Perinuclear region of a cell with some nuclear incorporation of Alexa–dUTP. (B) Merged (left) and individual color images of a p24CA-negative RTC. Note the presence (top right) of a dUTP-negative, p24CA-positive particle. (C) Merged and individual images of p24CA-positive RTCs. (D) p24CA-positive and -negative RTCs associate with microtubules. Microtubules (blue) were stained in addition to p24CA, and images were collected in four color spectra. (E) Shows the same two RTCs without microtubules, and (F) shows the p24CA (blue) staining profile. Bars: (A) 5 μm; (B–F) 2 μm.
Electron microscopic analysis of RTCs
Our studies demonstrate that it is possible to identify complexes that contain both GFP–Vpr and fluorescent nucleotides, and we propose that they represent steps in the HIV life cycle known to take place after delivery of the viral core into the cytoplasm. Because biophysical studies suggested these complexes are rather large, we surmised that we should be able to visualize RTCs by EM (Miller et al., 1997; Fassati and Goff, 2001). To this end, we adapted a method of correlative EM which was developed to visualize the cytoskeleton (Svitkina and Borisy, 1998). The technique allows the imaging of individual cells at both the light and electron microscopic levels. Key to this method is the growth of cells on coverslips that are gold-embossed with the image of an EM locator grid. The grid can be visualized by both light and electron microscopes, allowing relocation of individual cells using both methods. Because this method utilizes platinum replicas for the EM analysis, only very flat regions of the cells can be efficiently visualized. Therefore, we used human primary fibroblasts for this study. Because native HIV cannot infect cells without CD4 expression, we used envelope-defective HIV that we pseudotyped with the vesicular stomatitis virus envelope glycoprotein (VSV-G), which allows infection of primary human fibroblasts. Although VSV-G directs cellular entry by a distinct mechanism (CD4 and coreceptor fusion at the plasma membrane for gp120 as opposed to fusion out of acidified endosomes for VSV-G), the contents of viral particles delivered to the cytoplasm are derived from the same HIV structural proteins, and it is generally believed that the postfusion events of the delivered genome are similar to the normal HIV life cycle. Cells were injected with fluorescent dUTP, infected with VSV-G–pseudotyped, GFP–Vpr-labeled HIV for 4 h, extracted with detergent in the presence of taxol, and treated with gelsolin to deplete the actin cytoskeleton before fixation. Cells were imaged on a fluorescent microscope and then further processed for EM by platinum rotary shadowing. This technique permits the unambiguous identification of the unique microtubule architecture of individual cells, allowing precise localization of associated complexes (Fig. 9). An example of the correlation of the fluorescent (Fig. 9 A) and EM image (Fig. 9 B) is shown in Fig. 9 C. A high magnification EM image of the structure correlating with the RTC is shown in Fig. 9 D. This RTC appears to be a closed cylinder ∼100 nm by 400 nm, with an interesting cauliflower-like protrusion at one end. Also apparent is a stalk-like structure linking the RTC to the microtubule, similar to that seen tethering HSV-1 capsids to microtubules (Sodeik et al., 1997).
Figure 9.
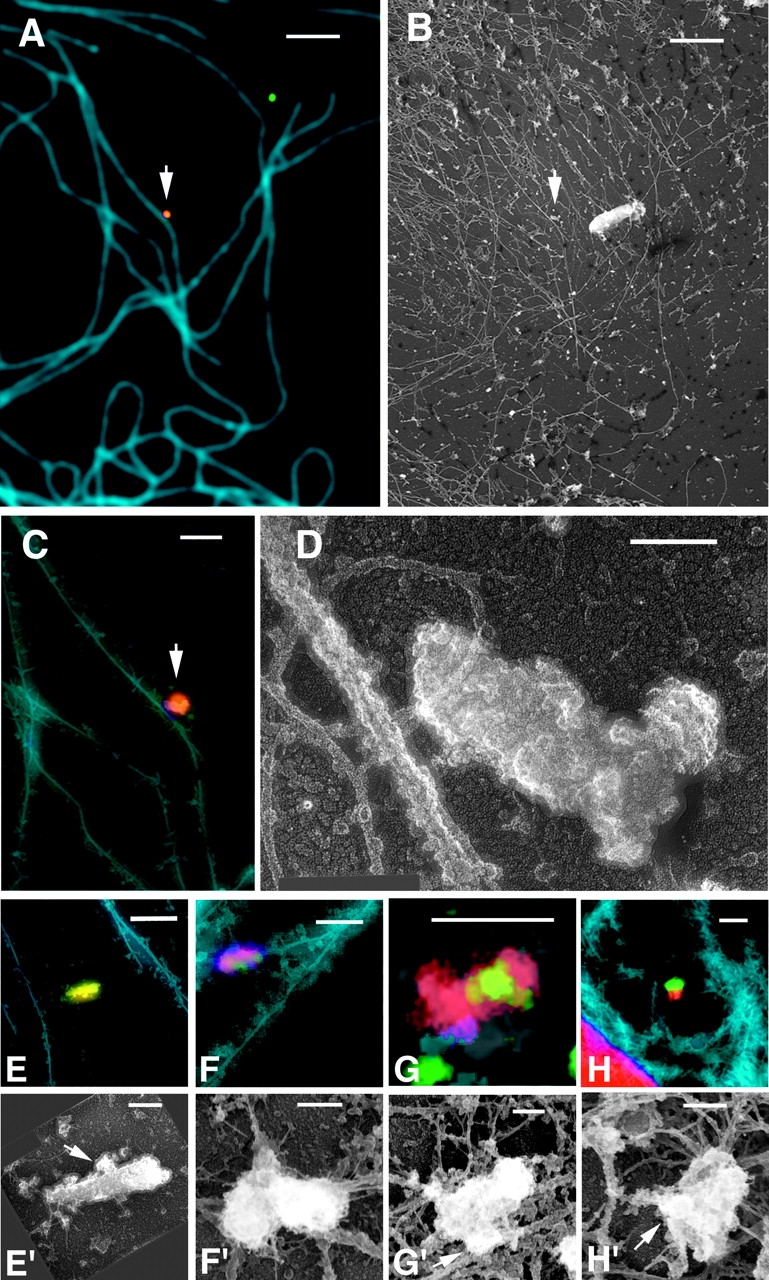
Electron microscopic analysis of cytoplasmic RTC structure. Fibroblasts were injected with Alexa–dUTP (red) and infected with GFP–Vpr-labeled VSV-G pseudotyped HIV (MOI ∼50–100, green) for 4 h. Cell membranes were extracted with 1% Triton X-100 containing taxol, actin was depleted by gelsolin treatment, and the cells were washed, fixed, and immunostained with an antitubulin mAb (blue). Cells were imaged with a fluorescence microscope and then processed for Pt rotary shadowing and relocated on the electron microscope. Microtubules with associated RTC (arrow) in immunofluorescent (A) and electron microscopic (B) images were aligned and overlayed (C). (D) High magnification image of the RTC in A–C. Immunofluorescent (E–H) and EM images (E'–H') of four RTCs, three of which (F–H) appear to be tethered to microtubules. Bars: (A and B) 10 μm; (C and E–H) 1 μm; (D and E'–H') 0.1 μm.
Due to the complexity of the cytoskeleton in the interior of cells, it is necessary to image structures in peripheral regions of the cytoplasm. This presents a significant problem for our analysis because the RTCs tend to move inward during infection. Nevertheless, we have identified several peripheral RTCs associated with microtubules following this procedure and have succeeded in acquiring several EM images of intracellular RTC structures (Fig. 9, D and E′–H′). Although the length of the RTCs varied from 400 to 700 nm, they all had a diameter of 100 ± 26 nm. The RTCs identified show similar size and structure with some variation, possibly due to the nature of the imaging technique (the quality of the image is dependent on several variables, including the orientation of the particle to the plane of focus) or possibly reflecting different stages of RTC maturation.
Discussion
Fluorescent imaging of intracellular HIV
Currently little is known about the intracellular path that the HIV genome takes after it enters the cytoplasm following envelope-mediated fusion with the plasma membrane. To analyze the behavior of the viral genome complex, we have developed several methods that allow the in situ identification of viral components. Key among these methods is exploitation of the property of HIV Vpr to package into virions and remain associated with the viral genome in the cytoplasm of infected cells. By tethering GFP to Vpr, we can detect virions and intracellular particles by fluorescent microscopy. We have confirmed by five different criteria that the GFP signal marks genuine HIV-derived complexes: sedimentation of GFP–Vpr with viral proteins, colocalization with p24CA, p17MA, and virion membranes, and the association of reverse transcriptase activity in infected cells. The composition of the HIV-derived complexes is consistent with the composition of bona fide viral particles. p17MA and p24CA are virtually always found associated with extracellular GFP–Vpr as is expected for intact virions. The intracellular complexes with reverse transcriptase activity all contain p17MA, which is known to remain associated with the viral genome after the completion of reverse transcription (Miller et al., 1997).
An important consideration in efforts to observe cytoplasmic complexes containing the HIV genome is the ability to distinguish productive from nonproductive binding events. HIV is considered to be relatively inefficient at infecting cells, with infectivity to particle ratios reported to be as low as 1 in 60,000 (Kimpton and Emerman, 1992; Piatak et al., 1993). However, more appropriate measures of infectivity, that is the fraction of particles which bind to the cell that cause an infectious event, suggest a ratio up to 1 in 10 for retroviruses (Andreadis et al., 2000). Even at this level of infectivity, it was necessary for us to distinguish between productive and nonproductive entry. For this reason, we focused on two types of GFP-tagged particles: those that had lost a membrane dye, suggesting they had fused with the cellular membrane, and those which had incorporated deoxynucleotides. In both cases, these particles represent a subset of HIV that had entered the cytoplasm of the target cells to form viral complexes which are competent to complete the infectious cycle.
Our time-lapse studies of internalized viral particles suggested both microtubule-dependent and microtubule-independent cytoplasmic movement. Since disruption of both actin and microtubule was necessary to completely halt motility the microtubule-independent movement may use actin. Previous studies have found that disrupting the actin network inhibits infection with HIV, and it was proposed that actin-based motility is important either for cytoplasmic entry (Iyengar et al., 1998) or for establishment of an infectious viral complex (Bukrinskaya et al., 1998). However, further study is required to determine if the microtubule-independent movement is directed through interaction with the actin cytoskeleton or by some other mechanism.
Role for cytoplasmic dynein in intracellular motility of HIV
Our studies suggest that HIV RTCs use the microtubule network for long range movement within cells. Most of the GFP–Vpr-labeled particles were found to be associated with microtubules. Over 80% (16 of 19) of the GFP–Vpr-labeled complexes that were also labeled with fluorescent nucleotides were associated with microtubules in Triton X-100–extracted cells (unpublished data). We documented movement of GFP–Vpr-labeled HIV along fluorescently labeled microtubules using time-lapse microscopy. Further, preliminary analysis suggests that the DiD-negative, GFP-labeled particles can move at burst velocities on the order of 1 μ/s, a rate consistent with that known for microtubule based motors (Presley et al., 1998). We visually demonstrated that GFP–Vpr-labeled complexes extracted from infected cells with saponin could bind to fluorescently labeled microtubules generated in vitro (unpublished data). Finally, attachments between the RTC and microtubules were detected ultrastructurally using correlative EM. Association with microtubules was apparently not cell type specific because the interaction was detected in Hos, HeLa, and primary fibroblast cells. The likely function of the microtubule-based motility is to transport the HIV genome from the cell periphery to the nucleus.
Other viruses use dynein-mediated transport along microtubules to gain access to the nucleus. To determine the role of dynein in the movement of HIV particles along microtubules, we inhibited its motor function by microinjection of an inhibitory monoclonal antibody. After infection, GFP–Vpr-labeled particles defined as entering the cytoplasm either by the loss of membrane label or by the incorporation of fluorescent nucleotides remained in the periphery of injected cells relative to adjacent uninjected cells. In cells with inhibited dynein function, the particles were clustered at points most distant from the nucleus of the infected cell. Preliminary studies suggest that the mislocalization was due to a reversal of the dominant, microtubule minus end–directed movement seen in uninjected cells (unpublished data). Studies of Ad-2 mobility in living cells revealed a similar pattern when dynein function was inhibited by overexpression of p50 dynamitin (Suomalainen et al., 1999). The Ad-2 mislocalization is thought to be a consequence of plus end microtubule activity associated with the motor complex used by this virus. Additional evidence for a plus end–directed activity lies in the observation that Ad-2 oscillates on the microtubules between short range plus end and minus end movements, very similar to the type of motility we have observed with HIV. This oscillation is not seen with Ad-5, and disruption of dynein in that case results in cessation of movement, not with plus end–directed motion (Leopold et al., 2000). Our studies suggest that HIV uses a motor complex with similarities to the one used by Ad-2.
It has been reported previously that disruption of the microtubule network by nocodazole treatment inhibits HIV infection by approximately twofold (Bukrinskaya et al., 1998). We have obtained similar results in this type of study (unpublished data). Nocodazole treatment during HSV and Ad-2 infection has also been shown to decrease the efficiency but not prevent infection (Sodeik et al., 1997; Suomalainen et al., 1999). Several possibilities have been proposed to explain this incomplete inhibition, including entry proximal to the nucleus, transport along nocodazole-resistant microtubules, or movement by Brownian motion (Mabit et al., 2002).
Initiation of reverse transcription in cytoplasmic HIV containing p24CA
There has been much speculation about the cytoplasmic fate of the conical capsid that contains the HIV genome inside virions. In some models, the capsid dissolves immediately after membrane fusion. Others propose that the capsid remains intact until the genome reaches a nuclear pore. The conical core is a relatively unstable complex that is sensitive to all but the most mild detergent treatments. Recently, methods have been developed which allow the isolation of large amounts of HIV cores from virions (Kotov et al., 1999; Accola et al., 2000; Wilk et al., 2001), although intact cores have not been isolated from infected cells. Our in situ analyses identified two distinct species of GFP–Vpr-labeled complexes associated with reverse transcriptase activity, one lacking p24CA and the other containing significant amounts of p24CA, suggesting that the capsid is intact in the latter complexes. Structural studies of the HIV capsid have suggested the presence of pores large enough to allow entry of nucleotides into the capsid (Li et al., 2000). Further, the ability to facilitate reverse transcription within intact virions by exposure to high concentrations of deoxynucleotides along with polyanions also suggests that reverse transcription within intact capsids is possible (Zhang et al., 1996). Previous biochemical studies have not identified significant amounts of p24CA associated with the RTC. However the conditions typically used to generate lysates for RTC and PIC purification may confound this analysis because the HIV capsid easily disassembles in the presence of detergents or possibly in the low salt conditions of hypotonic lysis (Ganser et al., 1999; Kotov et al., 1999; Accola et al., 2000; Wilk et al., 2001). This study provides the first evidence of reverse transcription within an intact HIV capsid in the cytoplasm of an infected cell, suggesting that the capsid remains intact for at least a portion of the process from the initiation of reverse transcription to maturation of the PIC. Furthermore, the p24CA containing RTCs are associated with microtubules (Fig. 9), suggesting that this association with the microtubule network can occur before loss of the capsid. Alternatively, the p24CA-associated RTC may reflect dead end events due to a failure of capsid dissolution after fusion.
Ultrastructure of cytoplasmic RTCs
To gain insight into the morphology of the cytoplasmic RTC, we adapted a correlative electron microscopic technique used previously to study the cytoskeleton. This method uses the alignment of fluorescent and platinum replicas of detergent extracted cells. Unfortunately, this method only allows visualization a short distance into the cell, and therefore, only RTC in the periphery could be imaged. Using EM, we find structures overlapping with the fluorescently visualized RTC. They are typically cylindrical in shape with varying length and a very similar diameter. Previous biophysical characterization of cytoplasmic HIV RTC derived from acutely infected cells suggests that the composition of the RTC is dynamic over time (Fassati and Goff, 2001); while the density of these complexes remains constant, the sedimentation velocity of complexes containing HIV proviral DNA increased at later time points. This observation suggests that protein–DNA complexes with a range of sizes and/or shapes are found in the cytoplasm at different times during reverse transcription. The large size of the RTCs identified in the periphery by correlative EM is most consistent with the early, larger complexes detected in the previous studies. Alternatively, the detergent treatment of the infected cells required for our method may lead to an increase in the size of complexes detected. In either case, the structures we propose to be RTCs are consistent with previous biophysical descriptions. Further study, including immunogold staining for viral proteins and analysis of complexes generated after entry mediated by the HIV envelope will clarify if the observed structures are bona fide cytoplasmic RTCs. In most cases the RTC cylinder is overlapping a microtubule or connected to a microtubule by a stalk-like projection. The observed connections between the identified complexes and microtubules is consistent with our model that intracellular HIV complexes are using the microtubule network to move within the cytoplasm.
Our observations of the composition and trafficking of intracellular HIV complexes suggests the following scenario. After entering the cytoplasm, the HIV genome uses some aspect of the actin cytoskeleton to move within the peripheral regions of the cell. This is consistent with evidence that actin can be used to gain access to the microtubule network (Taunton, 2001). It is also supported by previous reports that an intact actin cytoskeleton is necessary for efficient infectivity (Bukrinskaya et al., 1998). The infectious viral particle must next make its way to the microtubule network, where it can initiate reverse transcription even before losing the majority of its capsid protein. At some point after interaction with the microtubule network, the conical capsid dissociates, yet the RTC maintains its interaction with microtubules. This interaction is likely mediated by tethering with a cellular motor complex which has both minus end– and plus end–directed motor activities, as is proposed for Ad-2. Minus end–directed movement of the RTC along the microtubule network ultimately leads to the microtubule organizing center, very near the nuclear membrane, where the mature RTC can then enter through nuclear pore complexes in order to integrate into the host DNA.
Materials and methods
Cells and antibodies
MAGI cells are a HeLa cell line stably transfected with human CD4 and HIV–long terminal repeat promoter–β-galactosidase genes maintained in complete DME medium as described (Kimpton and Emerman, 1992). HOST4R5 are a human osteosarcoma cell line expressing hCD4 and hCCR5 (provided by Dr. Ned Landau, The Salk Institute) also maintained in complete DME medium. GHOST cells additionally express an HIV–long terminal repeat promoter–GFP construct. Human primary fibroblasts (American Type Culture Collection no. CRL-2522) are maintained in complete RPMI medium. mAb AG3.0 recognizes HIV gag p24CA and the p55GAG precursor (National Institutes of Health AIDS Research and Reference Reagent Program). mAb anti-HIV p17 (Capricorn Products) recognizes HIV gag p17MA. Antidynein intermediate chain mAb (anti-IC74) (Chemicon International) is prepared for microinjection by sequential concentration and dilution in PBS in a microcon concentrator (Millipore, Inc.) to remove azide. The ability of the antibody to retard nuclear accumulation of HIV particles was titered and found to require ∼1 mg/ml for optimal effect. Isotype-matched control antibody (IgG2b; R & D Systems) was used at the same concentration. Alexa-594–dUTP (Molecular Probes) is diluted in PBS or the appropriate antibody to 0.5 mM. Microtubules are immunostained with anti–α-tubulin mAb (Sigma-Aldrich). Actin fibers are stained with Texas red–labeled phalloidin (Molecular Probes). All secondary antibodies are fluorescent-labeled donkey anti–mouse antibodies (Jackson ImmunoResearch Laboratories). Microinjection was performed using an Eppendorf Transjector model 5246. Cells growing on a coverslip in a dish containing media with 50 mM Hepes, pH 7.5, are placed on a warming stage on the microscope, microinjected into the cytoplasm, and then returned to a CO2 incubator to recover for a short time before further manipulation. Rhodamine-labeled tubulin (5 mg/ml; Cytoskeleton) is injected 2 h before infection to allow incorporation into the microtubule network.
Virus production
GFP–Vpr-labeled HIV is produced by CaPO4 transfection of 293T cells with the proviral constructs pLAI or pLAI-Yu (Yu2 env in the pLAI backbone) (Vodicka et al., 1998) and the plasmid peGFPC3 (CLONTECH Laboratories, Inc.) containing the entire Vpr-coding region fused to the COOH terminus of eGFP (GFP–Vpr). Cells are washed at 16–20 h posttransfection, medium is replaced at 36 h, and supernatants containing labeled virus is collected at 2–4-h intervals for two or three harvests. For pseudotyped HIV, an env-deleted pLAI provirus, VSV-G expression plasmid, and the GFP–Vpr plasmid are cotransfected. Virus is collected as above and concentrated by ultracentrifugation for 2 h at 23,000 rpm through a 20% sucrose pad and resuspended in PBS at 50–100-fold concentration (Bartz and Vodicka, 1997). Purification of virus by gradient centrifugation was performed using an Optiprep (Nycomed) density gradient as described (Dettenhofer and Yu, 1999). DiD (DiIC18; Molecular Probes) labeling was achieved by addition of DiD (10 μM) after the initial wash, rinsing away unincorporated dye the next morning, and collecting supernatants as above. All virus preparations were assayed for infectivity using MAGI indicator cells, and the GFP–Vpr incorporation was assessed by p24CA staining of virions bound to glass using mAb AG3.0.
Immunofluorescence
Cells are grown on glass coverslips, rinsed with PBS, and fixed with 3.7% formaldehyde (Polysciences) in 0.1 M Pipes, pH 6.8. Antibodies are added at predetermined dilutions in SB (PBS, 10% normal donkey serum [Jackson ImmunoResearch Laboratories], 0.1% Triton X-100) for 20 min at RT. Coverslips are rinsed extensively and stained with labeled (AMCA-, Cy3- or Cy5-) donkey anti–mouse antibodies (Jackson ImmunoResearch Laboratories) in SB. Coverslips are then mounted onto glass slides with Gel Mount (Biomedia) containing an antifade reagent. Dried slides are imaged on an Olympus IX70 epifluorescent microscope fitted with an automated stage (Applied Precision Inc.), and images are captured in z-series on a CCD digital camera. Out of focus light is digitally removed using the Softworks deconvolution software (Applied Precision Inc.). Live cell video microscopy is performed using a heated open or closed cell chamber along with an objective heater (Bioptechs Inc.), and cells are maintained on coverslips in medium supplemented with 50 mM Hepes, pH 7.5.
Quantification of nuclear migration
Particle location is determined by measuring the distances to the nearest edge of both the nucleus and the cell periphery using the Deltavision Softworks program and is expressed as the fraction of the distance to the edge divided by the total distance so that particles at the periphery have values approaching 0 and nuclear particles approaching 1. For Figs. 3 and 5, the image z-series were first projected as a single image using the three-dimensional reconstruction feature of the software. As a result, it is not possible to measure particles directly over the nucleus so these particles were excluded from the analysis. For Fig. 7, RTCs that are over the nucleus are generally obscured by the strong nuclear Alexa–dUTP signal.
Correlative EM
Cytoskeletal samples are prepared as described (Svitkina and Borisy, 1998) with modifications. Briefly, primary fibroblast cells are grown on glass coverslips patterned by evaporation of gold onto an EM locator grid. Cells are injected with Alexa-594–dUTP (Molecular Probes), infected with GFP–Vpr-labeled, VSV-G pseudotyped HIV at high titer (MOI ∼50–100), and incubated for 4 h at 37°C. Coverslips are then extracted with 1% Triton X-100, 4% PEG mol wt 40 kD (SERVA Biochemicals), and 10 mM taxol (Sigma-Aldrich) in PEM buffer (80 mM Pipes, pH 6.8, 1 mM EGTA, 1 mM MgCl2). Actin is depleted by incubation in G' buffer containing taxol and gelsolin (3 mg/ml; provided by S. Choe, The Salk Institute) at RT for 1 h. Cells are rinsed and fixed with 2% glutaraldehyde and immunostained with anti–α-tubulin and Cy5 donkey anti–mouse secondary. Injected cells are imaged on an epifluorescent microscope, and the locations of candidate cells are noted with respect to the grid pattern. Samples are prepared for EM analysis by desiccation in ethanol and dried in a critical point dryer. They are then coated with platinum in a rotary vacuum evaporator followed by carbon coating to maintain structure. Candidate cells are mounted on formvar-coated EM grids and examined on a JEOL 1220 electron microscope. The unique microtubule structure is used to digitally align the immunofluorescent and EM images using Adobe Photoshop® software.
Online supplemental material
Figs. 3 and 4 time-lapse images are compiled into Quicktime videos available at http://www.jcb.org/cgi/content/full/jcb.200203150/DC1. Video 1 shows the movement of HIV particles in living cells depicted in Fig. 3. HeLa/CD4/CCR5 cells were infected with GFP–Vpr-labeled HIV (green), stained with MitoTracker (red), and mounted in a live cell chamber for observation at 37°C. 14 0.5-μm optical z-sections were acquired every 5 mn for 95 min (20 frames) and rendered in single three-dimensional volume views. The video is displayed at 1 s/frame. Individual HIV particles appear as several spots in some cases due to short rapid movement during acquisition of the z-series. Video 2 shows microtubule-dependent and -independent movement of HIV particles in living cells as depicted in Fig. 4. Hos/CD4 cells were microinjected with rhodamine-tubulin and incubated for 1 h at 37°C to label microtubules (blue), spinfected with GFP–Vpr (green), and DiD (red)-labeled HIV for 1 h at 1,200 g, 23°C. The cells were washed and placed in medium supplemented with 50 mM Hepes and 0.1 μM taxol. Images were collected every minute for 14 min (15 frames) in the three color spectra at 37°C. Video 1 shows the full frame images displayed at 1 s/frame, showing the movement of many particles near microtubules and in areas devoid of microtubules. Video 2 is a close-up of a region, showing microtubule-dependent (white arrow) and -independent (yellow arrow) movement.
Supplemental Material
Acknowledgments
We thank Derek Ko and Heather Feltman for technical assistance, Dr. Ned Landau and the AIDS Reference and Reagent Program for providing cells and antibodies, and Drs. Bellur Prabhakar, Mark Resnick, and Primal de Lanerolle for critical reading of the manuscript. We also thank Larry and Arthur Kramer and the James B. Pendleton Trust for ongoing support of the development of this technology.
This work was supported by National Institutes of Health R01 grant no. AI47770-04 to T. Hope and a grant from the University of Washington Center for AIDS Research to M. Vodicka.
The online version of this article contains supplemental material.
Footnotes
Abbreviations used in this paper: Ad, adenovirus; GFP–Vpr, GFP fused to the NH2 terminus of HIV-1 Vpr; HIV, human immunodeficiency virus; HSV, herpes simplex virus; MOI, multiplicity of infection; MTOC, microtubule-organizing center; PIC, HIV-1 preintegration complex; RTC, reverse transcription complex; VSV-G, vesicular stomatitis virus envelope glycoprotein.
References
- Accola, M.A., A. Ohagen, and H.G. Gottlinger. 2000. Isolation of human immunodeficiency virus type 1 cores: retention of Vpr in the absence of p6(gag). J. Virol. 74:6198–6202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreadis, S., T. Lavery, H.E. Davis, J.M. Le Doux, M.L. Yarmush, and J.R. Morgan. 2000. Toward a more accurate quantitation of the activity of recombinant retroviruses: alternatives to titer and multiplicity of infection. J. Virol. 74:1258–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartz, S.R., and M.A. Vodicka. 1997. Production of high-titer human immunodeficiency virus type 1 pseudotyped with vesicular stomatitis virus glycoprotein. Methods. 12:337–342. [DOI] [PubMed] [Google Scholar]
- Bukrinskaya, A., B. Brichacek, A. Mann, and M. Stevenson. 1998. Establishment of a functional human immunodeficiency virus type 1 (HIV-1) reverse transcription complex involves the cytoskeleton. J. Exp. Med. 188:2113–2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen, B.R. 2001. Journey to the center of the cell. Cell. 105:697–700. [DOI] [PubMed] [Google Scholar]
- Dettenhofer, M., and X.F. Yu. 1999. Highly purified human immunodeficiency virus type 1 reveals a virtual absence of Vif in virions. J. Virol. 73:1460–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fassati, A., and S.P. Goff. 2001. Characterization of intracellular reverse transcription complexes of human immunodeficiency virus type 1. J. Virol. 75:3626–3635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganser, B.K., S. Li, V.Y. Klishko, J.T. Finch, and W.I. Sundquist. 1999. Assembly and analysis of conical models for the HIV-1 core. Science. 283:80–83. [DOI] [PubMed] [Google Scholar]
- Goosney, D.L., M. de Grado, and B.B. Finlay. 1999. Putting E. coli on a pedestal: a unique system to study signal transduction and the actin cytoskeleton. Trends Cell Biol. 9:11–14. [DOI] [PubMed] [Google Scholar]
- Iyengar, S., J.E. Hildreth, and D.H. Schwartz. 1998. Actin-dependent receptor colocalization required for human immunodeficiency virus entry into host cells. J. Virol. 72:5251–5255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimpton, J., and M. Emerman. 1992. Detection of replication-competent and pseudotyped human immunodeficiency virus with a sensitive cell line on the basis of activation of an integrated beta-galactosidase gene. J. Virol. 66:2232–2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotov, A., J. Zhou, P. Flicker, and C. Aiken. 1999. Association of Nef with the human immunodeficiency virus type 1 core. J. Virol. 73:8824–8830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leopold, P.L., G. Kreitzer, N. Miyazawa, S. Rempel, K.K. Pfister, E. Rodriguez-Boulan, and R.G. Crystal. 2000. Dynein- and microtubule-mediated translocation of adenovirus serotype 5 occurs after endosomal lysis. Hum. Gene Ther. 11:151–165. [DOI] [PubMed] [Google Scholar]
- Li, S., C.P. Hill, W.I. Sundquist, and J.T. Finch. 2000. Image reconstructions of helical assemblies of the HIV-1 CA protein. Nature. 407:409–413. [DOI] [PubMed] [Google Scholar]
- Luby-Phelps, K. 2000. Cytoarchitecture and physical properties of cytoplasm: volume, viscosity, diffusion, intracellular surface area. Int. Rev. Cytol. 192:189–221. [DOI] [PubMed] [Google Scholar]
- Mabit, H., M.Y. Nakano, U. Prank, B. Saam, K. Dohner, B. Sodeik, and U.F. Greber. 2002. Intact microtubules support adenovirus and herpes simplex virus infections. J. Virol. 76:9962–9971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, M.D., C.M. Farnet, and F.D. Bushman. 1997. Human immunodeficiency virus type 1 preintegration complexes: studies of organization and composition. J. Virol. 71:5382–5390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller, B., U. Tessmer, U. Schubert, and H.G. Krausslich. 2000. Human immunodeficiency virus type 1 Vpr protein is incorporated into the virion in significantly smaller amounts than gag and is phosphorylated in infected cells. J. Virol. 74:9727–9731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Doherty, U., W.J. Swiggard, and M.H. Malim. 2000. Human immunodeficiency virus type 1 spinoculation enhances infection through virus binding. J. Virol. 74:10074–10080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxton, W., R.I. Connor, and N.R. Landau. 1993. Incorporation of Vpr into human immunodeficiency virus type 1 virions: requirement for the p6 region of gag and mutational analysis. J. Virol. 67:7229–7237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piatak, M., Jr., M.S. Saag, L.C. Yang, S.J. Clark, J.C. Kappes, K.C. Luk, B.H. Hahn, G.M. Shaw, and J.D. Lifson. 1993. High levels of HIV-1 in plasma during all stages of infection determined by competitive PCR. Science. 259:1749–1754. [DOI] [PubMed] [Google Scholar]
- Presley, J.F., C. Smith, K. Hirschberg, C. Miller, N.B. Cole, K.J. Zaal, and J. Lippincott-Schwartz. 1998. Golgi membrane dynamics. Mol. Biol. Cell. 9:1617–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh, S.P., P. Tungaturthi, M. Cartas, B. Tomkowicz, T.A. Rizvi, S.A. Khan, V.S. Kalyanaraman, and A. Srinivasan. 2001. Virion-associated HIV-1 Vpr: variable amount in virus particles derived from cells upon virus infection or proviral DNA transfection. Virology. 283:78–83. [DOI] [PubMed] [Google Scholar]
- Sodeik, B. 2000. Mechanisms of viral transport in the cytoplasm. Trends Microbiol. 8:465–472. [DOI] [PubMed] [Google Scholar]
- Sodeik, B., M.W. Ebersold, and A. Helenius. 1997. Microtubule-mediated transport of incoming herpes simplex virus 1 capsids to the nucleus. J. Cell Biol. 136:1007–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suomalainen, M., M.Y. Nakano, S. Keller, K. Boucke, R.P. Stidwill, and U.F. Greber. 1999. Microtubule-dependent plus- and minus end–directed motilities are competing processes for nuclear targeting of adenovirus. J. Cell Biol. 144:657–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svitkina, T.M., and G.G. Borisy. 1998. Correlative light and electron microscopy of the cytoskeleton of cultured cells. Methods Enzymol. 298:570–592. [DOI] [PubMed] [Google Scholar]
- Taunton, J. 2001. Actin filament nucleation by endosomes, lysosomes and secretory vesicles. Curr. Opin. Cell Biol. 13:85–91. [DOI] [PubMed] [Google Scholar]
- Vodicka, M.A., D.M. Koepp, P.A. Silver, and M. Emerman. 1998. HIV-1 Vpr interacts with the nuclear transport pathway to promote macrophage infection. Genes Dev. 12:175–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilk, T., I. Gross, B.E. Gowen, T. Rutten, F. de Haas, R. Welker, H.G. Krausslich, P. Boulanger, and S.D. Fuller. 2001. Organization of immature human immunodeficiency virus type 1. J. Virol. 75:759–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, H., G. Dornadula, and R.J. Pomerantz. 1996. Endogenous reverse transcription of human immunodeficiency virus type 1 in physiological microenvironments: an important stage for viral infection of nondividing cells. J. Virol. 70:2809–2824. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.