

Abstract
Unlike most kinesins, mitotic centromere–associated kinesin (MCAK) does not translocate along the surface of microtubules (MTs), but instead depolymerizes them. Among the motile kinesins, refinements that are unique for specific cellular functions, such as directionality and processivity, are under the control of a “neck” domain adjacent to the ATP-hydrolyzing motor domain. Despite its apparent lack of motility, MCAK also contains a neck domain. We found that deletions and alanine substitutions of highly conserved positively charged residues in the MCAK neck domain significantly reduced MT depolymerization activity. Furthermore, substitution of MCAK's neck domain with either the positively charged KIF1A K-loop or poly-lysine rescues the loss of MT-depolymerizing activity observed in the neckless MCAK mutant. We propose that the neck, analogously to the K-loop, interacts electrostatically with the tubulin COOH terminus to permit diffusional translocation of MCAK along the surface of MTs. This weak-binding interaction may also play an important role in processivity of MCAK-induced MT depolymerization.
Keywords: Kin I; XKCM1; kinesins; processivity; diffusional motility
Introduction
Members of the kinesin superfamily transport cargo along the surface of microtubules (MTs)* using the power of an ATP-hydrolyzing motor domain (Goldstein and Philp, 1999; Woehlke and Schliwa, 2000). Mitotic centromere–associated kinesin (MCAK) is a member of the Kin I subfamily of kinesin-related proteins that shares high homology within the motor domain with other members of the kinesin superfamily (Wordeman and Mitchison, 1995; Vale and Fletterick, 1997). However, although most kinesins translocate along the surface of MTs, MCAK and its homologues depolymerize MTs from either end (Walczak et al., 1996; Desai et al., 1999; Hunter and Wordeman, 2000; Kinoshita et al., 2001; Maney et al., 2001). Kinetic analysis suggests that processivity and diffusional motility may contribute to MCAK's rapid rate of MT depolymerization (unpublished data). The novel ability to modulate MT dynamics suggests that these kinesins may be important contributors to cell cycle progression (Zhai et al., 1996) and tumorgenesis (Lakshmi et al., 1993).
The direction of transport and the affinity for MTs are molecular refinements that adapt motile kinesins for specific cellular functions within the realm of intracellular transport, cell division, and MT dynamics (Goldstein and Philp, 1999; Woehlke and Schliwa, 2000). The “neck” domain, a region adjacent to the motor domain (head), is responsible for conferring directionality and processivity to the core motor domain (Case et al., 1997; Henningsen and Schliwa, 1997; Endow and Waligora, 1998; Romberg et al., 1998). MCAK's neck domain is highly conserved within the Kin I subfamily, but is very divergent from other kinesin-related proteins (Vale and Fletterick, 1997; Maney et al., 2001). A truncated MCAK protein, consisting of the neck domain (A182-D246) plus the core motor domain (P247-E581), shows MT depolymerization activity similar to that of the full-length (FL) MCAK (Maney et al., 2001). However, the neck itself does not possess depolymerizing activity (Maney et al., 1998), nor does it confer depolymerization activity to a motile kinesin because a chimera consisting of the MCAK neck fused to the motor domain of a conventional kinesin (KIF5B) exhibits no MT depolymerization activity when expressed in cultured cells (Maney et al., 2001). Finally, the neck domain is not involved in dimerization because the A182-E581 fragment of MCAK, which includes the neck, is a monomer (Maney et al., 2001). What, then, is the role of the neck domain in the MT depolymerization activity of MCAK?
The electrostatic interactions between a positively charged neck domain and the negatively charged COOH terminus of tubulin have been shown to be important for the processivity of conventional kinesins. The addition of positive charges to the neck of a conventional kinesin increases its processivity by perhaps tethering the kinesin near the MT surface (Thorn et al., 2000). Similarly, the positively charged K-loop in the motor domain of the KIF1 single-headed kinesin provides a flexible attachment site for binding to MTs in the weakly bound state (Okada and Hirokawa, 2000; Rogers et al., 2001). Given the high percentage of positively charged amino acids in the MCAK neck region and their strong conservation among Kin I kinesins (Fig. 1 A), it is reasonable to suggest that the neck might be anchoring MCAK to MTs. Here, we used mutational analysis and chimeras to test whether the MCAK neck functions as an electrostatic tether. We found that although the removal of positive charges from the neck region significantly diminished the MT depolymerization activity of MCAK, the insertion of the positively charged K-loop into “neckless” MCAK completely restored its MT depolymerization activity. We propose that the neck, analogous to the K-loop, may interact electrostatically with the negatively charged MT lattice. This interaction may serve to target MCAK to MT ends by diffusional motility. The weak electrostatic interaction between MCAK's neck and MT ends may also play an important role in processivity of MCAK-induced MT depolymerization.
Figure 1.

The structural analysis of the MCAK neck. (A) Sequence alignment of the neck domain of seven Kin I kinesins: CgMCAK, C. griseus (residues A182-D246; GenBank/EMBL/DDBJ accession no. U11790); HsMCAK, Homo sapiens (GenBank/EMBL/DDBJ accession no. U63743); RnKRP2, Rattus norvegicus (GenBank/EMBL/DDBJ accession no. U44979); XKCM1, Xenopus laevis (GenBank/EMBL/DDBJ accession no. U36485); HsKIF2, Homo sapiens (GenBank/EMBL/DDBJ accession no. CAA69621); MmKIF2, Mus musculus (GenBank/EMBL/DDBJ accession no. D12644); XKIF2, X. laevis (GenBank/EMBL/DDBJ accession no. U36486). Identical residues are shaded in black, similar ones are shaded in gray. The 64-amino acid neck domain sequences are 50% identical and 75% similar. The + symbols indicate highly conserved positively charged amino acids in the neck domain. The arrows show residues that were substituted for alanine. The borders of deletions in the neck domain are indicated by flanking residue numbers above the neck alignment. (B) Two sides of a helical diagram of a highly charged hydrophilic helix (residues R183–Q215) found in the CgMCAK neck. Positively charged amino acids are white, negatively charged residues are black, and the other residues are gray.
Results and discussion
Quantitation of MT depolymerization activity in vivo
To estimate the effect of the deletions in the MCAK neck domain on the MT depolymerization activity, we devised a quantitative in vivo MT depolymerization assay. In brief, EGFP–MCAK fusion constructs were transfected into cultured cells, fixed, and stained for tubulin (Fig. 2) . Digital images were acquired using a cooled CCD camera. The transfected cells chosen for quantitation displayed similar EGFP–MCAK fusion protein expression levels (Figs. 3 and 4) . The average pixel intensity (the mean gray value) over the entire cellular area was measured and corrected for noncellular background. Cells transfected with a control EGFP construct showed normal levels of MT polymer and were assigned an MT polymer value of 100%. EGFP-transfected cells displayed a mean tubulin fluorescence value of 1,902 gray-scale units, which represented the sum of free tubulin and MT polymer fluorescence (Fig. 3 A). Expression of FL MCAK resulted in the loss of all MTs at the analyzed level of expression with only a lightly stained background of unpolymerized tubulin remaining (Fig. 2 D, arrow). Such cells showed a mean tubulin fluorescence value of 734 gray-scale units, which was designated as 0% of the MT polymer (Fig. 3 A). Consequently, the mean intensity of tubulin fluorescence in the FL MCAK–transfected cells was less than half of that in the EGFP-transfected cells.
Figure 2.

Depolymerization of MTs by MCAK and MCAK mutants. (A–C) EGFP fluorescence, (D–F) tubulin staining of the same cells. A cell transfected with full-length (FL) MCAK (A and D) displays a loss of MT polymer and low intensity unpolymerized tubulin staining (arrow). A cell transfected with the neckless MCAK mutant (B and E; Δ A182-D218 MCAK) exhibits a normal network of cytoplasmic MTs (arrowhead) as a consequence of the significantly decreased MT depolymerization activity of neckless MCAK. (C and F) Cells transfected with the A182-K-loop-D218 MCAK construct, the K-loop insertion into the neckless Δ A182-D218 MCAK mutant. The insertion of the KIF1A K-loop (NKNKKKKK) into the neckless MCAK restores the MT depolymerization activity to the level of FL MCAK. Arrow in F illustrates a cell with MT polymer loss similar to that in FL MCAK–transfected cells (D, arrow). All panels are the same magnification. Bar, 10 μm.
Figure 3.
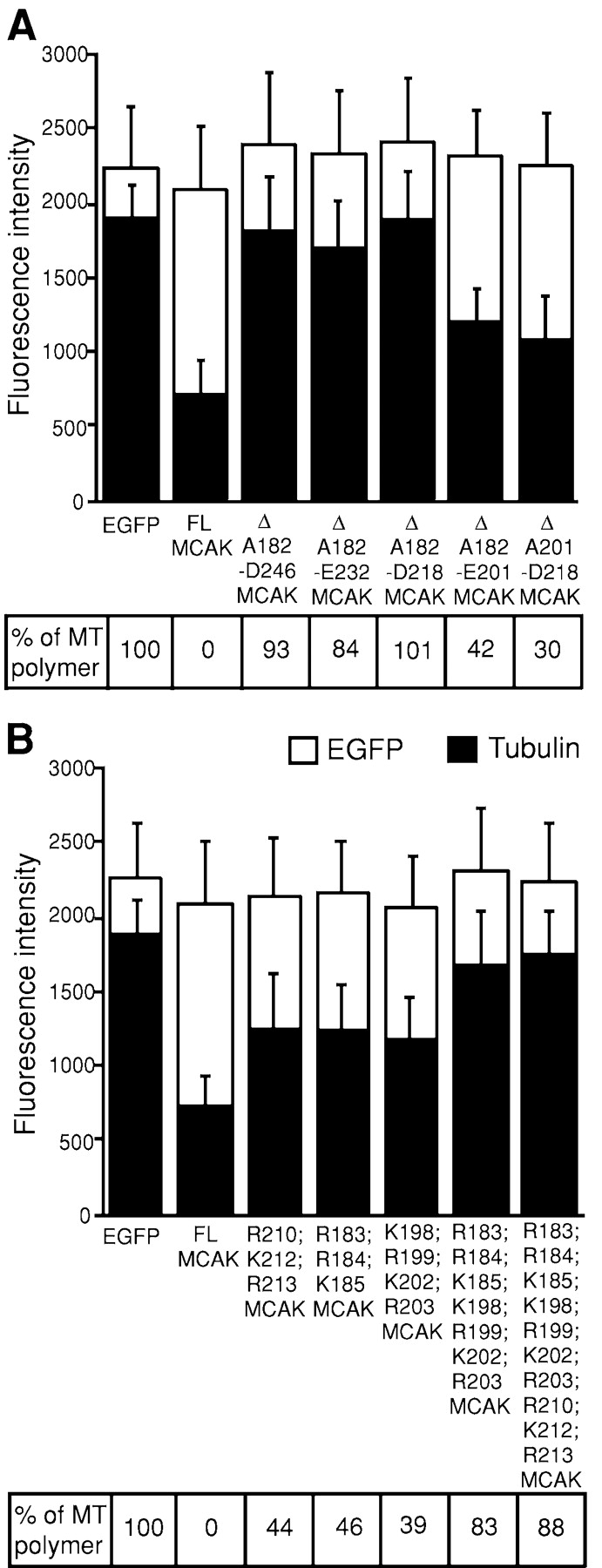
Removal of positive charges in the neck domain significantly reduced MT depolymerization activity of MCAK. (A) The effect of deletions in the neck |region. Each Δ symbol represents a deletion of the MCAK neck region with borders indicated by flanking amino acid numbers. (B) The effect of alanine substitutions in the neck region. The residues changed to alanine are shown for each MCAK mutant. The average pixel intensity over the cell area is plotted on the y axis in gray-scale units. White bars show the mean EGFP fluorescence. Black bars represent the mean tubulin fluorescence. Error bars are SDs. EGFP indicates cells transfected with the EGFP control; FL MCAK designates cells transfected with FL wild-type MCAK. The percentage of MT polymer left in the transfected cells is shown.
Figure 4.
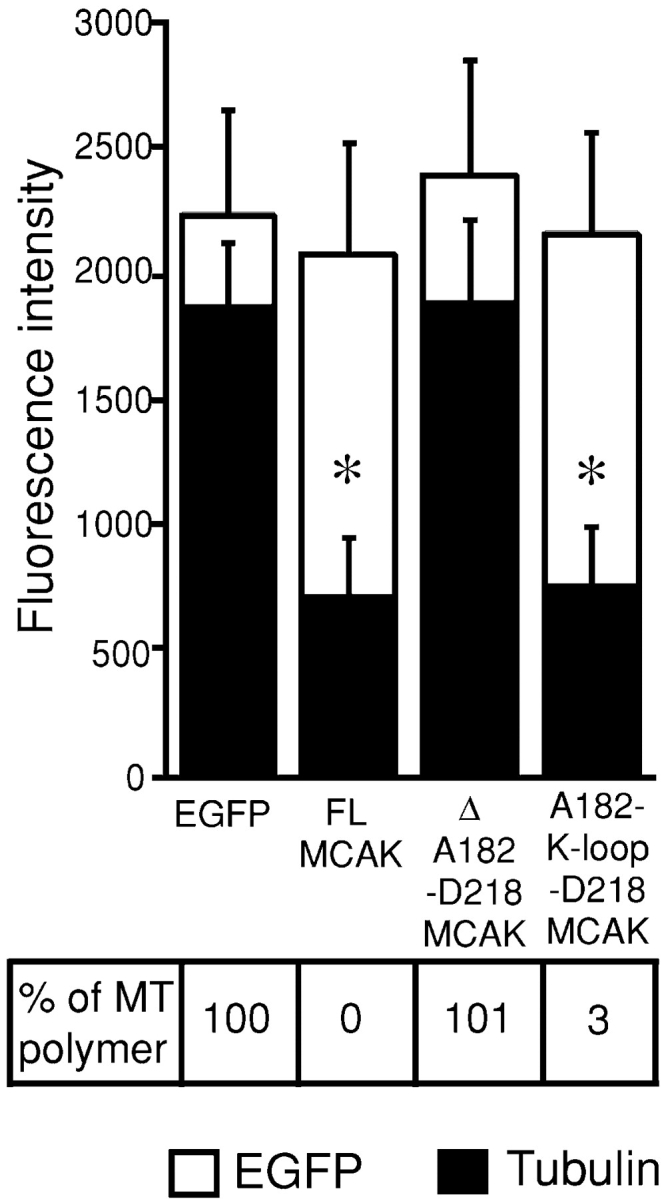
Insertion of KIF1A K-loop into the neckless MCAK fully restores MT depolymerization activity. y and x axis categories are the same as described in Fig. 3 legend. Error bars are SDs. A182-K-loop-D218 MCAK represents cells transfected with the KIF1A K-loop insertion into the neckless MCAK mutant, A182-NKNKKKKK-D218 MCAK. Asterisks label two samples, FL MCAK and A182-K-loop-D218 MCAK, whose means are statistically similar (P = 0.92) to each other as determined using the t test. The percentage of MT polymer left in the transfected cells is indicated.
The low level of free (unpolymerized) tubulin seen in cells transfected with FL MCAK might be a result of a tubulin autoregulation mechanism. Increasing the concentration of free tubulin by depolymerizing MTs with MT-destabilizing drugs, e.g., nocodazole, leads to a rapid decrease in the level of tubulin mRNAs and an eventual decrease in the unassembled tubulin level (Cleveland et al., 1981; Gonzalez-Garay and Cabral, 1996). Similar to MCAK-mediated MT depolymerization, we found that treatment with a low concentration of nocodazole (1 μM) for 12 h produced cells with a low intensity free-tubulin background and no MTs. The mean tubulin fluorescence intensity of these cells was 798 ± 169 gray-scale units (n = 88) as determined by the in vivo depolymerization assay described in this report. The mean tubulin intensity of cells treated with 1 μM nocodazole for 15 min was 1,645 ± 189 (n = 41). These cells displayed almost no MT polymer and bright free-tubulin staining due to the fact that 15 min was not long enough for autoregulation to take place and decrease the level of free tubulin. Nontreated cells with a normal network of cytoplasmic MTs showed the mean tubulin intensity equal to 1,870 ± 301 (n = 77).
The removal of positive charges from the neck domain dramatically reduced MT depolymerization activity of MCAK
As expected from previous studies (Maney et al., 2001), cells transfected with neckless MCAK (Δ A182-D246) displayed a normal MT network. The mean tubulin fluorescence of the Δ A182-D246 MCAK mutant was similar to that of the EGFP control and equaled 1,825 gray-scale units (Fig. 3 A). Cells transfected with MCAK mutants containing progressively smaller deletions in the neck domain, Δ A182-E232 and Δ A182-D218 (Fig. 2 E, arrowhead), also showed normal levels of MT polymer that were similar to that of the EGFP-transfected cells (Fig. 3 A). Hence, these neckless MCAK mutants exhibited severely impaired MT depolymerization activity. Smaller deletions in the neck region (Δ A182-E201 and Δ E201-D218) diminished MT depolymerization activity to a lesser degree. (Fig. 3 A). Next, we performed alanine substitutions of the highly conserved positively charged residues in the neck domain (Fig. 1, arrows) to test whether the loss of positive charges in this region affects the depolymerization activity of MCAK. Alanine substitutions of 3–4 positively charged amino acids resulted in some reduction of MT depolymerization activity (more than 50% of MTs were depolymerized in the transfected cells). Substitutions of 7 to 10 amino acids resulted in much greater inhibition of the depolymerization activity (<20% of MTs were depolymerized; Fig. 3 B). Therefore, the total sum of removed positive charges was of greater importance than the positions of the substituted residues.
In summary, we identified Δ A182-D218 as the smallest neck deletion that would reduce MT depolymerizing activity to the level of the EGFP control (Fig. 3 A). This region contains 13 (36%) positively charged residues and 7 (19%) negatively charged residues (Fig. 1). The net charge of A182-D218 region is positive and equals +6. Alanine substitutions of only 7 positive residues rendered the net charge of A182-D218 segment negative and resulted in greatly diminished MT depolymerization activity (Fig. 3 B). The A182-D218 region contains a highly charged hydrophilic helix (residues R183-Q215) as predicted by SSpro2 server (http://promoter.ics.uci.edu/BRNN-PRED [Baldi et al., 1999]). The helical wheel representation of this helix shows that the positively charged residues lie mostly on one side of the helix, whereas the opposite side contains most of the negatively charged residues found in this neck region (Fig. 1 B).
KIF1A K-loop insertion into neckless MCAK rescues MT depolymerization activity
We have shown that elimination of the positive charges in the MCAK neck region, A182-D218, either by deletions or alanine substitutions inhibits MT depolymerization activity of MCAK. Conversely, Niederstrasser et al. (2002) showed that the interaction between the COOH terminus of tubulin (C-hook) and MCAK is required for MT depolymerization because subtilisin cleavage of the C-hook abolishes depolymerization of MTs by the MCAK homologue, XKCM1. Furthermore, Okada and Hirokawa (2000) have shown that the positively charged K-loop of KIF1A interacts with the negatively charged C-hook and serves as an electrostatic tether to support the diffusion of KIF1A along MT protofilament. The positively charged neck of MCAK might interact with the negatively charged C-hook of tubulin analogously to the KIF1A K-loop. We tested this hypothesis by inserting the K-loop of KIF1A into the MT depolymerization deficient neckless MCAK. We found that MCAK with the A182-D218 neck segment replaced by the KIF1A K-loop, NKNKKKKK, (abbreviated as A182-K-loop-D218 MCAK) depolymerized MTs with the same efficiency as FL MCAK (Figs. 2 F and 4). Moreover, mitotic cells transfected with the A182-NKNKKKKK-D218 MCAK mutant displayed the same mitotic defects (unpublished data) described for overexpression of FL MCAK, namely bundling and depolymerization of spindle MTs, which in turn resulted in prometaphase arrest and an elevated mitotic index in transfected cells (Maney et al., 1998).
None of the MCAK mutations described in this report interfered with centromere or centrosome binding. The nuclear/cytoplasmic localization of the mutants was identical to that of wild-type FL MCAK (unpublished data). All MCAK mutants showed the ability to bind MTs in vivo when cells were extracted before fixation in the absence of nucleotide (unpublished data) in a manner identical to FL MCAK (Maney et al., 1998). This implies that all mutated MCAKs were functional proteins with motor domains able to bind MT lattice in the nucleotide-free state, similar to conventional kinesins.
In vitro MT depolymerization activity of MCAK neck mutants
The rescue of the MT depolymerization–deficient neckless MCAK by K-loop insertion suggests that the MCAK neck is affecting the interactions between motor and MTs rather than regulating the depolymerization activity. To test this hypothesis further, we replicated our depolymerization assays in vitro. We expressed and purified the following proteins: core motor plus neck (A182-S583); core motor plus alanine-substituted neck (A182-Ala-S583 with substituted residues indicated by arrows in Fig. 1 A); core motor plus truncated neck (D218-S583); core motor plus K-loop in substitution for the neck (A182-K-loop-D218-S583); core motor plus poly-lysine (5K) in substitution for the neck (A182–5K-D218-S583); and core motor domain of MCAK (I253-S583; Fig. 5 A). The purified proteins were added to the taxol-stabilized MTs and were incubated at 24 ± 1°C for 10 min in the presence of 1 mM ATP. The depolymerization of tubulin polymer was measured as the percentage of tubulin released in the supernatant after subtraction of tubulin in supernatant of the no-motor control reaction and normalizing the supernatant and pellet fractions. Fig. 5 B shows the supernatants and pellets of the in vitro depolymerization reactions containing 100 nM active motor proteins and 1,500 nM taxol MTs. Although the core motor plus neck (A182-S583) depolymerizes 92 ± 4% of tubulin polymer, the core motor plus alanine-substituted neck, A182-Ala-S583 depolymerized only 10 ± 4% of tubulin polymer (Fig. 5 B). The truncated neck plus core motor (D218-S583) lacks the positively charged helix found in the FL neck (Fig. 1 B). This protein depolymerized only 13 ± 5% of tubulin polymer. Therefore, removal of positive charges from the neck either by alanine substitutions or deletions dramatically suppresses the MT depolymerization activity of this kinesin, but does not eliminate it completely in vitro. Although analogous neckless MCAK mutants (Δ A182-D246, Δ A182-E232, and Δ A182-D218) expressed to high levels in vivo displayed no detectable depolymerization activity (Fig. 2 E, 3A), in corroboration with our in vitro data, extremely high levels of expression showed a slightly disassembled MT network (unpublished data).
Figure 5.
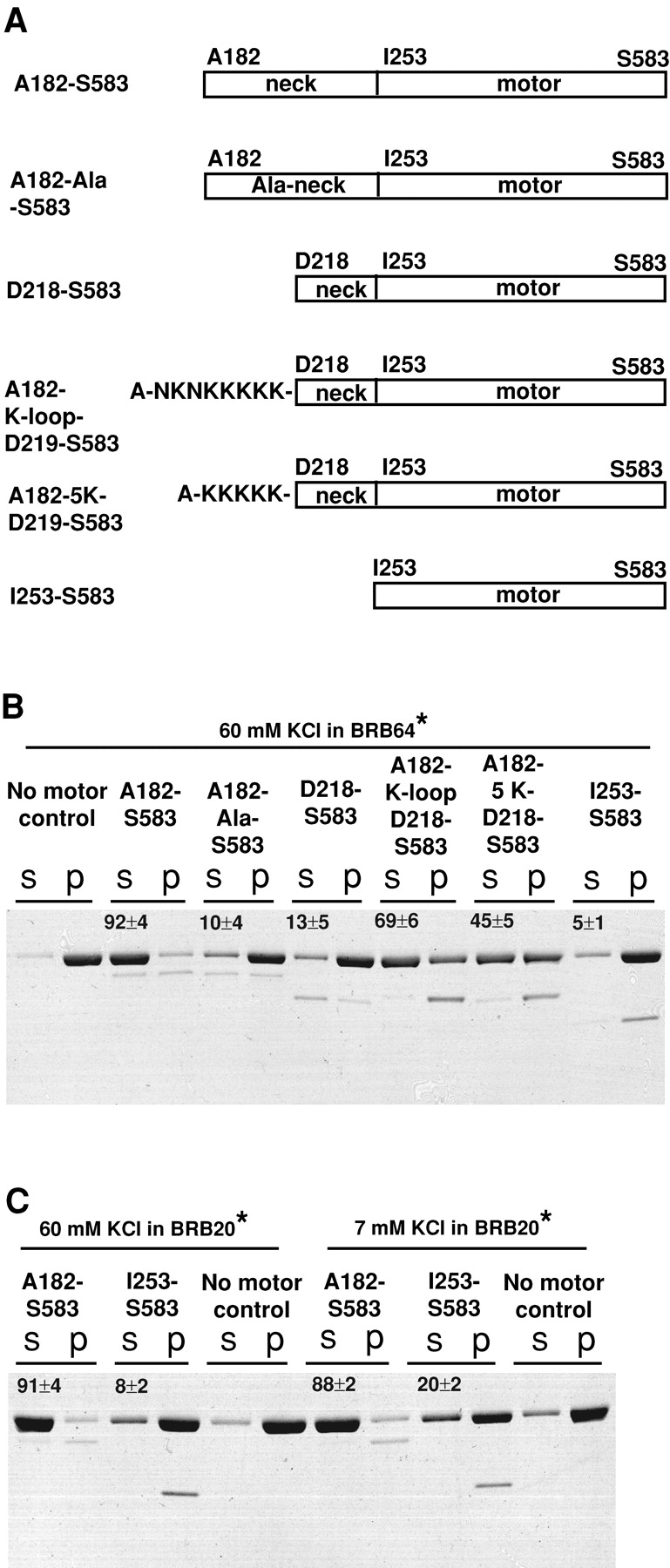
Depolymerization activity of MCAK mutants in vitro . (A) Diagram illustrating the truncated MCAK mutants used for in vitro depolymerization assays: core motor plus neck (A182-S583); core motor plus alanine-substituted neck (A182-Ala-S583 with R183, R184, K185, K198, R199, K202, R203, R210, K212, and R213 substituted to alanine); core motor plus truncated neck (D218-S583); core motor plus K-loop (NKNKKKKK) in substitution for the neck (A182-K-loop-D218-S583); core motor plus poly-l-lysine (KKKKK) in substitution for the neck (A182–5K-D218-S583); and core motor (I253-S583). B compares the depolymerization activity of the truncated MCAK mutants described above. C compares the depolymerization activity of the core motor plus neck (A182-S583) and the core motor domain of MCAK (I253-S583) in 60 mM KCl, BRB20 versus 7 mM KCl, BRB20. For both B and C, the percentage of tubulin in the supernatant of no-motor control reactions was subtracted from the percentage of tubulin in supernatant of the motor-containing reactions. The numbers are percentages of tubulin released in supernatant of the motor-containing reactions. The average of at least three experiments ± SD is shown for each supernatant fraction. In each case, the top band on the gel is tubulin and the bottom band is the added motor. In all assays, 100 nM of active motor protein was added to 1,500 nM taxol-stabilized MTs in the presence of 1 mM ATP. *Refer to Materials and methods for exact buffer composition.
Also, we substituted the positively charged helix found in the A182-D218 region of CgMCAK neck (Fig. 1 B) with the positively charged K-loop of KIF1A by linking the K-loop to the NH2 terminus of truncated neck plus core motor construct, D218-S583. This construct exhibited fivefold-increased depolymerization activity over D218-S583 (Fig. 5 B). However, although the K-loop rescues the depolymerization activity of the neckless mutant 100% in vivo (Fig. 4), it rescues only 75% of the depolymerization activity of D218-A182 construct in vitro (Fig. 5 B). Therefore, the MCAK neck plus motor (A182-S583) is still the most optimal construct for MT depolymerization, which might be one of the reasons why it is so highly conserved. It is likely that the K-loop rescues the depolymerization activity because it contains positively charged residues. We have further confirmed that charge is an important functional property of the neck by constructing the poly-lysine mutant, A182-KKKKK-D218-S583 (abbreviated as A182–5K-D218-S583), which contains poly-lysine peptide instead of K-loop in place of A182-D218 neck region. Fig. 5 B shows that the poly-lysine peptide substitution rescues the depolymerization activity of D218-S583, but to a lesser degree than the K-loop substitution, perhaps due to fewer positive charges found in the poly-lysine peptide (+5) than in K-loop (+6).
The rescue of the MT depolymerization–deficient neckless MCAK by K-loop insertion suggests that the MCAK neck might function analogously to the K-loop. The K-loop of KIF1A mediates diffusional motility (Okada and Hirokawa, 2000; Rogers et al., 2001). Similarly, we propose that the neck of MCAK may support diffusional motility to transport MCAK along the MT protofilament toward MT ends where Kin I–mediated MT depolymerization occurs (Desai et al., 1999). The neck of MCAK may also use diffusional motility to confer accelerated kinetics or processivity to the core motor domain (unpublished data).
Moores et al. (2002) have clearly shown that a conserved core motor domain of a Plasmodium falciparum Kin I kinesin (pKinI) can disassemble MTs. However, the assay conditions (extremely low ionic strength) were chosen to maximize electrostatic interactions. Maximizing the electrostatic interactions and raising the stoichiometry of Kin I/tubulin (≥1:10) obviates the need for processivity or diffusional motility despite the fact that it is essential for depolymerization under physiological conditions. We have duplicated the low ionic strength conditions used by Moores et al. (2002). The decrease in salt concentration from 60 to 7 mM KCl in BRB20 buffer produced a twofold increase of tubulin polymer depolymerized by the core motor (Fig. 5 C). This shows that the core motor of MCAK (I253-S583) is also a definite but enfeebled depolymerizer. Direct comparison of the neck plus motor (A182-S583) with the conserved core motor (I253-S583) shows that the neck plus motor is a tremendously more effective depolymerizer than the core motor (Fig. 5, B and C).
In summary, we demonstrated that deletions or alanine substitutions of positively charged residues of A182-D218 neck region drastically reduced the MT depolymerization activity. Insertion of the positively charged KIF1A K-loop or poly-lysine rescued the neckless MCAK mutants. We propose that the neck acts as an electrostatic tether. This electrostatic tether may increase the rate of MT depolymerization by either increasing the on-rate of MCAK to MT ends (perhaps by mediating diffusional motility) or decreasing the off-rate of MCAK from MTs (perhaps by conferring processivity to the core motor domain; unpublished data). The data presented here do not allow us to distinguish between diffusional targeting and processivity. Mediation of diffusional targeting and processivity are two distinct and not mutually exclusive hypotheses for the contribution of the neck to the MT depolymerization activity of MCAK.
Materials and methods
Constructs
pTRE2-EGFP-CgMCAK was made by the subcloning of NheI-BspEI EGFP cDNA fragment (from pEGFP-C1; CLONTECH Laboratories, Inc.) and the BspEI-NotI Cricetulus griseus MCAK cDNA fragment (from pOPRSVICAT-GFP-MCAK; Maney et al., 1998) into the pTRE2 vector (CLONTECH Laboratories, Inc.) digested with NheI and NotI. The gene expression level controlled by TRE promoter is similar to the high expression levels in the traditional CMV promoter–driven systems (Yin et al., 1996). The deletion, alanine substitution, and K-loop insertion constructs were prepared by overlap PCR using specially designed mutagenic primers and PfuTurbo® DNA polymerase (Stratagene). The mutations were confirmed by DNA sequencing. Constructs for bacterially expressed proteins were made by amplifying the coding sequences of MCAK mutants from the constructs in pTRE2 vector with PfuTurbo® DNA polymerase (Stratagene), and then subcloning the resulting fragments into pET-28a+ (Novagen) digested with Nco1 and Not1.
Cell transfection and immunofluorescence
CHO AA8 Tet-Off™ cells (CLONTECH Laboratories, Inc.) were grown in 90% αMEM, 10% FBS, and 100 μg/ml G418 at 37°C, 5% CO2. 1 d before transfection, cells were plated onto 12-mm coverslips at low density (2 × 104 cells/cm2). Transfections were done with ExGen 500 transfection reagent (MBI Fermentas). Cells were cultured for 24 h after transfection and fixed with 1% PFA in precooled methanol for 10 min. The cells were then incubated with mouse anti–tubulin DM1α (Sigma-Aldrich) at 1:50 dilution for 1 h and Texas red anti–mouse antibodies (Jackson ImmunoResearch Laboratories) at 1:100 dilution in PBS plus 0.1% Triton X-100 and 1% BSA for 1 h. Cells were washed with PBS, stained with DAPI, and mounted in Vectashield® mounting medium (Vector Laboratories). Analysis was done with a microscope (model FX-A; Nikon) equipped with 60×/1.4 NA Plan Apo oil objective. The digital images were acquired with a cooled CCD camera (SenSys; Photometrics) controlled by QED camera software (QED Imaging, Inc.).
Quantitation of MT depolymerization activity in vivo
To quantify the microtubule (MT) depolymerization activity, we collected digital images of the transfected cells at the same exposure for each set of GFP and MT images and below saturation level of the camera (the 4,096 gray-value maximum). All images were saved as 12-bit TIFF images for quantification by NIH Image 1.62 software and Microsoft Excel. Mean EGFP or tubulin fluorescence intensities over the entire cellular area were measured as the average gray value within the area. Mean fluorescence over the cell-free area in each image was subtracted from mean fluorescence over the cellular area to correct for background fluorescence. All analyzed cells were in interphase and were free of aggregates of overexpressed protein. 20–90 transfected cells were measured for each construct with >500 cells quantified overall. To quantify the MT polymer left in the transfected cells, we assigned an MT polymer value of 0% to the cells transfected with full-length (FL) MCAK, which displayed no MTs and a mean tubulin fluorescence intensity equal to 734 gray-scale units. Similar to nontransfected cells, EGFP-transfected cells showed normal levels of MT polymer, which was given a 100% value. The mean tubulin fluorescence of EGFP-transfected cells was equal to 1,902 gray-scale units. To quantify the percentage of MT polymer left in the cells transfected with mutant MCAK constructs, we used the following formula: {[(the mean tubulin fluorescence intensity of mutant-transfected cells – 734)/1,168] × 100%} where 1168 is the difference between EGFP control and FL MCAK mean gray values.
In vitro MT depolymerization assays
The bacterially expressed COOH-terminally 6his-tagged proteins were purified as described in Maney et al. (2001). The active motor concentration was determined as the concentration of nucleotide-binding sites because there is one ATP-binding site per each monomeric motor protein analyzed in this study in vitro. The concentration of nucleotide-binding sites was measured radiometrically as described by Coy et al. (1999). The depolymerization of taxol-stabilized MTs, polymerized from bovine tubulin (Cytoskeleton, Inc.), was performed as described in Maney et al. (2001). In brief, for the assay shown in Fig. 5 A, 100 nM of the active motor proteins in 20 μl of the elution buffer (250 mM imidazole, pH 7.0, 300 mM KCl, 0.2 mM MgCl2, 0.01 mM Mg-ATP, 1 mM DTT, and 20% glycerol) was mixed with 1,500 nM taxol-stabilized MTs in 80 μl of BRB80 (80 mM Pipes, pH 6.8, 1 mM EGTA, and 1 mM MgCl2), 12.5 μM taxol, 1 mM DTT, and 1.25 mM Mg-ATP incubated at 24 ± 1°C for 10 min, and then centrifuged at 30 psi for 10 min. For the assay shown in Fig. 5 B, 100 nM of the active motor proteins in 2.3 μl of the elution buffer was mixed with 1,500 nM taxol-stabilized MTs in 97.7 μl of BRB20 (20 mM Pipes, pH 6.8, 1 mM EGTA, and 1mM MgCl2), 10.2 μM taxol, 1 mM DTT, 10 μg/ml BSA, and 1.02 mM Mg-ATP with or without addition of KCl up to 60 mM, incubated at 24 ± 1°C for 10 min, and then centrifuged at 30 psi for 10 min. Supernatants and pellets were assayed for the presence of tubulin on Coomassie-stained SDS-polyacrylamide gels (Novex). Gels were calibrated and quantified using NIH Image 1.62 software. The percentage of tubulin in the supernatant of no-motor control reactions was subtracted from the percentage of tubulin in supernatant of the motor-containing reactions. The percentages of tubulin released in supernatant of the motor-containing reactions were then normalized with the percentages of tubulin in the corresponding pellet fractions.
Acknowledgments
We thank Andy Hunter, Ayana Moore, and Kathleen Rankin for many discussions and comments on the manuscript. We also thank Ayana Moore (University of Washington School of Medicine, Seattle, WA) for the pTRE2-GreenLantern-CgMCAK plasmid.
This work was supported by the National Institutes of Health grant (GM53654A) and the Department of Defense grant (DAMD17-01-1-0450) to L. Wordeman.
Footnotes
Abbreviations used in this paper: FL, full-length; MCAK, mitotic centromere–associated kinesin; MT, microtubule.
References
- Baldi, P., S. Brunak, P. Frasconi, G. Soda, and G. Pollastri. 1999. Exploiting the past and the future in protein secondary structure prediction. Bioinformatics. 15:937–946. [DOI] [PubMed] [Google Scholar]
- Case, R.B., D.W. Pierce, N. Hom-Booher, C.L. Hart, and R.D. Vale. 1997. The directional preference of kinesin motors is specified by an element outside of the motor catalytic domain. Cell. 90:959–966. [DOI] [PubMed] [Google Scholar]
- Cleveland, D.W., M.A. Lopata, P. Sherline, and M.W. Kirschner. 1981. Unpolymerized tubulin modulates the level of tubulin mRNAs. Cell. 25:537–546. [DOI] [PubMed] [Google Scholar]
- Coy, D.L., M. Wagenbach, and J. Howard. 1999. Kinesin takes one 8-nm step for each ATP that it hydrolyzes. J. Biol. Chem. 274:3667–3671. [DOI] [PubMed] [Google Scholar]
- Desai, A., S. Verma, T.J. Mitchison, and C.E. Walczak. 1999. Kin I kinesins are microtubule-destabilizing enzymes. Cell. 96:69–78. [DOI] [PubMed] [Google Scholar]
- Endow, S.A., and K.W. Waligora. 1998. Determinants of kinesin motor polarity. Science. 281:1200–1202. [DOI] [PubMed] [Google Scholar]
- Goldstein, L.S., and A.V. Philp. 1999. The road less traveled: emerging principles of kinesin motor utilization. Annu. Rev. Cell Dev. Biol. 15:141–183. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Garay, M.L., and F. Cabral. 1996. alpha-Tubulin limits its own synthesis: evidence for a mechanism involving translational repression. J. Cell Biol. 135:1525–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henningsen, U., and M. Schliwa. 1997. Reversal in the direction of movement of a molecular motor. Nature. 389:93–96. [DOI] [PubMed] [Google Scholar]
- Hunter, A.W., and L. Wordeman. 2000. How motor proteins influence microtubule polymerization dynamics. J. Cell Sci. 113:4379–4389. [DOI] [PubMed] [Google Scholar]
- Kinoshita, K., I. Arnal, A. Desai, D.N. Drechsel, and A.A. Hyman. 2001. Reconstitution of physiological microtubule dynamics using purified components. Science. 294:1340–1343. [DOI] [PubMed] [Google Scholar]
- Lakshmi, M.S., C. Parker, and G.V. Sherbet. 1993. Metastasis associated MTS1 and NM23 genes affect tubulin polymerisation in B16 melanomas: a possible mechanism of their regulation of metastatic behaviour of tumours. Anticancer Res. 13:299–303. [PubMed] [Google Scholar]
- Maney, T., A.W. Hunter, M. Wagenbach, and L. Wordeman. 1998. Mitotic centromere-associated kinesin is important for anaphase chromosome segregation. J. Cell Biol. 142:787–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maney, T., M. Wagenbach, and L. Wordeman. 2001. Molecular dissection of the microtubule depolymerizing activity of mitotic centromere-associated kinesin. J. Biol. Chem. 276:34753–34758. [DOI] [PubMed] [Google Scholar]
- Moores, C.A., M. Yu, J. Guo, C. Beraud, R. Sakowicz, and R.A. Milligan. 2002. A mechanism for microtubule depolymerization by KinI kinesins. Mol. Cell. 9:903–909. [DOI] [PubMed] [Google Scholar]
- Niederstrasser, H., H. Salehi-Had, E.C. Gan, C. Walczak, and E. Nogales. 2002. XKCM1 acts on a single protofilament and requires the C terminus of tubulin. J. Mol. Biol. 316:817–828. [DOI] [PubMed] [Google Scholar]
- Okada, Y., and N. Hirokawa. 2000. Mechanism of the single-headed processivity: diffusional anchoring between the K-loop of kinesin and the C terminus of tubulin. Proc. Natl. Acad. Sci. USA. 97:640–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers, K.R., S. Weiss, I. Crevel, P.J. Brophy, M. Geeves, and R. Cross. 2001. KIF1D is a fast non-processive kinesin that demonstrates novel K-loop-dependent mechanochemistry. EMBO J. 20:5101–5113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romberg, L., D.W. Pierce, and R.D. Vale. 1998. Role of the kinesin neck region in processive microtubule-based motility. J. Cell Biol. 140:1407–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorn, K.S., J.A. Ubersax, and R.D. Vale. 2000. Engineering the processive run length of the kinesin motor. J. Cell Biol. 151:1093–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vale, R.D., and R.J. Fletterick. 1997. The design plan of kinesin motors. Annu. Rev. Cell Dev. Biol. 13:745–777. [DOI] [PubMed] [Google Scholar]
- Walczak, C.E., T.J. Mitchison, and A. Desai. 1996. XKCM1: a Xenopus kinesin-related protein that regulates microtubule dynamics during mitotic spindle assembly. Cell. 84:37–47. [DOI] [PubMed] [Google Scholar]
- Woehlke, G., and M. Schliwa. 2000. Walking on two heads: the many talents of kinesin. Nat. Rev. Mol. Cell Biol. 1:50–58. [DOI] [PubMed] [Google Scholar]
- Wordeman, L., and T.J. Mitchison. 1995. Identification and partial characterization of mitotic centromere-associated kinesin, a kinesin-related protein that associates with centromeres during mitosis. J. Cell Biol. 128:95–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin, D.X., L. Zhu, and R.T. Schimke. 1996. Tetracycline-controlled gene expression system achieves high-level and quantitative control of gene expression. Anal. Biochem. 235:195–201. [DOI] [PubMed] [Google Scholar]
- Zhai, Y., P.J. Kronebusch, P.M. Simon, and G.G. Borisy. 1996. Microtubule dynamics at the G2/M transition: abrupt breakdown of cytoplasmic microtubules at nuclear envelope breakdown and implications for spindle morphogenesis. J. Cell Biol. 135:201–214. [DOI] [PMC free article] [PubMed] [Google Scholar]