

Abstract
In mammalian cells, the Golgi apparatus undergoes extensive fragmentation during apoptosis. p115 is a key vesicle tethering protein required for maintaining the structural organization of the Golgi apparatus. Here, we demonstrate that p115 was cleaved during apoptosis by caspases 3 and 8. Compared with control cells expressing native p115, those expressing a cleavage-resistant form of p115 delayed Golgi fragmentation during apoptosis. Expression of cDNAs encoding full-length or an NH2-terminal caspase cleavage fragment of p115 had no effect on Golgi morphology. In contrast, expression of the COOH-terminal caspase cleavage product of p115 itself caused Golgi fragmentation. Furthermore, this fragment translocated to the nucleus and its expression was sufficient to induce apoptosis. Most significantly, in vivo expression of the COOH-terminal fragment in the presence of caspase inhibitors, or upon coexpression with a cleavage-resistant mutant of p115, showed that p115 degradation plays a key role in amplifying the apoptotic response independently of Golgi fragmentation.
Keywords: Golgi apparatus; vesicular transport factor p115; apoptosis; caspases; Golgi matrix protein
Introduction
In most eukaryotic cells, the Golgi apparatus exists as a series of flattened membrane stacks arranged in a perinuclear lace-like reticulum. Recently, several proteins that maintain the characteristic morphology of the Golgi apparatus during interphase have been characterized (Linstedt and Hauri, 1993; Barroso et al., 1995; Nakamura et al., 1995; Barr et al., 1997; Lowe et al., 2000; Weide et al., 2001). A tightly membrane-associated peripheral Golgi protein, GM130, was shown to bind p115/transcytosis-associated protein, a vesicle tethering protein localized to the Golgi apparatus and vesicular tubular clusters. In turn, p115 interacts with a high molecular weight vesicle-associated protein, designated giantin (Nakamura et al., 1997; Lesa et al., 2000; Moyer et al., 2001). According to one model, by linking GM130 and giantin, p115 tethers vesicles to the Golgi apparatus to facilitate membrane fusion and allow maintenance of the Golgi architecture (Seemann et al., 2000). Considerable evidence has accumulated demonstrating that GM130 is phosphorylated during mitosis (Lowe et al., 1998b, 2000; Garcia-Mata et al., 2000). Phosphorylated GM130 can no longer interact with p115, thereby abolishing Golgi vesicle tethering and fusion (Nakamura et al., 1997). It has been proposed that continual budding of vesicles in the absence of fusion leads to mitotic fragmentation of the Golgi apparatus (Lowe et al., 1998a). Although other models for Golgi apparatus disassembly have been proposed (Zaal et al., 1999), very recent evidence (Puthenveedu and Linstedt, 2001) suggests that p115, but not giantin and GM130, is essential for maintaining the architecture of the Golgi apparatus.
Various agents induce fragmentation of the Golgi apparatus, including those causing apoptosis (Takizawa et al., 1993; Monier et al., 1998; de Figueiredo et al., 1999; Sesso et al., 1999; Siddhanta et al., 2000). Apoptosis is an organized form of cell death characterized by cell shrinkage, nuclear condensation, and formation of apoptotic bodies (Strasser et al., 2000). Many of these changes result from cleavage of organelle proteins by caspases, a family of cysteine proteases activated during the apoptotic response, all of which have an absolute requirement for cleavage after an aspartic acid residue (Thornberry et al., 1997). Among the organelles affected during apoptosis is the Golgi apparatus, which fragments into small vesiculo-tubular elements (Sesso et al., 1999). Golgin-160, a member of a family of high molecular weight Golgi-associated coiled-coil proteins implicated in maintaining the Golgi structure, was shown to be cleaved by caspases 2, 3, and 7 during apoptosis (Mancini et al., 2000). Most recently, GRASP65, a protein involved in the stacking of Golgi cisternae, was also shown to be cleaved by caspase-3 during the apoptotic response (Lane et al., 2002). Expression of a golgin-160 mutant lacking the caspase-2 cleavage site or a GRASP65 construct mutated at three caspase-3 sites delayed, but did not inhibit, apoptotic Golgi fragmentation (Mancini et al., 2000; Lane et al., 2002), suggesting that multiple factors regulating Golgi structure are affected during apoptosis.
The morphology of apoptotic and mitotic Golgi fragments is similar (Sesso et al., 1999); it is therefore possible that they are generated by similar mechanisms. To investigate this possibility, we have analyzed the role of GM130 and p115 during apoptotic Golgi fragmentation. Here we show that in contrast to mitosis, no change in GM130 phosphorylation was detected in apoptotic cells. Instead, the level of GM130 decreased significantly and p115 underwent selective proteolytic cleavage via caspases 3 and 8. A stable cell line expressing a cleavage-resistant form of p115 delayed Golgi fragmentation during apoptosis. Furthermore, expression of a region of p115 corresponding to a COOH-terminal apoptotic cleavage fragment was sufficient to disrupt the structure of the Golgi apparatus. Strikingly, this fragment also translocated into the nucleus and activated the apoptotic program. Our data suggest that caspase-mediated proteolysis of key vesicle tethering factors contributes to Golgi breakdown during apoptosis and may act to propagate downstream apoptotic signals.
Results
Fragmentation of the Golgi apparatus during apoptosis is independent of GM130 phosphorylation
Phosphorylation of GM130 at serine 25 has been shown to be a key step in regulating the structure of the Golgi apparatus during mitosis (Lowe et al., 1998b). We therefore tested whether the mechanisms of mitotic and apoptotic Golgi fragmentation might be similar. Given that staurosporine, a general protein kinase inhibitor, induces apoptosis and Golgi fragmentation (Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200208013/DC1), it was unlikely that GM130 would be phosphorylated. However, to eliminate this possibility, an antibody (PS25) that is highly specific for the phosphorylated form of GM130 was employed for immunofluorescence microscopy. As previously demonstrated (Lowe et al., 2000), PS25 immunostaining was present in mitotic Golgi fragments, but not in the Golgi apparatus of interphase cells. In sharp contrast to mitotic cells, the PS25 antibody failed to stain apoptotic cells treated with either staurosporine or etoposide (Fig. 1) . Identical results were obtained when the PS25 antibody was used for Western blot analysis (unpublished data).
Figure 1.
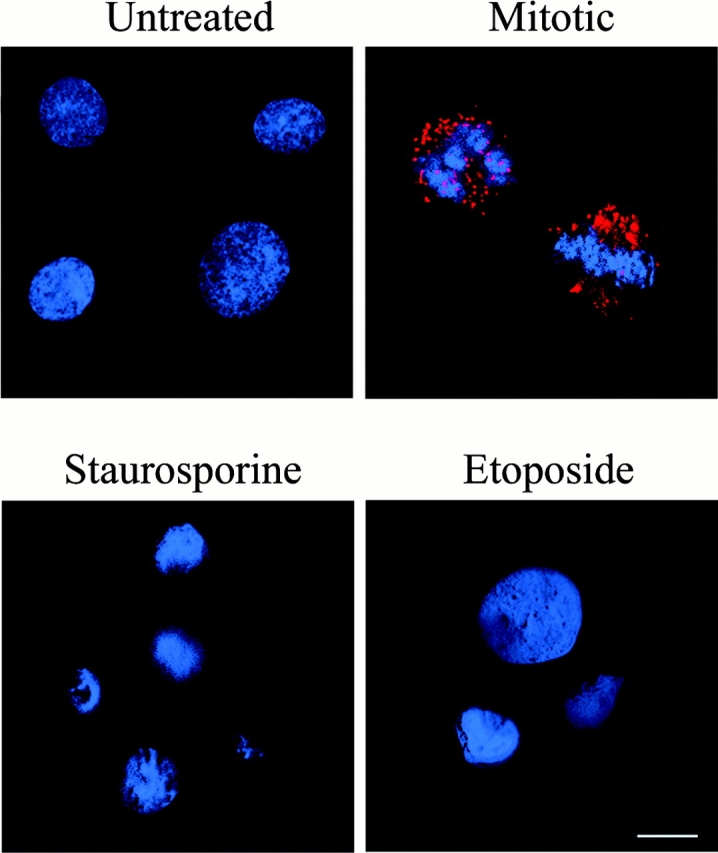
Fragmentation of the Golgi apparatus during apoptosis is independent of GM130 phosphorylation. Apoptotic NRK cells were induced with staurosporine for 12 h or etoposide for 24 h. The cells were then processed for immunofluorescence microscopy and examined using rabbit anti-PS25 antibodies specific for the phosphorylated form of GM130 (red) (Lowe et al., 2000). Nuclei were stained with Hoechst 33238 (blue). As a positive control, NRK cells were synchronized at mitosis using aphidicolin (Lowe et al., 2000). Note the appearance of phosphorylated GM130 only in mitotic cells. Bar, 10 μm.
p115 and GM130 levels decrease during apoptosis
The above result did not exclude the possibility that Golgi fragmentation involved alternative modifications other than phosphorylation of vesicle tethering factors. Because many proteins involved in the maintenance of cellular structure are cleaved by caspases during apoptosis, we reasoned that GM130 and p115 might undergo proteolysis during apoptosis. If this generated nonfunctional fragments of these factors, it could result in the Golgi breakdown seen in apoptosis. To test this idea, apoptosis was induced in COS7 cells and, at various times, total cell extracts were analyzed by Western blots using antibodies to GM130 and p115 (Fig. 2 A). A significant decrease in both GM130 and p115 levels was observed during the time course. Importantly, a polypeptide of ∼90,000 D (90 kD) that cross-reacted with the p115 antibody was evident in apoptotic cell lysates (Fig. 2 A). This 90-kD polypeptide was also recognized by three independent p115 monoclonal antibodies in lysates from apoptotic COS7 cells (unpublished data). Moreover, an antibody that specifically recognizes the COOH terminus of p115 revealed a polypeptide of ∼30,000 D (30 kD) in apoptotic cell lysates (Fig. 2 A). The time-dependent appearance of these polypeptides corresponded to decreases in the level of full-length p115 during apoptosis (Fig. 2 B), suggesting a precursor–product relationship. Significantly, even though staurosporine and etoposide induce apoptosis by different modes of action, identical p115-immunoreactive polypeptides were generated. The appearance of these polypeptides also coincided with the cleavage of poly(ADP-ribose) polymerase (PARP)* (Fig. 2 A), a classic marker of apoptosis. In contrast, the level of the Golgi SNARE protein Vti1a remained constant throughout the entire apoptosis time course (Fig. 2 A). The above results suggested that the p115-immunoreactive polypeptides (90 and 30 kD) represent apoptotic cleavage fragments of p115. However, it might be argued that the proteolysis of p115 was a consequence of nonphysiological effects induced by staurosporine and etoposide. To exclude this possibility, apoptosis was induced in HeLa cells by activation of the Fas receptor using agonistic anti-Fas antibodies (Fig. 2 C). In agreement with the above data, ligation of the Fas receptor resulted in p115 cleavage and a significant decrease in its level (Fig. 2 C, lanes 2 and 3), indicating that the foregoing results are indeed relevant physiologically.
Figure 2.




The levels of GM130 and p115 decrease during apoptosis. p115 is cleaved by caspases into two fragments. (A) COS7 cells were incubated with staurosporine or etoposide. At the indicated times, cell lysates were prepared for Western blotting using antibodies to GM130, p115, PARP, or the Golgi SNARE, Vtila. p115 antibodies revealed two polypeptides of 90 and 30 kD in apoptotic lysates. (B) Quantitation of GM130 and p115 breakdown and the appearance of the 90- and 30-kD polypeptides in staurosporine- and etoposide-treated cells. Note that the appearance of the 90- and 30-kD polypeptides coincided with the decrease in full-length p115, suggesting a precursor product relationship. (C) HeLa cells were treated with activating anti-Fas antibodies and cycloheximide (C.+F.). Cell lysates were prepared 24 or 48 h after treatment and analyzed by Western blotting using an antibody to p115. Unlike control samples treated with cycloheximide alone, the p115 levels of cells incubated with both anti-Fas antibodies and cycloheximide were decreased. (D) Lysates were prepared from COS7 cells treated with staurosporine or etoposide for the indicated times in the presence of z-VAD-fmk or z-YVAD-fmk. Lysates were analyzed by Western blotting using an antibody to p115. Generation of the 90-kD polypeptide in apoptotic cell lysates was inhibited in the presence of the apoptotic caspase inhibitor z-VAD-fmk.
p115 is cleaved by caspases 3 and 8 during apoptosis
To determine if the p115-immunoreactive polypeptides resulted from the cleavage activity of caspases, COS7 cells were preincubated with caspase inhibitors before apoptosis induction. At the indicated times (Fig. 2 D), cell lysates were analyzed by Western blotting using anti-p115 antibodies. In cells preincubated with the general caspase inhibitor z-VAD-fmk, the formation of the 90- (Fig. 2 D) and 30-kD (unpublished data) p115 polypeptides was completely abrogated. In contrast, the polypeptides were clearly visible in control samples preincubated with DMSO alone or with z-YVAD-fmk (Fig. 2 D). z-YVAD-fmk has weak inhibitory effects on the apoptotic caspases but it is a potent inhibitor of caspases involved in the immune system (caspases 1 and 4). These results strongly suggest that the 90- and 30-kD polypeptides are caspase cleavage fragments of p115 generated during apoptosis.
To confirm that p115 was a direct target of apoptotic caspases, mRNA encoding full-length human p115 was translated in vitro and the products were incubated with various purified caspases (Fig. 3 A). Treatment with caspase-3 generated p115 fragments of 90, 75, 50, and 30 kD, whereas incubation with caspase-8 yielded only the 90- and 30-kD fragments. In contrast, caspases 2 and 7 did not cleave p115 in vitro, even after prolonged incubation. Similarly, caspases 3 and 8, but not caspases 2 and 7, cleaved in vitro–translated bovine p115 to generate the same proteolytic fragments as human p115 (Fig. 3 B). Note that the 90- and 30-kD fragments reached maximum levels after only 10 min of incubation, whereas the 75- and 50-kD polypeptides were slowly generated over a period of 90–120 min (Fig. 3 C). The 90- and 30-kD fragments generated in vitro corresponded in size to the p115-derived polypeptides present in apoptotic COS7 cell lysates (Fig. 2 A). Together, these data suggest that in vivo, the 90- and 30-kD fragments are derived from the main caspase cleavage site in p115 (see Discussion).
Figure 3.

p115 is cleaved by caspases-3 in vitro and in vivo at residue TEKD 757 to generate the 90- and 30-kD fragments. (A) In vitro–translated human p115 mRNA was incubated with various caspases for 1 h at 37°C. p115 was cleaved by purified caspase-3 into four fragments of 90, 75, 50, and 30 kD. Caspase-8 generated only the 90- and 30-kD fragments. (B) In vitro–translated bovine p115 mRNA was incubated with the same caspases as human p115. Numbers at the top indicate the concentration (nM) of caspase in each reaction. (C) In vitro–translated human p115 mRNA was incubated with caspase-3 for the indicated times (min). For wild-type p115 (left), the 90- and 30-kD fragments were rapidly generated after 10 min of incubation. In contrast, the 75- and 50-kD fragments required prolonged incubation before reaching a plateau. For the p115 TEKD757Ala mutant (right), the appearance of the 90- and 30-kD fragments was completely abrogated, whereas the 75- and 50-kD fragments were unaffected. Asterisks indicate an incomplete translation product of p115. (D) COS7 cells were transfected with NH2-terminal FLAG constructs of wild-type p115 or the p115 TEKD757Ala mutant. 24 h after transfection, apoptosis was induced with staurosporine or etoposide for the indicated times. Lysates were prepared and analyzed by Western blotting using polyclonal FLAG antibodies. Wild-type p115, unlike the p115 TEKD757Ala mutant, was cleaved to generate the 90-kD fragment. (E) MCF-7 cells were treated with staurosporine, etoposide, cycloheximide (C.), or agonistic anti-Fas antibodies plus cycloheximide (C.+F.) for the indicated times. Cell lysates were prepared and Western blot analysis was performed using antibodies to p115 and BID.
The 90- and 30-kD fragments are generated by cleavage of p115 at position TEKD757
We used site-directed mutagenesis to identify caspase cleavage sites on p115. Based on our in vivo and in vitro data, the 90- and 30-kD fragments were likely to be products of the most prominent p115 caspase cleavage site. Western blot analysis of apoptotic cells transfected with an NH2-terminal FLAG-tagged p115 construct indicated that the 90-kD fragment contained the NH2 terminus (Fig. 3 D, lanes 1–3). Therefore, we analyzed the human p115 sequence for potential caspase cleavage sites toward the COOH terminus that would result in an NH2-terminal fragment of ∼90 kD. Three such sites, SEED635, REQD663, and TEKD757, were identified and their aspartic acid residues mutated separately to alanine. The mRNA encoding each alanine mutant was then translated in vitro and the products incubated with caspase-3. Two of the mutants, SEED635Ala and REQD663Ala, generated proteolytic fragments identical to those of native p115, suggesting that these sites were not targets of caspase-3 cleavage (unpublished data). Significantly, caspase-3 failed to release the 90- and 30-kD fragments in the third p115 mutant, TEKD757Ala (Fig. 3 C). This mutant also failed to generate these fragments when incubated with caspase-8 (unpublished data). In addition, production of the 75- and 50-kD fragments was not affected in this mutant. To confirm that p115 was also cleaved at position TEKD757 in vivo, COS7 cells were transiently transfected with either the wild type or the TEKD757Ala mutant, both of which were FLAG tagged at the NH2 terminus. Apoptosis was then induced using staurosporine or etoposide (Fig. 3 D). In contrast to wild-type p115 (lanes 1–3), Western blot analysis of the mutant p115 using anti-FLAG antibodies showed that it was not cleaved in vivo (lanes 4–6). Together these in vitro and in vivo data indicate that the TEKD757 cleavage site was responsible for generating the 90- and 30-kD cleavage fragments of p115.
To further elucidate the role of caspase-3 in mediating p115 cleavage, we used MCF-7 cells, which are devoid of this enzyme (Janicke, et al., 1998) (Fig. 3 E). As expected, these cells were quite resistant to apoptosis because of the lack of caspase-3. The levels of p115 in apoptotic MCF-7 cell lysates were compared with those of BID, an early apoptotic signaling molecule cleaved by caspase-8. Although BID cleavage is detected early in cells treated with agonistic anti-Fas antibodies, its cleavage occurred only after prolonged incubation in cells treated with staurosporine and etoposide. The kinetics of p115 and BID cleavage (Fig. 3 E) were similar in staurosporine- and etoposide-treated cells. However, BID cleavage was more efficient than p115 in MCF-7 cells stimulated by Fas receptor ligation. These data suggest that in addition to caspase-3, other caspases may be involved in p115 cleavage in vivo.
Cleavage of p115 contributes to Golgi fragmentation during apoptosis
Given the importance of p115 in maintaining normal Golgi structure (Shorter and Warren, 1999) and in membrane transport to and within the Golgi apparatus (Alvarez et al., 1999; Seemann et al., 2000), we reasoned that its cleavage was at least partly responsible for the fragmentation seen during apoptosis. If this were the case, expression of a cleavage-resistant form of p115 would be expected to delay fragmentation of the Golgi apparatus. To test this idea, stable COS7 cell lines expressing low levels of wild-type p115 or the TEKD757Ala mutant were generated and treated with etoposide for 24 h. After treatment, cells were stained with an antibody to GM130 and the number of cells displaying a fragmented Golgi staining pattern was quantified (Fig. 4 ; Table I). The Golgi apparatus of untreated COS7 cells exhibited a perinuclear localization where staining was tight and reticular (Fig. 4 A, left). After etoposide treatment for 24 h, cells displaying two distinct stages of Golgi fragmentation were clearly visible. In stage one, Golgi fragmentation was at an intermediate phase, where the Golgi staining is loose but maintains a reticular and perinuclear distribution (Fig. 4 A, middle). In stage two, no perinuclear staining was evident and Golgi fragments appeared as punctate structures dispersed throughout the cell (Fig. 4 A, right). When compared with control cells expressing wild-type p115, cells expressing the TEKD757Ala mutant had a modest effect in delaying the progression from normal to stage one of Golgi fragmentation (Fig. 4 B; Table I). In contrast, the transition from stage one to stage two of Golgi fragmentation was significantly delayed by more than twofold (Fig. 4 B; Table I). These results strongly support our hypothesis that caspase-3 cleavage of p115 is a key step in the fragmentation of the Golgi apparatus during apoptosis.
Figure 4.

Classification of Golgi morphology during apoptosis. (A) Untreated COS7 cells (left) were fixed and stained with a monoclonal antibody against GM130 (green). Nuclei were stained with Hoechst 33238 (blue). Note the tight perinuclear Golgi staining of untreated cells. After etoposide treatment for 24 h, two populations of cells were distinguished. In one population (middle), the Golgi apparatus is at an early stage of fragmentation where staining is loose, but still reticular and perinuclear. In the second population, the Golgi apparatus is completely dispersed and the resulting membrane fragments stain as punctate structures (right). (B) COS7 cells transfected with either wild-type p115 or the p115 TEKD757Ala mutant were treated with etoposide for 24 h. Cells were then stained and processed as above. Expression of the p115 TEKD757Ala mutant delayed both stages of Golgi fragmentation compared with control cells expressing wild-type p115. Bar, 10 μm.
Table I. Percentage of transfected COS7 cells displaying various Golgi morphologies after etoposide treatment for 24 h.
| Transfected construct | Tight and reticular | Loose and reticular | Dispersed and punctate |
|---|---|---|---|
| % | % | % | |
| Wild-type p115 | 18.4 | 42.9 | 38.7 |
| TEKD757Ala p115 | 24.0 | 58.3 | 17.3 |
Results are the averages of duplicate samples from two separate experiments. In each case, 1,400 cells were counted by two independent observers. The expression levels of the wild-type and TEKD757Ala p115 were identical, as determined by Western blot analysis (unpublished data).
Expression of the COOH-terminal caspase cleavage fragment of p115 disrupts the Golgi apparatus and induces apoptosis
We reasoned that if p115 is required to maintain the normal Golgi structure, then expression of the p115 caspase cleavage fragments may have a dominant negative effect on the endogenous protein, resulting in Golgi fragmentation. To determine if cleavage at TEKD757 was by itself sufficient to disrupt the structural integrity of the Golgi apparatus, full-length p115 as well as two truncation mutants corresponding to the NH2- and COOH-terminal p115 caspase cleavage fragments (i.e., p1151–757 and p115758–962, respectively) were expressed as FLAG-tagged proteins in COS7 cells. At various times after transfection, the cells were examined by immunofluorescence microscopy (Fig. 5) . Transfected cells were identified by staining with a monoclonal antibody to the FLAG epitope, and the Golgi apparatus was monitored using a polyclonal antibody against TGN38. At 20 h after transfection, the FLAG staining for full-length p115 colocalized tightly with the Golgi apparatus. There was also a minor level of diffuse staining throughout the cytoplasm (Fig. 5). Similarly, the NH2-terminal p1151–757 construct had an intracellular localization identical to that of full-length p115, with the exception of a slightly more diffuse cytoplasmic staining; however the bulk of its staining colocalized with the Golgi apparatus (Fig. 5). This is consistent with previous observations showing that the COOH-terminal acidic domain is not required for the localization of p115 to the Golgi apparatus and vesiculo-tubular clusters (Nelson et al., 1998). Importantly, in cells expressing this mutant, the Golgi apparatus had a normal morphology at 20 h after transfection, as monitored by TGN38 staining (Fig. 5; Table II). Identical results were obtained when giantin was used as the Golgi marker (unpublished data). This indicates that either p115 is not required for maintenance of Golgi structure or the p1151–757 mutant failed to act as a dominant negative and therefore did not disrupt the normal function of endogenous p115. In marked contrast to the expression of full-length p115 or the p1151–757 mutant, nearly every cell expressing the COOH-terminal p115758–962 mutant displayed a fragmented Golgi staining pattern (Fig. 5; Table II). This result strongly suggests that the p115758–962 mutant competed with endogenous p115 for binding to critical factors required for maintaining the normal Golgi architecture. Strikingly, most cells (∼78%) expressing the p115758–962 construct were apoptotic, as determined by chromatin condensation and cell shrinkage (Table II). In contrast, expression of neither native p115 nor p1151–757 induced apoptosis (Fig. 5; Table II). Quite unexpectedly, in addition to its dispersed cytoplasmic distribution, p115758–962 was highly enriched in apoptotic cell nuclei, where its staining colocalized perfectly with local DNA densities. Examination of >250 cells revealed that 76% of all p115758–962-transfected cells had significant levels of the protein in the nucleus. In sharp contrast, no nuclear localization of the expressed protein was present in any of the cells transfected with either the full-length p115 or the p1151–757 construct at all time points examined.
Figure 5.
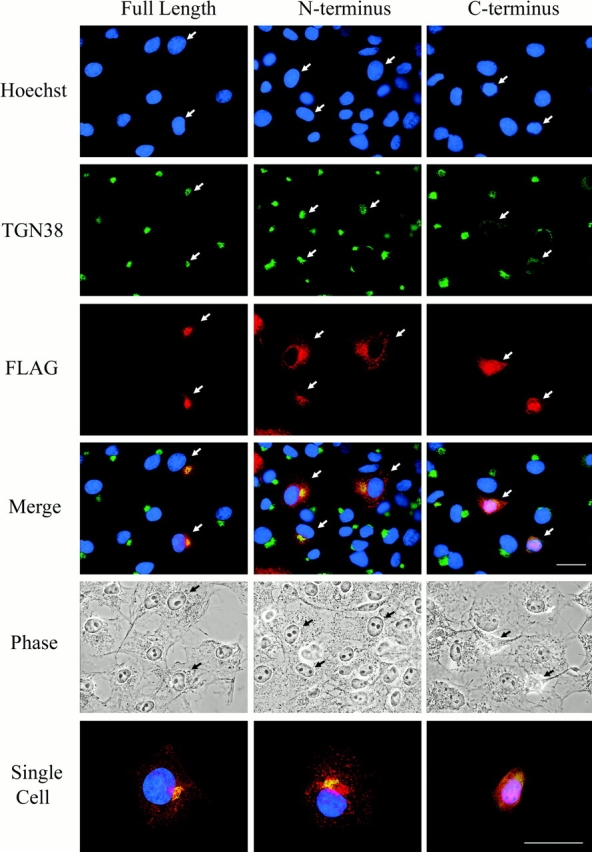
The COOH-terminal cleavage fragment of p115 induces Golgi fragmentation and apoptosis. COS7 cells were transfected with FLAG-tagged full-length p115, NH2-terminal fragment (p1151–757), or COOH-terminal fragment (p115758–962). Cells were fixed 20 h after transfection and stained with a TGN38 (green) antibody, a FLAG (red) antibody, and Hoechst 33238 (blue). Full-length p115 and p1151–757 localized predominantly to the Golgi region and their expression did not affect Golgi morphology or cell viability in transfected cells. In contrast, p115758–962 staining was diffuse in the cytoplasm and concentrated in the nucleus. Most significantly, expression of p115758–962 induced Golgi fragmentation and apoptosis, as indicated by TGN38 and Hoechst staining, respectively. The “single cell” panels show enlarged merged images of a representative cell transfected with each construct. Arrows indicate transfected cells. Bars, 10 μm.
Table II. Fragmentation of the Golgi apparatus and apoptosis in COS7 cells transfected with cDNAs encoding full-length or p115 fragments.
| Time after transfection
|
||||
|---|---|---|---|---|
| 20 h
|
33 h
|
|||
| Transfected constructs | Golgi fragmented | Apoptotic | Golgi fragmented | Apoptotic |
| % | % | % | % | |
| Full-length p115 | 8.0 | 6.0 | 14.0 | 11.3 |
| NH2-terminal fragment | 4.6 | 4.1 | 13.8 | 10.6 |
| COOH-terminal fragment | 81.2 | 77.6 | 86.5 | 79.2 |
Results are the averages of duplicate samples from two separate experiments. In each case, 150 cells were counted by two independent observers.
To ensure that the cells expressing the COOH-terminal p115758–962 fragment were indeed apoptotic, HeLa cells were transfected with either the NH2- or COOH-terminal p115 fragments and then stained for the presence of several apoptotic markers (Fig. 6) . Transfected cells were identified by coexpression of a GFP cDNA. Apoptotic cells were identified by staining with polyclonal antibodies specific for the cleaved p17 fragment of caspase-3 or the cleaved p85 fragment of PARP. These antibodies do not stain uncleaved forms of these proteins in healthy cells. There was no staining of either cleaved PARP or activated caspase-3 in cells expressing the NH2-terminal p1151–757 fragment (Fig. 6, A and B, top). Furthermore, nuclear and cellular morphology were normal, as indicated by Hoechst staining and phase contrast microscopy, respectively. In sharp contrast, most cells expressing the p115 COOH-terminal p115758–962 fragment displayed cell shrinkage and chromatin condensation, as shown above (Fig. 5; Fig. 6, A and B, bottom). Most significantly, these cells also stained positive for cleaved PARP and activated caspase-3, indicating that they were indeed apoptotic (Fig. 6, A and B, bottom). These results strongly support our hypothesis that the COOH-terminal p115 fragment is a potent mediator of apoptosis.
Figure 6.

Expression of the COOH-terminal caspase cleavage fragment of p115 induces caspase activation and apoptosis. (A) HeLa cells were cotransfected with vectors expressing GFP and either the NH2- or COOH-terminal caspase cleavage fragment of p115. At 24 h after transfection, cells were processed for immunofluorescence microscopy and stained with Hoechst 33238 (blue) and a polyclonal antibody that recognizes only the p85 caspase cleavage fragment of PARP (red). Transfected cells were identified by GFP fluorescence (green). Cells expressing the p115 NH2-terminal cleavage fragment did not stain for cleaved PARP, had normal nuclear staining, and were morphologically indistinguishable from untransfected cells (top). In sharp contrast, cells expressing the COOH-terminal cleavage fragment of p115 were positive for cleaved PARP and displayed additional apoptotic features including chromatin condensation and cell shrinkage (bottom). (B) HeLa cells were transfected and processed as above. Fixed cells were stained with Hoechst 33238 (blue) and a polyclonal antibody that specifically recognizes the p18 subunit of activated caspase-3 but not procaspase-3 (red). Transfected cells (green) expressing the COOH-terminal (bottom), but not the NH2-terminal (top), caspase cleavage fragment of p115 stained for the activated caspase-3 antibody. Arrows indicate transfected cells. Bar, 10 μm.
Because expression of the p115 COOH-terminal fragment itself induced apoptosis, it could be argued that the Golgi fragmentation resulted from apoptosis rather than an independent, dominant negative effect on endogenous p115. In addition, the appearance of the p115 COOH-terminal fragment in the nucleus could have resulted from its free diffusion through a more permeable nuclear membrane during apoptosis (Faleiro and Lazebnik, 2000). To exclude these possibilities, COS7 cells were incubated with the caspase inhibitor z-VAD-fmk before transfection of the p115 COOH-terminal fragment in order to inhibit apoptosis (Fig. 7 A, bottom). In the presence of z-VAD-fmk, the COOH-terminal fragment still translocated to the nucleus but failed to induce apoptosis. Furthermore, 76.5% of these cells displayed complete vesiculation of the Golgi apparatus even though apoptosis was inhibited (Table III). In contrast, >90% of the cells transfected with the COOH-terminal fragment alone were apoptotic and displayed Golgi fragmentation (Fig. 7 A, top; Table III). These data indicate that the translocation of the p115 COOH-terminal fragment into the nucleus and its ability to disrupt normal Golgi structure occur independently of apoptosis.
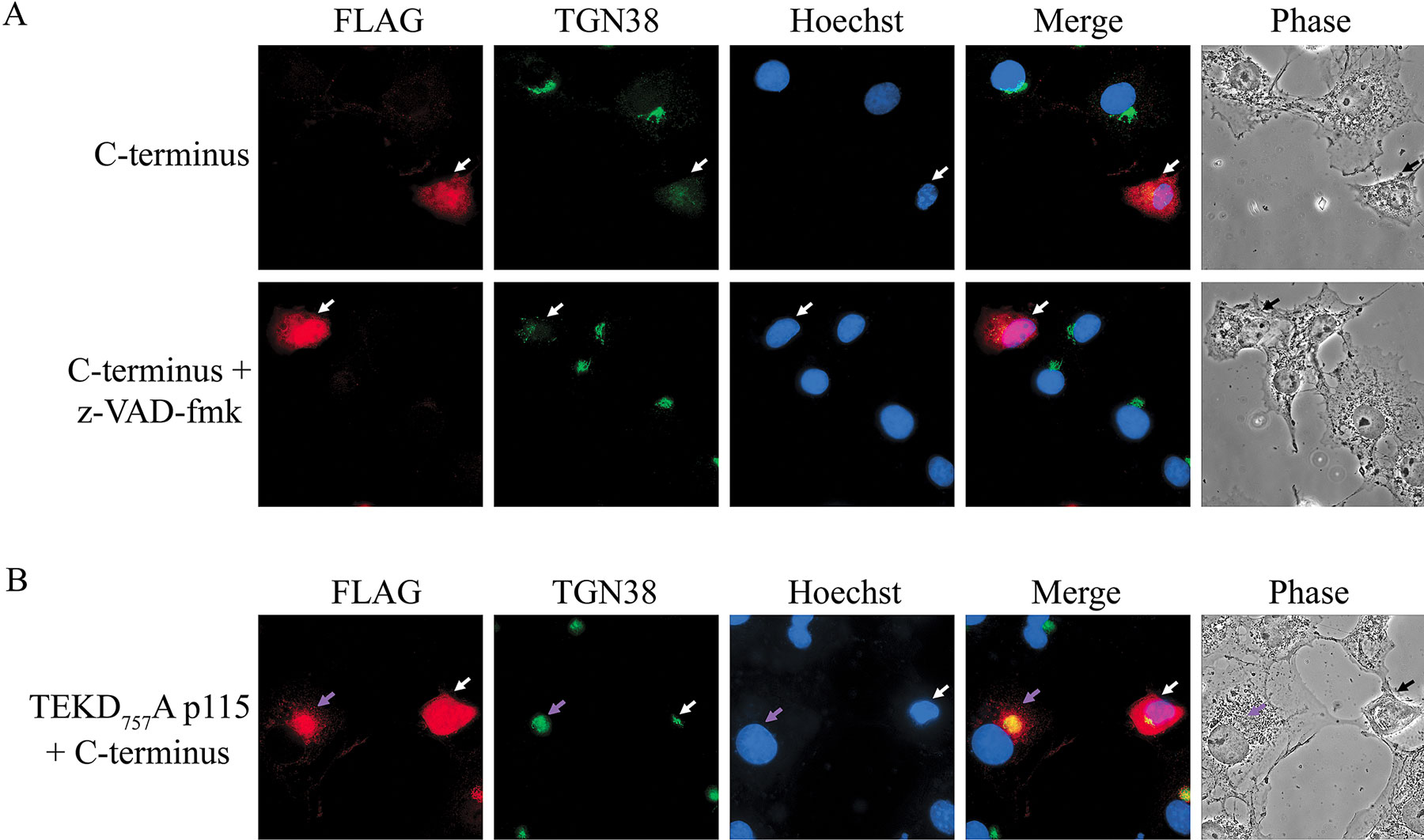
Figure 7.

The COOH-terminal caspase cleavage fragment of p115 induces Golgi fragmentation and apoptosis independently. (A) COS7 cells were pretreated with DMSO alone (top) or z-VAD-fmk (bottom) and then transfected with a FLAG-tagged construct of the p115 COOH-terminal fragment. 20 h after transfection, cells were fixed and stained with a FLAG (red) antibody, a GM130 (green) antibody, and Hoechst 33238 (blue). Transfected cells pretreated with z-VAD-fmk were not apoptotic but displayed extensive Golgi fragmentation and nuclear localization of the COOH-terminal fragment. (B) COS7 cells were cotransfected with FLAG-tagged versions of the p115 TEKD757Ala mutant and the COOH-terminal p115 fragment in a 10:1 ratio. Cells were fixed 20 h after transfection and stained as above. Cotransfected cells that were apoptotic (arrows) but displayed normal Golgi morphology were clearly present. (C) Cells were transfected with a FLAG-tagged construct of the COOH-terminal fragment lacking the acidic region (p115 TEKD757–933) and stained as above. Note that the transfected cell on the left (arrow) is in the early stages of apoptosis and the cell on the right (arrow) is at a later stage. Arrows indicate transfected (A and C) or cotransfected (B) cells. Bar, 10 μm.
Table III. The COOH-terminal fragment of p115 induces Golgi fragmentation and apoptosis independently in COS7 cells.
| Transfected constructs | Cell morphology | Intact Golgi | Fragmented Golgi |
|---|---|---|---|
| % | % | ||
| COOH-terminal fragment alone | Apoptotic | 7.0 | 93.0 |
| COOH-terminal fragment + z-VAD-fmk | Normal | 23.5 | 76.5 |
| COOH-terminal fragment + TEKD757Ala p115 | Apoptotic | 40.5 | 59.5 |
Results are the averages of duplicate samples from two separate experiments. In each case, 100–200 cells were counted by two independent observers.
It was possible that the apoptosis induced by expression of the p115 COOH-terminal fragment resulted indirectly from disruption of normal Golgi function. To eliminate this possibility, COS7 cells were cotransfected with cDNAs encoding the full-length p115 TEKD757Ala uncleavable mutant and the p115 COOH-terminal fragment in a 10:1 ratio. This ensured that cells expressing the COOH-terminal fragment (identified by FLAG staining in the nucleus) also coexpressed the uncleavable mutant. Our rationale was that the uncleavable mutant, which slows the rate of Golgi fragmentation (Table I), should protect against fragmentation of the Golgi apparatus induced by the p115 COOH-terminal fragment. However, apoptosis would still occur in transfected cells if cell death were not a result of Golgi fragmentation. Our data demonstrate that this was indeed the case (Fig. 7 B; Table III). 24 h after transfection, ∼80% of the cotransfected cells were apoptotic. However, only 59.5% of these apoptotic cells exhibited a fragmented Golgi apparatus, compared with >90% of cells expressing the COOH-terminal fragment alone, i.e., Golgi breakdown was significantly delayed. Identical results were obtained when TGN38 was used as a Golgi marker instead of GM130 (Fig. S2, available online at http://www.jcb.org/cgi/content/full/jcb.200208013/DC1).
To define which region of the p115 COOH-terminal fragment was responsible for inducing apoptosis and/or Golgi fragmentation, a truncated form of the molecule was generated. Our initial construct removed the last 29 residues of the COOH-terminal fragment, generating a molecule corresponding to residues 757–933 of p115; this deleted the acidic domain implicated in giantin–GM130 interactions (Nakamura et al., 1997; Linstedt et al., 2000). When this truncated fragment was expressed in COS7 cells (Fig. 7 C), it behaved identically to the complete COOH-terminal fragment, i.e., it localized to the nucleus, induced apoptosis, and led to Golgi fragmentation. Together, our data demonstrate that p115 plays a major role in the maintenance of the architecture of the Golgi apparatus, and its cleavage during apoptosis contributes to Golgi fragmentation and activation of downstream apoptotic signals.
Discussion
Here, we have shown that the Golgi apparatus fragments into distinct clusters of small vesicular and tubular structures during apoptosis. In contrast to mitosis, GM130 was not phosphorylated, although its level was diminished significantly. Strikingly, p115, a Golgi-associated polypeptide implicated in mediating several vesicle transport and docking steps (Waters et al., 1992; Sztul et al., 1993; Barroso et al., 1995), was cleaved by caspases in a time-dependent manner that correlated with fragmentation of the organelle and the onset of apoptosis. Preliminary data from our laboratory indicate that GM130 is also cleaved by caspases into several fragments in vitro. Our data demonstrate that the same Golgi proteins (p115 and GM130) can be differentially modified to effect quite distinct changes in the structural integrity of the Golgi apparatus. In mitosis, phosphorylation regulates the reversible breakdown of the organelle to ensure partitioning into daughter cells, whereas, in apoptosis, proteolytic cleavage results in irreversible Golgi fragmentation and eventually cell death.
In vitro experiments using purified caspases demonstrated that p115 was a potent substrate for caspases 3 and 8. Both of these enzymes cleaved p115 to generate two fragments of 90 and 30 kD. Several lines of evidence suggest that these two polypeptides were derived from either the major or only caspase cleavage site in p115 in vivo. First, in apoptotic cell lysates, the 90- and 30-kD polypeptides were detected in vivo using four different p115 antibodies (Fig. 2 A). In contrast, the 75- and 50-kD fragments were detected only in vitro (Fig. 3) and no immunoreactive species of these sizes were observed in vivo using any of the p115 antibodies. Second, incubation of in vitro–synthesized bovine or human p115 with caspase-8 generated only the 90- and 30-kD fragments (Fig. 3, A and B). Third, formation of the 90- and 30-kD fragments was evident upon incubation of p115 with 7–25 nM caspases 3 and 8, whereas generation of the other polypeptides required ∼50–100 nM caspases (Fig. 3 B). Fourth, time course experiments indicated that the generation of the 90- and 30-kD fragments reached a plateau at ∼10 min of incubation with purified caspase-3, whereas production of the 75- and 50-kD fragments required ∼180 min of incubation (Fig. 3 C). Finally, the full-length p115 TEKD757Ala mutant was not cleaved when expressed in apoptotic COS7 cells (Fig. 3 D), further supporting the idea that the 90- and 30-kD fragments are the only bona fide p115 cleavage fragments in vivo.
Site-directed mutagenesis of p115 demonstrated that cleavage at TEKD757 released the 90- and 30-kD cleavage fragments. We hypothesized that cleavage of p115 at this site contributes to apoptotic Golgi fragmentation. To test this hypothesis directly, two approaches can be used (both of which we have used) to evaluate the physiological significance of this caspase cleavage (Roy and Nicholson, 2000). The first approach involves expressing a cleavage-resistant mutant of p115 to determine if it delays Golgi fragmentation during apoptosis. Our data demonstrate that this was the case; expression of the p115 TEKD757Ala mutant delayed fragmentation of the Golgi apparatus when compared with expression of the native polypeptide (Table I). Although Golgi fragmentation was delayed, the kinetics of apoptosis were not altered significantly in these cells. However, because the apoptotic response involves multiple pathways and components that are functionally redundant, it is unlikely that inactivating one downstream signaling molecule would delay or inhibit apoptosis. Furthermore, similar to many caspases and Bcl-2 family members (Earnshaw et al., 1999; Strasser et al., 2000), the requirement of the COOH-terminal p115 fragment in apoptosis may be both cell type and stimulus dependent.
The second approach is to express the caspase cleavage fragments in healthy cells and demonstrate that they can disrupt the normal Golgi structure directly by acting in a dominant negative manner. Expression of the two p115 fragments generated by cleavage at TEKD757 indicated that the COOH-terminal fragment, p115758–962, disrupted the Golgi structure and, surprisingly, also translocated to the nucleus and induced apoptosis. These effects were not a consequence of overexpression because neither full-length p115 nor p1151–757 expressed at similar levels to the COOH-terminal fragment had an effect on Golgi structure or cell viability. Indeed, even 33 h after transfection, when expression of both full-length p115 and p1151–757 reached extreme levels, the percentage of transfected cells with apoptotic features remained <20% (Table II). These results strongly support the idea that p115 is required for maintaining normal Golgi structure and its cleavage at TEKD757 contributes to apoptotic signaling and fragmentation of the Golgi apparatus.
Considerable evidence has implicated the COOH-terminal acidic region of p115 in mediating vesicle tethering at the Golgi apparatus (Nelson et al., 1998; Dirac-Svejstrup et al., 2000). It was shown to be the binding site for both giantin and GM130 (Linstedt et al., 2000). Furthermore, this domain also possesses the phosphorylated serine (residue 941/942) that regulates p115 affinity for GM130 and giantin (Sohda et al., 1998; Dirac-Svejstrup et al., 2000). Recent observations (Puthenveedu and Linstedt, 2001) have also demonstrated that the Golgi undergoes COPI-dependent fragmentation after microinjection of antibodies against p115. In this context, our data are consistent with the idea that the p115 COOH-terminal fragment has two independent functions: one in disrupting Golgi structure by acting as a dominant negative of p115, and the other in translocating to the nucleus and inducing apoptosis. In support for this idea, we demonstrated that Golgi fragmentation and apoptosis induced by the COOH-terminal fragment could be uncoupled in coexpression and caspase inhibitor studies (Fig. 7, A and B). Furthermore, a deletion mutant of the extreme COOH-terminal acidic domain was able to effect nuclear translocation, Golgi fragmentation, and apoptosis (Fig. 7 C). These data suggested that in addition to the acidic stretch, other motifs in the COOH-terminal region contribute to the maintenance of Golgi structure and that the acidic domain was not required to induce apoptosis. Like p115, it is possible that additional cellular proteins that normally have functions unrelated to apoptosis may become pro-apoptotic factors once they are cleaved. For example, the neuronal microtubule-associated protein tau is also a substrate for caspase-3. Importantly, the percentage of cells expressing tau cleavage fragments that were apoptotic increased two- to threefold when compared with cells expressing the full-length tau protein (Fasulo et al., 2000).
In the caspase-9–dependent apoptotic pathway, the mitochondrion is considered to be the central organelle responsible for sensing and inducing the apoptotic response through molecules such as cytochrome c, apoptosis-inducing factor, and proteins of the Bcl-2 family (Desagher and Martinou, 2000). By interacting with other pro-apoptotic molecules after its release from the mitochondria, cytochrome c forms part of the apoptosome complex, which ultimately leads to caspase activation and cell death (Zou et al.,1999). Previous studies have suggested that the Golgi apparatus may also play a role in apoptosis because pro-apoptotic molecules such as caspase-2 and several death receptors were shown to be concentrated in this organelle (An and Dou, 1996; Bennett et al., 1998; Mancini et al., 2000; Zhang et al., 2000). Our data provide further evidence in support of a role for the Golgi apparatus in the apoptotic response.
An unexpected observation was that the p115758–962 fragment localized efficiently to the nucleus in apoptotic cells (Fig. 5). This was quite surprising because the p115 polypeptide lacks an obvious nuclear localization signal and, to date, has not been reported to have a nuclear distribution. We investigated this further by making use of an algorithm that predicts protein subcellular localization based on differences in amino acid composition (Reinhardt and Hubbard, 1998). Significantly, the nonapoptosis-inducing NH2-terminal fragment of p115 was predicted to be cytoplasmic with 55% reliability, whereas the COOH-terminal p115758–962 fragment was predicted to be nuclear with 94% reliability. In addition, a recent report demonstrated that a subset of golgin-160 caspase cleavage fragments also translocated to the nucleus (Hicks and Machamer, 2002). Whether these fragments are capable of eliciting the apoptotic response is not known. One attractive model is that the NH2 terminus of full-length p115 normally impedes the nuclear translocation of its COOH-terminal domain in healthy cells. However, once the NH2 terminus is cleaved during apoptosis, the COOH-terminal fragment translocates to the nucleus, where it may interact with and activate pro-apoptotic factors, thereby propagating or potentiating the upstream apoptotic signal. Experiments to identify possible nuclear targets of the p115758–962 fragment are currently in progress.
Materials and methods
Antibodies and reagents
Affinity-purified rabbit anti–phospho-GM130 antibody (PS25) and rabbit anti-GM130 antibody were provided by Martin Lowe (University of Manchester, Manchester, UK) and Graham Warren (Yale University School of Medicine, New Haven, CT), respectively. A polyclonal antibody specific for the COOH terminus of p115 was a gift from Adam Linstedt (Carnegie Mellon University, Pittsburgh, PA). Polyclonal antibodies to p115 were also provided by Elizabeth Sztul (University of Alabama, Birmingham, AL). Polyclonal TGN38 antibody was a gift from Sharon Milgram (University of North Carolina, Chapel Hill, NC). Monoclonal anti-GM130, anti-p115, and anti-Vtila antibodies were purchased from Transduction Laboratories. Antibodies against cleaved PARP and activated caspase-3 were purchased from Promega. An antibody recognizing full-length PARP was purchased from Cell Signaling. Alexa® Fluor goat anti–mouse and anti–rabbit antibodies were purchased from Molecular Probes, Inc. An agonistic anti-Fas antibody (clone CH11) was purchased from Upstate Biotechnology. The pSG5-FLAG–human p115 construct was provided by Yukio Ikehara (Fukuoka University School of Medicine, Fukuoka, Japan). The pBluescriptII-bovine p115 plasmid and four different anti-p115 monoclonal antibodies were gifts from Gerry Waters (Princeton University, Princeton, NJ). Caspases 2, 3, 7, and 8 were purified as previously described (Garcia-Calvo et al., 1999) and were gifts of Nancy A. Thornberry (Merck Research Laboratories, Rahway, NJ). A plasmid encoding procaspase-3 was provided by Tom Gilmore (Boston University, Boston, MA). Staurosporine, etoposide, and cycloheximide were purchased from Sigma-Aldrich. z-VAD-fmk and z-YVAD-fmk caspase inhibitors were purchased from Calbiochem.
Apoptosis induction
COS7, HeLa, and normal rat kidney (NRK) cells were grown in DME supplemented with 10% FCS. Pheochromocytoma (PC12) cells were grown in F-10 medium containing 2.5% FCS and 15% equine serum. 0.5–1 μM staurosporine or 80–100 μM etoposide was added to the growth medium for the indicated times at 37°C to induce apoptosis. Fas ligation was induced as shown previously (Nguyen et al., 2000) using 0.5 μg/ml agonistic anti-Fas antibody in the presence of 10 μg/ml cycloheximide for the indicated times at 37°C.
Immunofluorescence and electron microscopy
Cells were grown on coverslips and treated as indicated. Samples were fixed and processed for immunofluorescence microscopy as previously described (Austin and Shields, 1996). Images were processed using IPLab and Adobe Photoshop®. Hoechst staining was performed after secondary antibody incubation for 10 min in PBS. For PS25 staining, samples were fixed in 100% methanol for 6 min at −20°C before antibody incubations. Cells treated as indicated were fixed and processed for electron microscopy as previously described (Siddhanta et al., 2000).
Lysate preparation and Western blotting
After induction of apoptosis, cells were lysed as previously described (Roy and Nicholson, 2000) and 30 μg of each sample was loaded on a 10% SDS polyacrylamide gel after which the polypeptides were transferred to an Immobilon-P membrane (Millipore Corp.). Membranes were incubated with the appropriate antibodies and the bands visualized by ECL (Amersham Biosciences). For the caspase inhibitor studies, cells were preincubated with 0.1 mM z-VAD-fmk or z-YVAD-fmk for 1 h at 37°C before apoptosis induction.
In vitro caspase cleavage reactions
Human and bovine p115 cDNAs were transcribed and translated using the in vitro TNT-coupled reticulocyte lysate system (Promega). 5 μl of the translation product was incubated with the indicated concentration of purified caspases for 1 h at 37°C. Reactions were performed in buffer A (100 mM Hepes, pH 7.0, 10% sucrose, 0.1% CHAPS, 10 mM DTT) for caspases 3, 7, and 8, or buffer B (100 mM Mes, pH 6.5, 10% sucrose, 0.1% CHAPS, 10 mM DTT) for caspase-2. Caspase-7 reactions were performed in buffer A supplemented with 5 mM CaCl2; SDS gel loading buffer was added to each reaction at the end of the incubation. The samples were boiled and loaded on a 10% SDS polyacrylamide gel. The gel was dried and analyzed by fluorography.
Vector construction and transfection
The p115 D757A mutant was generated using the QuikChange site-directed mutagenesis kit (Stratagene) using primer C1 (5′-CAATCATAGAGGCCTTTTCAGTCAGCTGGC-3′) and primer C2 (5′-GCCAGCTGACTGAAAAGGCCTCTATGATTG-3′). All mutations were confirmed by automated DNA sequencing. To generate constructs expressing wild-type p115 and truncated p115 fragments, PCR was performed using the pSG5-FLAG–p115 plasmid as the template. Primers A (5′-CACGGATCCAATTTCCTCCGCGGGGTAATGGGG-3′) and B (5′-GTGCTCGAGTTATCAGATATGATCTAGATCCTTGCCAGGATC-3′) were used to generate the full-length coding region of human p115. Primers A and C (5′-GTGCTCGAGTTATCAATCCTCTTGGTCTCCAGATTCAAGTTC-3′) and primers D (5′-CACGGATCCTCTATGATTGAAAATATGAAATCTTCCC-3′) and B were used to generate the NH2-terminal and COOH-terminal p115 truncation mutants, respectively. All primers incorporate a BamHI site at the 5′ end and an XhoI site at the 3′ end of each construct (underlined). PCR products were digested with BamHI and XhoI and then purified using the QIAquick PCR purification kit (QIAGEN). The purified inserts were FLAG tagged at the NH2 terminus by subcloning into the pCMV-Tag2B mammalian expression vector (Stratagene). 1 μg of each plasmid was transfected into COS7 cells growing on coverslips using the Effectene transfection reagent (QIAGEN).
Online supplemental material
Supplemental figures are available online at http://www.jcb.org/cgi/content/full/jcb.200208013/DC1. Fig. S1 shows the fragmentation of the Golgi apparatus into vesicular and tubular structures in cells treated with staurosporine and etoposide. Fig. S2 demonstrates that the results of Fig. 7 are identical when TGN38 was used as a Golgi marker instead of GM130.
Supplemental Material
Acknowledgments
We thank Dr. Duncan Wilson for very helpful suggestions with the manuscript and Drs. Brian Burke, Tom Gilmore, Yukio Ikehara, Adam Linstedt, Martin Lowe, Sharon Milgram, Elizabeth Sztul, Nancy Thornberry, Graham Warren, and Gerry Waters for gifts of reagents.
This work was supported by National Institutes of Health grant DK-21860 to D. Shields.
The online version of this article includes supplemental material.
Footnotes
Abbreviations used in this paper: NRK, normal rat kidney; PARP, poly(ADP-ribose) polymerase.
References
- Alvarez, C., H. Fujita, A. Hubbard, and E. Sztul. 1999. ER to Golgi transport: requirement for p115 at a pre-Golgi VTC stage. J. Cell Biol. 147:1205–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An, B., and Q.P. Dou. 1996. Cleavage of retinoblastoma protein during apoptosis: an interleukin 1 β-converting enzyme-like protease as candidate. Cancer Res. 56:438–442. [PubMed] [Google Scholar]
- Austin, C.D., and D. Shields. 1996. Formation of nascent secretory vesicles from the trans-Golgi network of endocrine cells is inhibited by tyrosine kinase and phosphatase inhibitors. J. Cell Biol. 135:1471–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr, F.A., M. Puype, J. Vandekerckhove, and G. Warren. 1997. GRASP65, a protein involved in the stacking of Golgi cisternae. Cell. 91:253–262. [DOI] [PubMed] [Google Scholar]
- Barroso, M., D.S. Nelson, and E. Sztul. 1995. Transcytosis-associated protein (TAP)/p115 is a general fusion factor required for binding of vesicles to acceptor membranes. Proc. Natl. Acad. Sci. USA. 92:527–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett, M., K. Macdonald, S.W. Chan, J.P. Luzio, R. Simari, and P. Weissberg. 1998. Cell surface trafficking of Fas: a rapid mechanism of p53-mediated apoptosis. Science. 282:290–293. [DOI] [PubMed] [Google Scholar]
- de Figueiredo, P., R.S. Polizotto, D. Drecktrah, and W.J. Brown. 1999. Membrane tubule-mediated reassembly and maintenance of the Golgi complex is disrupted by phospholipase A2 antagonists. Mol. Biol. Cell. 10:1763–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desagher, S., and J.C. Martinou. 2000. Mitochondria as the central control point of apoptosis. Trends Cell Biol. 10:369–377. [DOI] [PubMed] [Google Scholar]
- Dirac-Svejstrup, A.B., J. Shorter, M.G. Waters, and G. Warren. 2000. Phosphorylation of the vesicle tethering protein p115 by a casein kinase II–like enzyme is required for Golgi reassembly from isolated mitotic fragments. J. Cell Biol. 150:475–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earnshaw, W.C., L.M. Martins, and S.H. Kaufmann. 1999. Mammalian caspases: structure, activation, substrates, and functions during apoptosis. Annu. Rev. Biochem. 68:383–424. [DOI] [PubMed] [Google Scholar]
- Faleiro, L., and Y. Lazebnik. 2000. Caspases disrupt the nuclear–cytoplasmic barrier. J. Cell Biol. 151:951–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fasulo, L., G. Ugolini, M. Visintin, A. Bradbury, C. Brancolini, V. Verzillo, M. Novak, and A. Cattaneo. 2000. The neuronal microtubule-associated protein tau is a substrate for caspase-3 and an effector of apoptosis. J. Neurochem. 75:624–633. [DOI] [PubMed] [Google Scholar]
- Garcia-Calvo, M., E.P. Peterson, D.M. Rasper, J.P. Vaillancourt, R. Zamboni, D.W. Nicholson, and N.A. Thornberry. 1999. Purification and catalytic properties of human caspase family members. Cell Death Differ. 6:362–369. [DOI] [PubMed] [Google Scholar]
- Garcia-Mata, R., Y. Gao, C. Alvarez, and E.S. Sztul. 2000. The membrane transport factor p115 recycles only between homologous compartments in intact heterokaryons. Eur. J. Cell Biol. 79:229–239. [DOI] [PubMed] [Google Scholar]
- Hicks, S.W., and C.E. Machamer. 2002. The NH2-terminal domain of golgin-160 contains both Golgi and nuclear targeting information. J. Biol. Chem. 277:35833–35839. First published on July 18, 2002; 10.1074/jbc.M206280200 [DOI] [PubMed] [Google Scholar]
- Janicke, R.U., M.L. Sprengart, M.R. Wati, and A.G. Porter. 1998. Caspase-3 is required for DNA fragmentation and morphological changes associated with apoptosis. J. Biol. Chem. 273:9357–9360. [DOI] [PubMed] [Google Scholar]
- Lane, J.D., J. Lucocq, J. Pryde, F.A. Barr, P.G. Woodman, V.J. Allan, and M. Lowe. 2002. Caspase-mediated cleavage of the stacking protein GRASP65 is required for Golgi fragmentation during apoptosis. J. Cell Biol. 156:495–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesa, G.M., J. Seemann, J. Shorter, J. Vandekerckhove, and G. Warren. 2000. The amino-terminal domain of the Golgi protein giantin interacts directly with the vesicle-tethering protein p115. J. Biol. Chem. 275:2831–2836. [DOI] [PubMed] [Google Scholar]
- Linstedt, A.D., and H.P. Hauri. 1993. Giantin, a novel conserved Golgi membrane protein containing a cytoplasmic domain of at least 350 kDa. Mol. Biol. Cell. 4:679–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linstedt, A.D., S.A. Jesch, A. Mehta, T.H. Lee, R. Garcia-Mata, D.S. Nelson, and E. Sztul. 2000. Binding relationships of membrane tethering components. The giantin N terminus and the GM130 N terminus compete for binding to the p115 C terminus. J. Biol. Chem. 275:10196–10201. [DOI] [PubMed] [Google Scholar]
- Lowe, M., N. Nakamura, and G. Warren. 1998. a. Golgi division and membrane traffic. Trends Cell Biol. 8:40–44. [DOI] [PubMed] [Google Scholar]
- Lowe, M., C. Rabouille, N. Nakamura, R. Watson, M. Jackman, E. Jamsa, D. Rahman, D.J. Pappin, and G. Warren. 1998. b. Cdc2 kinase directly phosphorylates the cis-Golgi matrix protein GM130 and is required for Golgi fragmentation in mitosis. Cell. 94:783–793. [DOI] [PubMed] [Google Scholar]
- Lowe, M., N.K. Gonatas, and G. Warren. 2000. The mitotic phosphorylation cycle of the cis-Golgi matrix protein GM130. J. Cell Biol. 149:341–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancini, M., C.E. Machamer, S. Roy, D.W. Nicholson, N.A. Thornberry, L.A. Casciola-Rosen, and A. Rosen. 2000. Caspase-2 is localized at the Golgi complex and cleaves golgin-160 during apoptosis. J. Cell Biol. 149:603–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monier, S., P. Chardin, S. Robineau, and B. Goud. 1998. Overexpression of the ARF1 exchange factor ARNO inhibits the early secretory pathway and causes the disassembly of the Golgi complex. J. Cell Sci. 111(Pt. 22):3427–3436. [DOI] [PubMed] [Google Scholar]
- Moyer, B.D., B.B. Allan, and W.E. Balch. 2001. Rab1 interaction with a GM130 effector complex regulates COPII vesicle cis-Golgi tethering. Traffic. 2:268–276. [DOI] [PubMed] [Google Scholar]
- Nakamura, N., C. Rabouille, R. Watson, T. Nilsson, N. Hui, P. Slusarewicz, T.E. Kreis, and G. Warren. 1995. Characterization of a cis-Golgi matrix protein, GM130. J. Cell Biol. 131:1715–1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura, N., M. Lowe, T.P. Levine, C. Rabouille, and G. Warren. 1997. The vesicle docking protein p115 binds GM130, a cis-Golgi matrix protein, in a mitotically regulated manner. Cell. 89:445–455. [DOI] [PubMed] [Google Scholar]
- Nelson, D.S., C. Alvarez, Y.S. Gao, R. Garcia-Mata, E. Fialkowski, and E. Sztul. 1998. The membrane transport factor TAP/p115 cycles between the Golgi and earlier secretory compartments and contains distinct domains required for its localization and function. J. Cell Biol. 143:319–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen, M., D.G. Breckenridge, A. Ducret, and G.C. Shore. 2000. Caspase-resistant BAP31 inhibits fas-mediated apoptotic membrane fragmentation and release of cytochrome c from mitochondria. Mol. Cell. Biol. 20:6731–6740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puthenveedu, M.A., and A.D. Linstedt. 2001. Evidence that Golgi structure depends on a p115 activity that is independent of the vesicle tether components giantin and GM130. J. Cell Biol. 155:227–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhardt, A., and T. Hubbard. 1998. Using neural networks for prediction of the subcellular location of proteins. Nucleic Acids Res. 26:2230–2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy, S., and D.W. Nicholson. 2000. Criteria for identifying authentic caspase substrates during apoptosis. Methods Enzymol. 322:110–125. [DOI] [PubMed] [Google Scholar]
- Seemann, J., E.J. Jokitalo, and G. Warren. 2000. The role of the tethering proteins p115 and GM130 in transport through the Golgi apparatus in vivo. Mol. Biol. Cell. 11:635–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sesso, A., D.T. Fujiwara, M. Jaeger, R. Jaeger, T.C. Li, M.M. Monteiro, H. Correa, M.A. Ferreira, R.I. Schumacher, J. Belisario, et al. 1999. Structural elements common to mitosis and apoptosis. Tissue Cell. 31:357–371. [DOI] [PubMed] [Google Scholar]
- Shorter, J., and G. Warren. 1999. A role for the vesicle tethering protein, p115, in the post-mitotic stacking of reassembling Golgi cisternae in a cell-free system. J. Cell Biol. 146:57–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddhanta, A., J.M. Backer, and D. Shields. 2000. Inhibition of phosphatidic acid synthesis alters the structure of the Golgi apparatus and inhibits secretion in endocrine cells. J. Biol. Chem. 275:12023–12031. [DOI] [PubMed] [Google Scholar]
- Sohda, M., Y. Misumi, A. Yano, N. Takami, and Y. Ikehara. 1998. Phosphorylation of the vesicle docking protein p115 regulates its association with the Golgi membrane. J. Biol. Chem. 273:5385–5388. [DOI] [PubMed] [Google Scholar]
- Strasser, A., L. O'Connor, and V.M. Dixit. 2000. Apoptosis signaling. Annu. Rev. Biochem. 69:217–245. [DOI] [PubMed] [Google Scholar]
- Sztul, E., M. Colombo, P. Stahl, and R. Samanta. 1993. Control of protein traffic between distinct plasma membrane domains. Requirement for a novel 108,000 protein in the fusion of transcytotic vesicles with the apical plasma membrane. J. Biol. Chem. 268:1876–1885. [PubMed] [Google Scholar]
- Takizawa, P.A., J.K. Yucel, B. Veit, D.J. Faulkner, T. Deerinck, G. Soto, M. Ellisman, and V. Malhotra. 1993. Complete vesiculation of Golgi membranes and inhibition of protein transport by a novel sea sponge metabolite, ilimaquinone. Cell. 73:1079–1090. [DOI] [PubMed] [Google Scholar]
- Thornberry, N.A., T.A. Rano, E.P. Peterson, D.M. Rasper, T. Timkey, M. Garcia-Calvo, V.M. Houtzager, P.A. Nordstrom, S. Roy, J.P. Vaillancourt, et al. 1997. A combinatorial approach defines specificities of members of the caspase family and granzyme B. Functional relationships established for key mediators of apoptosis. J. Biol. Chem. 272:17907–17911. [DOI] [PubMed] [Google Scholar]
- Waters, M.G., D.O. Clary, and J.E. Rothman. 1992. A novel 115-kD peripheral membrane protein is required for intercisternal transport in the Golgi stack. J. Cell Biol. 118:1015–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weide, T., M. Bayer, M. Koster, J.P. Siebrasse, R. Peters, and A. Barnekow. 2001. The Golgi matrix protein GM130: a specific interacting partner of the small GTPase rab1b. EMBO Rep. 2:336–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaal, K.J., C.L. Smith, R.S. Polishchuk, N. Altan, N.B. Cole, J. Ellenberg, K. Hirschberg, J.F. Presley, T.H. Roberts, E. Siggia, et al. 1999. Golgi membranes are absorbed into and reemerge from the ER during mitosis. Cell. 99:589–601. [DOI] [PubMed] [Google Scholar]
- Zhang, X.D., A.V. Franco, T. Nguyen, C.P. Gray, and P. Hersey. 2000. Differential localization and regulation of death and decoy receptors for TNF-related apoptosis-inducing ligand (TRAIL) in human melanoma cells. J. Immunol. 164:3961–3970. [DOI] [PubMed] [Google Scholar]
- Zou, H., Y. Li, X. Liu, and X. Wang. 1999. An APAF-1.cytochrome c multimeric complex is a functional apoptosome that activates procaspase-9. J. Biol. Chem. 274:11549–11556. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}