

Abstract
Ñeurotransmitter release requires the direct coupling of the calcium sensor with the machinery for membrane fusion. SNARE proteins comprise the minimal fusion machinery, and synaptotagmin I, a synaptic vesicle protein, is the primary candidate for the main neuronal calcium sensor. To test the effect of synaptotagmin I on membrane fusion, we incorporated it into a SNARE-mediated liposome fusion assay. Synaptotagmin I dramatically stimulated membrane fusion by facilitating SNAREpin zippering. This stimulatory effect was topologically restricted to v-SNARE vesicles (containing VAMP 2) and only occurred in trans to t-SNARE vesicles (containing syntaxin 1A and SNAP-25). Interestingly, calcium did not affect the overall fusion reaction. These results indicate that synaptotagmin I can directly accelerate SNARE-mediated membrane fusion and raise the possibility that additional components might be required to ensure tight calcium coupling.
Keywords: SNARE; fusion; synaptotagmin; calcium; exocytosis
Introduction
Interneuronal communication and information processing in the brain is mediated by the quantal release of neurotransmitters triggered by the influx of calcium into the nerve terminal. These calcium-induced events occur on the submillisecond timescale, thus suggesting that the calcium sensor is tightly coupled to the machinery that fuses neurotransmitter-containing synaptic vesicles with the plasma membrane. SNAREs, a family of compartment-specific transmembrane proteins, initiate membrane fusion when a v-SNARE on a transport vesicle pairs with its cognate t-SNARE on the target membrane, forming SNAREpins (trans SNARE complexes) (Söllner et al., 1993; Weber et al., 1998). In the case of neurotransmitter release, the synaptic vesicle-bound v-SNARE VAMP 2 interacts with its cognate t-SNARE, consisting of syntaxin 1 and SNAP-25, on the presynaptic plasma membrane. Subsequent stepwise SNARE assembly into a four helix bundle brings the two membranes in close apposition and drives lipid bilayer mixing (Sutton et al., 1998; Weber et al., 1998; Nickel et al., 1999; Chen et al., 2001). Regulatory components, such as the calcium sensor, may alter the kinetics of SNARE complex assembly at distinct stages by trapping or accelerating reaction intermediates.
The prime candidate for a calcium sensor in the central nervous system is synaptotagmin I, a type I transmembrane protein localized on synaptic vesicles (Brose et al., 1992; Südhof and Rizo, 1996). Gene deletions of synaptotagmin I in Mus musculus, Caenorhabditis elegans, and Drosophila melangaster all demonstrate a marked loss of calcium-evoked fusion (Littleton et al., 1993; Nonet et al., 1993; Broadie et al., 1994; Geppert et al., 1994). In addition, replacement of endogenous synaptotagmin I in mice with a mutant version that has a twofold lower affinity for calcium in the presence of phospholipid membranes causes a concomitant decrease in the calcium sensitivity of neurotransmitter release (Fernández-Chacon et al., 2001). These results provide compelling in vivo evidence that synaptotagmin I is the major calcium sensor for fast synaptic exocytosis. At the molecular level, synaptotagmin I is characterized by two conserved cytoplasmic calcium-binding domains, C2A and C2B, which interact in a calcium-dependent manner with the synaptic t-SNAREs, acidic lipids, and phosphoinositides (Bennett et al., 1992; Brose et al., 1992; Schiavo et al., 1996, 1997; Davis et al., 1999; Gerona et al., 2000; Earles et al., 2001). In addition, the C2B domain of synaptotagmin I is responsible for the homo- and heterooligomerization of synaptotagmins (Osborne et al., 1999; Desai et al., 2000) of which there are 13 known mammalian isoforms (Kelly, 1995; Augustine, 2001; Südhof, 2002). Recent electrophysiological studies of dense core vesicle fusion in PC12 cells have shown that overexpression of synaptotagmins with different calcium affinities differentially modulate fusion pore kinetics (Wang et al., 2001). These experiment further link synaptotagmin I to the actual fusion process.
Despite strong evidence that synaptotagmin I couples calcium sensitivity to vesicle fusion, the molecular mechanisms by which it acts remain elusive. Two main hypotheses exist about how synaptotagmin I might regulate membrane fusion: (1) synaptotagmin I acts as a clamp, preventing fusion of docked vesicles until an influx of calcium releases the clamp, and (2) synaptotagmin I stimulates fusion upon the influx of calcium. In addition, there are variations of these models that require additional components to control calcium-triggered exocytosis (Kelly, 1995).
Deconvolution of the mechanism of action of synaptotagmin I in vivo has been hindered by the degeneracy of synaptotagmin isoforms and the difficulty of separating the molecular functions of synaptotagmin I from those of other accessory proteins in the complex cellular environment. Thus, we used a simplified in vitro model of membrane fusion that allowed us to examine the role of synaptotagmin I in a defined and accessible system (Weber et al., 1998). This approach has been used previously to establish that SNAREs are the minimal machinery for membrane fusion (Weber et al., 1998) and to show that SNARE-mediated fusion is specific and topologically restricted (Fukuda et al., 2000; McNew et al., 2000; Parlati et al., 2000). In addition, this assay has demonstrated that the NH2-terminal regulatory domain (NRD)* of syntaxin I not only regulates t-SNARE assembly but also effects SNAREpin formation (Parlati et al., 2000). However, the relatively slow fusion kinetics of the in vitro system, in comparison with those of regulated exocytosis in vivo, indicate that additional components may be required to accelerate the overall reaction. In this article, we test the effect of synaptotagmin I on SNARE-mediated fusion, and we examine its putative regulatory roles using the in vitro fusion assay.
Results
Sytg I reconstituted into v-SNARE vesicles accelerates SNARE-dependent fusion
We monitored membrane fusion using a fluorescent resonance energy transfer–based lipid mixing assay (Struck et al., 1981). Briefly, when two fluorescent lipids, nitro-2,1,3-benzoxadiazole 1,2-dipalmitoyl-SN-glycero-3 (NBD)–phosphatidylethanolamine (PE) and lissamine rhodamine B 1,2-dipalmitoyl-SN-glycero-3–PE (rhodamine-PE) are incorporated at appropriate concentrations (0.8 mol%) into donor vesicles, the fluorescence signal emitted by NBD-PE is quenched by rhodamine-PE. Fusion of the labeled donor vesicles with unlabeled acceptor vesicles dilutes the fluorescent lipids causing a distance-dependent increase in NBD-PE fluorescence over time. Finally, the NBD signal is normalized to the maximum fluorescence signal obtained by infinite dilution in the presence of detergent and plotted as the percentage of the total fluorescence versus time. In the following experiments, VAMP 2 (v-SNARE) was reconstituted into labeled donor liposomes, and coexpressed syntaxin 1 and SNAP-25 (t-SNAREs) were incorporated into unlabeled acceptor liposomes (Weber et al., 1998).
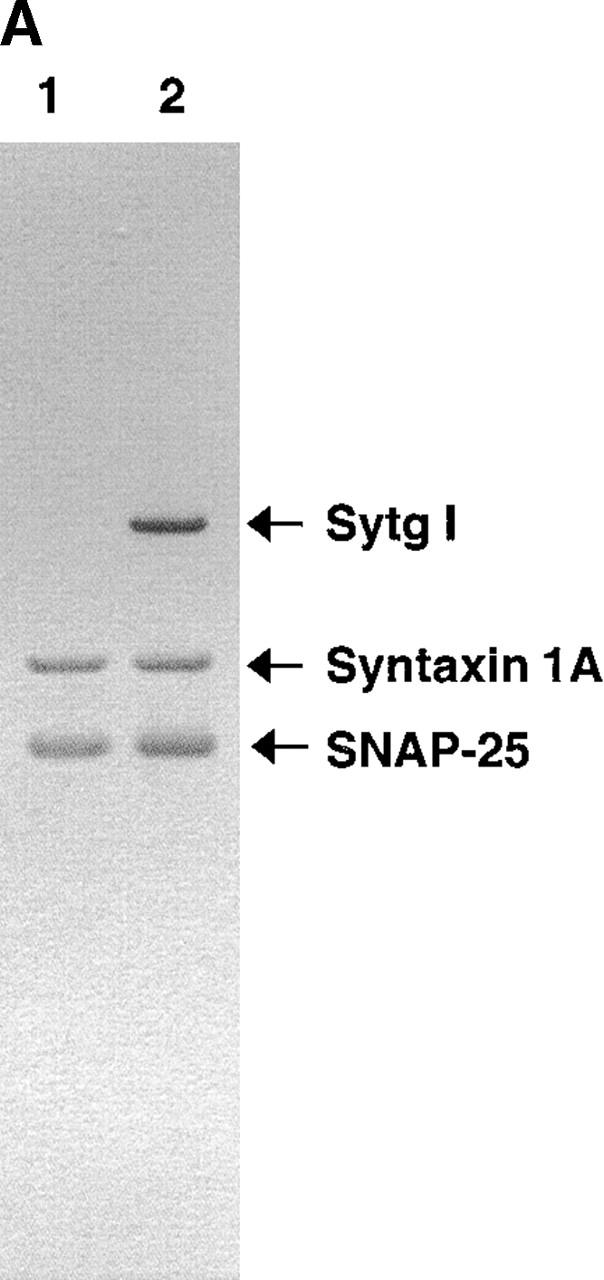
To facilitate synaptotagmin I expression and reconstitution into liposomes, we used a modified NH2-terminal truncated synaptotagmin I (Sytg I), which begins at aa 57 (see Material and methods for details). Donor liposomes containing reconstituted VAMP 2, VAMP 2 and Sytg I (Sytg I–VAMP 2), and Sytg I alone (Fig. 1 A) were prepared using standard protocols (Weber et al., 2000). These liposomes were mixed with acceptor t-SNARE liposomes in the presence of buffer or the cytosolic domain of VAMP 2 (cd–VAMP 2) without any calcium addition, and the fusion activity was monitored for 2 h at 37°C. Sytg I–VAMP 2 liposomes showed a remarkable increase in fusion activity when compared with liposomes containing only VAMP 2 (Fig. 1 B). Sytg I enhanced the fusion kinetics of VAMP 2 liposomes in a dose-dependent manner as demonstrated by the increased initial fusion rates (Fig. 1 C). At the highest concentration of Sytg I there is an approximate twofold excess of Sytg I over t-SNARE on a liposome to liposome basis. However, there is twofold more VAMP 2 than Sytg I in the v-SNARE liposomes, and the total amount of t-SNARE in the fusion assay is sixfold higher than Sytg I. For technical reasons, we could not incorporate more Sytg I without lowering the amount of VAMP 2 molecules, thus the observed fivefold stimulation of the initial fusion rate by synaptotagmin I must be considered as a minimal number. Soluble cd–VAMP 2 inhibited the stimulatory effect of Sytg I, confirming that the observed membrane fusion is SNARE dependent (Fig. 1 B). Furthermore, liposomes containing Sytg I but lacking VAMP 2 did not fuse with t-SNARE liposomes, although Sytg I interacts with t-SNAREs (Fig. 1 B and see Fig. 6 B) (Gerona et al., 2000). These results clearly show that synaptotagmin I by itself does not have any fusogenic activity and that the cross-linking of liposomes via Sytg I–t-SNARE binding interactions is insufficient to induce fusion.
Figure 1.



Incorporation of Sytg I into labeled VAMP 2 liposomes accelerates fusion. (A) Reconstituted donor liposomes. Donor liposomes containing VAMP 2 alone (lane 1), Sytg I–VAMP 2 (lane 2), and Sytg I alone (lane 3) were analyzed by SDS-PAGE and stained with Coomassie blue. The positions of VAMP 2 and Sytg I are marked by arrows. (B) Fusion in the presence of Sytg I. Unlabeled acceptor liposomes (45 μl) containing syntaxin 1A/SNAP 25 (t-SNARE) were prewarmed to 37°C and mixed at time = 0 with prewarmed donor liposomes (5 μl) labeled with rhodamine and NBD lipids in the presence and absence of the cytosolic domain of VAMP 2 (cd–VAMP 2, aa 1–94, added in approximately equimolar amounts to the t-SNARE). The increase in NBD fluorescence was monitored for 2 h at 37°C, and the results were normalized to the maximum NBD fluorescence signal after addition of detergent (dodecylmaltoside). (C) Dose-dependent stimulation of fusion by Sytg I. Donor liposomes containing a constant amount of VAMP 2 but increasing amounts of Sytg I were incubated with acceptor t-SNARE liposomes, and the fusion was monitored and analyzed as described. Linear regression analysis (Cricket Graph III, curve fit) was performed using the first eight data points (first 14 min), and the initial rate of fusion (Δ % total fluorescence/Δ time) was plotted against the total amount of reconstituted Sytg I–VAMP 2.
Figure 6.

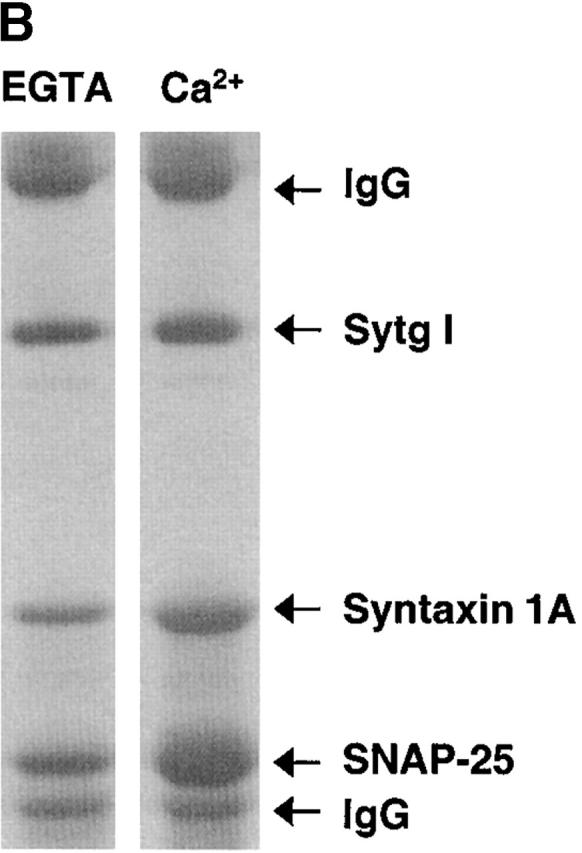
Sytg I–dependent acceleration of SNARE-mediated fusion is insensitive to calcium. (A) Kinetic profiles of membrane fusion of labeled Sytg I–VAMP 2 liposomes with unlabeled t-SNARE liposomes with 1 mM EGTA in the presence or absence of 2 mM CaCl2. The assay was performed as in the Fig. 1 legend. (B) Binding of t-SNARE (Syntaxin 1A/ SNAP 25) to Sytg I. Full-length t-SNARE (1 μM) was incubated with immobilized Sytg I in the presence of either 1 mM EGTA (EGTA) or 1 mM CaCl2 (Ca2+). t-SNARE binding was assayed by SDS-PAGE and with Coomassie blue staining. Controls lacking Sytg I failed to bind to t-SNARE (unpublished data). The positions of IgG, Sytg I, syntaxin 1A, and SNAP 25 are marked by arrows.
Soluble cd–Sytg I stimulates SNARE-dependent fusion
One possible explanation for the effect of Sytg I on fusion is that the t-SNARE binding activity of Sytg I potentiates vesicle binding, and therefore, the stimulatory effect on fusion is simply a consequence of the additional docking sites provided by Sytg I. To determine if this is the case, we constructed a cytosolic domain of synaptotagmin I (cd–Sytg I) that started immediately after the transmembrane region at aa 82. Notably, this construct differs from previously published soluble constructs, which start at aa 95 or 96 (Davis et al., 1999; Osborne et al., 1999). We then studied the ability of cd–Sytg I to accelerate fusion in an assay that is more sensitive to stimulatory components. For this purpose, we incorporated t-SNAREs into labeled donor liposomes and VAMP 2 into unlabeled acceptor liposomes. In addition, we reduced the amount of VAMP 2 incorporated into the acceptor liposomes ∼15-fold compared with the standard assay. This reduces the number of copies of VAMP 2 per liposome from ∼375 in the standard assay to ∼25 in the modified assay, a number comparable to the VAMP 2 concentration in synaptic vesicles (Jahn and Südhof, 1994; Weber et al., 1998).
The low concentration of v-SNAREs incorporated into the acceptor liposomes resulted in no observable fusion in the absence of cd–Sytg I within the 2-h incubation period (Fig. 2) . However, the addition of 9 μM cd–Sytg I significantly stimulated fusion. This stimulation was SNARE dependent, since it was inhibited by the presence of cd–VAMP 2. It should be noted that the overall concentration of synaptotagmin I in this assay is ∼10 fold higher and the ratio of synaptotagmin to t-SNARE is ∼30-fold higher than in the fusion assay shown in Fig. 1 B. A Sytg I construct starting at aa 95 gave similar results when added to the fusion assay (unpublished data). We also tested the membrane-spanning Sytg I construct in this modified assay and found comparable results to those seen in the standard assay (unpublished data). In summary, both the membrane anchored Sytg I and its cytosolic domain stimulated fusion. Our results using the cytosolic domain of synaptotagmin I exclude the possibility that the stimulatory effect is merely due to the presence of additional vesicle docking sites.
Figure 2.

Soluble cd–Sytg I accelerates SNARE-dependent fusion. Unlabeled acceptor liposomes (45 μl) containing VAMP 2 were prewarmed to 37°C and mixed at time = 0 with prewarmed labeled donor liposomes (5 μl) containing t-SNARE in the presence and absence of the cytosolic domain of Sytg I (cd–Sytg I, ∼10 μM final concentration) and cd–VAMP 2 (see Fig. 1B legend). Note that in assays treated with cd–VAMP 2, the donor liposomes were pretreated with cd–VAMP 2 for 15 min before their mixing with acceptor liposomes. The increase in NBD fluorescence was monitored, and the results were normalized as before.
The stimulatory effect of Sytg I on fusion is topologically restricted
Given that synaptotagmin I, which is localized on synaptic vesicles, cycles through the plasma membrane after exocytosis, Sytg I might also stimulate fusion when localized at the plasma membrane. To test this possibility, we reconstituted Sytg I into t-SNARE–containing unlabeled acceptor liposomes, which represent the plasma membrane in our assay (Fig. 3 A). In this topological arrangement, Sytg I did not stimulate the fusion reaction (Fig. 3 B), suggesting that the cis configuration of synaptotagmin I and t-SNARE does not allow a productive interaction. Thus, synaptotagmin I stimulates fusion only when present in trans to the t-SNARE (i.e., in the opposite membrane to the t-SNARE), demonstrating that the appropriate localization of Sytg I is crucial to its ability to enhance fusion.
Figure 3.

Sytg I reconstituted into t-SNARE liposomes does not accelerate fusion. (A) Acceptor liposomes containing t-SNARE (lane 1) or Sytg I and t-SNARE (lane 2) were analyzed by SDS-PAGE and stained with Coomassie blue. The positions of syntaxin 1A, SNAP 25, and Sytg I are marked by arrows. (B) Acceptor liposomes (45 μl) were prewarmed to 37°C and mixed at time = 0 with prewarmed labeled donor liposomes (5 μl) containing VAMP 2. The increase in NBD fluorescence was monitored, and the results were normalized as before.
Sytg I accelerates fusion-committed docking
To further understand how Sytg I regulates fusion, we analyzed the effect of Sytg I on different stages of the overall reaction. In the in vitro fusion assay, the following steps can be resolved (Fig. 4 A). First, t-SNARE complex activation involves the functional removal of the NH2-terminal domain of syntaxin 1A (NRD; Fig. 4 A) (Parlati et al., 1999). Subsequently, SNAREpins are formed between liposomes and “zip up” into a partially assembled helical bundle. This reaction is initially reversible and then becomes resistant to treatment with cd–VAMP 2 and neurotoxins (Weber et al., 1998; Melia et al., 2002). Low temperature (4°C) blocks membrane fusion allowing us to accumulate these SNAREpin intermediates (Weber et al., 1998). Finally, lipid bilayers fuse, and SNAREpins are fully assembled into cis-SNARE complexes in which membrane-spanning regions reside in the same membrane and their cytoplasmic domains form a “fully zipped up” four helical bundle.
Figure 4.


Cleavage of the NH 2 -terminal domain of syntaxin 1A does not effect Sytg I–dependent acceleration. (A) Steps in the in vitro fusion assay. First, the NRD (oval) of syntaxin 1A (represented here by the medium gray cylinder connected to the oval) moves to form an activated t-SNARE complex (SNAP 25 is represented by the two connected light gray cylinders). Next, VAMP 2 (single cylinder) interacts with the activated complex to form a fusion-competent docked SNARE complex. Finally, membrane fusion occurs accompanied by formation of the cis-SNARE complex. (B) Kinetic profiles of membrane fusion in the presence of Sytg I with t-SNARE missing the NRD. Unlabeled acceptor liposomes containing thrombin cleavable t-SNARE (N–t-SNARE) were treated with thrombin (tc) or buffer (Parlati et al., 1999). Fusion assays were performed as described in the Fig. 1 legend.
To test whether the stimulatory effect of synaptogmin I is comparable to that caused by the removal of NH2-terminal domain of syntaxin 1A, we used a t-SNARE construct containing syntaxin 1A with a thrombin-cleavable NH2-terminal domain (N–t-SNARE) (Parlati et al., 1999). Acceptor liposomes containing N–t-SNARE were prepared and treated with either thrombin (tc–N–t-SNARE) or AEBSF-inactivated thrombin (N–t-SNARE) immediately before addition into the fusion assay. As expected, cleavage of the NH2-terminal domain of syntaxin 1 increased the fusion efficiency in presence of VAMP 2 liposomes (Fig. 4 B) (Parlati et al., 1999). However, when the fusion kinetics of VAMP 2, tc-N–t-SNARE liposomes were compared with those of Sytg I–VAMP 2, N–t-SNARE liposomes, we observed that the NRD removal has a much less prominent effect than that caused by Sytg I (Fig. 4 B). Furthermore, cleavage of the inhibitory NH2-terminal domain of syntaxin 1A did not further enhance the initial kinetics of Sytg I–VAMP 2 liposome fusion, although it modestly increased the final fusion signal (Fig. 4 B). These results indicate that synaptotagmin I has a function that exceeds the mere removal of the NRD of syntaxin 1A and suggests a role in facilitating a later stage of SNARE complex assembly.
To examine if Sytg I affects the rate of SNARE assembly between liposomes, we captured docked complexes at 4°C (Weber et al., 1998) in the presence and absence of Sytg I and tested at which time they become resistant to the addition of cd–VAMP 2. This assay measures how far SNAREpin assembly has proceeded and indicates the time at which docked liposomes become fusion committed. Donor liposomes containing either VAMP 2 alone or Sytg I–VAMP 2 were incubated with acceptor t-SNARE liposomes at 4°C for varying amounts of time before cd–VAMP 2 was added (Fig. 5 A). All reactions were kept on ice until the final time point when the reactions were warmed to 37°C and fusion was allowed to take place. Liposomes containing Sytg I attained a fusion-committed docked state much faster than liposomes without Sytg I (Fig. 5, C and B, respectively). In fact, after only 1 h at 4°C Sytg I–VAMP 2 liposomes display the same signal that VAMP 2 liposomes attain after an overnight incubation. In contrast to VAMP 2 liposomes, which have a basal fusion activity of 5% of the total fusion signal at 4°C overnight, Sytg I–VAMP 2 liposomes showed an increased fusion potential at this low temperature (16% of the total fusion signal [unpublished data]). Thus, we conclude that Sytg I promotes the formation of fusion-committed SNAREpins. It may also catalyze SNARE-mediated membrane mixing, but that distinction is hard to make without the ability to examine individual fusion events.
Figure 5.



Sytg I accelerates fusion-committed docking. (A) Acceptor t-SNARE liposomes (45 μl) were mixed with donor liposomes (5 μl) containing VAMP 2 with and without Sytg I at 4°C. After time equals x, cd–VAMP 2 (equimolar to t-SNARE) was added, and the reaction was allowed to incubate at 4°C until the final time point. The mixture was then warmed to 37°C, and the reaction is monitored as described in Fig. 1 legend. (B) Kinetic profiles of fusion from a time course of functionally docked VAMP 2 liposomes. The assay was performed as outlined in A. Note that the normalized data ignores the preassay fusion and an initial drop in fluorescence, which is due to temperature effects on the fluorescent probes (Chapman et al., 1995a), is typically observed. In addition, please note that the time course for SNAREpin formation is concentration dependent. (C) Kinetic profiles of fusion from a time course of functionally docked VAMP 2–Sytg I liposomes. The assay was performed as outlined in A.
The stimulatory effect of Sytg I is calcium independent
So far, our experiments indicate that Sytg I stimulates fusion in absence of external calcium. However, since synaptotagmin I is the prime candidate for a neuronal calcium sensor, we analyzed the effect of calcium on the fusion reaction in the presence of Sytg I. To our surprise, neither the addition of EGTA nor an effective calcium concentration of ∼900 μM altered our basic results (Fig. 6 A). Compared with EGTA, calcium reproducibly caused a minor reduction of the initial fusion kinetics and a small increase in the extent of final fusion of the Sytg I–containing liposomes; however, the significance of these minor changes remains to be determined. We confirmed that our Sytg I construct retains the basic calcium-dependent properties, such as calcium-dependent liposome binding and oligomerization (unpublished data). We also reexamined the binding of Sytg I to t-SNARE. Due to the calcium-dependent oligomerization of the synaptotagmin construct, we prebound Sytg I to protein A beads using the anti-Sytg monoclonal antibody M48 and then added t-SNARE in the presence or absence of calcium. At saturating concentrations of t-SNARE (1 μM), significant binding between Sytg I and t-SNARE was observed in the absence and presence of calcium; however, calcium still increased the overall interactions (Fig. 6 B). We obtained similar results with the cd–Sytg I construct starting at aa 82 and with another anti-Sytg monoclonal antibody CI. 41.1 (unpublished data). These results are in agreement with data previously obtained with full-length native synaptotagmin I (Chapman et al., 1995b; Gerona et al., 2000). This largely calcium-independent synaptogamin I–t-SNARE interaction may provide the basis for the calcium-independent stimulatory effect on fusion that we observed.
Discussion
Synaptotagmin I plays a critical role in calcium-evoked neurotransmitter release, and accumulating evidence indicates that it functions as the major calcium sensor in neurons. SNARE complex formation, the process underlying exocytosis, is highly regulated in neuronal systems with synaptotagmin I forming part of the regulatory machinery. Herein we demonstrate that synaptotagmin I profoundly accelerates SNARE mediated membrane fusion in a topologically restricted manner, even in the absence of calcium.
SNARE complex formation in neurons begins with the controlled formation of the t-SNARE, a process which is negatively regulated by the NRD and other syntaxin binding proteins such as Munc 18-1 (Dulubova et al., 1999; Misura et al., 2000; Fisher et al., 2001). The NRD block must be released to allow t-SNARE formation, a process most likely mediated by a component such as Munc 13 (Betz et al., 1997). In the process of t-SNARE formation, the NRD block on SNAREpin formation seen in our fusion assay is most likely relieved. After t-SNARE formation on the plasma membrane, SNARE complexes assemble into a four-helix bundle in a zippering reaction that may involve multiple steps, a theory supported by several in vivo studies (Hua and Charlton, 1999; Xu et al., 1999). SNARE complex formation starts at the membrane-distal end of the SNARE motif and may be slowed down by the inherent instability of the membrane-proximal domains of the t-SNARE (Fiebig et al., 1999; Melia et al., 2002) and the increasingly repulsive forces between the lipid bilayers. Although membrane fusion will eventually proceed, the rate may be too slow to ensure fast regulated exocytosis, and additional components accelerating the reaction may be required (Weber et al., 1998; Fasshauer et al., 2002).
Our data indicate that synaptotagmin I could be such an accelerating component. Synaptotagmin I accelerates the initial rate of fusion at least by a factor of five (Fig. 1 C). These results are consistent with the recent observation that synaptotagmin I promotes the assembly of cytoplasmic SNARE domains in vitro (Littleton et al., 2001). Synaptotagmin overcomes the block caused by the NRD (Fig. 4 B) and, more significantly, accelerates SNAREpin formation (Fig. 5). Accordingly, synaptotagmin I converts the initially reversible SNAREpin into a fusion-committed state that can no longer be inhibited by the addition of cd–VAMP 2. In the absence of synaptotagmin I and at low temperatures, the initial v-liposome–t-liposome interaction occurs within minutes, but it takes several hours or an overnight incubation for the reversible SNAREpin interaction to become resistant to neurotoxins or to the inhibitory cytoplasmic domain of VAMP 2 (Weber et al., 1998). The latter reaction, which is greatly enhanced by synaptotagmin I, may involve the membrane-proximal domain of the t-SNARE. Indeed, it has been shown that synaptotagmin I interacts with the membrane-proximal part of the syntaxin 1 SNARE motif and the COOH-terminus of SNAP-25 (Chapman et al., 1995b; Kee and Scheller, 1996; Davis et al., 1999; Gerona et al., 2000). Induction or stabilization of helical confirmations in these membrane-proximal domains could facilitate SNAREpin zipping and thereby accelerate fusion. Interestingly, it has been shown that a COOH-terminal VAMP-2 peptide that binds to the membrane-proximal region of the t-SNARE structures these domains and accelerates fusion (Melia et al., 2002). Structural analysis of a synaptotagmin I–t-SNARE complex will be required to understand the reaction mechanism in detail.
The observed topological restriction of the stimulatory effect has important physiological consequences (Fig. 3). The inherent physicochemical properties of synaptotagmin I ensure that only the vesicle carrying the calcium sensor is subjected to accelerated fusion and therefore calcium regulation. In other words, the presence of synaptotagmin I on the plasma membrane would not affect the fusion of vesicles lacking synaptogamin I (e.g., constitutive transport vesicles), thus adding an addition level of control over which vesicles are primed for calcium-dependent fusion.
However, surprisingly the stimulatory effect of synaptotagmin was calcium independent. At first glance, this result seems to directly contradict the concept that synaptotagmin I is either an inhibitory clamp (hypothesis 1) or a calcium-dependent promoter of fusion (hypothesis 2). Indeed, the stimulatory effect of synaptotagmin I is incompatible with the clamp hypothesis. However, we cannot completely exclude that calcium might further stimulate the reaction. After calcium influx into the nerve terminal, the first synaptic vesicle fuses within less than 1 ms, indicating that SNAREpins are already preassembled. If synaptotagmin I–accelerated SNAREpin assembly is the rate-limiting step in our fusion assay, we would not be able to detect any faster reactions that may follow. We attempted to overcome this potentially rate-limiting step by accumulating prefusion intermediates in presence of synaptotagmin at 4°C and then adding calcium during the warm-up phase. Even under these conditions we could not detect a significant stimulatory effect of calcium (unpublished data). This raises the possibility that synaptotagmin I and SNAREs alone are not sufficient to mediate calcium-regulated exocytosis, and additional components, such as lipids or proteins (perhaps even a different synaptotagmin isoform), may be required. Indeed, genetic evidence has indicated that additional components are necessary (Kelly, 1995). In summary, our data demonstrates that synaptogamin I plays an important accelerating role in SNAREpin assembly and membrane fusion. Future experiments will reveal whether the addition of further components to the reconstituted assay will confer calcium sensitivity.
Materials and methods
All lipids were from Avanti with the exception of [3H]-1,2-dipalmitoyl phosphatidylcholine ([3H]-DPPC), which is from Amersham Pharmacia Biotech. The anti–synaptotagmin I monoclonal antibody CI.41.1 was from Synaptic Systems.
Plasmid constructions
Construction of membrane-anchored synaptotagmin I.
The cDNA encoding full-length synaptotagmin I was amplified by PCR using a rat brain λ-GT11 cDNA library (CLONTECH Laboratories, Inc.) and the following oligonucleotides: (a) GGGGGATCCATGGTGAGTGCCAGTCATCC and (b) GGGGAGCTCTTACTTCTTGACAGCCAGCATGG and cloned into PCR-Script (Stratagene) according to the manufacturer's instructions. This construct was digested with PstI and EcoRI, and a double-stranded oligonucleotide of phosphorylated oligonucleotides (c) GGTAGAGGAGGAGGTTGATGCCATGCTGGCTGTCAAGAAGGAGCTCCTCGAGG and (d) AATTCCTCGAGGAGCTCCTTCTTGACAGCCAGCATGGCATCAACCTCCTCCTCTAC- CTGCA was inserted by ligation yielding pTW25, thus removing the stop codon. The coding sequence for synaptotagmin I was excised by digestion of pTW25 with NcoI and XhoI. This fragment was ligated into pET-28b (Novagen) digested with the same enzymes yielding pTW27 encoding full-length synaptotagmin I with a COOH-terminal his6 tag. To remove the luminal domain, pTW27 was digested with NcoI and KpnI. The PCR product of oligonucleotides (e) AGATCTCCATGGGTCCGTGGGCCTTAATAGCTATAGCCATAGTTGCGGTCC and (f) CTAATTCCGAGTAGGGTACCTTGAAAGTAAATTGTTC and template pTW27 was digested with the same enzymes and then ligated into the cut pTW27 yielding pTW70. This construct encodes synaptotagmin I (under the control of a T7 promoter) without its lumenal domain (aa 1–56) but with a COOH-terminal his6 tag. In addition, the protein encoded is an isoform in which calcium-dependent oligomerization is abolished (Desai et al., 2000) and contains an amino acid mutation at aa 188 (Glu to Asp), which correlates to a conserved residue in all other synaptotagmins (note that numbering follows that of the whole protein as defined in Perin et al. [1990]). To restore calcium-dependent oligomerization, we mutated the aspartate at aa 374 to a glycine residue (Desai et al., 2000) using site-directed mutagenesis (QuikChange kit; Stratagene) and the following primers: (g) GGCAAGAACGACGCATC- GGCAAAGTCTTCGTTGGTTAC and (h) GTAACCAACGAAGACTTTGCCGATGGCGTCGTTCTTGCC to yield plasmid pLM1.
We then introduced the following mutations into pLM1: (1) Cys 75 to Ala using primers (a) GTCCTTTTAGTCGTAACCTG GCCTTTTGTGTCTGTAAGAAATG and (b) CATTTCTTACAGACACAAAAGGCGCAGGTTACGACTAAAAGGAC, (2) Cys 79 to Ile using primers (a) CCTGCGCCTTTTGTGTCATTAAGAAATGTTTGTTC and (b) GAACAAACATTTCTTAATGACACAAAAGGCGCAGG, (3) Cys 82 to Leu using primers (a) CCTTTTGTGTCATTAAGAAATTGTTGTTCAAAAAGAAAAAC and (b) GTTTTTCTTTTTGAACAACAATTTCTTAATGACACAAAAGG, (4) Cys 74 to Ser using primers (a) GTCCTTTTAGTCGTAACCTCCGCCTTTTGTGTCATTAAG, and (b) CTTAATGACACAAAAGGCGGAGGTTACGACTAAAAGGAC, and (5) Cys 77 to Ser using primers (a) GTCGTAACCTCCGCCTTTTCTGTCATTAAGAAATTGTTG and (b) CAACAATTTCTTAATGACAGAAAAGGCGGAGGTTACGAC, to yield the final plasmid pLM6 encoding a synaptotagmin I capable of calcium-dependent oligomerization with no cysteines in the transmembrane region. These transmembrane domain cysteines have been shown to form disulfide bonds in lipid bilayers in vitro when recombinant protein is used (Bai et al., 2000; Fukuda and Mikoshiba, 2000). However, this does not correspond to the condition in vivo, since native synaptotagmin I does not form these disulfide bonds due to the stoichiometric palmitoylation of these cysteines (Veit et al., 1996; Bai et al., 2000). Thus, we mutated the cysteines in the transmembrane domain to the C. elegans sequence and substituted serine for the two remaining Cys residues (aa 74 and 77) to create pLM6.
Construction of cytoplasmic synaptotagmin I plasmids.
To generate a plasmid that encodes the cytoplasmic synaptotagmin I domain starting at aa Cys 82, the pLM1 template and the following primers were used for PCR: (a) GGGCATATGTGTTTGTTCAAAAAGAAAAACAAGAAGAAGGGGAAGGAAAAGGGAGGAAAGAACGC and (b) TTTCTCGAGCTTCTTGACAGCCAGCATGGCATCAACCTCCTCCTCTA. The PCR product was digested with NdeI and XhoI and ligated into the pET 24 vector, which codes for a COOH-terminal his6 Tag yielding the plasmid pLM7.
The plasmid pLM8, encoding a soluble synaptotagmin I beginning at Lys 95, was made in an identical manner to pLM7 but with the following primers: (a) GGGCATATGAAGGGAGGAAAGAACGCCATTAAC and (b) TTTCTCGAGCTTCTTGACAGCCAGCATGGCATC.
Protein expression and purification
To obtain Sytg I, pLM6 was transformed into BL21 DE3 pLysS tuner cells (Novagen). The cells of 1 liter of overnight preculture in superbroth containing 50 μg/ml kanamycin and 35 μg/ml chloramphenicol were used to start 4 × 2–liter cultures in superbroth containing 50 μg kanamycin. The cultures were induced with 0.5 mM IPTG when an optical density (OD600 nm) of 0.8 was reached. After 3 h, the bacteria were sedimented by centrifugation, washed once in D-PBS (2.67 mM KCl, 1.47 mM KH2PO4, 138 mM NaCl, 8.10 mM Na2HPO4 · 7H20), and resuspended in breaking buffer (25 mM Hepes · KOH, pH 7.4, 400 mM KCl, 5 mM β-mercaptoethanol, 1 mM MgCl2, 0.01 mM CaCl2, 10% glycerol). To this cell suspension was added a protease inhibitor cocktail (final concentrations: 1.2 μg/ml leupeptin, 2 μg/ml antipain, 20 μg/ml turkey trypsin inhibitor, 10 μg/ml benzamidine, 5 μg/ml pefabloc SC, 8.2 TIC/L aprotinin, 5 μg/ml chymostatin, 2.5 μg/ml pepstatin) and 1/4 vol 20% (wt/vol) Triton X-100. The suspension was then passed three times through an Avestin cell disrupter at >5,000 psi, and the resulting mixture was centrifuged for 1 h at 35,000 rpm in a Ti45 rotor (Beckman Coulter). The supernatant was incubated for 1.5 h at 4°C with 3 ml Ni-NTA agarose equilibrated in breaking buffer. The beads were washed twice in breaking buffer containing 1% Triton X-100. The beads were then extensively washed (∼10 column vol) with buffer A (25 mM Hepes · KOH, pH 7.4, 100 mM KCl, 5 mM β-mercaptoethanol, 1% octyl-β-d-glucopyranoside (βOG), 10% glycerol) with 20 mM imidazole to remove nonspecifically bound proteins. Elution of the desired protein from the Ni-NTA beads was accomplished using a linear gradient from 20 mM to 1 M imidazole in buffer A.
For the expression of cd–Sytg I, BL21DE3 (Novagen) were transformed with construct pLM7. Two 1-liter cultures of LB containing 50 μg/ml kanamycin were inoculated with 100 ml each of overnight precultures of transformed cells (2 × 100 ml LB media containing 50 μg/ml kanamycin). Cells were grown to an OD (600 nm) of 0.6 and induced with 1 mM IPTG. After 3 h, cells were pelleted, washed with PBS, and resuspended in breaking buffer containing protease inhibitor cocktail. Protein purification procedures were the same as for Sytg I with the following exceptions: (a) no detergent (Triton X-100 or βOG) was used and (b) the gradient was from 50 mM imidazole to 500 mM imidazole. Expression and purification of protein from construct pLM8 was performed in an identical manner to cd–Sytg I.
Full-length t-SNARE complex (mouse his6-SNAP 25 and rat syntaxin 1A) was expressed and purified from vector pTW34 as described previously (Weber et al., 1998). Thrombin-cleavable (tc) t-SNARE complex (mouse his6-SNAP 25 and tc-syntaxin 1A) was expressed and purified from vector pTW69 as described previously (Parlati et al., 1999). Full-length mouse VAMP-2 was expressed and purified from vector pTW2 as described previously (Weber et al., 1998).
Lipid mixtures
Donor lipid mix.
Donor lipid mix contains 83.3 mol% 1-palmitoyl-2-oleoyl-SN-glycero-3-phosphatidylcholine (POPC), 15.1 mol% 1,2-dioleoyl-SN-glycero-3-phosphatidylserine (DOPS), 0.8 mol% R-PE, 0.8 mol% NBD-PE and trace amounts of [3H]-DPPC, and 3 mM total lipid.
Acceptor lipid mix.
Acceptor lipid mix contains mol% POPC, 15 mol% DOPS and trace [3H]-DPPC, and 15 mM total lipid.
POPC only lipid mix (for lipid binding assay).
POPC only lipid mix contains 100 mol% POPC and trace amounts of [3H]-DPPC, and 15 mM total lipid.
POPC/DOPS lipid mix (for lipid binding assay).
POPC/DOPS lipid mix contains 75 mol% POPC, 25 mol% DOPS and trace amounts of [3H]-DPPC, and 15 mM total lipid.
Protein reconstitution into liposomes and thrombin cleavage of t-SNARE liposomes
Liposomes were formed in the presence of VAMP 2 (0.7–1 mg/ml), Sytg I (1 mg/ml), and t-SNARE (1.5–3 mg/ml) in various combinations using the previously described technique of dilution and dialysis followed by a Nycodenz gradient (Weber et al., 1998) with the donor and acceptor lipid mixes defined above. Note that for unlabeled v-SNARE liposomes, the quantity of VAMP2 used was much lower (0.09 mg/ml). Protein amounts in reconstituted liposomes were determined using Coomassie blue–stained SDS-PAGE with protein standards and Quantity One Quantitation Software (Bio-Rad Laboratories). The NH2-terminal domain of syntaxin was removed by thrombin cleavage as described previously (Parlati et al., 1999).
Fusion assays
Fusion reactions and data analysis were performed as described previously (Weber et al., 1998) with the following exceptions: (a) In all cases, 45 μl of acceptor (unlabeled) and 5 μl of donor (labeled) liposomes were used, (b) unless otherwise noted, both acceptor and donor liposomes were prewarmed to 37°C before mixing, and (c) to minimize quenching, 10 ml of 2.5% (wt/vol) dodecylmaltoside instead of Triton X-100 was added at the end of the fusion reaction.
Lipid binding assay
For each vesicle preparation (POPC only or POPC/DOPS), 100 μl of lipid solution (see POPC only and POPC/DOPS lipid mixtures above) was dried down in a 10 × 75 glass test tube by a stream of nitrogen, and trace amounts of chloroform were removed under vacuum for 1 h. The dried lipids were resuspended in 500 ml reconstitution buffer A (R buffer, 25 mm Hepes · KOH, pH 7.4), 100 mM KCl) and subjected to seven cycles of freezing (liquid N2) and thawing (warm water). The lipid mixture was then extruded through a 50 nm polycarbonate membrane using Liposofast™-Basic (Avestin). The extruded liposomes were centrifuged at 25,000 rpm for 20 min in a Ti 100.3 (Beckman Coulter) rotor to pellet any aggregates, and the liposome-rich supernatant was harvested and used for the binding assay.
Sytg I beads or control beads were prepared by the immunoprecipitation of Sytg I (110.6 mg) or buffer (control beads) with monoclonal antibody M48 (10 μl, ascites fluid) and immobilization onto protein G beads (100 μl; Amersham Pharmacia Biotech) in the presence of R buffer. Beads were diluted to give a final concentration of 20%.
For each sample, 50 μl of 20% beads (Sytg I or control) were transferred to a 1.5 ml Eppendorf tube, pelleted, and the supernatant was removed. To these beads were added the appropriate liposomes (POPC only or POPC/DOPS, 100,000 cpm, ∼1 mM final lipid concentration), EGTA (2 mM final concentration), where appropriate calcium in the form of calcium chloride (2.102 mM final concentration, 100 μM effective concentration), and R buffer to give a final volume of 100 μl. The mixtures were incubated 30 min at RT with rotation. The beads were then pelleted, washed with 3 × 1 ml R buffer (containing either 100 μM EGTA-buffered calcium or 2 mM EGTA as appropriate), and solubilized in 100 μl 10% SDS solution. The mixture was transferred to scintillation vials containing Scintiverse (10 ml; Fischer), and the 3H radioactivity was counted (5 min per vial; Beckman Coulter LS6001C).
t-SNARE binding assay
Sytg I (107 μg) was immobilized on protein A beads (100 μl beads; Amersham Pharmacia Biotech) by immunoprecipitation with monoclonal antibody CI.41.1 (10 μl) in buffer T (20 mM Tris · HCl, pH 7.4, 150 mM NaCl, 0.5% Triton X-100) with 1 mg/ml BSA. The beads were diluted to give a final concentration of 20%, split into two pools, and washed with buffer T containing 1 mg/ml BSA and either 1 mM EGTA or 1 mM CaCl2 (Ca). Beads were again diluted to 20% in the appropriate buffer. For each sample, 50 μl of 20% beads (Ca or EGTA) were transferred to a 1.5 ml Eppendorf tube, and full-length t-SNARE complex (30 μg) was added. Reaction volume was adjusted to a final volume of 500 μl (1 μM final concentration t-SNARE). The mixtures were incubated at 4°C for 1 h with shaking, pelleted, and washed 3 × 1 ml with buffer T containing either Ca or EGTA. The beads were then pelleted, supernatant was removed, and 15 μl of 1 × SDS PAGE sample buffer (50 mM Tris · HCl, pH 6.8, 2% SDS, 0.1% bromphenol blue, 10% glycerol, 100 mM DTT) was added. Samples were heated at 95°C for 10 min and analyzed by SDS-PAGE. The experiment was also done using cd–Sytg I and monoclonal antibody M48 in an identical manner.
Acknowledgments
We thank Dr. Thomas Weber (Mount Sinai School of Medicine, New York, NY) for his generous gift of pTW70. In addition, we thank Stéphanie Bourdelle for help with the completion of the article and Dr. Oleg Varlamov for critical reading of the article.
Research was supported by postdoctoral fellowships from the Jane Coffin Childs Memorial Fund (to L.K. Mahal) and the Portuguese Foundation for Science and Technology (to S.M. Sequeira).
Footnotes
Abbreviations used in this paper: NBD, nitro-2,1,3-benzoxadiazole 1,2-dipalmitoyl-SN-glycero-3; NRD, NH2-terminal regulatory domain; PE, phosphatidylethanolamine.
References
- Augustine, G.J. 2001. How does calcium trigger neurotransmitter release? Curr. Opin. Neurobiol. 11:320–326. [DOI] [PubMed] [Google Scholar]
- Bai, J., C.A. Earles, J.L. Lewis, and E.R. Chapman. 2000. Membrane-embedded synaptotagmin penetrates cis or trans target membranes and clusters via a novel mechanism. J. Biol. Chem. 275:25427–25435. [DOI] [PubMed] [Google Scholar]
- Bennett, M.K., N. Calakos, and R.H. Scheller. 1992. Syntaxin: a synaptic protein implicated in docking of synaptic vesicles at presynaptic active zones. Science. 257:255–259. [DOI] [PubMed] [Google Scholar]
- Betz, A., M. Okamoto, F. Benseler, and N. Brose. 1997. Direct interaction of the rat unc-13 homologue Munc13-1 with the N terminus of syntaxin. J. Biol. Chem. 272:2520–2526. [DOI] [PubMed] [Google Scholar]
- Broadie, K., H.J. Bellen, A. DiAntonio, J.T. Littleton, and T.L. Schwarz. 1994. Absence of synaptotagmin disrupts excitation-secretion coupling during synaptic transmission. Proc. Natl. Acad. Sci. USA. 91:10727–10731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brose, N., A.G. Petrenko, T.C. Südhof, and R. Jahn. 1992. Synaptotagmin: a calcium sensor on the synaptic vesicle surface. Science. 256:1021–1025. [DOI] [PubMed] [Google Scholar]
- Chapman, C.F., Y. Liu, G.J. Sonek, and B.J. Tromberg. 1995. a. The use of exogenous fluorescent probes for temperature measurements in single living cells. Photochem. Photobiol. 62:416–425. [DOI] [PubMed] [Google Scholar]
- Chapman, E.R., P.I. Hanson, S. An, and R. Jahn. 1995. b. Ca2+ regulates the interaction between synaptotagmin and syntaxin 1. J. Biol. Chem. 270:23667–23671. [DOI] [PubMed] [Google Scholar]
- Chen, Y.A., S.J. Scales, and R.H. Scheller. 2001. Sequential SNARE assembly underlies priming and triggering of exocytosis. Neuron. 30:161–170. [DOI] [PubMed] [Google Scholar]
- Davis, A.F., J. Bai, D. Fasshauer, M.J. Wolowick, J.L. Lewis, and E.R. Chapman. 1999. Kinetics of synaptotagmin responses to Ca2+ and assembly with the core SNARE complex onto membranes. Neuron. 24:363–376 (erratum published 24:1049). [DOI] [PubMed] [Google Scholar]
- Desai, R.C., B. Vyas, C.A. Earles, J.T. Littleton, J.A. Kowalchyck, T.F. Martin, and E.R. Chapman. 2000. The C2B domain of synaptotagmin is a Ca(2+)-sensing module essential for exocytosis. J. Cell Biol. 150:1125–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulubova, I., S. Sugita, S. Hill, M. Hosaka, I. Fernandez, T.C. Südhof, and J. Rizo. 1999. A conformational switch in syntaxin during exocytosis: role of munc18. EMBO J. 18:4372–4382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earles, C.A., J. Bai, P. Wang, and E.R. Chapman. 2001. The tandem C2 domains of synaptotagmin contain redundant Ca2+ binding sites that cooperate to engage t-SNAREs and trigger exocytosis. J. Cell Biol. 154:1117–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fasshauer, D., W. Antonin, V. Subramaniam, and R. Jahn. 2002. SNARE assembly and disassembly exhibit a pronounced hysteresis. Nat. Struct. Biol. 9:144–151. [DOI] [PubMed] [Google Scholar]
- Fernández-Chacon, R., A. Konigstorfer, S.H. Gerber, J. Garcia, M.F. Matos, C.F. Stevens, N. Brose, J. Rizo, C. Rosenmund, and T.C. Südhof. 2001. Synaptotagmin I functions as a calcium regulator of release probability. Nature. 410:41–49. [DOI] [PubMed] [Google Scholar]
- Fiebig, K.M., L.M. Rice, E. Pollock, and A.T. Brunger. 1999. Folding intermediates of SNARE complex assembly. Nat. Struct. Biol. 6:117–123. [DOI] [PubMed] [Google Scholar]
- Fisher, R.J., J. Pevsner, and R.D. Burgoyne. 2001. Control of fusion pore dynamics during exocytosis by Munc18. Science. 291:875–878. [DOI] [PubMed] [Google Scholar]
- Fukuda, M., and K. Mikoshiba. 2000. Calcium-dependent and -independent hetero-oligomerization in the synaptotagmin family. J. Biochem. (Tokyo). 128:637–645. [DOI] [PubMed] [Google Scholar]
- Fukuda, R., J.A. McNew, T. Weber, F. Parlati, T. Engel, W. Nickel, J.E. Rothman, and T.H. Söllner. 2000. Functional architecture of an intracellular membrane t-SNARE. Nature. 407:198–202. [DOI] [PubMed] [Google Scholar]
- Geppert, M., Y. Goda, R.E. Hammer, C. Li, T.W. Rosahl, C.F. Stevens, and T.C. Südhof. 1994. Synaptotagmin I: a major Ca2+ sensor for transmitter release at a central synapse. Cell. 79:717–727. [DOI] [PubMed] [Google Scholar]
- Gerona, R.R., E.C. Larsen, J.A. Kowalchyk, and T.F. Martin. 2000. The C terminus of SNAP25 is essential for Ca(2+)-dependent binding of synaptotagmin to SNARE complexes. J. Biol. Chem. 275:6328–6336. [DOI] [PubMed] [Google Scholar]
- Hua, S.-Y., and M.P. Charlton. 1999. Activity-dependent changes in partial VAMP complexes during neurotransmitter release. Nat. Neurosci. 2:1078–1083. [DOI] [PubMed] [Google Scholar]
- Jahn, R., and T.C. Südhof. 1994. Synaptic vesicles and exocytosis. Annu. Rev. Neurosci. 17:219–246. [DOI] [PubMed] [Google Scholar]
- Kee, Y., and R.H. Scheller. 1996. Localization of synaptotagmin-binding domains on syntaxin. J. Neurosci. 16:1975–1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly, R.B. 1995. Neural transmission. Synaptotagmin is just a calcium sensor. Curr. Biol. 5:257–259. [DOI] [PubMed] [Google Scholar]
- Littleton, J.T., M. Stern, K. Schulze, M. Perin, and H.J. Bellen. 1993. Mutational analysis of Drosophila synaptotagmin demonstrates its essential role in Ca(2+)-activated neurotransmitter release. Cell. 74:1125–1134. [DOI] [PubMed] [Google Scholar]
- Littleton, J.T., J. Bai, B. Vyas, R. Desai, A.E. Baltus, M.B. Garment, S.D. Carlson, B. Ganetzky, and E.R. Chapman. 2001. Synaptotagmin mutants reveal essential functions for the C2B domain in Ca2+-triggered fusion and recycling of synaptic vesicles in vivo. J. Neurosci. 21:1421–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNew, J.A., T. Weber, F. Parlati, R.J. Johnston, T.J. Melia, T.H. Söllner, and J.E. Rothman. 2000. Close is not enough: SNARE-dependent membrane fusion requires an active mechanism that transduces force to membrane anchors. J. Cell Biol. 150:105–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melia, T.J., T. Weber, J.A. McNew, L.E. Fisher, R.J. Johnston, F. Parlati, L.K. Mahal, T.H. Söllner, and J.E. Rothman. 2002. Regulation of membrane fusion by conformational switching of the membrane-proximal coil of the t-SNARE during zippering of SNAREpins. J. Cell Biol. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misura, K.M., R.H. Scheller, and W.I. Weis. 2000. Three-dimensional structure of the neuronal-Sec1-syntaxin 1a complex. Nature. 404:355–362. [DOI] [PubMed] [Google Scholar]
- Nickel, W., T. Weber, J.A. McNew, F. Parlati, T.H. Söllner, and J.E. Rothman. 1999. Content mixing and membrane integrity during membrane fusion driven by pairing of isolated v-SNAREs and t-SNAREs. Proc. Natl. Acad. Sci. USA. 96:12571–12576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonet, M.L., K. Grundahl, B.J. Meyer, and J.B. Rand. 1993. Synaptic function is impaired but not eliminated in C. elegans mutants lacking synaptotagmin. Cell. 73:1291–1305. [DOI] [PubMed] [Google Scholar]
- Osborne, S.L., J. Herreros, P.I. Bastiaens, and G. Schiavo. 1999. Calcium-dependent oligomerization of synaptotagmins I and II. Synaptotagmins I and II are localized on the same synaptic vesicle and heterodimerize in the presence of calcium. J. Biol. Chem. 274:59–66. [DOI] [PubMed] [Google Scholar]
- Parlati, F., T. Weber, J.A. McNew, B. Westermann, T.H. Söllner, and J.E. Rothman. 1999. Rapid and efficient fusion of phospholipid vesicles by the alpha- helical core of a SNARE complex in the absence of an N-terminal regulatory domain. Proc. Natl. Acad. Sci. USA. 96:12565–12570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parlati, F., J.A. McNew, R. Fukuda, R. Miller, T.H. Söllner, and J.E. Rothman. 2000. Topological restriction of SNARE-dependent membrane fusion. Nature. 407:194–198. [DOI] [PubMed] [Google Scholar]
- Perin, M.S., V.A. Fried, G.A. Mignery, R. Jahn, and T.C. Südhof. 1990. Phospholipid binding by a synaptic vesicle protein homologous to the regulatory region of protein kinase C. Nature. 345:260–263. [DOI] [PubMed] [Google Scholar]
- Schiavo, G., Q.M. Gu, G.D. Prestwich, T.H. Söllner, and J.E. Rothman. 1996. Calcium-dependent switching of the specificity of phosphoinositide binding to synaptotagmin. Proc. Natl. Acad. Sci. USA. 93:13327–13332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiavo, G., G. Stenbeck, J.E. Rothman, and T.H. Söllner. 1997. Binding of the synaptic vesicle v-SNARE, synaptotagmin, to the plasma membrane t-SNARE, SNAP-25, can explain docked vesicles at neurotoxin-treated synapses. Proc. Natl. Acad. Sci. USA. 94:997–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Söllner, T., S.W. Whiteheart, M. Brunner, H. Erdjument-Bromage, S. Geromanos, P. Tempst, and J.E. Rothman. 1993. SNAP receptors implicated in vesicle targeting and fusion. Nature. 362:318–324. [DOI] [PubMed] [Google Scholar]
- Struck, D.K., D. Hoekstra, and R.E. Pagano. 1981. Use of resonance energy transfer to monitor membrane fusion. Biochemistry. 20:4093–4099. [DOI] [PubMed] [Google Scholar]
- Südhof, T.C. 2002. Synaptotagmins: why so many? J. Biol. Chem. 277:7629–7632. [DOI] [PubMed] [Google Scholar]
- Südhof, T.C., and J. Rizo. 1996. Synaptotagmins: C2-domain proteins that regulate membrane traffic. Neuron. 17:379–388. [DOI] [PubMed] [Google Scholar]
- Sutton, R.B., D. Fasshauer, R. Jahn, and A.T. Brunger. 1998. Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 A resolution. Nature. 395:347–353. [DOI] [PubMed] [Google Scholar]
- Veit, M., T.H. Söllner, and J.E. Rothman. 1996. Multiple palmitoylation of synaptotagmin and the t-SNARE SNAP-25. FEBS Lett. 385:119–123. [DOI] [PubMed] [Google Scholar]
- Wang, C.-T., R. Grishanin, C.A. Earles, P.Y. Chang, T.F.J. Martin, E.R. Chapman, and M.B. Jackson. 2001. Synaptotagmin modulation of fusion pore kinetics in regulated exocytosis of dense-core vesicles. Science. 294:1111–1115. [DOI] [PubMed] [Google Scholar]
- Weber, T., B.V. Zemelman, J.A. McNew, B. Westermann, M. Gmachl, F. Parlati, T.H. Söllner, and J.E. Rothman. 1998. SNAREpins: minimal machinery for membrane fusion. Cell. 92:759–772. [DOI] [PubMed] [Google Scholar]
- Weber, T., F. Parlati, J.A. McNew, R.J. Johnston, B. Westermann, T.H. Söllner, and J.E. Rothman. 2000. SNAREpins are functionally resistant to disruption by NSF and alphaSNAP. J. Cell Biol. 149:1063–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, T., B. Rammner, M. Margittai, A.R. Artalejo, E. Neher, and R. Jahn. 1999. Inhibition of SNARE complex assembly differentially affects kinetic components of exocytosis. Cell. 99:713–722. [DOI] [PubMed] [Google Scholar]