

Abstract
p53 is a transcription factor that induces growth arrest or apoptosis in response to cellular stress. To identify new p53-inducible proapoptotic genes, we compared, by differential display, the expression of genes in spleen or thymus of normal and p53 nullizygote mice after γ-irradiation of whole animals. We report the identification and characterization of human and mouse Scotin homologues, a novel gene directly transactivated by p53. The Scotin protein is localized to the ER and the nuclear membrane. Scotin can induce apoptosis in a caspase-dependent manner. Inhibition of endogenous Scotin expression increases resistance to p53-dependent apoptosis induced by DNA damage, suggesting that Scotin plays a role in p53-dependent apoptosis. The discovery of Scotin brings to light a role of the ER in p53-dependent apoptosis.
Keywords: transactivation; p53-binding site; cell death; cancer; 3p21
Introduction
Mutations that inactivate the p53 tumor suppressor protein are the most common genetic aberrations known to occur in human cancers. The best described biological functions of p53 are the induction of cell cycle arrest and apoptosis in response to cellular stresses (May and May, 1999; Oren, 1999; Vogelstein et al., 2000). Therefore, p53 prevents damaged cells from proliferating and thus protects against neoplastic transformation. Thus, p53 functions as a “guardian of the genome” (Lane, 1992).
p53 can act as a transcription factor that, in response to cellular stress, binds DNA in a sequence-specific manner and induces the expression of genes containing a p53 binding-site element in their promoter or introns (El-Deiry et al., 1992; Funk et al., 1992; Bourdon et al., 1997). Only a few genes have been shown to be directly transactivated, in vivo, by wild-type (wt)* p53 after cellular stress. Identification of transcriptional targets of p53 is critical in discerning pathways by which p53 affects global cellular outcomes such as growth arrest and cell death. The identification of the cyclin-dependent kinase inhibitor WAF1 as a p53-responsive gene helps to explain how p53 can induce cell cycle arrest. In contrast, the biochemical basis of p53-dependent apoptosis is still unclear. Depending on the experimental models used, p53 transcriptional activity has been reported to be required (Attardi et al., 1996; Yonish-Rouach et al., 1996; Jimenez et al., 2000) or dispensable (Caelles et al., 1994; Haupt et al., 1995) for p53-dependent apoptosis. Recently, several p53-inducible genes have been identified that encode proteins with apoptotic potential (Bax, CD95/Fas, Noxa, Pidd, P53AIP, and PUMA) (Miyashita and Reed, 1995; Muller et al., 1998; Lin et al., 2000; Munsch et al., 2000; Nakano and Vousden, 2001; E. Oda et al., 2000; K. Oda et al., 2000; Yu et al., 2001). However, it remains to be seen whether one or a subset of the newly identified genes play a key role in p53-dependent apoptosis.
To increase the likelihood of identifying new proapoptotic genes induced by p53 under physiological conditions, we used the normal and p53 nullizygote (p53−/−) mouse model as a source of differentially expressed mRNA. We report here the identification and characterization of the mouse and the human Scotin gene, a novel p53-inducible proapoptotic gene.
Results
Isolation of a novel p53-regulated gene by differential display
Previous studies have shown that cells from thymus or spleen undergo massive p53-dependent apoptosis after γ-irradiation in normal mice but not in p53−/− mice (Clarke et al., 1993; Lowe et al., 1993). Therefore, this model can be used to identify proapoptotic genes induced by p53, in vivo, after γ-irradiation of the entire animal instead of cellular models. Cellular models are generally established from tumor or immortalized cells that might have lost or reduced proapoptotic gene expression as an adaptation to in vitro culture. Hence, we have compared by differential display the expression of genes in the spleen or thymus of homozygote (p53+/+) and p53 nullizygote (p53−/−) mice after γ-irradiation of whole animals.
Two female mice, one p53−/− and the other p53+/+ from the same litter (6 wk old), were γ-irradiated for 1 min at a dose of 5 Gy/min. The spleen and thymus were removed 3 h after irradiation. After total RNA extraction from spleens, we compared by a differential display method (Liang and Pardee, 1992; Zhao et al., 1996) only expression of RNA from p53+/+ and p53−/− irradiated mice to identify genes specifically induced by p53 in response to irradiation. The screening resulted in the isolation of 112 differentially expressed DNA fragments. 46 fragments among the most differentially expressed were cloned and sequenced. Most sequences did not correspond to previously identified genes.
We analyzed the levels of 10 of the most differentially expressed mRNAs by Northern blot and semi-quantitative RT-PCR to confirm differential expression, comparing levels after irradiation in spleens from normal or p53−/− mouse. The mRNA corresponding to clone 105.9 displayed stronger and more consistent expression after ionizing radiation in the wt mouse than in p53−/− mouse (Fig. 1, A and B) , suggesting that the differential expression was p53-dependent. Therefore, clone 105.9 was chosen for further study and was named Scotin.
Figure 1.
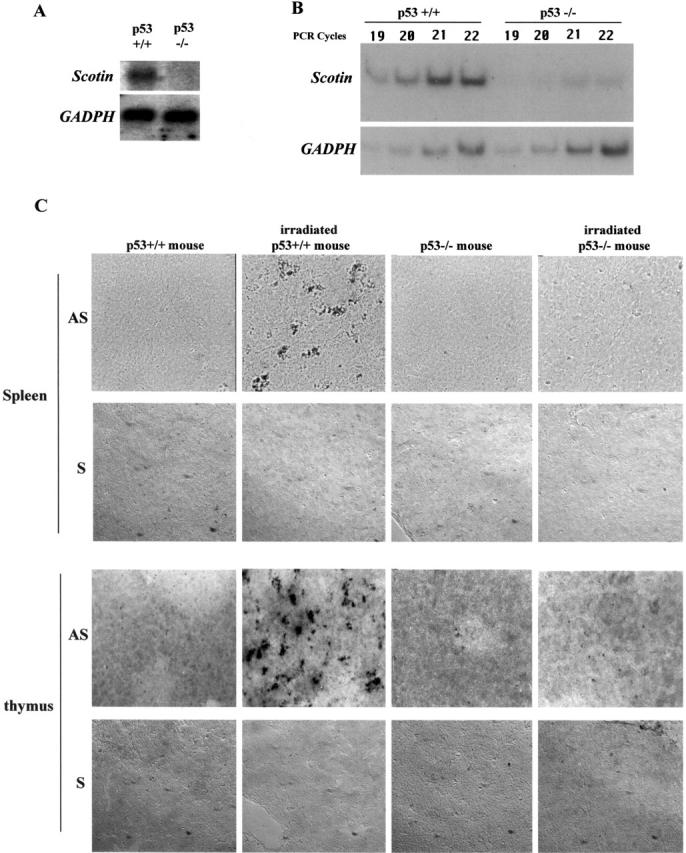
Scotin mRNA is induced after γ-irradiation in the spleen and thymus of normal mouse but not in p53 −/− mouse. p53 nullizygote (−/−) mice as well as wt (p53+/+) litter mates were exposed to 5 Gy γ-irradiation. Total RNA was extracted 3 h later from the spleen of each mouse. (A) Northern blot. 10 μg of total RNA was analyzed by hybridization with a mouse Scotin probe. After autoradiography, the blot was stripped and rehybridized with rat GAPDH probe. (B) Semi-quantitative RT-PCR. 0.5 μg of total RNA was analyzed by RT-PCR by incorporating [33P]dATP and using Scotin- or GAPDH-specific primers as described in Materials and methods. PCR reactions were stopped after different cycles to assess the linear amplification. PCR products were electrophoresed on 8% polyacrylamide gel before autoradiography. (C) In situ hybridization. Two p53+/+ male mice and two p53−/− male mice were exposed to 5Gy of whole body γ-irradiation. Spleen and thymus were removed 3 h after irradiation. Sections were incubated with a digoxigenin-labeled antisense (AS) Scotin RNA probe or with a digoxigenin-labeled sense (S) Scotin RNA probe. Sections were incubated with antidigoxigenin antibody conjugated to alkaline phosphatase. Scotin mRNA was then visualized by the addition of a precipitating substrate (NBT/BCIP).
To confirm the in vivo differential expression of Scotin mRNA, we performed an in situ hybridization analysis (Fig. 1 C). Spleens and thymus were resected after irradiation along with the same organs from nonirradiated mice of the same genotype as controls. Cryosections of spleen and thymus were hybridized with an antisense (AS) digoxigenin-labeled Scotin RNA probe. Fig. 1 C shows that Scotin mRNA was strongly induced only in the spleen and in the thymus of irradiated wt mice. No induction of Scotin mRNA could be detected in the spleen or thymus of p53−/− mice after γ-irradiation. Hybridization with the sense digoxigenin-labeled Scotin RNA probe performed in the same conditions gave no signal, confirming that the in situ hybridization was specific for Scotin mRNA. Together, these results indicate that Scotin gene expression is induced, in vivo, in a p53-dependent manner in response to ionizing radiation.
Scotin gene is directly transactivated by p53
Using a genome walking kit PCR, we isolated a fragment of 2,980 bp corresponding to the mouse Scotin promoter. On the basis of the statistics of known p53 target genes and using a computer approach (Bourdon et al., 1997), we identified a potential p53-responsive element composed of nine decamers located between positions 768 and 592 (Fig. 2 A) in the mouse Scotin promoter. We performed electrophoretic mobility shift assay (EMSA) to examine whether p53 could bind a double-stranded oligonucleotide corresponding to the core (Fig. 2 A, S1) of the putative p53-binding site from the Scotin promoter. We observed a mobility shift when purified wt p53 was incubated with the S1-binding site. Moreover, addition of an anti-p53 antibody (pAb 421) to the reaction mixture induced a supershift, confirming the presence of p53 in the protein–DNA complex (Fig. 2 B). To assess the specificity of binding of p53 to the S1-binding site, we added to the reaction mixture 50-fold molar excess of unlabeled wt S1-binding site or of unlabeled mutant S1-binding site. The S1 mutant-binding site is mutated only in one decamer, leaving the other one intact. The unlabeled wt S1-binding site, but not the mutant S1-binding site, efficiently competed with the formation of the electrophoretically retarded p53–DNA complex, confirming the specificity of p53 binding to the S1 site from the Scotin promoter (Fig. 2 B).
Figure 2.

Scotin gene is directly transactivated by p53. (A) A potential p53-binding site was identified in the promoter of the Scotin gene. Nine decamers containing no more than three disparities compared with the consensus sequence (RRRCWWGYYY) were identified according to our previous analysis of known p53-binding sites (Bourdon et al., 1997). (R = G or A, W = T or A, Y = C or T). The boxed sequence (S1) indicates the pair of decamers with a minimal space between both decamers and containing the smallest number of disparities with the consensus sequence. The S1 oligonucleotide was studied by EMSA. The asterisk indicates the disparities with the consensus sequence. (B) p53 binding to the DNA element (S1) from the Scotin promoter. EMSA of double-stranded oligonucleotides. Equal amounts of 32P-labeled oligonucleotide S1 (130 fmol) were incubated with the purified baculovirus–produced wt human p53 (400 fmol). In the designated lanes, the p53 antibody pAb 421 (0.3 μg) was included. For competition assays, the unlabeled wt S1 oligonucleotides (50-fold molar excess), or the mutant (mut) S1 oligonucleotides (50-fold molar excess) were added to the reaction. (C) The Scotin promoter is induced by p53 in a dose-dependent manner. The entire Scotin promoter was subcloned upstream of the luciferase reporter gene in the promoter-less plasmid pGL3 basic. The plasmid PG13-Luc containing the luciferase reporter gene driven by a p53-responsive promoter was used as a positive control for p53 transcriptional activity. H1299 cells were cotransfected with the indicated reporter plasmid (1 μg/ml), a trace amount of pSVrenilla (0.02 μg/ml) as an internal control, and an increasing concentration of p53 expression vector driven by SV-40 early promoter (SVp53). (D) The wt p53-binding site from the Scotin promoter confers p53 responsiveness to a minimal heterologous promoter. A fragment of 760 bp containing the entire p53-binding site from the Scotin promoter was subcloned upstream of a minimal promoter fused to the luciferase reporter gene (Scot-Adluc). The minimal promoter derives from the adenovirus major late promoter, which is not responsive to p53. The S1 p53-binding site was mutated in 1 bp of one decamer by point mutagenesis PCR to generate Mut–Adluc plasmid as described in Materials and methods. H1299 cells were cotransfected with the indicated reporter plasmid (3 μg/ml), a trace amount of pSVrenilla (0.02 μg/ml) as an internal control, and an increasing concentration of p53 expression vector driven by SV-40 early promoter (SVp53). In reporter assays, the amount of SV-40 promoter was maintained constant at 0.7 μg/ml in each transfection by adding empty SV-40 expression vector. Luciferase activity was normalized to Renilla activity. Results shown are the average of four independent experiments performed in triplicate. SDs are indicated as error bars.
We further examined whether p53 could transactivate the Scotin promoter. The genomic fragment corresponding to the mouse Scotin promoter was subcloned upstream of the luciferase reporter gene to generate the Scotluc reporter plasmid. Cotransfection of different amounts of human wt p53 expression vector with the Scotluc plasmid into H1299 cells resulted in a significant increase in reporter luciferase activity in a p53 dose-dependent manner. As a positive control, we cotransfected into H1299 cells different amounts of human wt p53 expression vector with the PG13-luciferase reporter plasmid, which contains a synthetic p53 binding element. The Scotin promoter and PG13 promoter have a comparable responsiveness to p53 (Fig. 2 C).
To determine whether the S1 p53-binding site contained in the Scotin promoter can confer responsiveness to p53, the 760-bp fragment containing the entire wt p53-binding site was subcloned upstream from a minimal promoter, which drives the luciferase gene (AdLuc). As a control, the 760-bp fragment mutated only in 1 bp of the S1 p53-binding site was subcloned into the AdLuc plasmid. After transfection with various amounts of p53 expression vector, only the insert containing the wt p53-binding site confers responsiveness to p53 in a dose-dependent manner to a minimal promoter (Fig. 2 D). The Scotin gene is therefore a bona fide direct target of p53.
Scotin is conserved between mouse and human
We designed primers from the short sequence corresponding to the 3′end of mouse Scotin mRNA and performed a 5′/3′ rapid amplification of cDNA ends (RACE) PCR on mRNA extracted from thymus of irradiated p53+/+ mice. We obtained a sequence of 1,850 bp, which is consistent with the apparent size of Scotin mRNA observed in Northern blots. The ORF predicts a protein of 235 amino acid residues, containing in the NH2 terminus a putative signal sequence of 22 residues, and in the central part a putative transmembrane domain composed of 18 hydrophobic amino acids immediately followed by a domain rich in prolines. No further protein domain homologies to any known gene product have been identified. The presence of an in-frame stop codon within the 5′ untranslated region (UTR) supports the correct assignment of the first methionine of the ORF. (Fig. 3 A).
Figure 3.

Scotin is a putative transmembrane protein. (A) Diagram representing the primary structure of Scotin protein. (B) Human and mouse Scotin protein alignment, hydrophobic domain in solid box, and putative signal sequences in hashed box.
To obtain human Scotin cDNA, we searched in GenBank for human EST sequences homologous to mouse Scotin cDNA. We designed primers from the longest human Scotin EST sequence and performed a 5′/3′ RACE PCR on mRNA from human placenta. We obtained a complete cDNA with the apparent size of 2.2 kb. It contains one ORF and a relatively long 3′ UTR. The ORF predicts a protein of 240 amino acid residues sharing 72% homology (70% identity) with the mouse Scotin protein. Alignment of mouse and human Scotin protein shows that the signal sequence, the cysteines in the NH2 terminus, the transmembrane domain, and the proline/tyrosine domains are well conserved (Fig. 3 B).
We searched for the human Scotin gene in the human draft genome database. The Scotin gene is composed of six exons and maps to the chromosome 3p21.3, which is a very frequent breakpoint in many cancers (GenBank/EMBL/DDBJ accession no. AC104448). We also identified a Scotin pseudogene localized on chromosome Xq13.1–13.3 (GenBank/EMBL/DDBS accession no. AL139400). By RT-PCR, we could not detect transcripts expressed from the Scotin pseudogene.
Scotin protein expression is induced in a p53-dependent manner in response to cellular stress
Two affinity-purified rabbit pAbs (M105 and H105) were raised against a peptide corresponding to the COOH terminus of mouse or human Scotin protein, respectively. Their specificity was assessed by Western blot analysis on protein extracts from H1299 cells transfected with mouse or human Scotin expression vectors. Mouse and human anti-Scotin antibody detected only one protein with an apparent size of 25 kD, which is consistent with the expected size for Scotin (unpublished data).
To further characterize Scotin protein, it was essential to identify cell lines that could induce Scotin upon DNA damage. We treated primary mouse embryonic fibroblasts (MEF) from p53−/− and p53+/+ littermate mice or human primary fibroblast (MRC5) and several p53 positive or negative human tumors cell lines with UV-C light or with actinomycin D (60 ng/ml), a DNA intercalator. Actinomycin D used at 60 ng/ml does not prevent RNA polymerase II activity, but is a potent activator of p53 (Blattner et al., 1999; Nakano and Vousden, 2001). Proteins were extracted after treatment and analyzed by Western blot. Waf and p53 protein levels were used as an indication of p53 activation. Scotin protein clearly accumulates after UV irradiation or actinomycin D treatment in mouse p53+/+ MEF, human primary fibroblast, and human tumor cells expressing wt p53, but not in mouse p53−/− MEF or human tumor cell lines devoid of p53 expression (Fig. 4, A and B) .
Figure 4.
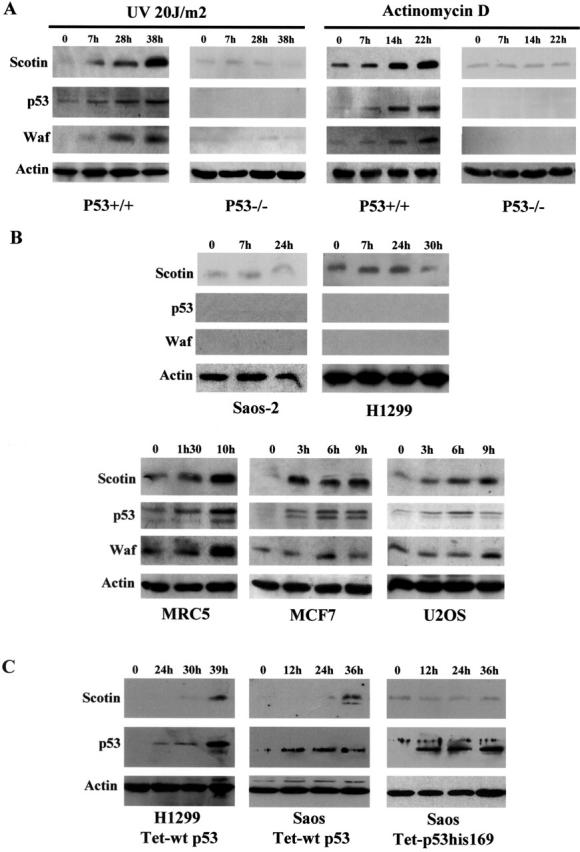
Scotin protein expression is induced in a p53-dependent manner. (A) Primary MEF expressing wtp53 induce Scotin after UV irradiation or Actinomycin D treatment. MEF (passage 2) from p53−/− and p53+/+ littermate mice were exposed to UV-C light (20 J/m2) or Actinomycin D (60 ng/ml), a potent p53 activator. Proteins were extracted at the indicated time after treatment and analyzed by Western blot. (B) Primary human fibroblasts and human tumor cell lines expressing functional p53 induce Scotin in response to Actinomycin D. The primary human fibroblast MRC5, the tumor cell lines (MCF7 and U2OS) expressing functional p53, and the tumor cell lines devoid of p53 expression (Saos-2 and H1299) were treated with 60 ng/ml of Actinomycin D. Proteins were extracted at the indicated time and analyzed by Western blot. (C) p53 expression is sufficient to induce human Scotin expression. Proteins were extracted at the indicated time after tetracycline induction from tetracycline-inducible p53 H1299 cells or Saos-2 cell lines described in Materials and methods. Membranes were probed in succession using anti-Scotin pAb, anti-p53 pAb, and anti-Waf mAb, as described in Materials and methods. To control loading and transfer efficiency, membranes were incubated with antiactin mAb.
To determine whether p53 expression is sufficient to induce Scotin expression, we used human p53-null cells (H1299 and Saos-2) that contain stable integrated p53 cDNA inducible by tetracycline. In both cell lines, Scotin was induced after the induction of wt p53, whereas no Scotin induction could be detected in the control mutant p53His169-tetracycline inducible Saos-2 cells (Fig. 4 C), indicating that wt p53 is necessary and sufficient to induce Scotin. However, Scotin induction is slower in such cell lines compared with induction after cellular stress, suggesting that cellular stress and p53 may act in synergy to induce Scotin.
Scotin protein is localized in the ER and the nuclear membrane
After exposure to UV-C irradiation, p53+/+ MEF were stained for Scotin using anti–mouse Scotin antibody. A bright ring around the nucleus was observed in cells treated with UV but not in control cells (Fig. 5 , a and b). Similar localization was observed in mouse NIH3T3 fibroblast and in human MCF7 cells (unpublished data). To determine the subcellular localization of endogenous Scotin protein, we treated MCF 7 cells with actinomycin D and costained them for human Scotin and human gp96/GRP94 protein. gp96 is a chaperone protein predominantly expressed in the ER (Koch et al., 1986; Li and Srivastava, 1993). As shown by confocal microscopy, gp96 and Scotin were colocalized in the ER after cellular stress (Fig. 5, c–e).
Figure 5.


Mouse and human Scotin proteins are located in the ER and the nuclear envelope. (a and b) MEF were incubated in the presence or absence of 60 ng/ml of actinomycin D and stained by indirect fluorescence (FITC) using anti–mouse Scotin antibody (a) nontreated MEF, and (b) treated MEF. (c–e) MCF7 cells treated with 60 ng/ml of actinomycin D were costained by indirect fluorescence for endogenous Scotin protein using (c) anti–human Scotin antibody (FITC) and (d) the monoclonal anti-gp96 antibody (Texas red); (e) merge. gp96/GRP94 is a chaperone protein exclusively located in the ER. (f and g) Immunostaining of H1299 cells transfected with mouse Scotin expression vectors, (f) 5 μg of AdScotin, (g) 10 μg of SVScotin, and stained by indirect fluorescence (FITC) using anti–mouse Scotin antibody. (h–j) Scotin is colocalized with gp96 in the ER and the nuclear membrane after ectopic expression. H1299 cells transfected with 10 μg of SVScotin expression vector were costained by indirect fluorescence (h) using rabbit polyclonal anti–mouse Scotin antibody (FITC) and (i) mouse monoclonal anti-gp96 antibody (Texas red); (j) merge. (k–m) Scotin and mitochondria have distinct staining patterns. H1299 cells transfected with 5 μg of SVScotin expression vector were costained by indirect fluorescence using (k) anti–mouse Scotin antibody (FITC) and (l) red MitoTracker® (red); (m) merge. Bars, 5 μm.
Because we planned to express Scotin by transient transfection to study its biological activity, it was essential to determine whether the subcellular localization of ectopic Scotin was identical to the endogenous. Human H1299 cells were transiently transfected with mouse Scotin expression vector. To mimic physiological expression levels as closely as possible, we used Scotin expression vectors driven either by the SV-40 promoter or the weak minimal major late promoter from adenovirus (Ad). A bright ring around the nucleus was observed in H1299 cells transfected with AdScotin vector. (Fig. 5 f). We observed the same staining pattern after transient transfection in Saos-2 and NIH3T3 cell lines (unpublished data). Moreover, as judged by immunostaining, transfection of AdScotin plasmid did not give rise, at the cellular level, to a strong overexpression of Scotin. Transfection of SVScotin vector gave rise to a stronger overexpression of Scotin in H1299 cells, revealing the characteristic staining pattern of the ER and of the nuclear membrane (Fig. 5 g). To assess the localization of Scotin protein after transfection, we costained H1299-transfected cells for Scotin and gp96 or mitochondria. As shown by confocal microscopy, the Scotin and gp96 proteins were colocalized, confirming that Scotin protein is localized in the ER and in the nuclear membrane after transfection (Fig. 5, h–j). However, the Scotin staining pattern is different from the mitochondria staining pattern (Fig. 5, compare k with l and m [merge]). Together, these results indicate that Scotin protein is mainly located in the ER and can be located in the nuclear membrane in cells overexpressing Scotin after transfection.
Scotin can promote apoptosis independently of p53
We noticed that induction of Scotin protein was coincident with cell death in cell lines expressing wt p53 after treatment with UV or actinomycin D. Moreover, no stable clone overexpressing wt Scotin could be obtained in H1299 and Saos-2 cells (cell lines devoid of p53) after cotransfection of a vector expressing the neomycin resistance gene with AdScotin or SVScotin vectors (unpublished data), suggesting that Scotin protein might prevent cell proliferation.
The ER can trigger cell signals leading to apoptosis in response to stresses (ER stress) that impair its functions, such as protein overexpression after transfection, misfolded protein, hypoxia, inhibition of glycosylation, and disruption of the ER calcium store (for review see Kaufman, 1999). Because Scotin is localized in the ER, it was essential to determine whether the cytotoxic effect of the Scotin protein was due to an intrinsic activity or due to an artifact of overexpression after transfection. To examine this, we constructed three different Scotin mutant proteins by deleting large regions of Scotin protein (Fig. 6 A). Because the Δpro Scotin mutant lost the epitope for the anti-Scotin antibody, all mutants and wt Scotin proteins were fused at the COOH-terminal end to the FLAG peptide. H1299 cells were transiently transfected with wt Scotin-FLAG expression vectors driven by SV-40 or Ad promoters (Fig. 6 A). Anti-FLAG and anti-Scotin antibodies gave identical patterns of staining, indicating that the subcellular localization of Scotin was not affected by the FLAG fusion. After transfection of H1299 cells with mutant or wt Scotin vectors, cells were stained by anti-FLAG antibody. The localization of the ΔCys Scotin mutant protein was similar to that of the wt Scotin protein (Fig. 6 B, compare c with a). The immunostaining pattern of the ΔN Scotin was similar to the immunostaining pattern obtained after transfection of wt Scotin expressed under the SV-40 promoter (Fig. 6 B, compare d with b), probably because the ΔN Scotin mutant was expressed at a comparable level. The results indicate that the subcellular localization of ΔN Scotin, ΔCys Scotin, and wt Scotin protein is similar and that the cysteine domain of Scotin is not required for the localization to the ER. The immunostaining pattern of the Δpro Scotin mutant protein is different from the immunostaining pattern observed with wt Scotin protein (Fig. 6 B, compare e with b), indicating that the proline-rich domain is required for the ER localization. We have not studied further the Δpro Scotin mutant because it was impossible to determine whether the lack of cytotoxicity of this mutant (unpublished data) was due to the loss of the ER localization or due to the deletion (100 aa) of an active domain.
Figure 6.
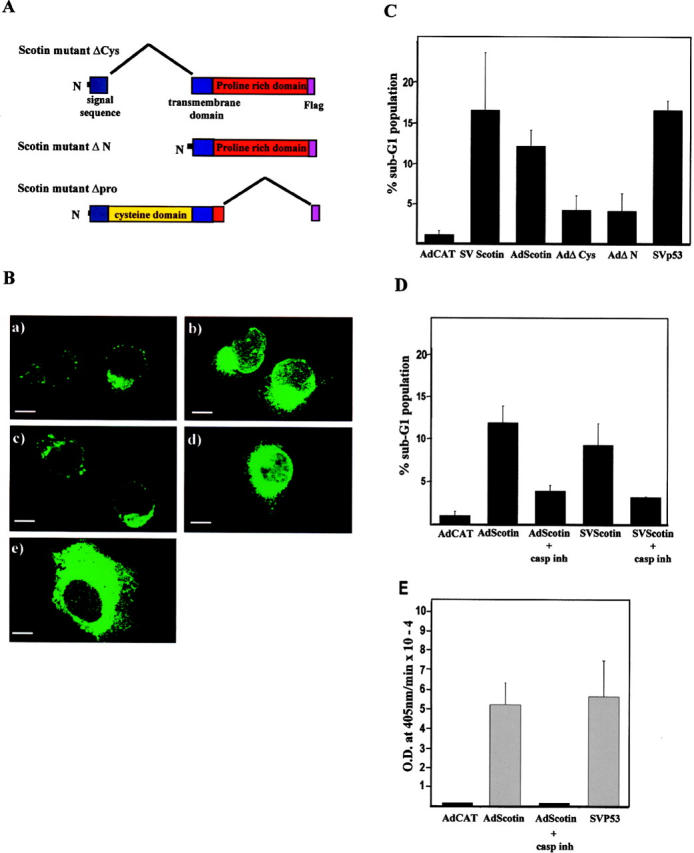
wt Scotin protein has an intrinsic proapoptotic activity. (A) Schematic representation of the Scotin mutants. (B) Scotin mutants deleted of the NH2 terminus are located in the ER. H1299 cells were transfected with 0.5 μg of AdScotin-FLAG (a), or SVScotin-FLAG (b), or AdΔCys (c, or AdΔN (d), or SVΔpro (e). Cells were stained with anti-FLAG (M2) mouse mAb followed by anti–mouse antibody conjugated to FITC. Bars, 5 μm. (C) wt Scotin protein induces apoptosis after transfection. The DNA content of each transfected population was determined by flow cytometry analysis. The percentage of sub-G1 DNA content represents the percentage of apoptotic cells 48 h after transfection. H1299 cells transfected with the following different expression vectors: 5 μg/ml AdCAT, or 10 μg/ml SVScotin, or 5 μg/ml AdScotin, or 2 μg/ml SVp53, or 5 μg/ml AdΔCys, or 5 μg/ml AdΔN. Histogram represents the average of at least three independent transfections. SDs are reported as error bars. (D) Scotin-mediated apoptosis is inhibited by caspase inhibitor. The DNA content of each transfected population was determined by flow cytometry analysis. H1299 cells transfected with 5 μg/ml of AdCAT, 5 μg/ml of SVScotin-FLAG, or 5 μg/ml of AdScotin-FLAG were incubated in presence or absence of the caspase inhibitor Z-VAD-FMK. Histogram represents the average of at least three independent transfections. SDs are reported as error bars. (E) Scotin induces caspase-3 activation. The caspase-3 activity was determined using the Colorimetric Caspase-3 Cellular Activity Assay kit. The H1299 cells were transfected with 5 μg/ml of AdCAT, 5 μg/ml of AdScotin-FLAG, or 2 μg/ml of SVp53 expression vectors. As indicated, cells were incubated in the presence or absence of the caspase inhibitor Z-VAD-FMK. Histogram represents the average of three independent transfections. SDs are reported as error bars.
To investigate the Scotin-mediated cell death, we performed a flow cytometry analysis as previously described to quantify apoptosis induced by p53 (Yonish-Rouach et al., 1994; Haupt et al., 1995; Deguin-Chambon et al., 2000). H1299 cells were transiently transfected with wt or mutant Scotin-FLAG expression vectors. The results of three independent experiments are presented Fig. 6 C. Transient transfection of SVp53 expression vector caused 16% of transfected cells to have a sub-G1 DNA content, which is indicative of apoptotic cells, in agreement with earlier reports (Yonish-Rouach et al., 1994; Haupt et al., 1995). The Scotin-expressing cells transfected with plasmid SVScotin-FLAG or AdScotin-FLAG also exhibited a significantly high fraction of cells with sub-G1 DNA content (16 and 12%, respectively). In contrast, only 4.5% of the total ΔCys or ΔN Scotin mutant–expressing cells contained a sub-G1 DNA content. This indicates that both truncated Scotin mutant proteins have lost most of the apoptotic activity. Therefore, Scotin-mediated cell death is due to an intrinsic activity and not due to disruption of the ER functions after overexpression of an ER-located protein.
Scotin-mediated cell death was inhibited when H1299 cells were incubated with the caspase inhibitor zVAD before transfection (Fig. 6 D). To determine whether Scotin could promote caspase 3 activation, Scotin expression vector was transfected into H1299 cells. Transfection of wt p53 or CAT expression vector into H1299 cells was used as positive and negative controls, respectively. Only Scotin and p53 induced caspase activation as assessed by caspase3 cellular activity assay. No caspase activity could be detected upon transfection of AdCAT vector (Fig. 6 E). This indicates that Scotin can induce apoptosis in a caspase-dependent manner.
Scotin protein is involved in apoptosis induced by DNA damage
To assess the role of Scotin in apoptosis under physiological conditions, NIH3T3 cells were stably transfected with an AS Scotin expression vector. As a control, NIH3T3 cells were stably transfected with pcDNA3 expression vector expressing a noncoding sequence unrelated to Scotin or other known genes. After selection in the presence of neomycin, the pooled populations of transfectant expressing the control or the Scotin AS vector were exposed to actinomycin D (60 ng/ml). Scotin protein level was analyzed by Western blot (Fig. 7 A). Scotin basal level was detectable and well-induced after treatment in control AS-expressing cells. However, Scotin was barely detectable in Scotin antisense-expressing cells (Scotin-AS) despite a strong activation of p53 after actinomycin D treatment. This indicates that Scotin AS expression vector strongly inhibited endogenous Scotin expression.
Figure 7.
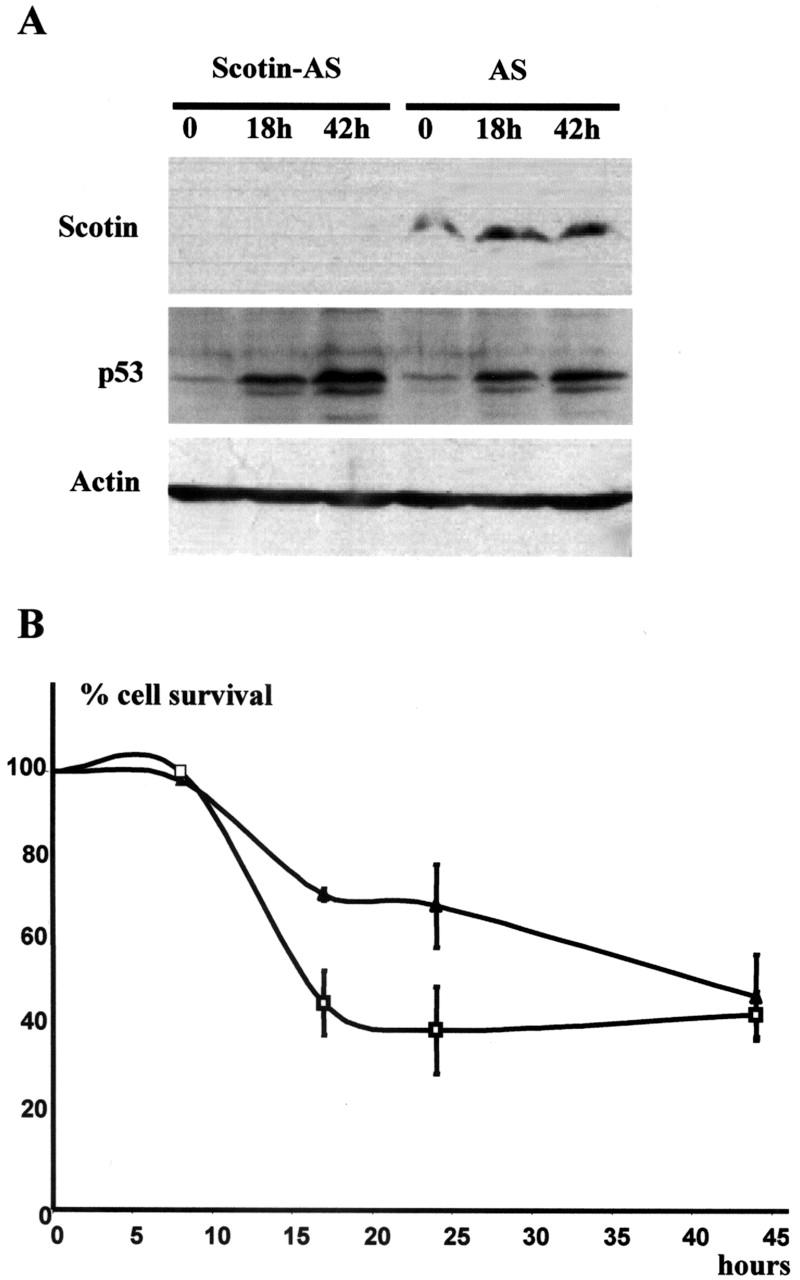
Scotin is involved in p53-dependent apoptosis. (A) Endogenous Scotin expression is inhibited in cells stably transfected with Scotin AS vector. 3T3 cells stably transfected with control antisense (AS) expression vector and 3T3 cells stably transfected with Scotin antisense (Scotin-AS) expression vectors were treated with actinomycin D (60 ng/ml). Proteins were extracted at the indicated time and analyzed by Western blot. Scotin expression was detected by using anti–mouse Scotin antibody. As a positive control, p53 induction was determined using CM5 anti–mouse p53 antibody. Protein loading was controlled by antiactin antibody. (B) Cells expressing a low level of Scotin protein are resistant to DNA damage. For the trypan blue exclusion assay, Scotin-AS cells (▴) or AS cells (□) were irradiated by UV-C (15 J/m2) and harvested at the indicated times. Viable cells were determined by trypan blue exclusion and counted using a hemocytometer. The percentage of cell survival was calculated as the number of viable cells in relation to the number of cells plated at the start of the trial. Each point is the average of at least three experiments. SDs are reported as error bars.
To determine if Scotin plays a role in the p53-dependent apoptosis induced by DNA damage, we treated with UVC (15 J/m2) the pooled populations expressing the control or the Scotin AS vector. The percentage of cell survival was determined by trypan blue exclusion analysis (Fig. 7 B). The DNA damage–mediated apoptosis is strongly impaired, but not abolished, by the inhibition of Scotin expression. This indicates that Scotin plays a role in the p53-dependent apoptosis under physiological conditions but, as previously reported for Noxa and PUMA (E. Oda et al., 2000; Nakano and Vousden, 2001), other proapoptotic genes might also be activated.
Discussion
Few proapoptotic genes directly induced, in vivo, by p53 have been described. This is probably due to the use of cellular p53 models, which being derived from tumors or immortalized primary cells, are likely to have lost some proapoptotic gene expressions as an adaptation to in vitro culture. It has been shown that thymus and spleen cells undergo a massive p53-dependent apoptosis after ionizing radiation in normal, but not in p53−/−, mice (Clarke et al., 1993; Lowe et al., 1993). This animal model can thus be used to identify new genes involved in p53-dependent apoptosis induced by irradiation. In the present study, we report the identification and characterization of mouse and human homologues of a novel gene, named Scotin. The Scotin gene is induced, in vivo, after ionizing radiation in a p53-dependent manner. p53 binds specifically to a p53-binding site contained in the Scotin gene promoter and directly transactivates the Scotin gene. By Western blot, we showed that human and mouse Scotin proteins are induced in a p53-dependent manner in response to DNA damage in primary and tumor cell lines. By using a tetracycline-inducible p53 system, we showed that wt p53 expression is sufficient to induce Scotin protein expression in absence of cellular stresses. However, we noticed that Scotin protein is constitutively expressed at a basal level independently of p53. Introduction of wt Scotin protein, but not of mutant Scotin protein, promotes apoptosis in a caspase-dependent manner in tumor cells devoid of p53. Inhibition of endogenous Scotin protein expression by AS reduced strongly, but not completely, the p53-dependent apoptosis induced by UV irradiation, suggesting that other proapoptotic pathways were activated. The generation of knockout mice for the Scotin gene will help to understand the contribution of the Scotin gene in p53-dependent apoptosis and whether loss of Scotin gene contributes to carcinogenesis.
Endogenous Scotin protein is localized in the ER in primary and in tumor cells. After transfection, Scotin is detected in the ER and in the nuclear envelope, but not in the Golgi apparatus (unpublished data) or the mitochondria. The fact that the endogenous Scotin protein is localized in the ER suggests that Scotin may trigger apoptosis from the ER, although we cannot rule out that a very small fraction of Scotin proteins, undetectable by immunostaining, can be located in other cellular compartments. Our results with truncated forms of Scotin proteins indicate that the proline-rich domain of Scotin is required for the localization of Scotin in the ER, whereas the NH2-terminal domain is dispensable. We have not studied further the Δpro Scotin mutant as >100 amino acids were deleted, the lack of cytotoxicity of this mutant (unpublished data) could be due to the loss of the ER localization or due to the deletion of an active domain. Further study of the proline domain will be needed to characterize the element(s) responsible for the localization of Scotin in the ER and to determine whether the localization of Scotin in the ER is required for its proapoptotic activity.
Evidence is emerging that the ER plays a major role in apoptosis (Ferri and Kroemer, 2001). As a protein-folding compartment, the ER is exquisitely sensitive to alterations in homeostasis that disrupt ER functions. ER stresses include ER calcium store depletion, inhibition of glycosylation, reduction of disulfide bonds, overexpression of mutant protein or protein subunits, expression of viral proteins, TNFα treatment, and hypoxia (for review see Kaufman, 1999). Sustained elevation of cytosolic [Ca2+] can induce cell death by apoptosis. The release of calcium from the ER upon proapoptotic signaling or after drug treatment triggers the opening of the calcium-sensitive mitochondrial permeability transition pore (PTP), allowing the release of cytochrome c from the mitochondria to the cytosol. The cytosolic cytochrome c binds Apaf-1 and procaspase-9, leading to caspase-9 activation, which processes and activates other caspases to orchestrate the programmed cell death (for review see Green and Reed, 1998). Whether Scotin can promote apoptosis in response to ER stress remains to be determined.
The human Scotin gene maps to chromosome 3p21.3, a region highly susceptible to aberrant chromosomal rearrangements and deletion in human cancer (Mitelman et al., 1997). The very frequent loss of not only one allele of chromosome arm 3p in both small lung cancer and nonsmall cell lung cancer (Kok et al., 1987; Naylor et al., 1987), but also in many types of human cancers such as breast, uterine and cervical carcinoma, renal cell carcinoma, ovarian, head and neck, and pancreatic islet cell carcinoma provides strong evidence for the existence of tumor suppressor genes in this chromosome region (Kok et al., 1997; Mitelman et al., 1997). Therefore, future studies will determine whether mutations of the Scotin gene occur in tumors and whether the Scotin gene is a bona fide tumor suppressor gene. In summary, Scotin displays all the characteristics expected of a gene that can contribute to p53-dependent apoptosis. The discovery of Scotin brings to light a role of the ER in p53-dependent apoptosis.
Materials and methods
Cell culture and cellular stress
All cell lines were purchased from American Type Culture Collection except p53+/+ and p53−/− MEF. All cell lines except H1299 cells were maintained at 37°C, 5% CO2 in DME supplemented with 10% heat-inactivated FCS. The H1299 cell line, a human lung carcinoma cell line devoid of p53, was routinely maintained at 37°C, 5% CO2 in RPMI 1640 medium supplemented with 10% FCS.
H1299Tetwtp53 was derived from H1299 cells that were stably transfected with a tetracycline-inducible vector encoding for wt human p53 (Gossen et al., 1995). H1299Tetwtp53 was maintained at 37°C, 5% CO2 in DME supplemented with 10% inactivated FCS, 0.4 mg/ml G418, 0.2 mg/ml hygromycin, and 0.5 μg/ml anhydrotetracycline. To induce p53 expression, cells were incubated with fresh medium containing no anhydrotetracycline. H1299Tetwtp53 cells were a gift from Dr. L. Debussche (Aventis, Vitry-sur-Seine, France). SaosTetwtp53 and SaosTetmutp53, a gift from Dr. C. Midgley (University of Dundee, Dundee, UK), were derived from Saos-2 (human osteosarcoma cell lines devoid of p53) that were stably transfected with a tetracycline-inducible vector encoding for wt or mutant His169 mouse p53. Cells were routinely maintained at 37°C, 5% CO2 in DME supplemented with 10% FCS and 0.5 mg/ml G418. To induce p53 expression, cells were incubated with fresh medium supplemented with 0.5 μg/ml anhydrotetracycline.
Cells expressing the Scotin AS were derived from NIH3T3 cells that were stably transfected with Scotin AS expression vector. Cells expressing the control AS vector were derived from NIH3T3 cells that were stably transfected with pcDNA3 expression vector. We used the pcDNA3 expression vector as a negative AS control because it contains, between the CMV promoter and the poly(A)+ signal, a noncoding sequence of 100 bp not homologous to any known genes. Both cells lines were selected for 3 wk in DME supplemented with 10% FCS and 1 mg/ml G418. Actinomycin D and UV-C (20 J/m2) treatments were performed as described previously (Blattner et al., 1999).
Differential display, RT-PCR and Northern blot
p53+/+ mice and p53−/− mice littermates were exposed for 1 min to 5 Gy of whole body γ-irradiation in a 137Cs γ-irradiator (model IBL 437C; CIS-US, Inc.). Spleen and thymus were resected 3 h after irradiation. After total RNA extraction, differential display was performed using the Delta™ RNA Fingerprinting kit (CLONTECH Laboratories, Inc.). Northern blot analysis was performed as described previously (Deguin-Chambon et al., 2000). The cloned differentially expressed fragments and a 1.3-kb PstI cDNA fragment corresponding to the rat GAPDH gene were used as radiolabeled probes for Northern blot analysis. Semi-quantitative RT-PCR analysis was performed using a poly-dT primer and the AMV reverse transcriptase, followed by PCR incorporating [33P]dCTP and using the mouse Scotin primers 5′-GCTGTATAGAGGGCCACATGTGTTCACT-3′ and 5′-AAAG- ACAGTGCAGGGAGAAACCAGAGTG-3′, or the mouse GAPDH primers 5′-TGGACTGTGGTCATGAGCCC-3′ and 5′-CAGCAATGCATCCTGCACC-3′. Scotin and GAPDH PCR products were analyzed on 8% PAGE/0.5% TBE before autoradiography.
In situ hybridization
One wt male mouse and one p53−/− male mouse were γ-irradiated (5 Gy). Spleens and thymus from untreated or treated mice were removed 3 h after radiation. Cryosections of spleen and thymus were fixed in 4% paraformaldehyde in PBS, washed in sterile PBS, and dehydrated. The AS–digoxigenin-labeled Scotin RNA was produced by T7 RNA polymerase. As a negative control, the sense digoxigenin-labeled Scotin RNA was produced by SP6 RNA polymerase. Sections were hybridized overnight at 60°C with digoxigenin-labeled Scotin RNA probe. After washing at 55°C in solution A (50% formamide, 2× SSC, 0.1% Tween 20), sections were washed in TBS, blocked for 1 h at RT with 10% FCS in TBS, and incubated overnight at 4°C with an antidigoxigenin antibody conjugated to alkaline phosphatase (Roche Molecular Biochemicals). The sections were washed with TBS and hybridized probe was revealed by hydrolysis of phosphatase substrate NBT/BCIP (Sigma-Aldrich).
Electrophoretic mobility shift assay
The binding of p53 to a DNA fragment derived from the promoter of Scotin was analyzed by EMSA as described previously (Bourdon et al., 1997). The potential p53-binding site S1 derived from the Scotin promoter 5′-TGTGTGGCCTTGTTAGGAACTTTGTCC-3′ and the mutant version of S1 5′-CCGTTCCCTGGAATTCTTAGGAACTTTGTCC-3′ were used as indicated. Point mutations are underlined.
Plasmids
The mouse Scotin cDNA was amplified from mouse thymus total RNA using the primers 5′-CGGCCGGGGCGGGGCAAG-3′ and 5′-CCCGGGAAGGACAGTGACATC-3′ in the 5′/3′ RACE PCR kit (Roche Molecular Biochemicals). The human Scotin cDNA was amplified from human placenta mRNA using the primers 5′-GGTGGACAGGAGCCCTGCTCACCT-3′ and 5′-ATCACTGAGGCTGTGGCGGCACTGC-3′ in the 5′/3′ RACE PCR kit. These sequence data are available from GenBank/EMBL/DDBJ under accession nos. AF520698, AF520699, AF520702, and AF520703.
Mouse Scotin expression vector (AdScotin) is a mouse Scotin expression vector plasmid driven by the minimal adenovirus major late promoter (Ad). The plasmid AdScotin-FLAG was generated by PCR to add in frame the FLAG peptide to the carboxyl end of Scotin (AdScotin-FLAG). SVScotin is a mouse Scotin expression vector plasmid driven by the SV-40 early promoter.
The PCR products containing the 5′UTR of mouse Scotin or the signal sequence were amplified from plasmid AdScotin-FLAG using the primer 5′-ACGACGTTGTAAAACGACGGCCAGAGAA-3′ with either the primer 5′-AGGCCGCGGGCGCAGCCATG-3′ or the primer 5′-CAGACCGCGGGGATCGAATT-3′. The SacII site present in the mouse Scotin cDNA was used to clone both PCR products into the plasmid AdScotin-FLAG to generate plasmids AdΔN and AdΔCys. The mutant deleted of the proline/tyrosine domain was made by PCR from AdScotin plasmid using the primers 5′-TATGTCAGGGTTCGGAGCGACCGTCGCCATTGG-3′ and 5′-CGCGCTCGAGCTACTTGTCATCGTCGTCCTTGTAATCCAGACAGCAG-3′. The BstXI site present in the mouse Scotin cDNA was used to clone the PCR product into the plasmid SVScotin to generate the plasmid SVΔpro.
The mouse Scotin promoter (2,900 bp; GenBank/EMBL/DDBJ accession nos. AF520700 and AF520701) was obtained by genome walking PCR (GenomeWalker™ PCR kit; CLONTECH Laboratories, Inc.) using the primer 5′-CTCATTCCAGAGACAGCTAGGGGAAGTG-3′, and then the primer 5′-CCTCCAGGTAACTGCTCACCCTGGCTCA-3′. The luciferase reporter plasmid driven by the mouse Scotin promoter (Scot-luc) was generated by cloning the mouse Scotin promoter into the promoter-less plasmid pGL3-basic (Promega). The pAdluc plasmid is a luciferase expression vector driven by the Ad promoter (Bourdon et al., 1997). The Scot-Adluc plasmid is composed of a fragment of 760 bp derived from the mouse Scotin promoter containing the p53-binding site and cloned upstream from the minimal promoter Ad. The Mut-Adluc plasmid contains the fragment of 760 bp mutated by PCR in 1 bp of the p53-binding site using the primers 5′-CCTTGTTAGGAACTTTCTCCCACATTGAGG-3′ and 5′-CCTCAATGTGGGAGAAAGTTC CTAACAAGG-3′ (mutation underlined).
The plasmid SVp53 is an expression vector of human wtp53 driven by the SV-40 early promoter (Nylander et al., 2000). The plasmid SVRenilla was purchased from Promega (pRL-SV-40 vector). The PCR product amplified from plasmid Ad-Scotin using the primers 5′-GCCCTCGAGCCTCCGGGTGCCCATG-3′ and 5′-GCGGAATTCGCGGGGGTGGAAAATCTG-3′ was cloned in the AS orientation into the pcDNA3 expression vector to generate the mouse Scotin AS plasmid (pScot-AS). All constructs were checked by sequencing.
Luciferase reporter assay and transfection
104 H1299 cells seeded per well of a 96-well plates were transfected in triplicate by calcium phosphate precipitate containing 1 μg/ml of luciferase reporter plasmid (Scot-luc, Scot-Adluc, Mut-Adluc, or PG13), 20 ng/ml of SV-Renilla luciferase (internal control), and increasing concentration of SVp53 as indicated in the legend (Fig. 2, C and D). For each transfection mix, the concentration of SV-40 promoter was balanced to 0.7 μg/ml with SV-40 empty expression vector, and the total DNA concentration was balanced to 20 μg/ml with pBS plasmid (Stratagene). Cells were incubated for 36 h at 37°C, washed, and lysed by adding 20 μl/ well of passive lysis buffer (Promega). The 96-well microplates were analyzed in a luminometer (MicroLumat LB96V; EG&G Berthold) using the Dual-Luciferase® Reporter Assay System (Promega).
FACScan™ analysis
8 × 105 H1299 cells were seeded in a 10-cm petri dish and transfected using calcium phosphate precipitate. After 48-h incubation at 37°C, cells were fixed in 70% ethanol and immunostained for Scotin or p53. DNA was stained by propidium iodide. Cells were analyzed by flow cytometry (FACScan™; Becton Dickinson) using a three-parameter analysis (Deguin-Chambon et al., 2000). Experiments with <2% transfection efficiency were discarded.
Caspase activity assay
The caspase-3 activity was determined 48 h after transfection using the Colorimetric Caspase-3 Cellular Activity Assay kit (Calbiochem). In all experiments using caspase inhibitor, the Z-VAD-FMK caspase inhibitor (Calbiochem) was added to a final concentration of 10 μM to the culture medium 4 h before transfection and maintained during the cell incubation.
Western blot
After treatment, cells were scraped and washed in PBS. Proteins were extracted and analyzed as described previously (Deguin-Chambon et al., 2000).
Immunofluorescence staining
3 × 105 cells seeded on 2-well glass chamber slides (LabTek) were transfected as described, fixed by 4% paraformaldehyde, and permeabilized with 0.1% Triton X-100 in PBS. Cells were incubated for 1 h at RT with primary antibody diluted in 10% FCS-DME. After washing with PBS, cells were incubated with fluorescein (FITC)-conjugated anti–mouse or anti–rabbit IgG (Jackson ImmunoResearch Laboratories) as appropriate and diluted in 10% FCS-DME. For double immunofluorescence staining, secondary antibodies were tested for minimal interspecies cross-reaction (Jackson ImmunoResearch Laboratories).
For mitochondrial staining, cells were incubated for 15 min at 37°C with 25 nM MitoTracker® red CMXRos (Molecular Probes), 1% final concentration of paraformaldehyde (diluted in PBS; pH 7.1) was added to the culture medium, and the cells were incubated for a further 30 min at 37°C. Cells were washed and treated for immunostaining.
Production and affinity purification of the mouse and human anti-Scotin antibodies
The peptide PYHESLAGASQPPYNPTYK, corresponding to the COOH end of mouse Scotin protein, or the peptide YHETLAGGAAAPYPASQPPK, corresponding to the COOH end of human Scotin protein, was conjugated to the carrier protein KLH and inoculated to a rabbit as described by Harlow and Lane (1999). The anti-Scotin antibodies were affinity-purified using a peptide column.
Antibodies
The anti-p53 rabbit sera (CM1 and CM5) were described previously (Midgley et al., 1992), and the anti–p53 DO-1 mouse mAb was described in Stephen et al. (1995). The mouse monoclonal anti-gp96/GRP94 antibody was purchased from StressGen Biotechnologies Corp. The mouse monoclonal antibody was purchased from Sigma-Aldrich (ANTI-FLAG® M2). The mouse monoclonal (F5) anti–mouse Waf antibody was purchased from Santa Cruz Biotechnology, Inc. The mouse monoclonal anti–human Waf antibody and IgM mouse monoclonal antiactin antibody (Actin Ab-1) were purchased from Calbiochem.
Acknowledgments
We thank Dr. Borek Vojtesek for production of the Scotin antibodies, Drs. K. Brown, K. McLeod, and S. Lorimore for providing p53+/+ and p53−/− mice, Dr. L. Debussche for the gift of the H1299wtp53 tetracycline–inducible cell line, Dr. C. Midgley for the gift of the SaosWtp53 tetracycline–inducible cell lines, Drs. D. Lunny and A. Steward for the help in the in situ hybridization experiments, and Drs. S. Ponnambalam and A. Prescott for helpful discussions. We would like to thank M. Coelho for his excellent technical assistance. We are very grateful to K. Hudson (AstraZeneca) for his support in this work and to our colleagues for helpful discussions and proofreading.
This work was funded by AstraZeneca and the Cancer Research Campaign (CRC). J.C. Bourdon is funded by the Association for International Cancer Research, and P.L. Robertson and K. Fernandes are funded by the Medical Research Council. D.P. Lane is a Gibb Fellow of the CRC.
J. Renzig's present address is Roche Diagnosis GmbH, Nonnenwald 2, 82372 Penzberg, Germany.
Footnotes
Abbreviations used in this paper: Ad, adenovirus; AS, antisense; EMSA, electrophoretic mobility shift assay; MEF, mouse embryonic fibroblast; p53−/−, p53 nullizygote; p53+/+, p53 homozygote; RACE, rapid amplification of cDNA ends; UTR, untranslated region; wt, wild-type.
References
- Attardi, L.D., S. Lowe, J. Brugarolas, and T. Jacks. 1996. Transcriptional activation by p53, but not induction of the p21 gene, is essential for oncogene-mediated apoptosis. EMBO J. 15:3693–3701. [PMC free article] [PubMed] [Google Scholar]
- Blattner, C., A. Sparks, and D.P. Lane. 1999. Transcription factor E2F-1 is upregulated in response to DNA damage in a manner analogous to that of p53. Mol. Cell. Biol. 19:3704–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourdon, J.C., V. Deguin-Chambon, J.C. Lelong, P. Dessen, P. May, B. Debuire, and E. May. 1997. Further characterisation of the p53 responsive element—identification of new candidate genes for trans-activation by p53. Oncogene. 14:85–94. [DOI] [PubMed] [Google Scholar]
- Caelles, C., A. Helmberg, and M. Karin. 1994. p53-dependent apoptosis in the absence of transcriptional activation of p53-target genes. Nature. 370:220–223. [DOI] [PubMed] [Google Scholar]
- Clarke, A.R., C. Purdie, D. Harrison, R. Morris, C. Bird, M. Hooper, and A.H. Wyllie. 1993. Thymocyte apoptosis induced by p53-dependent and independent pathways. Nature. 362:849–852. [DOI] [PubMed] [Google Scholar]
- Deguin-Chambon, V., M. Vacher, M. Jullien, E. May, and J.C. Bourdon. 2000. Direct transactivation of c-Ha-Ras gene by p53: evidence for its involvement in p53 transactivation activity and p53-mediated apoptosis. Oncogene. 19:5831–5841. [DOI] [PubMed] [Google Scholar]
- El-Deiry, W.S., S. Kern, J. Pietenpol, K. Kinzler, and B. Vogelstein. 1992. Definition of a consensus binding site for p53. Nat. Genet. 1:45–49. [DOI] [PubMed] [Google Scholar]
- Ferri, K.F., and G. Kroemer. 2001. Organelle-specific initiation of cell death pathways. Nat. Cell Biol. 3:E255–E263. [DOI] [PubMed] [Google Scholar]
- Funk, W.D., D. Pak, R. Karas, E. Wright, and J.W. Shay. 1992. A transcriptionally active DNA-binding site for human p53 protein complexes. Mol. Cell. Biol. 12:2866–2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gossen, M., S. Freundlieb, G. Bender, G. Muller, W. Hillen, and H. Bujard. 1995. Transcriptional activation by tetracyclines in mammalian cells. Science. 268:1766–1769. [DOI] [PubMed] [Google Scholar]
- Green, D.R., and J.C. Reed. 1998. Mitochondria and apoptosis. Science. 281:1309–1312. [DOI] [PubMed] [Google Scholar]
- Harlow, E., and D.P. Lane. 1999. Immunoaffinity purification. Using Antibodies: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York. 313–343.
- Haupt, Y., S. Rowan, E. Shaulian, K. Vousden, and M. Oren. 1995. Induction of apoptosis in HeLa cells by trans-activation-deficient p53. Genes Dev. 9:2170–2183. [DOI] [PubMed] [Google Scholar]
- Jimenez, G.S., M. Nister, J. Stommel, M. Beeche, E. Barcarse, X. Zhang, S. O'Gorman, and G.M. Wahl. 2000. A transactivation-deficient mouse model provides insights into Trp53 regulation and function. Nat. Genet. 26:37–43. [DOI] [PubMed] [Google Scholar]
- Kaufman, R.J. 1999. Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes Dev. 13:1211–1233. [DOI] [PubMed] [Google Scholar]
- Koch, G., M. Smith, D. Macer, P. Webster, and R. Mortara. 1986. Endoplasmic reticulum contains a common, abundant calcium-binding glycoprotein, endoplasmin. J. Cell Sci. 86:217–232. [DOI] [PubMed] [Google Scholar]
- Kok, K., J. Osinga, B. Carritt, M. Davis, A. van der Hout, A. van der Veen, R. Landsvater, L. de Leij, H. Berendsen, P. Postmus, S. Poppema, and C.H. Buys. 1987. Deletion of a DNA sequence at the chromosomal region 3p21 in all major types of lung cancer. Nature. 330:578–581. [DOI] [PubMed] [Google Scholar]
- Kok, K., S. Naylor, and C.H. Buys. 1997. Deletions of the short arm of chromosome 3 in solid tumors and the search for suppressor genes. Adv. Cancer Res. 71:27–92. [DOI] [PubMed] [Google Scholar]
- Lane, D.P. 1992. Cancer. p53, guardian of the genome. Nature. 358:15–16. [DOI] [PubMed] [Google Scholar]
- Li, Z., and P.Z. Srivastava. 1993. Tumor rejection antigen gp96/grp94 is an ATPase: implications for protein folding and antigen presentation. EMBO J. 12:3143–3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang, P., and A.B. Pardee. 1992. Differential display of eukaryotic messenger RNA by means of the polymerase chain reaction. Science. 257:967–971. [DOI] [PubMed] [Google Scholar]
- Lin, Y., W. Ma, and S. Benchimol. 2000. Pidd, a new death-domain-containing protein, is induced by p53 and promotes apoptosis. Nat. Genet. 26:122–127. [DOI] [PubMed] [Google Scholar]
- Lowe, S.W., E. Schmitt, S. Smith, B. Osborne, and T. Jacks. 1993. p53 is required for radiation-induced apoptosis in mouse thymocytes. Nature. 362:847–849. [DOI] [PubMed] [Google Scholar]
- May, P., and E. May. 1999. Twenty years of p53 research: structural and functional aspects of the p53 protein. Oncogene. 18:7621–7636. [DOI] [PubMed] [Google Scholar]
- Midgley, C.A., C. Fisher, J. Bartek, B. Vojtesek, D. Lane, and D.M. Barnes. 1992. Analysis of p53 expression in human tumours: an antibody raised against human p53 expressed in Escherichia coli. J. Cell Sci. 101:183–189. [DOI] [PubMed] [Google Scholar]
- Mitelman, F., F. Mertens, and B. Johansson. 1997. A breakpoint map of recurrent chromosomal rearrangements in human neoplasia. Nat. Genet. 15 Spec No:417–474. [DOI] [PubMed]
- Miyashita, T., and J.C. Reed. 1995. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell. 80:293–299. [DOI] [PubMed] [Google Scholar]
- Muller, M., S. Wilder, D. Bannasch, D. Israeli, K. Lehlbach, M. Li-Weber, S.L. Friedman, P.R. Galle, W. Stremmel, M. Oren, and P.H. Krammer. 1998. p53 activates the Cd95 (Apo-1/Fas) gene in response to DNA damage by anticancer drugs. J. Exp. Med. 188:2033–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munsch, D., R. Watanabe-Fukunaga, J.-C. Bourdon, S. Nagata, E. May, E. Yonish-Rouach, and P. Reisdorf. 2000. Human and mouse Fas (APO-1/CD95) death receptor genes each contain a p53-responsive element that is activated by p53 mutants unable to induce apoptosis. J. Biol. Chem. 275:3867–3872. [DOI] [PubMed] [Google Scholar]
- Nakano, K., and K.H. Vousden. 2001. PUMA, a novel proapoptotic gene, is induced by p53. Mol. Cell. 7:683–694. [DOI] [PubMed] [Google Scholar]
- Naylor, S.L., B. Johnson, J. Minna, and A.Y. Sakaguchi. 1987. Loss of heterozygosity of chromosome 3p markers in small-cell lung cancer. Nature. 329:451–454. [DOI] [PubMed] [Google Scholar]
- Nylander, K., J.-C. Bourdon, S. Bray, N. Gibbs, R. Kay, I. Hart, and P.A. Hall. 2000. Transcriptional activation of tyrosinase and TRP-1 by p53 links UV irradiation to the protective tanning response. J. Pathol. 190:39–46. [DOI] [PubMed] [Google Scholar]
- Oda, E., R. Ohki, H. Murasawa, J. Nemoto, T. Shibue, T. Yamashita, T. Tokino, T. Taniguchi, and N. Tanaka. 2000. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science. 288:1053–1058. [DOI] [PubMed] [Google Scholar]
- Oda, K., H. Arakawa, T. Tanaka, K. Matsuda, C. Tanikawa, T. Mori, H. Nishimori, K. Tamai, T. Tokino, Y. Nakamura, and Y. Taya. 2000. p53aip1, a potential mediator of p53-dependent apoptosis, and its regulation by Ser-46-phosphorylated p53. Cell. 102:849–862. [DOI] [PubMed] [Google Scholar]
- Oren, M. 1999. Regulation of the p53 tumor suppressor protein. J. Biol. Chem. 274:36031–36034. [DOI] [PubMed] [Google Scholar]
- Stephen, C.W., P. Helminen, and D.P. Lane. 1995. Characterisation of epitopes on human p53 using phage-displayed peptide libraries: insights into antibody-peptide interactions. J. Mol. Biol. 248:58–78. [DOI] [PubMed] [Google Scholar]
- Vogelstein, B., D. Lane, and A.J. Levine. 2000. Surfing the p53 network. Nature. 408:307–310. [DOI] [PubMed] [Google Scholar]
- Yonish-Rouach, E., J. Borde, M. Gotteland, Z. Mishal, A. Viron, and E. May. 1994. Induction of apoptosis by transiently transfected metabolically stable wt p53 in transformed cell lines. Cell Death Diff. 1:39–47. [PubMed] [Google Scholar]
- Yonish-Rouach, E., V. Deguin, T. Zaitchouk, C. Breugnot, Z. Mishal, J. Jenkins, and E. May. 1996. Transcriptional activation plays a role in the induction of apoptosis by transiently transfected wild-type p53. Oncogene. 11:2197–2205. [PubMed] [Google Scholar]
- Yu, J., L. Zhang, P. Hwang, K. Kinzler, and B. Vogelstein. 2001. PUMA induces the rapid apoptosis of colorectal cancer cells. Mol. Cell. 7:673–682. [DOI] [PubMed] [Google Scholar]
- Zhao, S., S. Ooi, F. Yang, and A.B. Pardee. 1996. Three methods for identification of true positive cloned cDNA fragment in differential display. Biotechniques. 20:400–404. [DOI] [PubMed] [Google Scholar]