

Abstract
Wnt signaling orchestrates morphogenetic processes in which changes in gene expression are associated with dramatic changes in cell organization within developing tissue/organss. Upon signaling, excess β-catenin not utilized at cell–cell junctions becomes stabilized, where it can provide the transcriptional activating domain for Lef/Tcf DNA binding proteins. In skin epithelium, forced stabilization of β-catenin in epidermis promotes hair follicle morphogenesis, whereas conditional removal of β-catenin in hair progenitor cells specifies an epidermal fate. We now report that a single protein, a stabilized version of β-catenin lacking the COOH-terminal transactivation domain, acts in epidermis to promote hair fates and in hair cells to promote epidermal fate. This reveals fundamental differences in ways that epidermal and hair cells naturally respond to β-catenin signaling. In exploring the phenotype, we uncovered mechanistic insights into the complexities of Lef1/Tcf/β-catenin signaling. Importantly, how a cell will respond to the transgene product, where it will be localized, and whether it can lead to activation of endogenous β-catenin/Tcf/Lef complexes is specifically tailored to skin stem cells, their particular lineage and their relative stage of differentiation. Finally, by varying the level of β-catenin signaling during a cell fate program, the skin cell appears to be pliable, switching fates multiple times.
Keywords: Wnt signaling; epidermis; hair follicle; β-catenin; morphogenesis
Introduction
Cells able to receive and transduce a Wnt signal respond by stabilizing any excess β-catenin not used in cell–cell junction formation and recruiting it to act as a transcriptional cofactor for the activation of genes regulated by the Lef1/Tcf family of DNA binding proteins (Huelsken and Birchmeier, 2001; Brantjes et al., 2002). In cells that make cell–cell junctions, β-catenin is naturally stabilized through its interaction with the adherens junction components, E-cadherin and α-catenin (Jou et al., 1995; Aberle et al., 1996; Gottardi and Gumbiner, 2001; Nagafuchi, 2001). Excess cytoplasmic β-catenin is phosphorylated at its NH2-terminal serine-threonine residues by the GSK3β kinase, and targeted for proteosome-mediated degradation by a complex that includes the tumor-suppressor gene product adenomatous polyposis coli (APC)* (Behrens et al., 1998; Henderson, 2000; Reinacher-Schick and Gumbiner, 2001; van Noort et al., 2002). When a cell's Wnt surface receptors are stimulated, they can activate a signal transduction cascade that culminates in the inhibition of GSK3β, leading to the accumulation of β-catenin. An artificial promoter Tcf-optimal-promoter (TOP) containing multimerized Lef1/Tcf binding sites, has been used in conjunction with various reporter genes to identify Wnt-responsive cells (Molenaar et al., 1996; DasGupta and Fuchs, 1999; for review see Brantjes et al., 2002).
Recent studies have implicated members of the Wnt signaling pathway in the epithelial–mesenchymal exchanges that specify morphogenesis of the hair follicle (for review see Hardy, 1992; Fuchs et al., 2001). In mouse, TOP-βgalactosidase (TOPGAL) activity was detected in embryonic skin at sites of hair follicle formation, and postnatally, TOPGAL was active in the stem cell compartment (bulge) at the beginning of each hair cycle and also in the precursor cells that differentiate into hairs (DasGupta and Fuchs, 1999). The studies of Kishimoto et al. (2000) suggest that Wnts maintain the inductive ability of mesenchyme to stimulate hair cell fate at the start of each cycle (Kishimoto et al., 2000).
Evidence that Wnt signaling is functionally important for hair follicle morphogenesis stems from transgenic mice engineered to express a constitutively stable and active form of β-catenin under the control of an epidermal keratin promoter (K14) (Gat et al., 1998). In epidermis, its expression spontaneously induced aberrant hair follicles throughout the interfollicular epidermis between the preexisting follicles (Gat et al., 1998). Similarly, in chick embryos, what appeared to be sustained activation of β-catenin led to excessive feather and scale morphogenesis (Noramly et al., 1999; Widelitz et al., 2000). Conversely, ablation of Lef1 gene expression in mouse skin or β-catenin expression in K14-expressing cells of mouse skin impairs hair follicle morphogenesis (van Genderen et al., 1994; Huelsken et al., 2001). Based on these findings, Wnt signaling seems to coax skin stem cells along a hair (epidermal appendage) cell fate. Consistent with this notion is the finding that the hair-specific keratin promoters are bona fide target genes of Lef/β-catenin signaling (Zhou et al., 1995; Merrill et al., 2001). Additionally, concomitant with the development of a hair placode is an upregulation in transcription of both Lef1 and β-catenin genes (Zhou et al., 1995; Noramly et al., 1999; Widelitz et al., 2000; Huelsken et al., 2001).
Although Wnt signaling seems to promote hair cell fates, an absence of or interference with Wnt signaling seems to favor epidermal and sebocyte cell fates. Thus, a K14-conditional loss of function mutation in the β-catenin gene locus resulted in a blockage of embryonic hair development if lost early and formation of epidermal-like cysts in place of hair follicles if lost later (Huelsken et al., 2001). In addition, transgenic mice expressing K14-ΔNLef1, unable to bind β-catenin, yielded epidermal and sebocyte differentiation at sites where postnatal hair cycling was expected (Merrill et al., 2001; Niemann et al., 2002).
The picture emerging from these studies suggests that setting the levels of β-catenin plays a crucial role in deciding if stem cells or their progeny will contribute to structures below the bulge (hair cell types) or above the bulge (sebaceous gland, upper outer root sheath [ORS], and epidermis). These findings have prompted us to wonder whether epidermal and hair follicle cells are truly equivalent with respect to their internal ability to translate a Wnt signal, or whether there might be important internal differences that might be overridden and obscured by overexpressing or ablating β-catenin in mice. If, as recent evidence implies, stem cell progeny migrate from the bulge and specify cell fates in different environments (Taylor et al., 2000; Oshima et al., 2001), do they maintain equivalency with respect to their ability to respond to a stabilized β-catenin signal, or does this chemistry change? Is it simply differences in Wnt/Wnt receptors, or are there distinct differences in the way hair and epidermal cells respond to increased β-catenin expression?
Placed in a broader context of cell lineage determination, the heart of this problem focuses on the extent to which the levels of β-catenin's varied interacting partners might differ in cells and how this impacts on the manner in which the cell responds to a Wnt signal and/or stabilized β-catenin. The skin becomes an interesting model system to tackle this problem, because not only is the hair versus epidermal fate manipulated by overexpression or underexpression of β-catenin, but in addition, at least some of β-catenin's associates, including Lef1/Tcf levels (Zhou et al., 1995; DasGupta and Fuchs, 1999; Merrill et al., 2001) and intercellular junction proteins (Nanba et al., 2000) are known to be differentially expressed in these two cell types.
To determine the extent to which hair and epidermal cells might differ in their internal ability to process and respond to a given level of β-catenin, we sought a strategy for manipulating β-catenin signaling in a way in which we would expect to see the same phenotypic outcomes in mouse epidermal and hair cells if they responded equivalently, but different outcomes if they harbored distinctly different means of receiving the same signal. Rather than stabilizing β-catenin and seeing hair follicle morphogenesis in epidermis as well as hair follicles (Gat et al., 1998), or ablating β-catenin and seeing epidermal differentiation in hair follicles as well as in epidermis (Huelsken et al., 2001), we searched for conditions in which epidermal cells responded in one way and hair cells in another. We settled on engineering mice to express a COOH terminally truncated form of the constitutively stabilized version of β-catenin that we had used earlier in generating our super-furry mice (Gat et al., 1998).
The COOH terminus of β-catenin is required for transactivation, and it binds the transcriptional activator p300/CBP as well as the chromatin remodeling factor Brg-1 (Molenaar et al., 1996; Orsulic and Peifer, 1996; van de Wetering et al., 1997; Huber et al., 1997; Hsu et al., 1998; Hecht et al., 1999, 2000; Barker et al., 2001). Indeed, β-catenin mutants lacking the COOH terminus can be deficient in signaling not only in vitro, but also in vivo (Orsulic and Peifer, 1996; Cox et al., 1999; Hecht et al., 1999; Vleminckx et al., 1999; Barker et al., 2001; Tutter et al., 2001). This said, prior studies on β-catenin signaling in Xenopus and in Drosophila had demonstrated that overexpression of membrane-tethered or NH2/COOH terminally truncated forms of β-catenin could potentiate signaling depending on its interacting partners within the cell. This led to the hypothesis that these forms of β-catenin might act by displacing endogenous β-catenin from intercellular junctions, freeing it to be utilized in transactivation (Miller and Moon, 1997; Cox et al., 1999).
By expressing a single protein with potentially dominant negative and dominant positive character, we find that different cells within the skin behave unidirectionally towards this protein. However, strikingly and ironically, whereas the follicle stem cells and their progeny respond in a dominant negative fashion to the transgene product, the epidermal cells respond in a dominant positive fashion. Moreover, we show that by changing transgene expression midstream in the process, the process becomes reversible, and cells can revert back to their original lineage. Our findings have important and broad implications for how cells respond to and translate Wnt/β-catenin signals and how cell type specificity and cell fate commitment is achieved.
Results
K14-ΔNΔCβcatenin interferes with the transactivation of target genes in keratinocytes in vitro
To engineer K14-ΔNΔCβcatenin encoding a stable form of β-catenin that lacks Tcf/Lef1-mediated signaling activity, we used the K14-ΔN87βcatenin transgene (Gat et al., 1998) and removed sequences encoding the COOH-terminal 98 amino acid residues of β-catenin (Fig. 1 A). We also added a COOH-terminal HA-epitope tag so we could easily monitor transgene expression in vitro and in vivo. Addition of this epitope was previously found to yield functional ΔNβcatenin (Gat et al., 1998; unpublished data). To verify that the transgene encoded a protein of the expected size, we transiently transfected K14-ΔNΔCβcatenin into mouse keratinocytes. 48 h after transfection, cells were processed for immunoblot analysis using a rat anti-HA monoclonal antibody. Only cells transfected with K14-ΔNΔCβcatenin showed a band of the correct size (∼66.5 kD), whereas mock-transfected cells displayed no expression (Fig. 1 B). The blot was probed with an anti-tubulin antibody to verify that the protein loadings were equivalent.
Figure 1.
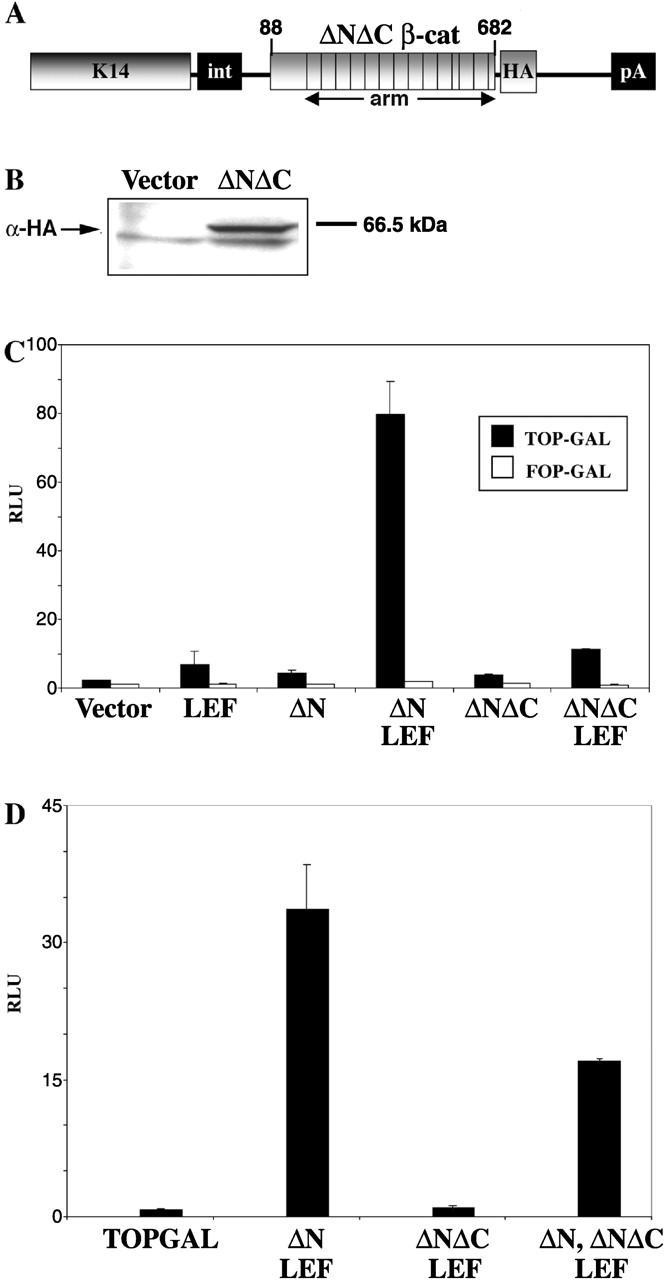
The K14-ΔNΔCβcatenin construct and in vitro transfection assays. (A) The K14-ΔNΔCβcatenin construct starting with amino acid 88 at the NH2 terminus of human β-catenin and ending at amino acid 682. The K14 expression construct comprises the K14 promoter at the very 5′-end. It also includes the β-globin intron, the COOH-terminal HA tag, and pA, the poly-adenylation signal, which is at the 3′ end of the construct. (B) Western blot using the anti-HA antibody on protein extracts derived from control K14 empty vector transfected and K14-ΔNΔCβcatenin–transfected mouse keratinocytes. Note that a 66.5-kD band exists only in extracts from cells transfected with ΔNΔCβcatenin. (C) Transient transfection assays with TOPGAL reporter. Mouse keratinocyte line, RD1, was transfected with pTOPGAL, pCMV-luciferase (as an internal control gene) and equimolar amounts of plasmids K14-ΔN87βcat, K14-ΔNΔCβ-cat, pK14-Lef1, or empty expression vector, as indicated (see Gat et al., 1998, for method). 36–48 h later, cells were lysed and protein extracts were assayed for β-galactosidase activity (test) and luciferase (to correct for transfection efficiency). Normalized activities of TOPGAL (black bars) represent an average of three experiments, with variations shown by error bars. FOPGAL (white bars) had no activity. Whereas Lef1 and ΔNβcat super-activated the TOPGAL reporter, Lef1 and ΔNΔCβ-cat failed to do so. (D) K14–ΔNΔCβcat acts like a dominant negative; same as C, but with equimolar amounts of K14-ΔNβcat and K14–ΔNΔCβcat added together with Lef1, which blocked the activation mediated by Lef1 and ΔNβcat alone.
To assay for the signaling activity of ΔNΔCβcatenin, we performed keratinocyte transfection assays with the TOPGAL reporter, driving β-galactosidase gene expression by an enhancer composed of multimerized Lef1/Tcf DNA binding sites (DasGupta and Fuchs, 1999). The luciferase equivalent of this transgene, TOPFLASH, has been widely used as a sensitive method to detect Lef1/Tcf/β-catenin–mediated signaling in a broad variety of cell types, including keratinocytes (Molenaar et al., 1996). We analyzed the behavior of the two β-catenins in response to Lef1, naturally expressed in hair precursor cells (Zhou et al., 1995), and Tcf3, naturally expressed in the stem cell compartment and ORS of the hair follicle (DasGupta and Fuchs, 1999; Merrill et al., 2001).
Similar to that seen previously for TOPFLASH (Gat et al., 1998), ΔNβcatenin and Lef1 on their own each had only a modest effect on TOPGAL reporter activity, but together, they strongly transactivated (Fig. 1 C). These effects were dependent on the presence of functional Lef1 binding sites, as evidenced by the lack of activation with FOPGAL mutant in the Lef1 binding sites (Fig. 1 C). In contrast, Lef1 and ΔNΔCβcatenin together exhibited only slightly higher levels than Lef1 alone, although the activity was consistently above background (Fig. 1 C). Thus, despite the fact that ΔNΔCβcatenin lacks the transactivating domain, some activation was still observed when it was coexpressed with Lef1 in keratinocytes. We will return to this point again later.
When equal amounts of ΔNβcatenin and ΔNΔCβcatenin were expressed together with Lef1, ΔNΔCβcatenin suppressed the activation of the TOPGAL reporter by twofold (Fig. 1 D). Similar inhibitory effects were observed with Tcf3, although because Tcf3 normally functions as a repressor, this was more difficult to demonstrate (unpublished data). The dominant negative effects of ΔNΔCβcatenin were expected, given that ΔNΔCβcatenin still contained the armadillo repeats, able to interact with the Tcf/Lef1 DNA binding proteins (Huber et al., 1997; Graham et al., 2000; Tutter et al., 2001). Thus, irrespective of which Lef1/Tcf family member ΔNΔCβcatenin was combined with, ΔNΔCβcatenin impaired the transactivation potential conferred to by stabilized β-catenin.
Expression of ΔNΔCβcatenin in the epidermis, follicle ORS, and stem cell compartment of transgenic mice
Given the ability of ΔNΔCβcatenin to act in a dominant negative fashion in interfering with the activity of functional Lef1/βcatenin complexes in keratinocytes in vitro, we then turned towards using this transgene to assess the consequences of diminished Wnt signaling on hair organogenesis and postnatal cycling. For purposes of comparison to the previously generated K14-ΔNβcatenin and K14-Cre conditional β-catenin–null mice, we used the K14 promoter to drive the expression of ΔNΔCβcatenin in the dividing cells of the epidermis, the ORS, and the stem cell compartment of hair follicles.
K14-ΔNΔCβcatenin mice were generated in the CD-1 strain, and PCR of tail DNAs confirmed the identity of mice that carried the transgene. Expression of ΔNΔCβcatenin protein was confirmed by immunoblot analysis of proteins isolated from whole newborn skin of transgenic and control littermate animals, as well as from primary keratinocytes derived K14-ΔNΔCβcatenin–expressing mice. In transgenic keratinocytes in vivo and in vitro, the transgenic protein of ∼66.5 kD was detected by the anti-HA antibody (Fig. 2 A). Immunofluorescence analysis using the HA antibody on frozen skin sections confirmed the expression of the transgene specifically in the basal epidermal layer (Fig. 2 B) and the follicle ORS and the bulge (Bu) (Fig. 2, B′ and C, schematic of follicle). Two independently derived mouse lines behaved similarly in this and all subsequent analyses, thereby attributing the observed effects to transgene expression and not chromosomal integration site.
Figure 2.
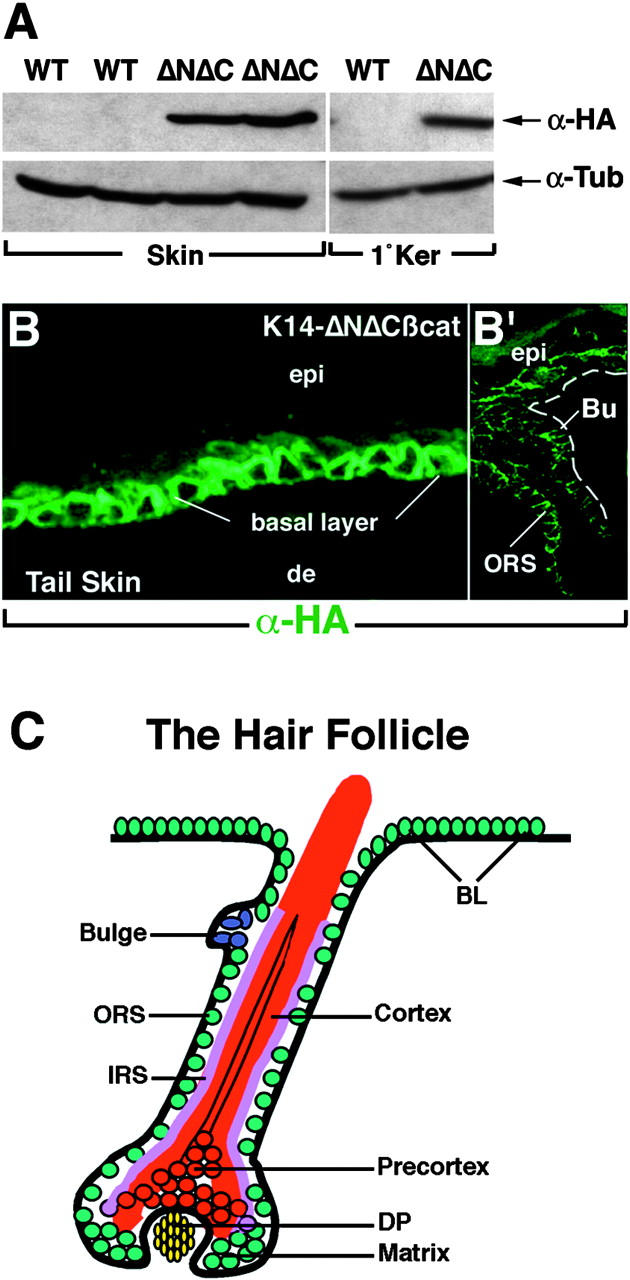
Stable expression of HA-tagged ΔNΔCβcatenin transgene in skin and primary keratinocytes. (A) Immunoblot analysis of K14-ΔNΔCβcatenin in transgenic mouse skin. Transgenic skin from a neonatal transgenic and control WT mouse was frozen in liquid nitrogen and ground to a powder. After extraction, proteins were resolved by electrophoresis through 8% polyacrylamide gels and subjected to immunoblot analysis using an anti-HA and anti–β-tubulin (loading control) antibody and ECL chemiluminescence. The same experiment was performed on protein extract isolated from primary keratinocytes derived from neonatal WT and transgenic mouse skin. For protocol for isolation of primary keratinocytes, and making protein extracts, see Materials and methods. (B) Immunostaining on frozen sections of neonatal tail skin (left) and head skin (right) with anti-HA antibody, depicting expression of the transgene in basal epidermal cells and the follicle ORS. Some staining is also observed in the stratified differentiated layers of ΔNΔCβcatenin expressing transgenic skin, perhaps due to the stability of the transgenic product. (C) The anatomy of the mature hair follicle. The mature follicle consists of an ORS contiguous with the basal layer of the epidermis (green cell layer), an inner root sheath that serves as the channel from which the hair exits the skin surface (pink), and the hair shaft itself (red). The hair is derived from the precortical cells, which along with the inner root sheath are descendents of proliferating matrix cells at the bulb of the follicle. Matrix cells are thought to maintain their proliferative state through sustained association with dermal papilla (yellow), encloaked by the matrix.
ΔNΔCβcatenin-expressing transgenic mice display some features resembling a loss-of-function phenotype: switch from a hair cell to epidermal cell fate
Despite strong transgene promoter activity by embryonic day 14.5, an overt phenotype did not emerge until 3 to 4 wk of postnatal development, corresponding to the approximate timing of the synchronized first postnatal hair cycle. This is the time when secondary hair germ normally emerges from the follicle bulge and grows downward to become the new follicle (Fig. 3 , A–C). In K14-ΔNΔCβcatenin mice, although the secondary hair germ emerged, a cyst-like structure developed rather than a hair follicle (Fig. 3, D–F). The cysts grew rapidly over the period of 3 to 4 d after initiation of postnatal cycling.
Figure 3.
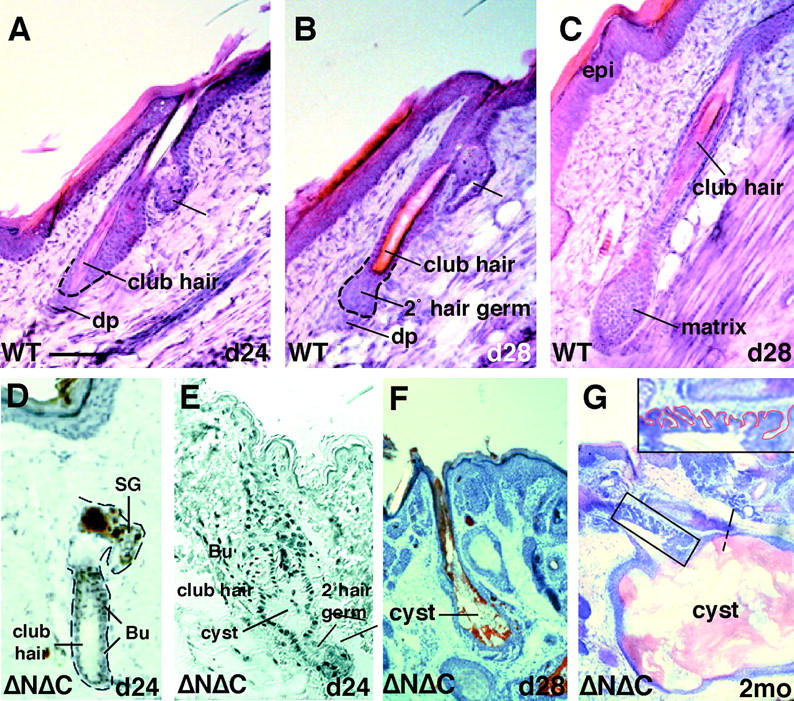
Formation of epidermal cysts in ΔNΔCβcatenin mice, at the start of postnatal hair cycle. (A–C and D–G) Hair cycle in WT and transgenic skin, respectively. (D–H) Progression of hair cycle and cyst formation in the skin of transgenic mice. Skin sections from day 24–28 ΔNΔCβcatenin transgenic line and control littermates (WT) were fixed in 4% PFA fixative, embedded in OCT compound and sectioned (10 μm), and stained with hematoxylin and eosin and in some cases with Oil Red O to mark cells of sebaceous lineage. Shown are brightfield images of representative sections of: (A) onset of anagen; (B) formation of secondary hair germ; and (C) formation of new hair matrix in WT tail follicles. (D) ΔNΔCβcat transgenic skin at the onset of anagen; (E) transgenic follicle displaying the formation of a small cyst-like structure; (F) early anagen transgenic follicle depicting the rapid formation of cyst, with Oil Red O staining in red sebaceous; (G) 2-mo-old skin follicle displaying late stage large cysts emanating from an old follicular structure. Higher magnification in inset shows epithelial invaginations emanating from the upper ORS.
Although many cysts consisted of cells resembling epidermis, some cysts displayed signs of the sebaceous lineage, readily identified by Oil Red O counterstaining of hematoxylin and eosin stained sections (Fig. 3 F). By 2 mo, cysts had become large and irregular in shape, extending into areas of the dermis that were much larger than the sphere occupied by their parent follicle (Fig. 3 G). Serial sectioning of skin at these later times often enabled us to trace the origin of the cells to a hair follicle, consistent with their origins visualized at earlier times (e.g., Fig. 3, E–G). These cysts eventually grew sufficiently large to yield a visible bump on the skin surface (unpublished data). At these later times, the upper ORS above the cyst often displayed numerous cellular extensions that were not seen within the cysts (Fig. 3 G and inset). The significance of this will be discussed later.
The outermost layer of cells surrounding the follicular cysts exhibited morphological features typical of the basal layer of the epidermis (Fig. 4 , compare A, epidermis, with B, cyst). Progressively more inward, the cyst resembled terminally differentiating epidermis. Large suprabasal-like cells were seen within the range between the outer basal-like layer and the middle of the cyst, whereas cells packed with keratohyalin granules were seen further inward. Enucleated squamous-like cells were found at the cyst center. Immunofluorescence with antibodies against a variety of epidermal differentiation markers confirmed the epidermal character of the cysts (Fig. 4, C and D). The basal-like cells at the periphery of the cyst were positive for K5 and K14, the suprabasal cells stained with antibodies against the spinous layer marker keratin 1 (K1), and the granular cells labeled with antibodies against filaggrin and loricrin, typically seen in the epidermal granular layer.
Figure 4.
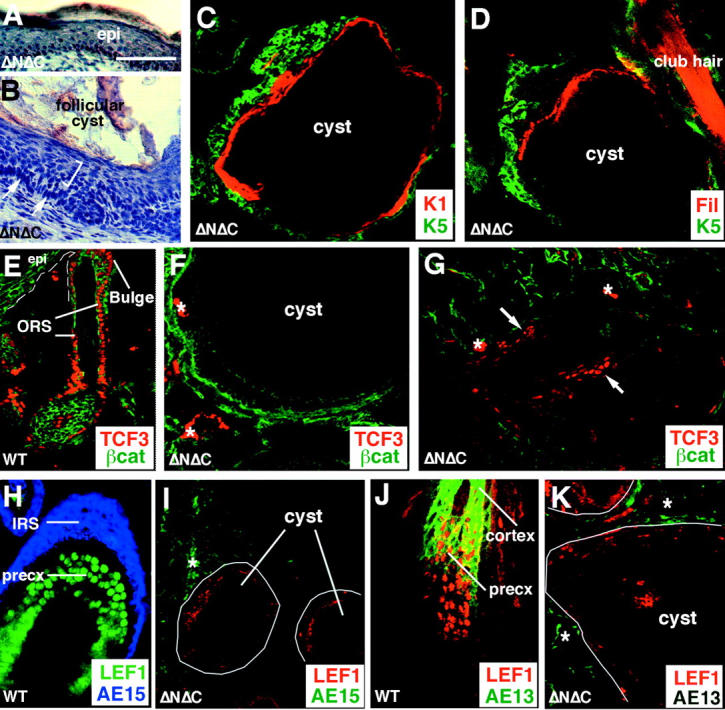
Epitheliod cysts in ΔNΔCβcatenin mice display epidermal characteristics and lose the expression of hair follicle markers. 2-mo-old tail skin from ΔNΔCβcatenin mice was harvested for H and E staining and immunofluorescence on the epitheliod cysts, formed in transgenic animals. The tissue was embedded in OCT, cut into 10-μm sections, and processed for staining purposes. Shown are bright field images of representative examples of (A) 40× magnification of H and E staining of transgenic interfollicular epidermis; and (B) the periphery of a cyst showing epidermal characteristics such as basal cells (arrows) and the supra-basal layers (straight bracket). (C and D) Confocal images of immunostaining for early and late differentiation markers, respectively. K1 and filaggrin are red and basal marker K5 is depicted in green. Filaggrin is not always expressed all around the epitheliod cyst. (E–K) Immunostaining for hair follicle markers in WT and transgenic skin follicles. (E) TCF3 staining in ORS of WT hair follicle in red. (F) Loss of TCF3 in the basal layer of the epidermal cysts. (G) remnants of some TCF3 positive cells in an early cyst, suggesting TCF3 expression is lost or fails to be maintained in the basal-like cells as the follicles develop into epitheliod cysts. (H) WT hair follicles expressing AE15, inner root sheath marker in blue, and LEF1 in the precortex in green. (J) AE13, pan-hair keratin marker in green, and LEF1 in red. (K) Representative example of loss of LEF1 (in red) and AE13 (in green) hair follicle markers in the epitheliod cysts formed in ΔNΔC transgenic mice. Note that the asterisks mark background, noncellular staining of mouse monoclonal antibodies on scar tissue found in transgenic skin. precx, precortex.
The cysts bore little if any resemblance to their hair follicle origins. Tcf3, the Lef1/Tcf member normally expressed in the nuclei of the lower ORS and the bulge of normal follicles (Fig. 4 E), was largely absent in the cysts (Fig. 4 F), although a few clusters of Tcf3-positive nuclei were detected at the periphery of some cysts (Fig. 4 G). Cysts were also devoid of staining with AE15 antibodies that recognize the trichohyalin granules of the inner root sheath (Fig. 4, compare H, wild-type [WT], with I, cyst), and most cysts failed to stain with AE13, an antibody specific for the hair keratins (Fig. 4, J, WT, and K, transgenic). Finally, the cysts exhibited a marked reduction in the hair-specific expression of Lef1 (Fig. 4, compare H and J, Lef11 expression in WT follicles, and I and K, loss of Lef1 expression). Taken together, the results demonstrate that the cysts were predominantly epidermal rather than hair follicle in nature.
Overall, the marked morphological and biochemical resemblance of these cysts to masses of differentiating epidermal cells paralleled the description of the cysts seen when β-catenin was conditionally ablated in K14-expressing cells of mice (Huelsken et al., 2001) and when transgenic mice were engineered to express a K14-ΔNLef1 transgene, encoding a form of Lef1 unable to associate with β-catenin (Merrill et al., 2001; Niemann et al., 2002). In contrast to the striking effects on the hair cycle, the skin epidermis of K14-ΔNΔCβcatenin mice was only modestly affected (Fig. 4 A). The patterns of K5, K1, and filaggrin expression in transgenic epidermis appeared largely normal (unpublished data), although the spinous layer and granular layers were somewhat thickened in some areas. The increased thickening was attributed to enhanced proliferation, as judged by staining with an antibody against Ki67, present in the nuclei of proliferating basal epidermal cells (unpublished data). These changes aside, the overall spatial and temporal pattern of epidermal differentiation was largely similar between WT and K14-ΔNΔCβcatenin transgenic skin, and in some areas, no major differences were noted (Fig. 3, D and E).
Expression of ΔNΔCβcatenin generates a failure to initiate Lef1/Tcf/βcatenin-mediated signaling in the bulge
Given the similarities between the epidermal cysts of ΔNΔCβcatenin and β-catenin–null skin, and the inhibitory effects of ΔNΔCβcatenin on Lef1/Tcf/β-catenin–regulated gene transcription in vitro, it seemed most likely that the cysts arose from a failure to activate this transcription pathway. To test this, we mated our previously generated TOPGAL transgenic mice on the background of the ΔNΔCβcatenin transgenic mice, and examined the double transgenic offspring for their ability to transactivate TOPGAL. In normal TOPGAL mice, a few cells within the bulge of many hair follicles expressed β-galactosidase at the start of the first postnatal hair cycle, when the mesenchymal dermal papilla was abutted against the epithelial stem cell compartment (Fig. 5 A; DasGupta and Fuchs, 1999). When bred against the background of our previously generated K14-ΔNβcatenin transgenic mice, very strong TOPGAL activity was observed in most bulge cells undergoing hair cycle initiation (Fig. 5 B). In striking contrast, the bulges at the equivalent stage of ΔNΔCβcatenin transgenic follicles were usually devoid of β-galactosidase activity (Fig. 5 C). The finding that the cells in the bulge compartment of ΔNΔCβcatenin expressing follicles were silent for TOPGAL activity and resulted in epidermal cyst formation provided compelling evidence that a loss of Tcf/Lef1/β-catenin–mediated signaling activity in the bulge leads or contributes to the acquisition of epidermal cell fates.
Figure 5.

Failure of TOPGAL activation in the bulge of ΔNΔCβcatenin transgenic follicles, at the onset of the first anagen. Mouse skin was harvested at the start of anagen, embedded in O>C>T compound and snap frozen for later use. Shown are representative examples of 10-μm sections that were fixed for 2 min in 0.1% glutaraldehyde and subsequently stained with X-gal staining solution. (A) TOPGAL activation of one cell (arrowhead) in the bulge region of WT anagen follicle. (B) Superactivation of TOPGAL in cells streaming out of the bulge in K14-ΔN87βcat transgenic mice. (C) Lack of TOPGAL activity in the bulge of ΔNΔCβcat transgenic follicles at the start of the new hair cycle.
ΔNΔCβcatenin-expressing transgenic mice display some features resembling a gain of function phenotype: switch from an epidermal cell to a hair cell fate
Whereas K14-ΔNΔCβcatenin transgenic animals displayed epidermal cysts characteristic of a loss-of-function phenotype of β-catenin, they displayed hair germ–like invaginations that resembled the gain-of-function features that we had seen in our K14-ΔNβcatenin transgenic mice (Gat et al., 1998; Widelitz et al., 2000; Noramly et al., 1999). Intriguingly and importantly, the gain-of-function phenotype was seen in cellular compartments that were mutually exclusive from the loss-of-function phenotype. Thus, in contrast to epidermal cysts, which emerged from secondary hair germ outgrowths of postnatal hair follicles, the hair germ–like invaginations appeared to be restricted to the interfollicular epidermis and the ORS (Fig. 6) . By 28 d of postnatal development, interfollicular epidermis was riddled with these projections, which displayed numerous secondary projections, leading to flower-like structures (Fig. 6, compare A, WT skin, with B, ΔNΔCβcatenin skin). These structures were accompanied by aberrant TOPGAL activation, reflective of functional Lef1/β-catenin complexes (Fig. 6 C). Moreover, similar to what was previously reported by Gat et al. (1998) in the ΔNβcatenin mice, the epithelial invaginations in the ΔNΔCβcatenin transgenic skin displayed elevated levels of Lef1 in both the newly forming hair germs as well as the interfollicular epithelia (Fig. 6, compare E, F, and G). Invaginations emanating from the ORS appeared to be largely restricted to the upper segment, i.e., above the epidermal cysts (Fig. 6 D). Thus, a clean boundary existed between the negative action of the transgene below the bulge and the positive action of the transgene above it.
Figure 6.
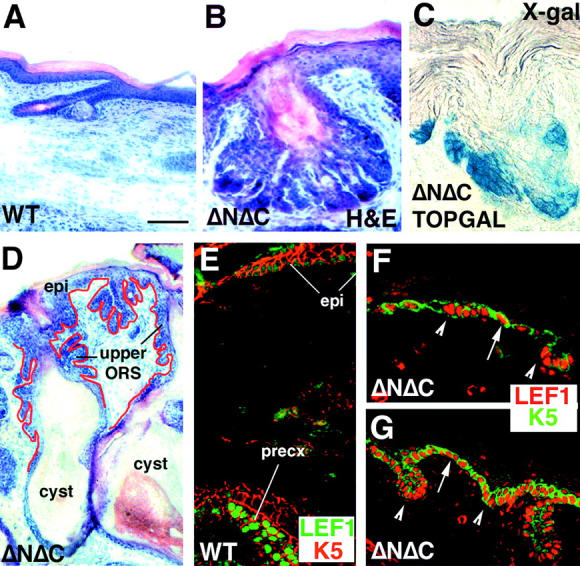
ΔNΔCβcat transgenic mice display gain-of-function β-catenin signaling phenotype in interfollicular epidermis. Tail skin sections from day 28 ΔNΔCβcatenin transgenic line and control littermates (WT) were fixed in 4% PFA fixative, embedded in OCT compound and sectioned (10 μm), and stained with hematoxylin and eosin. Shown are representative sections of A and B, tail skin follicles of WT and transgenic littermates, respectively. Hematoxylin and eosin staining revealed a phenotype in the interfollicular epidermis, reminiscent of that obtained in the ΔNβ-catenin transgenic mice. (C) X-gal staining in transgenic mice harboring both ΔNΔCβcatenin and TOPGAL. TOPGAL is activated in the de novo hair-like invaginations. (D) Multiple epithelial invaginations sprouting from the upper ORS of older follicles and the interfollicular epidermis of transgenic skin. (E) Confocal immunofluorescence image of WT 28-d tail skin, with Lef1 in green and β-catenin in red. The epidermis displays very low levels, if any, of Lef1 staining (compare staining intensity with the nuclear Lef1 in precortex). (F and G) 28-d ΔNΔCβcatenin transgenic tail skin stained for Lef1 in red and K5 in green. Arrowheads denote activation of Lef1 in hair placodes and hair germs. (F and G) Arrows denote ectopic expression of LEF1 in the basal layer of epidermis of 28-d transgenic animals.
Consistent with TOPGAL activation in these flower-like invaginations of epidermis and upper ORS, Lef1 expression was also detected. In WT skin, nuclear Lef1 is always strongest in the follicle precortex, where TOPGAL and hair-specific keratin genes are activated (Fig. 6 E; DasGupta and Fuchs, 1999; Merrill et al., 2001). In ΔNΔCβcatenin skin, early de novo hair germs often exhibited intense nuclear Lef1 antibody staining (Fig. 6 F), also seen at the leading front of more advanced downgrowths (Fig. 6 G). Both interfollicular and ORS growths exhibited this intense nuclear Lef1 staining accompanied by TOPGAL activation. As judged by antibody staining specific for Ki67, a proliferating nuclear antigen, these downgrowths were also accompanied by a marked increase in proliferation (unpublished data). Eventually, the mice developed pilomatricoma tumors which, in humans, have been shown to be a result of stabilizing mutations in β-catenin (Chan et al., 1999), and which in mice arise from constitutive stabilization of β-catenin (Gat et al., 1998). The downgrowths and the pilomatricomas stained for AE13, indicative of hair-keratin expression.
Overall, the effects of ΔNΔCβcatenin on epidermal cells was as a positive stimulator of β-catenin signaling, thereby activating the hair cell fate and eventually leading to hair cell tumors, or pilomatricomas in all transgenic mice. This was in marked contrast to the transgene's effects as an inhibitor of β-catenin signaling in the follicle precursor cells, where the outcome was epidermal cyst formation. The opposing effects were at least in part due to differences in transcriptional activation of Lef/Tcf/β-catenin–regulated genes, as revealed by the fact that the epidermal cysts at sites anticipated for hair follicles were TOPGAL negative, whereas the follicle-like structures at sites anticipated for epidermis were TOPGAL positive. Most remarkably, the opposing effects of the transgene were not random, but rather were displayed in a highly cell type–specific fashion.
ΔNΔCβcatenin is able to interact with E-cadherin and with members of the β-catenin degradation machinery
Previous studies have shown that ΔNΔCβcatenin can associate with Lef/Tcf family members, and our in vitro studies show that it can influence transactivation potential of Lef-regulated genes in keratinocytes. To assess how ΔNΔCβcatenin is capable of having positive effects on epidermal cells at the same time as it has negative effects on hair cells, it was necessary to confirm that this protein is also able to associate with candidate cytoplasmic partners, including the adherens junction protein E-cadherin and APC, the protein that targets β-catenin for the proteosome degradation pathway.
Cell lysates from skin and calcium-treated primary keratinocytes and from skin of WT and transgenic mice were first adjusted to have equal levels of E-cadherin in the starting lysate extracts for WT and transgenic (Fig. 7) . Anti–E-cadherin antibody was then used in a pulldown assay to bind E-cadherin complexes to protein G–conjugated Sepharose beads. These complexes contained both HA-tagged ΔNΔCβcatenin and endogenous β-catenin (Fig. 7). In skin and in primary keratinocytes, the overall E-cadherin pulled down in the assays was consistently higher in the transgenic versus the WT samples, despite the roughly comparable levels in the aliquots used for pulldown.
Figure 7.
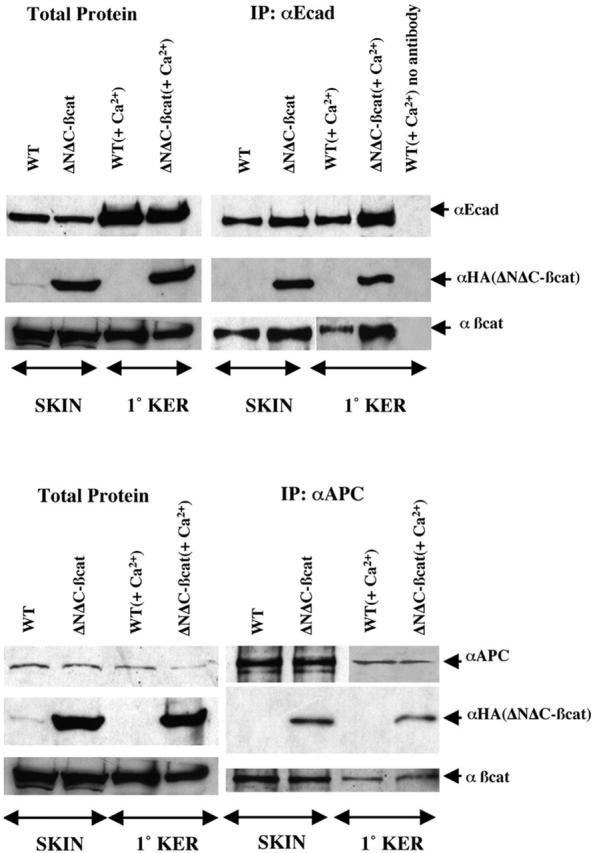
Biochemical interactions of ΔNΔCβcatenin with E-cadherin and APC. (Top) Interactions of ΔNΔCβcatenin with E-cadherin. Coimmunoprecipitation studies were performed to assay interaction between ΔNΔCβcatenin and E-cadherin. Total protein extracts were made from WT and ΔNΔCβcat transgenic skin and primary keratinocytes by solubilizing in strong lysis (RIPA) buffer. Shown on the right panel are results from Western blot analysis of α-E-cadherin immunoprecipitates, probed with anti-E-cadherin, anti-HA, and anti–β-catenin antibodies. On the left panel, the same antibodies were used to detect total protein levels, before the pulldown (input) with anti–E-cad antibody. The anti-E-cad antibody could successfully pull down E-cadherin and the proteins associated with it. Extracts to which anti–E-cad antibody was not added, failed to pull down the complex (lane 5, right panel). (Bottom) Interactions of ΔNΔCβcatenin with APC. Experiment was performed as in top, except that anti-APC antibody was used in place of anti–E-cad.
To assess whether ΔNΔCβcatenin might be capable of eliciting its positive effects on β-catenin signaling through stabilizing endogenous β-catenin, we next determined its ability to associate with APC, which normally interacts with endogenous β-catenin and targets it for phosphorylation and ubiquitin-mediated degradation. As shown in Fig. 7 (bottom), APC is expressed in keratinocytes and can immunoprecipitate ΔNΔCβcatenin as well as β-catenin. The complexities of the cells within the skin samples and the relative specificities of the antibodies precluded our ability to use immunoprecipitation and immunoblot assays to make further comments as to the relative levels of mutant and endogenous β-catenin proteins associated with cell junctions versus APC complexes.
The ratio of nuclear ΔNΔCβcatenin to endogenous β-catenin is lower in epidermal than in hair cells, and yet K14 promoter activity is higher in epidermal than in hair cells
Without a COOH-terminal activation domain, β-catenin is unable to function with Lef1/Tcf family members to transactivate genes. To understand how ΔNΔCβcatenin might be eliciting positive effects on interfollicular and upper ORS epithelium, we first examined the location of ΔNΔCβcatenin when transiently expressed in keratinocytes. To distinguish transgenic from endogenous β-catenin, we used an anti-HA antibody to recognize the epitope tag of the transgene product and an anti–β-catenin antibody directed against the COOH terminus of β-catenin to specifically recognize the endogenous β-catenin protein by immunofluorescence. The results are summarized in Fig. 8 . Whereas only ∼2–5% of primary keratinocytes were transfected, ∼40% of them exhibited nuclear as well as cytoplasmic localization of the transgene product (Fig. 8 A, ΔNΔCβcat in green). Interestingly, 100% of transfected cells exhibited nuclear localization of endogenous β-catenin (Fig. 8, A and B, endogenous β-catenin in red). Under the low-calcium conditions used here, endogenous β-catenin in untransfected and transfected cells was always cytoplasmic. However, in stark contrast to the nuclei of transfected cells, the nuclei of untransfected cells were negative for endogenous β-catenin. These findings suggest that by expressing ΔNΔCβcatenin protein in keratinocytes, endogenous β-catenin is translocated to the nucleus, a point which we confirmed by immunoblot analyses (unpublished data). Moreover, the data demonstrate at a molecular level that the transgene product can displace endogeous β-catenin from the cytoplasmic pool, and stimulate a process that results in its accumulation in the nucleus. The generation of nuclear β-catenin in transfected cells is likely to explain why our transfections with ΔNΔCβcatenin always gave a slight enhancement of TOPGAL activity when coexpressed with Lef1 (Fig. 1 C).
Figure 8.

β-Catenin translocates into the nucleus of cells transfected with ΔNΔCβcatenin. Shown are representative samples of immunofluorescence (confocal) images of mouse keratinocytes transfected with K14-ΔNΔCβcatenin (A and B) and of primary keratinocytes derived from ΔNΔCβcatenin–expressing transgenic mice (C and C′). Endogenous β-catenin staining is shown in red; and anti-HA staining in green. The anti-HA antibody detects the HA-tagged transgenic protein product whereas the anti–β-catenin antibody specifically detects the endogenous β-catenin, as it is directed toward the COOH terminus of the protein which is absent from the ΔNΔCβcatenin transgenic product. In mouse keratinocyte cell line, endogenous β-catenin translocated to the nucleus, only in cells transfected with K14-ΔNΔCβcatenin. The same phenomenon was seen in primary keratinocytes, derived from the K14-ΔNΔCβcatenin transgenic mice. (D–G) Increase in cytosolic and nuclear β-catenin in ΔNΔCβcatenin transgenic mice. Shown are confocal images of immunostaining for endogenous β-catenin in red and anti-HA in green. (D and E) Day 28 WT tail epidermis and hair bulb, respectively. Note the absence of cytoplasmic (and the predominantly membrane) staining for endogenous β-catenin in the WT epidermis (D). Also note nuclear β-catenin (arrowheads) staining in the precortical cells of the hair bulb (E). (F and G) Age-matched sections of transgenic epidermis and epithelial invaginations in ΔNΔCβcatenin transgenic skin. Note increased cytoplasmic β-catenin in transgenic epidermis (F, arrowheads) and nuclear β-catenin in hair-like invaginations (G, arrows; G′ depicts a high magnification view of G, arrowheads).
Next, we isolated primary mouse keratinocytes from newborn transgenic mice, and cultured them in high-calcium medium (1.2 mM) where they formed adherens junctions and more closely simulated in vivo epidermis. Immunofluorescence was performed to examine the localization of ΔNΔCβcatenin and endogenous β-catenin (Fig. 8, C and C′). Under these conditions, the transgene product (green) colocalized with endogenous β-catenin (red) and other adherens junction proteins, including E-cadherin and α-catenin (data not shown). Nuclear staining was primarily endogenous β-catenin (red). Nuclear β-catenin was also seen in transgenic keratinocytes where the junctions were not formed (Fig. 8, borders), but nuclear β-catenin was not detected in the wild-type keratinocytes (Fig. 8, A and B). Taken together, these results clearly demonstrate that expression of ΔNΔCβcatenin influences endogenous β-catenin localization and allows it to accumulate in the nucleus, even when cell–cell junctions are formed. Interestingly, as judged by immunofluorescence, the ratio of ΔNΔCβcatenin to endogenous β-catenin was significantly greater at the membrane and in the cytoplasm than in the nucleus.
In WT epidermis in vivo, β-catenin is normally restricted to cell–cell borders, and is not found in the cytoplasm or nucleus (Fig. 8 D). However, in the hair follicle, cytoplasmic and nuclear β-catenin are readily detected in the hair progenitor cells, referred to as precortex (Fig. 8 E). The precortex (Fig. 8, arrowheads) is where activation of Lef1/β-catenin regulated genes, including the hair-specific keratins and TOPGAL, typically occurs (DasGupta and Fuchs, 1999; Merrill et al., 2001).
The pattern of endogenous β-catenin was dramatically different in ΔNΔCβcatenin epidermis (Fig. 8 F). In addition to cell–cell border staining, which was seen with antibodies against both the HA-tagged ΔNΔCβcatenin gene product and endogenous β-catenin, cytoplasmic staining was also prominent, particularly for endogenous β-catenin (Fig. 8 F). In the TOPGAL-positive, flower-like downgrowths of ΔNΔCβcatenin skin, nuclear β-catenin was also detected (Fig. 8 G, arrowheads). In these downgrowths, the ratio of FITC (transgene product) to Texas red (endogenous β-catenin) was always higher at cell borders than in the nuclei (Fig. 8 G′, inset, higher magnification). The preferential detection of endogenous nuclear β-catenin was consistent with the presence of Lef1 and TOPGAL activity in these downgrowths.
Interestingly, in the early stages of epidermal cyst formation, we occasionally captured follicle-like structures where intense anti-HA staining, reflective of ΔNΔCβcatenin, was detected in the nuclei of the precortex (Fig. 9 E). At these early stages, HA nuclear staining was sometimes also seen in the bulge compartment (Fig. 9 F). These findings are consistent with the dominant negative action of ΔNΔCβcatenin in transcriptional regulation and with our failure to detect TOPGAL activity in the bulge or the precortex of the ΔNΔCβcatenin follicles. In epidermal cysts, peripheral basal-like cells showed intense border staining for the K14 promoter-driven transgene product, paralleling the intensity seen in the K14-positive epidermal basal layer and the ORS (Fig. 9 D, arrows). In these regions, some nuclei stained with HA antibodies (Fig. 9 D′′, inset, arrows; higher magnification of such HA-positive nuclei). The more central, spinous-like cells did not display nuclear HA staining, and in fact expressed considerably less ΔNΔCβcatenin, consistent with the switch from K5/K14 to K1/K10 and the downregulation of K14 promoter activity in these layers. These results provide additional evidence that expression of ΔNΔCβcatenin in the basal-like cells of developing cysts is high and able to block the transactivating functions of β-catenin.
Figure 9.

Plasticity of the epitheliod cysts formed in ΔNΔC transgenic skin. Differences in the levels of ΔNΔCβcat and endogenous β-catenin could influence cell fate decisions in the epitheliod cysts. (A). Hematoxylin and eosin combined with X-gal staining on epitheliod cysts formed in mice harboring both ΔNΔCβcatenin and TOPGAL. A few cysts display X-gal staining which coincides with activation of Lef1 (in red) and hair keratins, as judged by AE13 (pan-hair keratin antibody) staining in green in a similar section (B and C, confocal image). All sections were 10 μm in thickness and fixed in 4% PFA. Sections processed for X-gal staining were fixed in 0.1% glutaraldehyde. (C–F) Levels of endogenous β-catenin and ΔNΔCβcatenin complexes with Lef1 determines epidermal versus hair cell fates in epitheliod cysts. (C) Immunostaining for Lef1 (in red) and AE13 (in green) D demonstrate high levels of nuclear Lef1 (white arrowheads) in both ΔNΔCβcatenin–expressing basal cells and high β-catenin–expressing suprabasal cells. Moreover, cells with Lef1 and higher levels of endogenous β-catenin display AE13-positive staining (hair cortex marker), whereas the basal cells are not immunoreactive toward AE13 antibody. (D) Confocal immunofluorescence image of a cyst stained for endogenous β-catenin (in red) and the epitope HA (in green). Insets D′ and D′′ display diffuse nuclear βcat and nuclear HA staining, respectively. Note the reciprocal levels of staining between the HA-tagged transgenic ΔNΔCβcat protein and endogenous β-catenin in the epitheliod cysts. (D) Antibody against HA-tagged ΔNΔCβcatenin protein (in green) detected the transgene product in the basal cells of the cysts. However, staining for endogenous β-catenin (in red) showed lower levels in basal cells but higher staining in suprabasal cells that have much lower levels of ΔNΔCβcatenin. (E and F) Confocal images of early stages of cyst formation with E, an occasional precortex like structure with high levels of nuclear ΔNΔCβcat (as seen by HA antibody staining in green, arrowheads) and (F) diffuse nuclear HA staining in the bulge (Bu) of ΔNΔC transgenic follicle.
In occasional nuclei of the inner, spinous-like cells of epidermal cysts, endogenous β-catenin was detected (Fig. 9, D, arrowhead, and D′, endogenous β-catenin–positive nuclei at higher magnification). Interestingly, whenever we detected nuclear endogenous β-catenin in spinous like nuclei, we also detected AE13 (hair keratin) staining (Fig. 9 C, serial section of the cyst shown in D). These regions were also those where we saw the wisps of TOPGAL activity (Fig. 9, A and B). We also detected strong nuclear Lef1 staining in the wisps of TOPGAL/AE13-positive cells within the cysts (Fig. 9, A and B). These areas were relatively rare, but the correlation was always observed.
Interestingly, despite considerable β-catenin in the cytoplasm of spinous epidermal cells (e.g., Fig. 8 F), AE13 and TOPGAL staining was not seen in differentiating epidermis. Thus, although the epidermal cysts appeared morphologically and biochemically similar to epidermis, they retained a certain level of atypical plasticity, and were able to revert midstream from an epidermal to a hair program of terminal differentiation.
To summarize the data presented in Figs. 8 and 9, there were marked differences in the relative levels of ΔNΔCβcatenin and endogenous β-catenin in the nuclei of different epithelial cells within the skin of the transgenic mice. The ratio of nuclear ΔNΔCβcatenin to endogenous β-catenin seemed to be highest in hair follicle cells and lowest in epidermal basal cells and upper ORS cells. These differences correlated well with whether the transgene acted in a negative or positive fashion to influence TOPGAL activity. These differences also correlated well with the type of epithelial cell within the skin and its relative stage of differentiation along a particular lineage. The differences did not correlate with K14 promoter activity.
Discussion
Molecular insights into ΔNΔCβcatenin's loss of function phenotypes in the stem cells and hair lineage of the skin
Although ΔNΔCβcatenin has the potential to exert both activating and repressor effects (Miller and Moon, 1997; Cox et al., 1999), the protein behaved primarily as a repressor in mouse keratinocytes in vitro. Anticipating that ΔNΔCβcatenin's actions in skin keratinocytes in vivo would parallel those in vitro, we expected to see an abrogation of β-catenin–mediated nuclear signaling and a maintenance of adherens junctions in transgenic skin epithelium. In agreement with our expectations, the epidermal cysts that formed at the start of the first postnatal hair cycle were similar to those described by Huelsken et al. (2001), who used K14-Cre recombinase to conditionally ablate β-catenin gene expression in skin.
In β-catenin–null skin, plakoglobin is able to associate with E-cadherin to assemble adherens junctions (Aberle et al., 1996; Huber et al., 1997). In ΔNΔCβcatenin skin, ΔNΔCβcatenin and endogenous β-catenin both localized to adherens junctions. Despite these differences in armadillo proteins at cell–cell junctions, the β-catenin–null and ΔNΔCβcatenin mice displayed similar epidermal cysts, suggesting that the cysts arose from a failure to activate β-catenin/Lef1/Tcf–regulated downstream target genes, rather than changes in intercellular junctions per se. Consistent with this notion is our finding that TOPGAL activation was largely silent during the postnatal hair cycles that led to epidermal cyst formation at the expense of hair differentiation. These findings were in good agreement with, and extended those of, Huelsken et al. (2001) and Niemann et al. (2002).
Our in vitro and in vivo assays tell us that elevated levels of ΔNβcatenin, but not ΔNΔCβcatenin, can activate TOPGAL in the stem cell compartment of the hair follicle. The failure to transactivate β-catenin/Lef/Tcf–regulated genes in the bulge of K14-ΔNΔCβcatenin mice did not appear to affect the downgrowth of the secondary hair germs. However, the process of their subsequent development into hair follicle was severely perturbed.
The nuclear HA staining and reduced endogenous β-catenin staining of the precortical cells of the K14-ΔNΔCβcatenin secondary hair germs provided an explanation for the dominant negative effects of the transgene on hair cells. However, the strong HA staining could not be explained by K14 promoter activity, which is actually very low in matrix and precortical cells (Byrne et al., 1994; Wang et al., 1997). Moreover, the strong nuclear HA staining could not be explained by a simple canonical Wnt signaling mechanism, which should have increased nuclear levels of endogenous β-catenin without affecting levels of ΔNΔCβcatenin. Rather, the preferential concentration of nuclear ΔNΔCβcatenin over endogenous β-catenin seemed to be a reflection of a novel mechanism that either specifically stabilizes β-catenin through its armadillo repeats or preferentially translocates the truncated β-catenin to the nucleus. Although the molecular nature of the underlying mechanism remains to be explored, its existence is one that seemed to occur preferentially in the bulge, matrix and precortex, i.e., cells known to express members of the Tcf/Lef1 family of HMG proteins.
Molecular insights into ΔNΔCβcatenin's gain of function phenotypes in the epidermis and upper ORS of the skin
An important and revealing feature of our ΔNΔCβcatenin transgenic mice was the surprising existence of a gain-of-function phenotype within the interfollicular and upper ORS epithelium of the skin. By all criteria examined, these ΔNΔCβcatenin-expressing cells behaved analogously to the equivalent cells of the ΔNβcatenin mice. Thus, whereas ΔNΔCβcatenin elicited a dominant negative effect on keratinocytes in vitro and on skin epithelial stem cells and follicle cells in vivo, it elicited a positive effect on interfollicular and upper ORS epithelium. The interfollicular and upper ORS epithelium are thought to be derived from the stem cells in the bulge. How can we account for the remarkable difference between stem cells and their progeny in the way they respond to this single transgene protein?
Several key observations help us to understand this paradox. One important clue is that despite the inability of ΔNΔCβcatenin/Lef/Tcf complexes to transactivate Lef1/Tcf regulated genes on their own, TOPGAL was ectopically activated in the epidermis and the ORS at sites of hair germ like invaginations. Conversely, as detected through TOPGAL activity, normal Lef/Tcf/β-catenin regulated gene activity was impaired in the bulge and precortical cells of postnatal transgenic follicles. Because the transgene protein lacks a transactivation domain, its positive actions on TOPGAL activity must then be indirect. In this regard, our in vitro studies revealed endogenous β-catenin in the nuclei of ΔNΔCβcatenin–transfected cells, and in vivo, nuclear endogenous β-catenin was seen in areas where TOPGAL was activated.
How then does ΔNΔCβcatenin lead to generation of critical threshold levels of stabilized β-catenin in interfollicular epidermal cells and in the upper ORS, but not in the bulge or in the precortex? Our analyses left us to conclude that it is a property inherent to the character of each epithelial skin cell type that determines how the cell will respond to ΔNΔCβcatenin. Thus, ironically, hair follicle–like invaginations were restricted to transgenic upper ORS and epidermis, whereas epidermal transdifferentiation was restricted to transgenic cells that normally would adopt a hair cell fate.
At sites of epithelial downgrowth in the interfollicular epidermis or upper ORS, the pools of cytoplasmic and nuclear endogenous β-catenin seemed to increase. One way in which this might be achieved is that in epidermis, ΔNΔCβcatenin localizes strongly to cell–cell junctions, where it may displace endogenous β-catenin and raise its cytoplasmic pool. Localization of the transgene protein to cell–cell junctions sequesters it, physically restraining it from interfering in the nucleus. Intriguingly, cell–cell junctions are fewer in hair follicle precursor cells (Nanba et al., 2000), perhaps increasing the likelihood that ΔNΔCβcatenin will enter the nuclei of these cells and act to impair Lef1/Tcf gene activity.
Another contributing factor is likely to be ΔNΔCβcatenin's association with APC (for review see Bienz, 1999; Mimori-Kiyosue and Tsukita, 2001). Through ΔNΔCβcatenin-mediated sequestering of APC or other members of the β-catenin degradation machinery, endogenous β-catenin could be stabilized. The preferential concentration of endogenous β-catenin in the nuclei of cells at epithelial downgrowths suggests that ΔNΔCβcatenin may preferentially associate with either or both of these interacting partners.
Our transient transfection experiments with cultured keratinocytes demonstrate that even under conditions where cell–cell junctions do not form, endogenous βcatenin from the cytoplasm can translocate to the nucleus when cells express the transgene. This finding is compatible with a displacement mechanism that involves ΔNΔCβcatenin-mediated sequestration of either a component of the proteosome degradation machinery or a component that keeps endogenous β-catenin from entering the nucleus. APC has been implicated in both processes, and its ability to associate with transgenic and endogenous β-catenin makes it a good candidate for explaining these observations. Overall, our in vitro and in vivo observations corroborated similar studies in Xenopus (Miller and Moon, 1997) and in Drosophila (Cox et al., 1999), where positive actions of a membrane-tethered form of β-catenin also prompted investigators to posit that competition for E-cadherin and APC may displace endogenous β-catenin into the nucleus. A related parallel stems from altering E-cadherin levels, which also results in phenotypes consistent with sequestering signaling-competent β-catenin away from the nucleus (Orsulic et al., 1999; Ciruna and Rossant, 2001).
Another intriguing phenomenon that is likely relevant to our understanding of Lef1/Tcf/β-catenin action in skin epithelium is the elevated Lef1 mRNA and protein expression which occurs in epithelial invaginations transgenic for either ΔNβcatenin (Gat et al., 1998) or ΔNΔCβcatenin (present study). This feature suggests the existence of a positive feedback mechanism that could lead to Lef1 transcription when β-catenin levels are stabilized in epidermis. Because the Lef1 promoter contains functional Lef1/Tcf binding sites (Hovanes et al., 2001 and references therein), it is possible that displacement of endogenous β-catenin frees it to associate with small amounts of Lef1 which could then initiate this cycle of upregulated Lef1 gene expression. Further experiments will be necessary to fully understand this mechanism. In addition, recent studies on E-cadherin have also provided new insights into how altering the levels of adherens junction proteins may modulate Wnt-mediated signaling in morphogenetic processes (Orsulic et al., 1999; Ciruna and Rossant, 2001).
A number of other β-catenin interacting proteins have been identified in recent years, and a priori, some of these proteins may also be involved in accounting for the unexpected transactivating-like phenotype associated with ΔNΔCβcatenin in skin. Consistent with this notion are transcriptional repressor proteins such as XSox17, XSox3, and the Smad proteins, which have been shown to physically interact with β-catenin (Zorn et al., 1999; Schohl and Fagotto, 2002), and in this regard, the transgene protein might act to titrate out some of these repressors. In contrast, the repressor proteins reptin52 and pontin52 have recently been identified as COOH terminally interacting β-catenin–associated repressor proteins, which would render them insensitive to the transgene protein (Bauer et al., 2000; Etard et al., 2000; Takemaru and Moon, 2000). However, for these proteins to be involved, ΔNΔCβcatenin would have to function by relief of β-catenin/Tcf-mediated transcriptional repression, a mechanism previously invoked to explain certain positive phenotypes obtained with the ΔNΔCβcatenin mutant (Funayama et al., 1995; Miller and Moon, 1997). This seems unlikely for epidermis because: (a) the nuclear β-catenin seen in the activated cells was mostly endogenous rather than transgene product; (b) the epidermis and upper ORS do not normally express appreciable levels of Lef1/Tcf family members; and (c) the Lef/Tcf member activated at these sites was Lef1, acting primarily as a coactivator and not a repressor. However, repression might still be relieved if endogenous nuclear β-catenin competes for the binding of Tcf/Lef repressor proteins, e.g., Grg/Tle (Merrill et al., 2001).
Reconciling two opposing actions from the same transgene
Ironically, by expressing ΔNΔCβcatenin, the epidermal-like cells ended up generating hair follicle–like phenotypes, whereas the hair follicle–like cells ended up generating epidermal-like phenotypes. These findings imply that the behavior of ΔNΔCβcatenin in epithelial cells of the skin is highly tailored to the particular stem cell lineage and stage of differentiation of the skin cell. The final outcome of epithelial skin cell fate then appears to be dictated not only by the levels and location of β-catenin in a cell, but also the relative levels of other β-catenin interacting factors in the cell. An interesting point that supports this conclusion is that by downregulation of transgene expression in differentiating epidermal cysts, the former hair cells turned epidermal cells reverted back to their hair cell fate, activating hair keratin gene expression. Such reversibility of cell fates was not observed in the bona fide epidermis, indicating that suppression of nuclear β-catenin in a hair precursor cell required sustained transgene expression to be maintained, whereas epidermal precursor cells have some intrinsic mechanism to prevent this from happening.
Given the complexities of β-catenin's interacting partners, a detailed picture of how epidermal and hair follicle precursor cells regulate intracellular levels of β-catenin is beyond the scope of the present study. However, based on these new findings, we can already develop models that are consistent with our present knowledge of cellular differences in hair and epidermal precursor cells and that enable us to begin to envisage how such differences might influence the way in which these might respond to the transgene protein. In summary, the ability of ΔNΔCβcatenin to act in both positive and negative fashions in a highly specialized and cell differentiation–specific fashion has uncovered a fascinating new frontier underlying the complex ways in which different cells receive and respond to changes in β-catenin.
Materials and methods
Cell culture and transfections
For transient transfections, we used an immortalized keratinocyte cell line which arose spontaneously from an epidermal primary keratinocyte culture of WT murine skin. Transfections were performed using the lipid-based FuGENE6 reagent (Roche) according to manufacturer's protocol. Cells transfected with pTOPGAL/pFOPGAL were processed and β-galactosidase activity was determined using a Galacto-Lite assay kit (TROPIX, Inc.) and a luminometer (MGM Instruments). Transfections were normalized by Luciferase activity of the K14-Luciferase construct, which was cotransfected with all transfections. Cells were harvested 36–48 h after transfection, washed in PBS, lysed in 1× lysis buffer (passive lysis buffer for dual-luciferase-assay-kit) provided in the Promega kit, and assayed for Luciferase or β-galactosidase activity.
For analyses of cells where the transgene had integrated and was expressed at levels not lethal to the animal, we isolated primary keratinocytes from transgenic and control littermates and cultured them as previously described (Vasioukhin et al., 2000).
Extraction of proteins
For cultured cells: 100-mm plate of confluent cells was washed 2× in PBS. The cells were lysed with 500 μL either mild lysis buffer (1% Triton X-100 in PBS with 10 mM EDTA, 0.3 mg/mL freshly made PMSF, and protease inhibitor cocktail) or RIPA buffer (1% Triton X-100 in PBS with 10 mM EDTA, 150 mN NaCl, 1% sodium deoxycholate, 0.1% SDS and protease inhibitor cocktail; Roche]). Cells were scraped off the plate and transferred into a 1.5-mL eppendorf tube. The cell suspension was sonicated 3 × 15 s and centrifuged at 14,000 rpm at 4°C. The supernatant-soluble fraction was transferred into a fresh tube and equal volume of 2× sample buffer was added to it. The pellet was dissolved directly in 2× sample loading buffer and treated as the insoluble fraction.
For skin tissue: Frozen tissue was pulverized in a liquid Nitrogen-cooled Gevebesmascher and the powder scraped into a chilled microfuge tube. RIPA buffer (described above) was added to the tube and samples were processed as described above. Instead of adding the sample buffer, the protein extracts were stored at −80°C for future use in co-IPs and Western blotting.
Coimmunoprecipitation studies
Primary keratinocytes grown on tissue culture dishes were washed in ice-cold PBS. RIPA buffer was added to each plate and the cells scraped off and collected in a tube. Soluble fractions were isolated as described in the previous section. The soluble extracts were incubated with 50 μL protein-G Sepharose beads for 1 h at 4°C, to preclear the lysate. The samples were centrifuged for 1 min and transferred into a fresh microfuge tube. The protein samples were incubated with primary antibody (α-E-cadherin 5 μL and α-APC 10 μL) O/N at 4°C. The antibody–E-cadherin/antibody-APC complex, was retrieved by incubating with 50 μL protein-G Sepharose for 1 h, at 4°C. The immunoprecipitated E-cadherin or APC was washed three times with cold lysis buffer and immediately used for immunoblotting. The same protocol was followed for coimmunoprecipitation studies of skin protein extracts as described above.
For immunoblotting, the following primary antibodies were used: α-Ecad (1:1,000, Eccd2; Zymed); α-APC (1:1,000, C-20, Santa Cruz Biotechnology); α-HA (1:1,000; Roche Biochemicals). HRP-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories) and ECL (chemiluminescent solution) were used for detecting the bound antibody.
Generation of K14-ΔNΔCβcatenin construct
A PCR strategy was used to create the COOH-terminal deletion on the K14-ΔN87β-catenin construct. The PCR product was flanked by SpeI site (contained within the β-catenin cDNA) at the 5′-end and XbaI site at the 3′-end. It was first cloned into pCRII-Blunt, TOPO vector (Invitrogen) for sequence confirmation. The fragment was then excised by SpeI-XbaI double digestion and subcloned into the K14-ΔN87β-catenin construct cut with SpeI-XbaI (which eliminates the COOH terminus of β-catenin). Sequences of the primers used for the PCR reaction in mentioned in the list of primers. The primers used were SpeI-ΔCF.1 and ΔCR.1-HA-XbaI. cDNA sequence encoding the HA-tag was included in the reverse primer.
Immunohistochemistry
For immunofluorescence, 8–10-μm sections of frozen tissue were fixed in 4% PFA, treated with blocking solution (5% heat-inactivated normal goat serum, 1% BSA in 1× PBS), and incubated with the primary antibody (diluted in the blocking solution + 0.1% Triton X-100), as described previously (DasGupta and Fuchs, 1999). Fluorescein- or rhodamine-conjugated secondary antibodies (obtained from Jackson ImmunoResearch Laboratories) were used to detect primary bound antibody. Results were visualized under the Zeiss 410 confocal microscope or the Zeiss Axiophot (Zeiss) for fluorescent microscopy.
For immunofluorescence, the primary antibodies used were α-Ecadherin (1:1,000, Eccd2; Zymed); α-HA (1:100; Roche); α-β-catenin (1:1,000; 15γ8; Sigma-Aldrich); α-K1 (1:250; Babco); α-filaggrin (1:1,000; Babco); α-AE13 (1:5; a gift from Dr. T.T. Sun [New York University, New York, NY]); α-AE15 (1:5; a gift from Dr. T.T. Sun); α-Lef1 (1:250; lab generated); and α-Tcf3 (1:100; lab generated). Rhodamine- or fluorescein-conjugated secondary antibodies (1:100; Jackson ImmunoResearch Laboratories) were used to detect the primary antibodies bound to their respective targets.
Acknowledgments
We thank Dr. Bradley Merrill and Dr. Colin Jamora for their thoughtful comments on this manuscript and for their valuable suggestions during the course of these studies. We thank Dr. Uri Gat for his advice and suggestions in the initial phases of the study. E. Fuchs is an Investigator of the Howard Hughes Medical Institute.
This work was supported by a grant from the National Institutes of Health.
R. DasGupta's present address is Harvard Medical School, HHMI, Dept. of Genetics, 200 Longwood Ave., Boston, MA 02115.
Footnotes
Abbreviations used in this paper: APC, adenomatous polyposis coli; ORS, outer root sheath; TOP, Tcf-optimal-promoter; TOPGAL, TOP-βgalactosidase; WT, wild-type.
References
- Aberle, H., H. Schwartz, H. Hoschuetzky, and R. Kemler. 1996. Single amino acid substitutions in proteins of the armadillo gene family abolish their binding to alpha-catenin. J. Biol. Chem. 271:1520–1526. [DOI] [PubMed] [Google Scholar]
- Barker, N., A. Hurlstone, H. Musisi, A. Miles, M. Bienz, and H. Clevers. 2001. The chromatin remodeling factor Brg-1 interacts with beta-catenin to promote target gene activation. EMBO J. 20:4935–4943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer, A., S. Chauvet, O. Huber, F. Usseglio, U. Rothbacher, D. Aragnol, R. Kemler, and J. Pradel. 2000. Pontin52 and reptin52 function as antagonistic regulators of beta-catenin signalling activity. EMBO J. 19:6121–6130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrens, J., B.A. Jerchow, M. Wurtele, J. Grimm, C. Asbrand, R. Wirtz, M. Kuhl, D. Wedlich, and W. Birchmeier. 1998. Functional interaction of an axin homolog, conductin, with beta-catenin, APC, and GSK3beta. Science. 280:596–599. [DOI] [PubMed] [Google Scholar]
- Bienz, M. 1999. APC: the plot thickens. Curr. Opin. Genet. Dev. 5:595–603. [DOI] [PubMed] [Google Scholar]
- Brantjes, H., N. Barker, E.J. van, and H. Clevers. 2002. TCF: Lady Justice casting the final verdict on the outcome of Wnt signalling. J. Biol. Chem. 383:255–261. [DOI] [PubMed] [Google Scholar]
- Byrne, C., M. Tainsky, and E. Fuchs. 1994. Programming gene expression in developing epidermis. Development. 120:2369–2383. [DOI] [PubMed] [Google Scholar]
- Chan, E.F., U. Gat, J.M. McNiff, and E. Fuchs. 1999. A common human skin tumour is caused by activating mutations in beta-catenin. Nat. Genet. 21:410–413. [DOI] [PubMed] [Google Scholar]
- Ciruna, B., and J. Rossant. 2001. FGF signaling regulates mesoderm cell fate specification and morphogenetic movement at the primitive streak. Dev. Cell. 1:37–49. [DOI] [PubMed] [Google Scholar]
- Cox, R.T., L.M. Pai, J.R. Miller, S. Orsulic, J. Stein, C.A. McCormick, Y. Audeh, W. Wang, R.T. Moon, and M. Peifer. 1999. Membrane-tethered Drosophila armadillo cannot transduce wingless signal on its own. Development. 126:1327–1335. [DOI] [PubMed] [Google Scholar]
- DasGupta, R., and E. Fuchs. 1999. Multiple roles for activated LEF/TCF transcription complexes during hair follicle development and differentiation. Development. 126:4557–4568. [DOI] [PubMed] [Google Scholar]
- Etard, C., D. Wedlich, A. Bauer, O. Huber, and M. Kuhl. 2000. Expression of Xenopus homologs of the beta-catenin binding protein pontin52. Mech. Dev. 94:219–222. [DOI] [PubMed] [Google Scholar]
- Fuchs, E., B.J. Merrill, C. Jamora, and R. DasGupta. 2001. At the roots of a never-ending cycle. Dev Cell. 1:13–25. [DOI] [PubMed] [Google Scholar]
- Funayama, N., F. Fagotto, P. McCrea, and B.M. Gumbiner. 1995. Embryonic axis induction by the armadillo repeat domain of beta-catenin: evidence for intracellular signaling. J. Cell Biol. 128:959–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gat, U., R. DasGupta, L. Degenstein, and E. Fuchs. 1998. De novo hair follicle morphogenesis and hair tumors in mice expressing a truncated beta-catenin in skin. Cell. 95:605–614. [DOI] [PubMed] [Google Scholar]
- Gottardi, C.J., and B.M. Gumbiner. 2001. Adhesion signaling: how beta-catenin interacts with its partners. Curr. Biol. 11:R792–R794. [DOI] [PubMed] [Google Scholar]
- Graham, T.A., C. Weaver, F. Mao, D. Kimelman, and W. Xu. 2000. Crystal structure of a beta-catenin/Tcf complex. Cell. 103:885–896. [DOI] [PubMed] [Google Scholar]
- Hardy, M.H. 1992. The secret life of the hair follicle. Trends Genet. 8:55–61. [DOI] [PubMed] [Google Scholar]
- Hecht, A., C.M. Litterst, O. Huber, and R. Kemler. 1999. Functional characterization of multiple transactivating elements in beta-catenin, some of which interact with the TATA-binding protein in vitro. J. Biol. Chem. 274:18017–18025. [DOI] [PubMed] [Google Scholar]
- Hecht, A., K. Vleminckx, M.P. Stemmler, F. van Roy, and R. Kemler. 2000. The p300/CBP acetyltransferases function as transcriptional coactivators of beta-catenin in vertebrates. EMBO J. 19:1839–1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson, B.R. 2000. Nuclear-cytoplasmic shuttling of APC regulates beta-catenin subcellular localization and turnover. Nat. Cell Biol. 2:653–660. [DOI] [PubMed] [Google Scholar]
- Hovanes, K., T.W. Li, J.E. Munguia, T. Truong, T. Milovanovic, J. Lawrence Marsh, R.F. Holcombe, and M.L. Waterman. 2001. Beta-catenin-sensitive isoforms of lymphoid enhancer factor-1 are selectively expressed in colon cancer. Nat. Genet. 28:53–57. [DOI] [PubMed] [Google Scholar]
- Hsu, S.C., J. Galceran, and R. Grosschedl. 1998. Modulation of transcriptional regulation by LEF-1 in response to Wnt-1 signaling and association with beta-catenin. Mol. Cell. Biol. 18:4807–4818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber, A.H., W.J. Nelson, and W.I. Weis. 1997. Three-dimensional structure of the armadillo repeat region of beta-catenin. Cell. 90:871–882. [DOI] [PubMed] [Google Scholar]
- Huelsken, J., and W. Birchmeier. 2001. New aspects of Wnt signaling pathways in higher vertebrates. Curr. Opin. Genet. Dev. 11:547–553. [DOI] [PubMed] [Google Scholar]
- Huelsken, J., R. Vogel, B. Erdmann, G. Cotsarelis, and W. Birchmeier. 2001. beta-Catenin controls hair follicle morphogenesis and stem cell differentiation in the skin. Cell. 105:533–545. [DOI] [PubMed] [Google Scholar]
- Jou, T.S., D.B. Stewart, J. Stappert, W.J. Nelson, and J.A. Marrs. 1995. Genetic and biochemical dissection of protein linkages in the cadherin-catenin complex. Proc. Natl. Acad. Sci. USA. 92:5067–5071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishimoto, J., R.E. Burgeson, and B.A. Morgan. 2000. Wnt signaling maintains the hair-inducing activity of the dermal papilla. Genes Dev. 14:1181–1185. [PMC free article] [PubMed] [Google Scholar]
- Merrill, B.J., U. Gat, R. DasGupta, and E. Fuchs. 2001. Tcf3 and Lef1 regulate lineage differentiation of multipotent stem cells in skin. Genes Dev. 15:1688–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, J.R., and R.T. Moon. 1997. Analysis of the signaling activities of localization mutants of beta-catenin during axis specification in Xenopus. J. Cell Biol. 139:229–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimori-Kiyosue, Y., and S. Tsukita. 2001. Where is APC going? J. Cell Biol. 154:1105–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molenaar, M., M. van de Wetering, M. Oosterwegel, J. Peterson-Maduro, S. Godsave, V. Korinek, J. Roose, O. Destree, and H. Clevers. 1996. XTcf-3 transcription factor mediates beta-catenin-induced axis formation in Xenopus embryos. Cell. 86:391–399. [DOI] [PubMed] [Google Scholar]
- Nagafuchi, A. 2001. Molecular architecture of adherens junctions. Curr. Opin. Cell Biol. 13:600–603. [DOI] [PubMed] [Google Scholar]
- Nanba, D., Y. Hieda, and Y. Nakanishi. 2000. Remodeling of desmosomal and hemidesmosomal adhesion systems during early morphogenesis of mouse pelage hair follicles. J. Invest. Dermatol. 114:171–177. [DOI] [PubMed] [Google Scholar]
- Niemann, C., D.M. Owens, J. Hulsken, W. Birchmeier, and F.M. Watt. 2002. Expression of DeltaNLef1 in mouse epidermis results in differentiation of hair follicles into squamous epidermal cysts and formation of skin tumours. Development. 129:95–109. [DOI] [PubMed] [Google Scholar]
- Noramly, S., A. Freeman, and B.A. Morgan. 1999. beta-Catenin signaling can initiate feather bud development. Development. 126:3509–3521. [DOI] [PubMed] [Google Scholar]
- Orsulic, S., and M. Peifer. 1996. An in vivo structure-function study of armadillo, the beta-catenin homologue, reveals both separate and overlapping regions of the protein required for cell adhesion and for wingless signaling. J. Cell Biol. 134:1283–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orsulic, S., O. Huber, H. Aberle, S. Arnold, and R. Kemler. 1999. E-cadherin binding prevents b-catenin nuclear localization and b-catenin/LEF-1-mediated transactivation. J. Cell Sci. 112:1237–1245. [DOI] [PubMed] [Google Scholar]
- Oshima, H., A. Rochat, C. Kedzia, K. Kobayashi, and Y. Barrandon. 2001. Morphogenesis and renewal of hair follicles from adult multipotent stem cells. Cell. 104:233–245. [DOI] [PubMed] [Google Scholar]
- Reinacher-Schick, A., and B.M. Gumbiner. 2001. Apical membrane localization of the adenomatous polyposis coli tumor suppressor protein and subcellular distribution of the β-catenin destruction complex in polarized epithelial cells. J. Cell Biol. 152:491–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schohl, A., and F. Fagotto. 2002. Beta-catenin, MAPK and Smad signaling during early Xenopus development. Development. 129:37–52. [DOI] [PubMed] [Google Scholar]
- Takemaru, K.I., and R.T. Moon. 2000. The transcriptional coactivator CBP interacts with β-catenin to activate gene expression. J. Cell Biol. 149:249–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor, G., M.S. Lehrer, P.J. Jensen, T.T. Sun, and R.M. Lavker. 2000. Involvement of follicular stem cells in forming not only the follicle but also the epidermis. Cell. 102:451–461. [DOI] [PubMed] [Google Scholar]
- Tutter, A.V., C.J. Fryer, and K.A. Jones. 2001. Chromatin-specific regulation of LEF-1-beta-catenin transcription activation and inhibition in vitro. Genes Dev. 15:3342–3354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Wetering, M., R. Cavallo, D. Dooijes, M. van Beest, J. van Es, J. Loureiro, A. Ypma, D. Hursh, T. Jones, A. Bejsovec, et al. 1997. Armadillo coactivates transcription driven by the product of the Drosophila segment polarity gene dTCF. Cell. 88:789–799. [DOI] [PubMed] [Google Scholar]
- van Genderen, C., R.M. Okamura, I. Farinas, R.G. Quo, T.G. Parslow, L. Bruhn, and R. Grosschedl. 1994. Development of several organs that require inductive epithelial-mesenchymal interactions is impaired in LEF-1-deficient mice. Genes Dev. 8:2691–2703. [DOI] [PubMed] [Google Scholar]
- van Noort, M., J. Meeldijk, R. van Der Zee, O. Destree, and H. Clevers. 2002. Wnt signaling controls the phosphorylation status of b-catenin. J. Biol. Chem. In press. [DOI] [PubMed] [Google Scholar]
- Vasioukhin, V., C. Bauer, M. Yin, and E. Fuchs. 2000. Directed actin polymerization is the driving force for epithelial cell-cell adhesion. Cell. 100:209–219. [DOI] [PubMed] [Google Scholar]
- Vleminckx, K., R. Kemler, and A. Hecht. 1999. The C-terminal transactivation domain of beta-catenin is necessary and sufficient for signaling by the LEF-1/beta-catenin complex in Xenopus laevis. Mech. Dev. 81:65–74. [DOI] [PubMed] [Google Scholar]
- Wang, X., S. Zinkel, K. Polonsky, and E. Fuchs. 1997. Transgenic studies with a keratin promoter-driven growth hormone transgene: prospects for gene therapy. Proc. Natl. Acad. Sci. USA. 94:219–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widelitz, R.B., T.X. Jiang, J. Lu, and C.M. Chuong. 2000. beta-catenin in epithelial morphogenesis: conversion of part of avian foot scales into feather buds with a mutated beta-catenin. Dev. Biol. 219:98–114. [DOI] [PubMed] [Google Scholar]
- Zhou, P., C. Byrne, J. Jacobs, and E. Fuchs. 1995. Lymphoid enhancer factor 1 directs hair follicle patterning and epithelial cell fate. Genes Dev. 9:700–713. [DOI] [PubMed] [Google Scholar]
- Zorn, A.M., G.D. Barish, B.O. Williams, P. Lavender, M.W. Klymkowsky, and H.E. Varmus. 1999. Regulation of wnt signaling by Sox proteins: XSox17 alpha/beta and XSox3 physically interact with beta-catenin. Mol. Cell. 4:487–498. [DOI] [PubMed] [Google Scholar]