

Abstract
Communication between different signaling pathways enables cells to coordinate the responses to diverse environmental signals. Activation of the transmembrane growth factor precursors plays a critical role in this communication and often involves metalloprotease-mediated proteolysis. Stimulation of G protein–coupled receptors (GPCR) transactivates the EGF receptors (EGFRs), which occurs via a metalloprotease-dependent cleavage of heparin-binding EGF (HB-EGF). However, the metalloprotease mediating the transactivation remains elusive. We show that the integral membrane metalloprotease Kuzbanian (KUZ; ADAM10), which controls Notch signaling in Drosophila, stimulates GPCR transactivation of EGFR. Upon stimulation of the bombesin receptors, KUZ increases the docking and activation of adaptors Src homology 2 domain–containing protein and Gab1 on the EGFR, and activation of Ras and Erk. In contrast, transfection of a protease domain–deleted KUZ, or blocking endogenous KUZ by morpholino antisense oligonucleotides, suppresses the transactivation. The effect of KUZ on shedding of HB-EGF and consequent transactivation of the EGFR depends on its metalloprotease activity. GPCR activation enhances the association of KUZ and its substrate HB-EGF with tetraspanin CD9. Thus, KUZ regulates the relay between the GPCR and EGFR signaling pathways.
Keywords: signal crosstalk; bombesin; HB-EGF; shedding; tetraspanin
Introduction
Cells sense diverse extracellular signals with different types of receptors on the cell surface, and transduce these signals into the cells through distinct signal pathways. But there is also significant crosstalk between the signaling pathways. In many cases, crosstalk occurs at the level of sharing components in the pathway, such as MAP kinases and other intracellular kinases. Crosstalk also occurs directly between membrane receptors; for example, stimulation of G protein–coupled receptors (GPCRs)* leads to the activation of EGF receptors (EGFRs) (Daub et al., 1996; Carpenter, 1999; Pierce et al., 2001a). This transactivation of EGFR by GPCR occurs via a release of heparin-binding EGF (HB-EGF), which is blocked by metalloprotease inhibitor (Prenzel et al., 1999).
Metalloproteases regulate cell behavior by modifying both the macro- and microenvironment of cells during their normal growth and development (Werb, 1997; Werb and Yan, 1998). Misregulation of metalloprotease activities contributes to many pathological processes, including tumorigenesis (Sternlicht et al., 1999). The integral membrane metalloproteases with a disintegrin domain (ADAMs) cleave various membrane-bound proteins, including ligands, receptors, and ligand–receptor complexes (Black and White, 1998; Blobel, 2000). Among the well-studied ADAMs are tumor necrosis factor (TNF)-α–converting enzyme (TACE; ADAM17), which not only cleaves TNF-α but also converts pro-TGFα precursors to active TGFα (Peschon et al., 1998), and Kuzbanian (KUZ; ADAM10), which is a key regulator of Notch signaling pathways in Drosophila possibly because of its metalloprotease activity (Pan and Rubin, 1997; Qi et al., 1999). Thus, proteolysis by ADAMs can change the active state of surface molecular complexes, affecting the signaling pathways inside cells.
Metalloprotease-mediated release of EGFR ligands not only contributes to a normal development process, as revealed with the metalloprotease-deficient mice, but also plays important roles in the abnormal growth of tumor cells. Inhibition of metalloprotease-mediated EGFR ligand shedding reduces the proliferation and migration of breast cancer cells (Dong et al., 1999). Moreover, so-called autocrine growth of tumor cells is often EGFR dependent, and GPCR ligands, such as bombesin, also act as growth factors. In PC3 prostate cancer cells, the metalloprotease inhibitor BB94 inhibits bombesin- and phorbol ester–induced EGFR transactivation (Prenzel et al., 1999). Thus, metalloproteases are an integral part of this EGFR-dependent autocrine growth pathway. However, understanding the pathway from GPCR to EGFR activation has been limited by the lack of knowledge about the metalloprotease involved. Several metalloproteases are capable of cleaving EGFR ligands, but there is no evidence that they support the EGFR transactivation by GPCR. For example, ADAM9 cleaves HB-EGF when PKCδ is activated, but neither wild-type nor dominant-negative ADAM9 affects the EGFR transactivation (Izumi et al., 1998). Here we report that the metalloprotease KUZ (ADAM10), described initially as the regulator of Notch signaling, supports the GPCR-induced transactivation of the EGFR signaling pathway.
Results and discussion
To identify the metalloprotease that mediates the transactivation of EGFR, we transfected COS7 cells with mouse KUZ and other metalloprotease disintegrins (ADAMs), and examined EGFR activation after activating the transfected cells with GPCR ligands. Stimulation of COS7 cells with the GPCR ligands lysophosphatidic acid (LPA) and bombesin significantly increased EGFR activation, confirming that, indeed, there were strong interactions between the GPCR and EGFR signaling pathways (Fig. 1, A and B) . Transfection of KUZ further augmented GPCR-induced EGFR phosphorylation. The enhancement of EGFR phosphorylation in the presence of transfected KUZ was moderate but significant, ranging from a 1.5- to 3.2-fold increase over the transactivation mediated by the endogenous protease (Fig. 1 C). These results suggest that KUZ enhances EGFR transactivation by GPCR.
Figure 1.
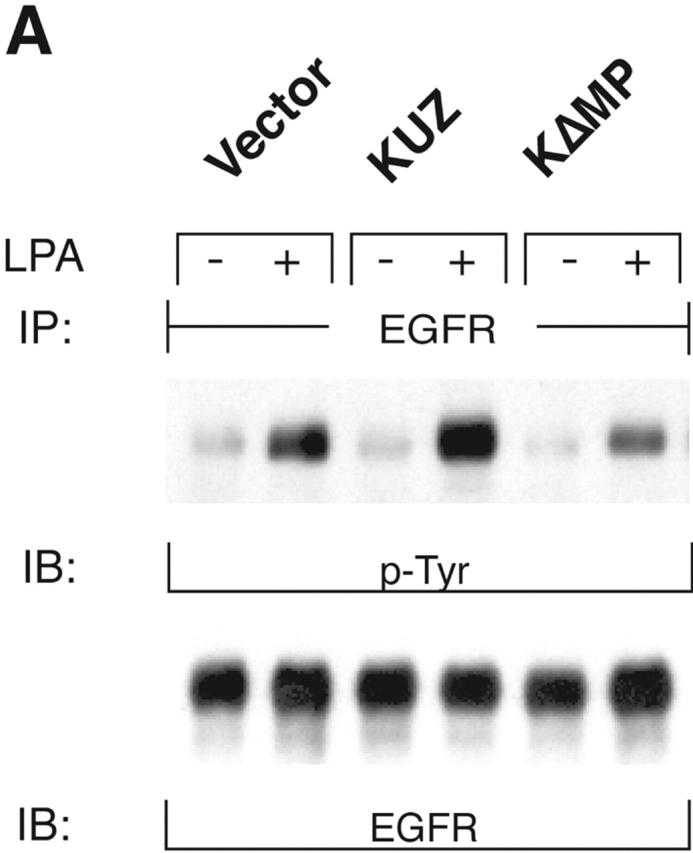
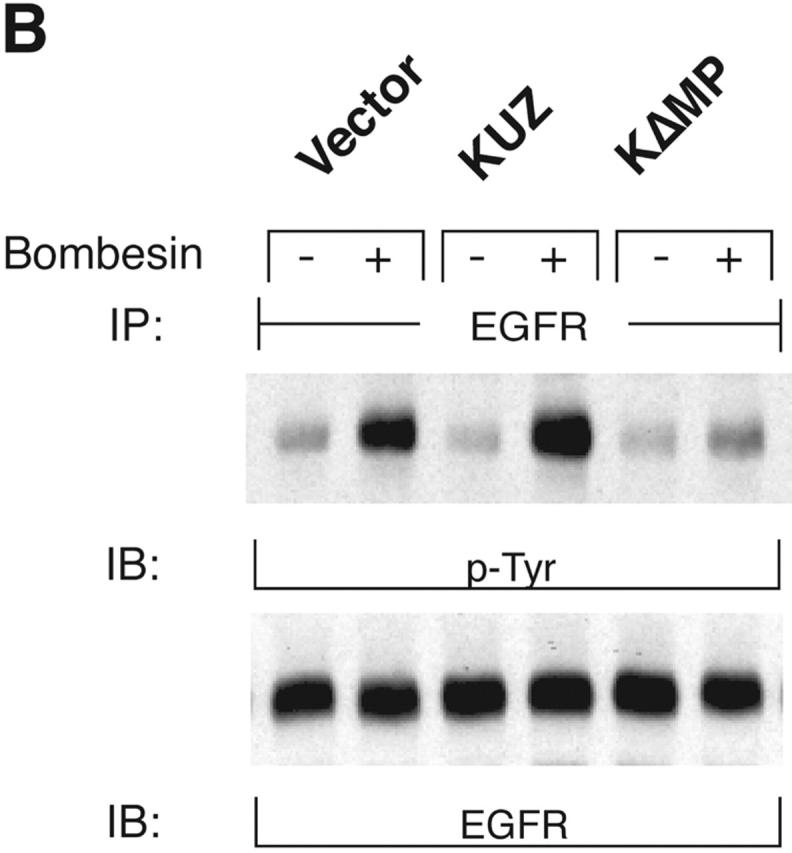

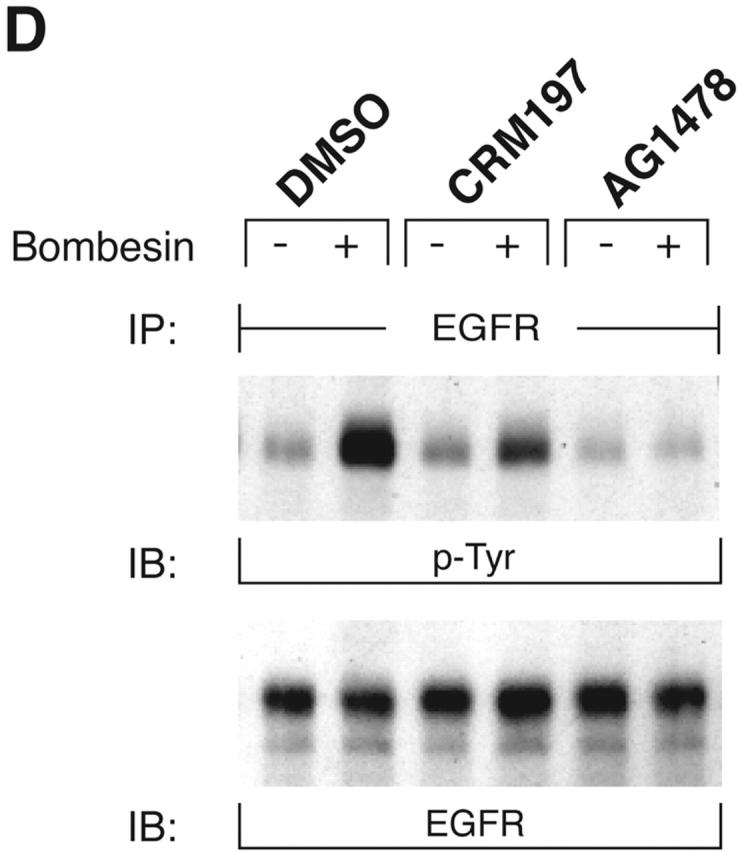
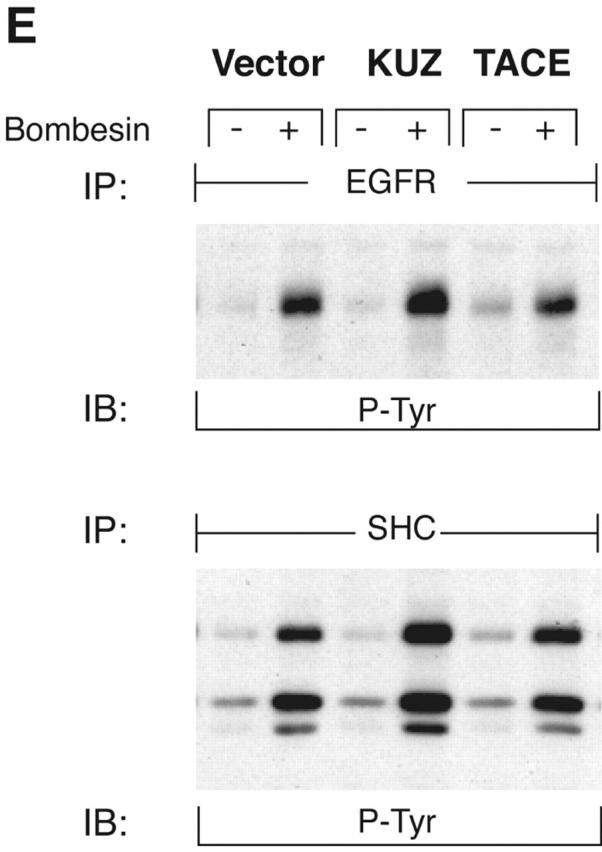
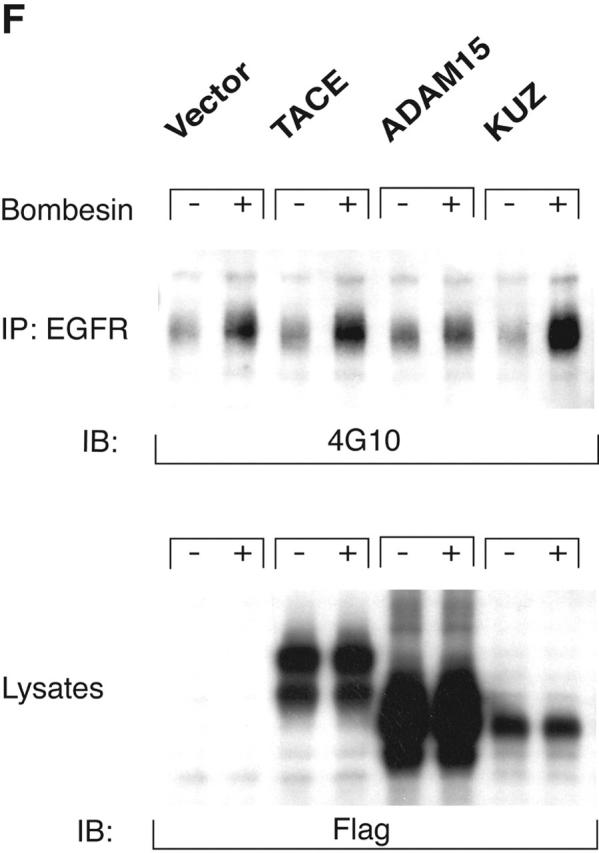

KUZ mediates GPCR transactivation of EGFR in COS7 cells. (A and B) KUZ, but not KΔMP, stimulates the LPA- and bombesin-induced EGFR phosphorylation. EGFR activation was detected in precipitated EGFR with antiphosphotyrosine antibody 4G10 and the amount of EGFR was detected with goat anti-EGFR antibody in the same blot. (C) Increase of EGFR phosphorylation induced by bombesin in the presence of exogenous KUZ or KΔMP. Phosphorylation was quantified with NIH Image 1.62 (n = 6 sets of experiments). (D) Bombesin-induced EGFR phosphorylation depends on HB-EGF and EGFR kinase. COS7 cells were pretreated for 20 min with CRM197 (lanes 3 and 4) or specific EGFR kinase inhibitor AG1487 (lanes 5 and 6) before treatment with bombesin. (E–G) KUZ is more effective than ADAM17 (TACE) or ADAM15 in mediating bombesin-induced EGFR transactivation. Transactivation of EGFR (E–G) and activation of the downstream adaptor SHC (E) are shown in cells transfected with equal amounts of plasmids containing (E) wild-type TACE and KUZ or (F) COOH-terminal Flag-tagged TACE, ADAM15, and KUZ. The amount of ADAMs expressed is shown in an immunoblot of cell lysates with anti-Flag antibody M2. Results (n = 3 sets of experiments) are quantified in G.
In contrast to the transfection of wild-type KUZ, transfection of COS7 cells with a KUZ mutant that lacks the metalloprotease domain (KΔMP) and acts as a dominant-negative mutation in Drosophila reduced the GPCR-induced EGFR phosphorylation. In fact, after bombesin treatment, the level of EGFR phosphorylation in the KΔMP-transfected cells was lower than in cells transfected with a vector plasmid, indicating that KΔMP blocks the endogenous metalloprotease-mediated EGFR transactivation in COS7 cells. In cells transfected with KUZ, the phosphorylation of EGFR in the presence of GPCR ligand was effectively inhibited by CRM197, an inactivated diphtheria toxin that specifically binds to HB-EGF, and by the EGFR kinase inhibitor AG1478, confirming that the effect of KUZ on increased EGFR phosphorylation is due to the activation of the EGFR ligand HB-EGF by GPCR signaling (Fig. 1 D). Thus, KUZ appears to function as an intermediate between activated GPCR and EGFR phosphorylation.
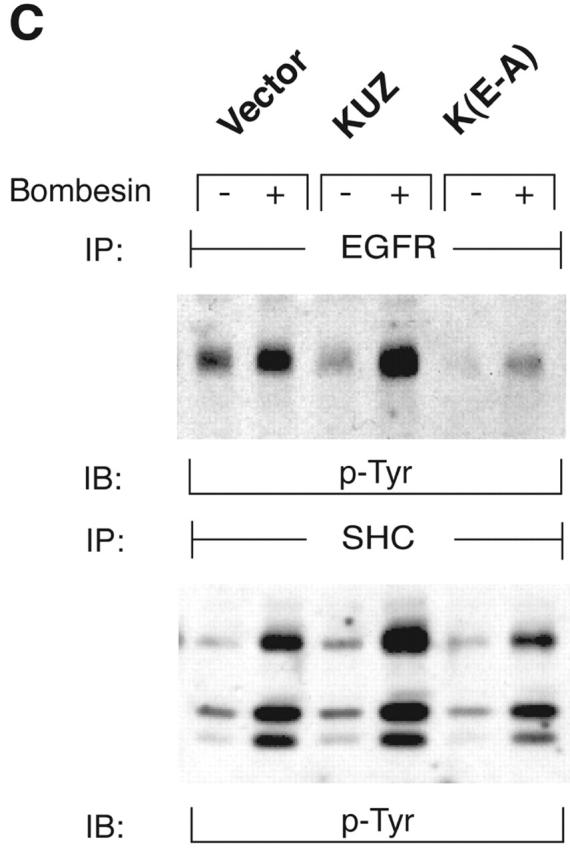
To test whether other ADAMs have the same effect on EGFR transactivation in COS7 cells, we transfected the same amount of plasmids carrying wild-type ADAM15, TACE (ADAM17), and KUZ and similar constructs with a Flag-tagged COOH terminus (Fig. 1, E–G). Bombesin induced a fourfold increase of EGFR phosphorylation in vector-transfected cells and an eightfold increase in cells transfected with wild-type KUZ, and increased phosphorylation of all three isoforms of the adaptor Src homology 2 domain–containing protein (SHC) (Fig. 1, E and G). However, transfection of neither TACE nor ADAM15 increased EGFR phosphorylation; in fact, they gave a reproducible decrease in phosphorylation (Fig. 1 G). The effect on EGFR transactivation did not correlate with the expression level of these ADAMs in COS7 cells. In fact, KUZ was expressed at lower levels than either ADAM17 or ADAM15 (Fig. 1 F). In COS7 cells, transfection of the evolutionarily related ADAM9 or a dominant-negative ADAM9 had no effect on EGFR transactivation (Izumi et al., 1998; Prenzel et al., 1999). Therefore, KUZ appeared to be uniquely effective as a mediator of EGFR transactivation in COS7 cells.
To assess the effects of KUZ on signaling pathways downstream of the transactivation of EGFR, we investigated the docking and phosphorylation of proteins known to associate with phosphorylated EGFR. Transfection of wild-type KUZ increased, and of KΔMP decreased, the bombesin-dependent phosphorylation of SHC (Fig. 2 A) and of another adaptor Gab1 (Fig. 2 B). KUZ, but not KΔMP, increased the amount of phosphorylated EGFR that coprecipitated with SHC upon bombesin treatment (unpublished data), indicating that KUZ facilitates the direct recruitment of SHC by activated EGFR. Thus, KUZ increases the activation of signaling components docked on activated EGFR upon stimulation of GPCR.
Figure 2.
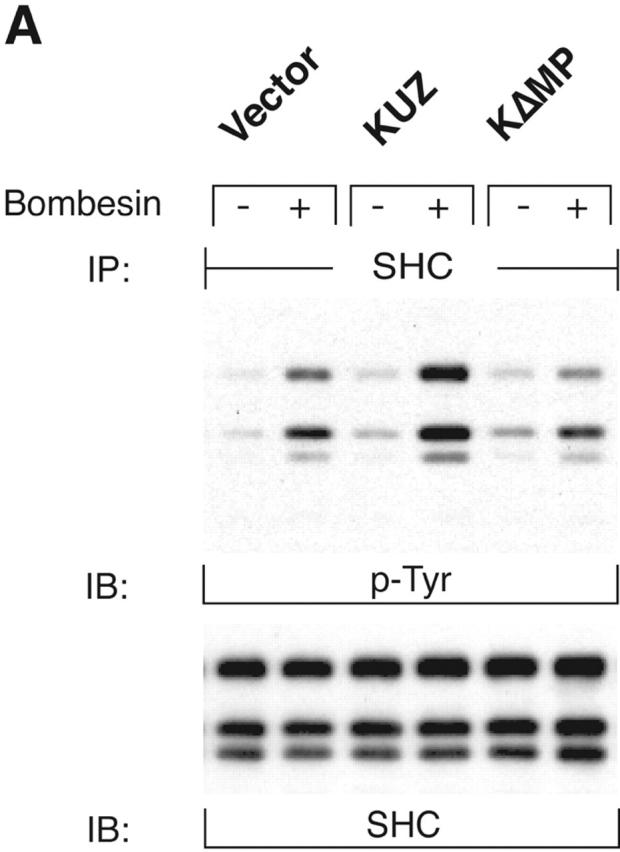

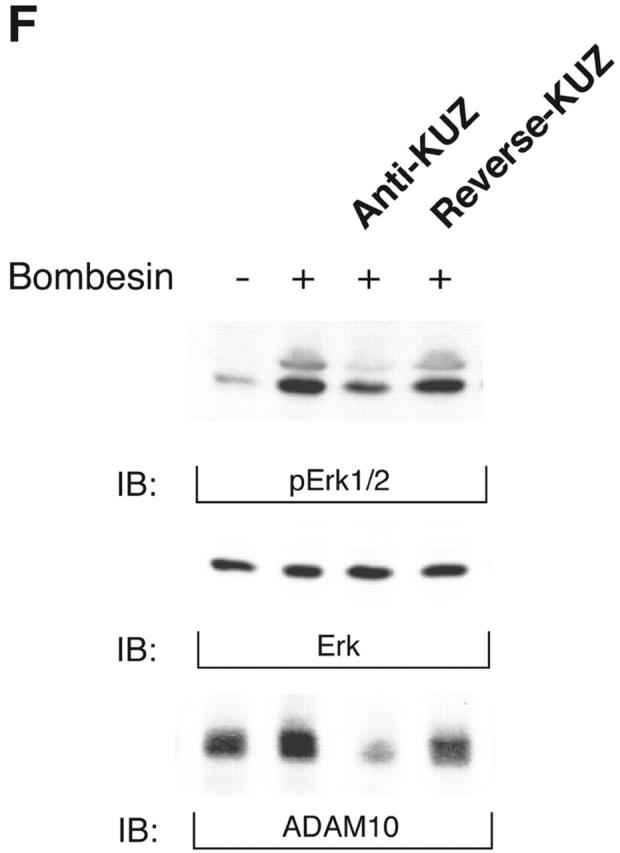
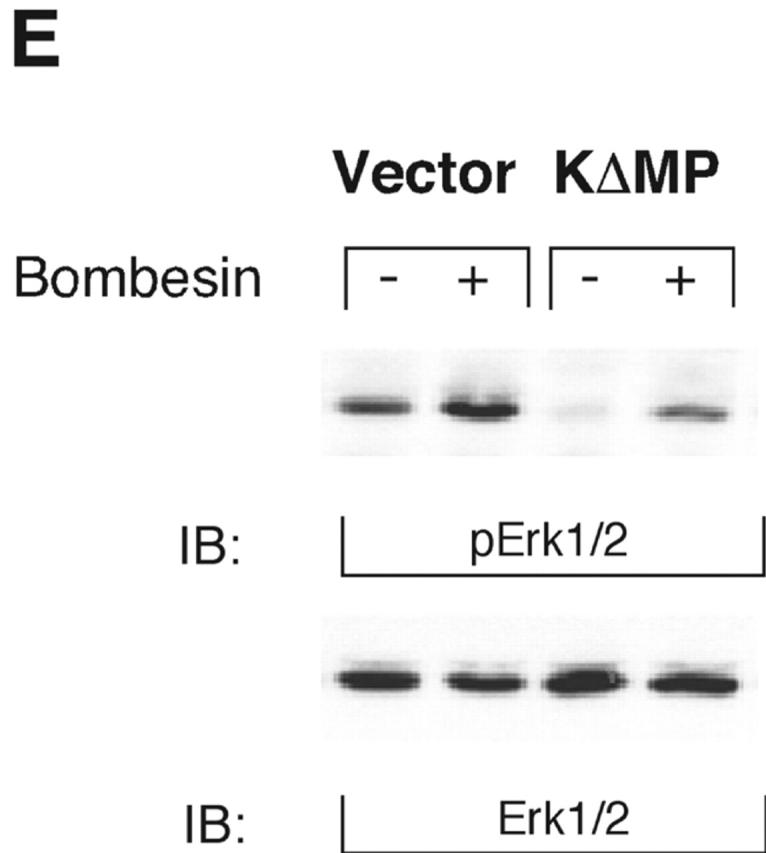
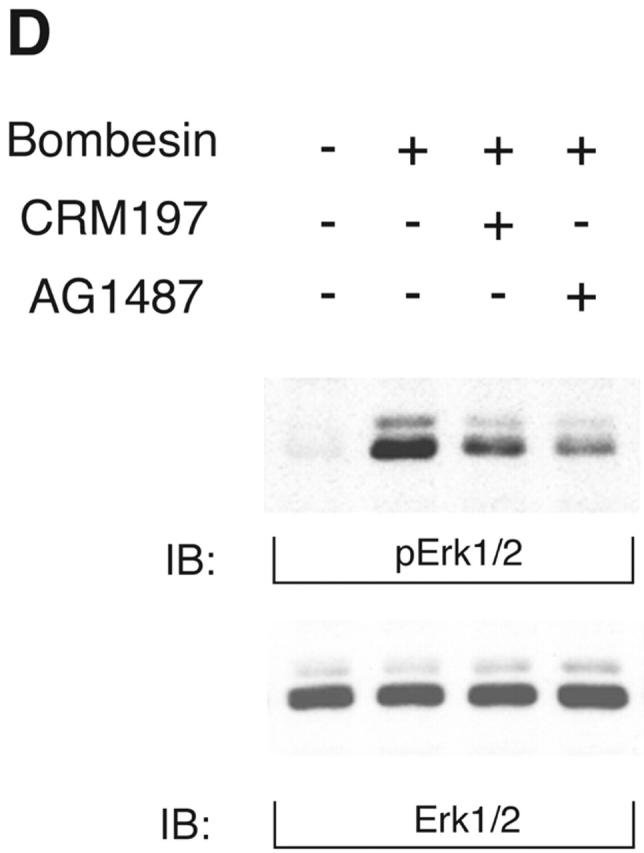

KUZ stimulates, and blocking endogenous KUZ inhibits, the transactivation of signaling pathways downstream of EGFR. (A) Activation of all three forms of SHC and (B) Gab1 phosphorylation by GPCR are stimulated by transfecting KUZ, but not protease-deleted KΔMP, into COS7 cells. (C) KUZ elevates GPCR-induced Ras activation in COS7 cells. The upper band, nonspecifically reactive to the anti-Ras antibody, is shown as the loading control. (D) Bombesin induces Erk1/2 activation in PC3 cells, which is partly due to the transactivation of EGFR and depends on HB-EGF release. PC3 cells were treated with CRM197 or AG1487 and then stimulated with bombesin. (E) Transfecting KΔMP suppresses Erk1/2 activation in PC3 cells. Active Erk1/2 was detected in bombesin-treated or untreated PC3 cells transfected with vector or KΔMP with anti–phospho-Erk1/2 antibody. (F) Bombesin-induced Erk1/2 activation can be inhibited by an antisense morpholino oligonucleotide against ADAM10. Erk1/2 phosphorylation was detected in lysates of PC3 treated with the morpholino anti-ADAM10 oligo (Anti-KUZ) and control oligo with the same base composition but in reverse order (Reverse-KUZ). The endogenous ADAM10 in antisense oligo–treated cells was detected with a polyclonal antibody to ADAM10 (KUZ).
GPCRs, such as bombesin receptor, activate growth signaling through both Ras-dependent and independent pathways. To test whether KUZ-mediated transactivation of EGFR is an integral part of the mechanism by which GPCRs activate the Ras pathway, we next examined the effect of KUZ on the activation of Ras, which links the activated EGFR to MAP kinase activation. Precipitation of activated Ras from the COS7 cell lysates with a fusion protein containing the Ras-binding domain of Raf1 showed that bombesin treatment significantly increased Ras activation (Fig. 2 C). The expression of KUZ, but not KΔMP, further elevated the increase of activated Ras. The results demonstrate that the metalloprotease KUZ contributes to the GPCR activation of EGFR, which leads to the activation of the Ras-dependent signaling pathway.
Antagonists of bombesin inhibit tumor growth in nude mice seeded with PC3 cells, and their effects are EGFR dependent, suggesting that EGFR is involved in bombesin-induced tumor growth (Plonowski et al., 2000; Heasley, 2001). Interestingly, in preliminary studies, we have found that the expression of KUZ increases with neoplastic progression in transgenic models of mouse prostate cancer (unpublished data). Moreover, ADAM10, the human homologue of KUZ, significantly increases with androgen treatment in PC3 cells, whereas TACE expression is inhibited (McCulloch et al., 2000). To assess whether endogenous KUZ was involved in GPCR transactivation of the EGFR signaling pathway in PC3 prostate cancer cells, we investigated the effect of KUZ on the activation of MAP kinase (Erk1/2). Inhibition of EGFR kinase with AG1478 and neutralization of HB-EGF with CRM197 both reduced Erk1/2 phosphorylation by bombesin, suggesting that transactivation of EGFR is required, at least in part, for bombesin to activate Erk signaling in PC3 cells (Fig. 2 D).
To determine whether PC3 cells require KUZ for this transactivation, we used two methods to inhibit endogenous KUZ and then tested the response of the cells to bombesin. First, transfecting KΔMP suppressed transactivation of Erk1/2 by bombesin (Fig. 2 E). Second, introduction of the specific antisense morpholino oligonucleotide against ADAM10 inhibited the bombesin-mediated transactivation of Erk1/2, whereas a control oligonucleotide with same nucleotide sequence but in a reverse orientation did not (Fig. 2 F), nor did the antisense oligonucleotides against ADAMs 9 and 15 and TACE (unpublished data). These results show that blocking KUZ inhibits transactivation, implicating endogenous KUZ as a critical player in GPCR transactivation of EGFR signaling.
The effect of KUZ on EGFR transactivation required its metalloprotease activity. KUZ was involved in shedding HB-EGF from COS7 cells. Stimulation of cells with bombesin increased the amount of shed HB-EGF in the medium. KUZ enhanced the ability of bombesin to release HB-EGF into the medium (Fig. 3 A). In contrast, KΔMP blocked the release of HB-EGF. These data show that KUZ is required to shed HB-EGF. Inhibition of metalloprotease, using the broad-spectrum inhibitor TAPI, also inhibited bombesin-induced EGFR and SHC phosphorylation (Fig. 3 B). A single E385A mutation causes the loss of KUZ metalloprotease activity. When the mutant K(E-A) was introduced into COS7 cells, EGFR and SHC activation did not increase upon bombesin treatment (Fig. 3 C). Therefore, metalloprotease activity of KUZ is responsible for EGFR transactivation.
Figure 3.


Metalloprotease activity of KUZ is responsible for the GPCR transactivation EGFR signaling pathway in COS7 cells. (A) Transfection of wild-type KUZ, but not KΔMP, stimulates the release of soluble HB-EGF into the medium. The HA-tagged HB-EGF in the medium of transfected cells was analyzed by collecting the heparin-binding proteins and blotting with the anti-HA antibody 12CA5. (B) Metalloprotease inhibitor TAPI blocks bombesin-induced transactivation of EGFR and SHC. Cells were preincubated with either TAPI or solvent DMSO before stimulation with bombesin. The immunoprecipitated EGFR or SHC was blotted with antiphosphotyrosine antibody 4G10 to reveal the activated EGFR and SHC. (C) Catalytically inactive KUZ (K[E-A]) does not support the EGFR and SHC transactivation. The total EGFR and SHC was equivalent in each lane (not depicted).
However, the ability to cleave HB-EGF alone is not sufficient to cause the transactivation; an additional signaling step is needed. Indeed, ADAM9 cleaves HB-EGF under the regulation of PKC, yet neither the wild-type nor the dominant-negative ADAM9 affects the GPCR-induced EGFR phosphorylation (Izumi et al., 1998; Prenzel et al., 1999). Stimulation of GPCR unleashes an array of signaling mediators that may activate KUZ to support the EGFR transactivation. Interestingly, only Gi- and Gq-coupled GPCRs are associated with the transactivation of EGFR, and these GPCRs activate p44/42 Erk. Whether Erk directly regulates the cleavage of pro-TGFα (Fan and Derynck, 1999) and c-Met (Nath et al., 2001) in response to extracellular signals is unresolved, because dominant-negative MEK1 does not block the transactivation resulting from HB-EGF shedding (Pierce et al., 2001b). However, inhibition of Src kinase has a profound effect on transactivation (Pierce et al., 2001b). It is conceivable that kinases, such as Src activated by GPCR, can phosphorylate the cytoplasmic domain of ADAMs. The mechanism by which this phosphorylation activates metalloprotease remains elusive because the cytoplasmic domain of TACE is dispensable in PMA-induced shedding (Reddy et al., 2000).
For shedding to occur in cis, KUZ would have to bind to its substrate pro–HB-EGF. We therefore hypothesized that GPCR affects KUZ activity by regulating the formation of such a complex. Indeed, we found that the antibody to CD9, a tetraspan transmembrane protein, precipitated not only HB-EGF, but also KUZ from cell lysates, suggesting that they coexist in the same molecular complex on the cell membrane (Fig. 4) . CD9 coprecipitated with two forms of HB-EGF, a long 24-kD form and a short 18-kD form. Whereas CD9 binding of the long form appeared to be constitutive, the short form–CD9 complex increased in the presence of transfected KUZ and with bombesin treatment. Therefore, GPCR regulates KUZ-dependent activation of HB-EGF by promoting the binding of KUZ to the molecular complex centered on CD9. Interestingly, the potency of HB-EGF in stimulating cell growth correlated with its binding to CD9. CD9 also associates with ADAM2 and regulates interaction of ADAM2 with α6β1 integrin (Chen et al., 1999; Shi et al., 2000).
Figure 4.
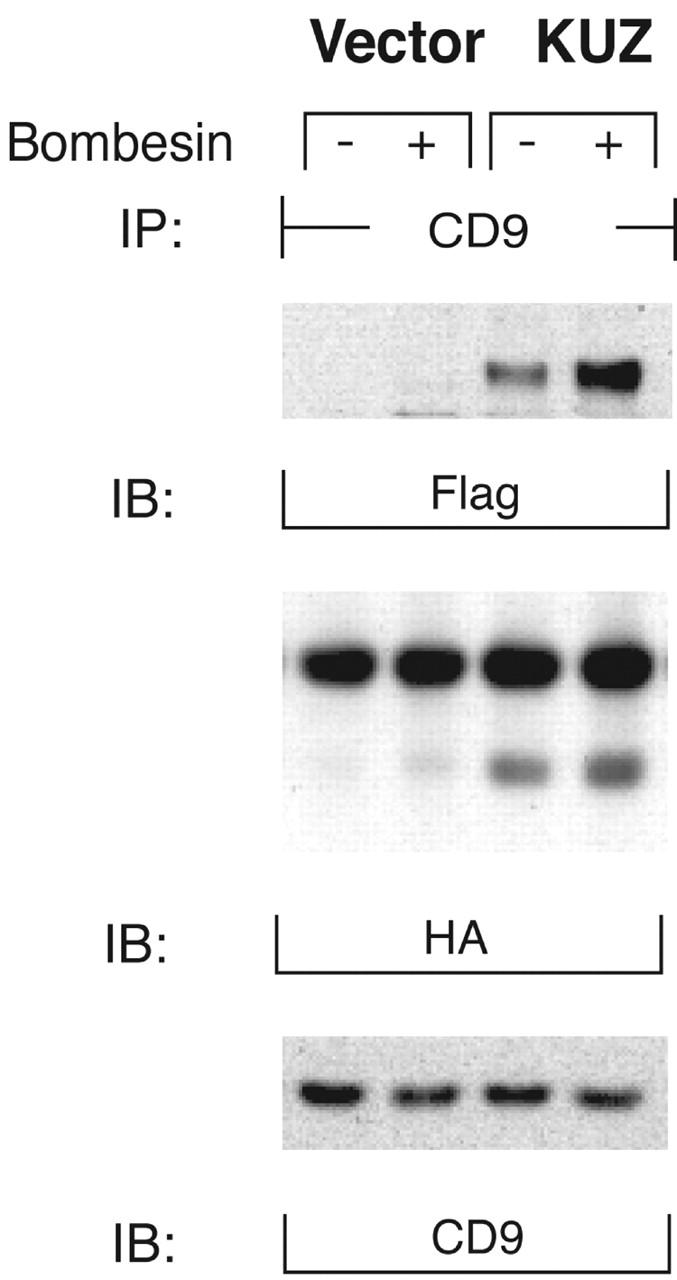
GPCRs regulate the formation of a complex of CD9 with KUZ and HB-EGF. COS7 cells were transfected with Flag-tagged KUZ and HA-tagged HB-EGF. CD9 was precipitated from CHAPS lysates of control or bombesin-treated cells with a polyclonal anti-CD9 antibody. Proteins from the immunoprecipitation were blotted with monoclonal antibodies against Flag epitope, HA epitope, and CD9, respectively.
We have shown that the metalloprotease KUZ defines a control point in the relay between the GPCR and the EGFR signaling pathways. The identification of KUZ as the mediator of transactivation reveals an evolutionarily conserved role of KUZ in coordinating cell behavior. EGFR transactivation occurs not only in the same cell where both KUZ and EGFR reside, but also affects the neighboring cells that sense released HB-EGF. By relaying the extracellular signal from GPCR to EGFR, KUZ not only propagates the signal laterally on the cell membrane, but also coordinates the response of surrounding cells with cleaved HB-EGF. Interestingly, two other well-defined KUZ functions also involve the precise control of signals between adjacent cells: in lateral inhibition where KUZ mediates cleavage of Notch and its ligand Delta (Pan and Rubin, 1997; Qi et al., 1999), and in interaction and cleavage of ephrin-A2 upon binding to its receptor Eph in the contacting cell (Hattori et al., 2000). In all cases, KUZ functions at the point of cell–cell contact. Elucidation of the mechanism of GPCR activation of KUZ increases our understanding of how metalloprotease-mediated shedding regulates signaling pathways.
Materials and methods
Cells and constructs
COS7 and PC3 cells were cultured in six-well plates with DME to 90% of confluence before transfection with plasmid DNAs premixed with Fugene6 (Roche). 12 h after transfection, cells were washed three times and incubated for an additional 24–36 h in the serum-free medium. Mouse KUZ (a gift from D.J. Pan, University of Texas Southwestern Medical School, Dallas, TX), protease-deleted KUZ, ADAM17 (TACE; a gift from R. Black, Immunex Corporation, Seattle, WA), and ADAM15 were tagged with the Flag epitope at COOH termini in the same expression vector.
EGFR activation assay
Transfected cells were treated with GPCR agonists (LPA, 10 μM; bombesin, 200 nM) for 5 min at 37°C before being washed with cold PBS and lysed in the lysis buffer (50 mM Hepes, pH 7.5, 150 mM NaCl, 1% Triton X-100, 1 mM EDTA, 10% glycerol, 10 mM sodium pyrophosphate, 2 mM sodium orthovanadate, 10 mM sodium fluoride, plus protease inhibitors). Cell lysates were cleared by spinning at 15,000 rpm for 10 min and diluted with an equal volume of dilution buffer (50 mM Hepes, pH 7.5, 150 mM NaCl, 0.1% Triton X-100, 1 mM EDTA, 10% glycerol, 10 mM sodium pyrophosphate, 2 mM sodium orthovanadate, 10 mM sodium fluoride). EGFRs were precipitated from lysates with a polyclonal antibody (Upstate Biotechnology) overnight at 4°C. The precipitates were blotted with an antiphosphotyrosine antibody (4G10) to detect the activated EGFR, and a polyclonal antibody against EGFR (Santa Cruz Biotechnology, Inc.) to detect the quantities of EGFR. In some experiments, cells treated with CRM197 (10 μg/ml), AG1487 (250 nM), or DMSO for 20 min were then stimulated with agonist for 5 min. Metalloprotease inhibitor TAPI was applied at 50 μM in DMSO for 40 min at 37°C before stimulation with bombesin.
Assay for SHC, Gab1, Ras, and Erk1/2 activation
COS7 cells transfected with KUZ, KΔMP, and vector for 24 h were serum starved for an additional 24 h before stimulation with bombesin for 5 min at 37°C. Adaptor protein SHC or Gab1 was immunoprecipitated with anti-SHC or anti-Gab1 polyclonal antibody (Transduction Lab). Activation of SHC or Gab1 was detected in immunoblots as the phosphorylation of SHC or Gab1 with antiphosphotyrosine antibody 4G10. The activated Ras was precipitated from the same set of cell lysates with the Ras-binding domain of Raf1 supplied in a reagent kit (Upstate Biotechnology). The amount of activated Ras in the cells was detected in the immunoblot with anti-Ras antibody. Phosphorylation of Erk1/2 was detected with the anti–phospho-ERK1/2 antibody (New England Biolabs, Inc.) in cell lysates.
Antisense experiment
Morpholino antisense oligonucleotide to human ADAM10 (AATTAACACTCTCAGCAACACCATC) and control morpholino oligonucleotide that has the same base composition but in reverse sequence (CTACCACAACGACTCTCACAATTAA) were synthesized by GeneTools, LLC. Oligos were introduced into PC3 cells with ethoxylated polyethylenimine as the delivery reagent according to the procedure recommended by the manufacturer. PC3 cells in a six-well plate were incubated with 1.5 ml of 1 μM oligo mixture per well for 3 h at 37°C. Cells were washed and incubated in serum-free medium for an additional 36 h before stimulation with bombesin and assaying for Erk1/2 phosphorylation. The endogenous ADAM10 levels in antisense oligo–treated cells were detected with a polyclonal antibody to ADAM10 (KUZ; Chemicon International, Inc.).
Shedding of HB-EGF
COS7 cells were transfected with vector, KUZ–Flag, KΔMP–Flag, and hemagglutinin (HA)-tagged HB-EGF (a gift from M. Klagsburn, Children's Hospital, Boston, MA). After bombesin treatment for 20 min, serum-free media was collected and cleared by spinning at 15,000 rpm for 10 min. Heparin-binding proteins were precipitated from the medium with heparin-agarose at 4°C overnight and immunoblotted with an anti-HA antibody (12CA5) to detect HB-EGF.
Immunoprecipitation of CD9
COS7 cells were transfected with KUZ–Flag and HA-tagged HB-EGF with Fugene 6. After stimulating cells with bombesin for 5 min, cell lysates were prepared in a buffer containing 50 mM Hepes, pH 7.5, 150 mM NaCl, 10 mM CHAPS, 1 mM EDTA, 10% glycerol, 10 mM sodium pyrophosphate, 2 mM sodium orthovanadate, 10 mM sodium fluoride, plus protease inhibitors. CD9 in cell lysates was precipitated with a polyclonal anti-CD9 antibody (4 μg/ml, Santa Cruz Biotechnology, Inc.) overnight, and blotted with monoclonal antibodies against CD9 (BD Biosciences) or HA epitope (12CA5; Roche).
Acknowledgments
We thank B. Wiseman (University of California, San Francisco) for critical reading of the manuscript and D.J. Pan, R. Black, and M. Klagsbrun for providing plasmid DNA for KUZ, TACE, and HA-tagged HB-EGF, respectively.
This work was supported by funds from the DOD Breast Cancer Program (DAMD17-9818193 to Y. Yan), the National Cancer Institute (CA72006 to Z. Werb), and the University of California San Francisco Prostate Cancer Center Development Program (to Z. Werb).
Y. Yan's present address is Millennium Pharmaceuticals, Inc., South San Francisco, CA 94080.
K. Shirakabe's present address is Department of Molecular Cell Biology, Medical Research Institute, Tokyo, 10-0062, Japan.
Footnotes
Abbreviations used in this paper: ADAM, metalloprotease with a disintegrin domain; EGFR, EGF receptor; GPCR, G protein–coupled receptors; HA, hemagglutinin; HB-EGF, heparin-binding EGF; KUZ, Kuzbanian; KΔMP, protease domain–deleted KUZ; LPA, lysophosphatidic acid; SHC, Src homology 2 domain–containing protein; TACE, TNF-α–converting enzyme; TNF, tumor necrosis factor.
References
- Black, R.A., and J.M. White. 1998. ADAMs: focus on the protease domain. Curr. Opin. Cell Biol. 10:654–659. [DOI] [PubMed] [Google Scholar]
- Blobel, C.P. 2000. Remarkable roles of proteolysis on and beyond the cell surface. Curr. Opin. Cell Biol. 12:606–612. [DOI] [PubMed] [Google Scholar]
- Carpenter, G. 1999. Employment of the epidermal growth factor receptor in growth factor–independent signaling pathways. J. Cell Biol. 146:697–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, M.S., K.S. Tung, S.A. Coonrod, Y. Takahashi, D. Bigler, A. Chang, Y. Yamashita, P.W. Kincade, J.C. Herr, and J.M. White. 1999. Role of the integrin-associated protein CD9 in binding between sperm ADAM 2 and the egg integrin α6β1: implications for murine fertilization. Proc. Natl. Acad. Sci. USA. 96:11830–11835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daub, H., F.U. Weiss, C. Wallasch, and A. Ullrich. 1996. Role of transactivation of the EGF receptor in signalling by G-protein-coupled receptors. Nature. 379:557–560. [DOI] [PubMed] [Google Scholar]
- Dong, J., L.K. Opresko, P.J. Dempsey, D.A. Lauffenburger, R.J. Coffey, and H.S. Wiley. 1999. Metalloprotease-mediated ligand release regulates autocrine signaling through the epidermal growth factor receptor. Proc. Natl. Acad. Sci. USA. 96:6235–6240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan, H., and R. Derynck. 1999. Ectodomain shedding of TGF-α and other transmembrane proteins is induced by receptor tyrosine kinase activation and MAP kinase signaling cascades. EMBO J. 18:6962–6972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattori, M., M. Osterfield, and J.G. Flanagan. 2000. Regulated cleavage of a contact-mediated axon repellent. Science. 289:1360–1365. [DOI] [PubMed] [Google Scholar]
- Heasley, L.E. 2001. Autocrine and paracrine signaling through neuropeptide receptors in human cancer. Oncogene. 20:1563–1569. [DOI] [PubMed] [Google Scholar]
- Izumi, Y., M. Hirata, H. Hasuwa, R. Iwamoto, T. Umata, K. Miyado, Y. Tamai, T. Kurisaki, A. Sehara-Fujisawa, S. Ohno, and E. Mekada. 1998. A metalloprotease-disintegrin, MDC9/meltrin-γ/ADAM9 and PKCδ are involved in TPA-induced ectodomain shedding of membrane-anchored heparin-binding EGF-like growth factor. EMBO J. 17:7260–7272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCulloch, D.R., M. Harvey, and A.C. Herington. 2000. The expression of the ADAMs proteases in prostate cancer cell lines and their regulation by dihydrotestosterone. Mol. Cell. Endocrinol. 167:11–21. [DOI] [PubMed] [Google Scholar]
- Nath, D., N.J. Williamson, R. Jarvis, and G. Murphy. 2001. Shedding of c-Met is regulated by crosstalk between a G-protein coupled receptor and the EGF receptor and is mediated by a TIMP-3 sensitive metalloproteinase. J. Cell Sci. 114: 1213–1220. [DOI] [PubMed] [Google Scholar]
- Pan, D., and G.M. Rubin. 1997. Kuzbanian controls proteolytic processing of Notch and mediates lateral inhibition during Drosophila and vertebrate neurogenesis. Cell. 90:271–280. [DOI] [PubMed] [Google Scholar]
- Peschon, J.J., J.L. Slack, P. Reddy, K.L. Stocking, S.W. Sunnarborg, D.C. Lee, W.E. Russell, B.J. Castner, R.S. Johnson, J.N. Fitzner, et al. 1998. An essential role for ectodomain shedding in mammalian development. Science. 282:1281–1284. [DOI] [PubMed] [Google Scholar]
- Pierce, K.L., L.M. Luttrell, and R.J. Lefkowitz. 2001. a. New mechanisms in heptahelical receptor signaling to mitogen activated protein kinase cascades. Oncogene. 20:1532–1539. [DOI] [PubMed] [Google Scholar]
- Pierce, K.L., A. Tohgo, S. Ahn, M.E. Field, L.M. Luttrell, and R.J. Lefkowitz. 2001. b. Epidermal growth factor receptor-dependent ERK activation by G protein-coupled receptors: a co-culture system for identifying intermediates upstream and downstream of HB-EGF shedding. J. Biol. Chem. 276:23155–23160. [DOI] [PubMed] [Google Scholar]
- Plonowski, A., A.V. Schally, J.L. Varga, Z. Rekasi, F. Hebert, G. Halmos, and K. Groot. 2000. Potentiation of the inhibitory effect of growth hormone-releasing hormone antagonists on PC-3 human prostate cancer by bombesin antagonists indicative of interference with both IGF and EGF pathways. Prostate. 44:172–180. [DOI] [PubMed] [Google Scholar]
- Prenzel, N., E. Zwick, H. Daub, M. Leserer, R. Abraham, C. Wallasch, and A. Ullrich. 1999. EGF receptor transactivation by G-protein-coupled receptors requires metalloproteinase cleavage of proHB-EGF. Nature. 402:884–888. [DOI] [PubMed] [Google Scholar]
- Qi, H., M.D. Rand, X. Wu, N. Sestan, W. Wang, P. Rakic, T. Xu, and S. Artavanis-Tsakonas. 1999. Processing of the notch ligand δ by the metalloprotease Kuzbanian. Science. 283:91–94. [DOI] [PubMed] [Google Scholar]
- Reddy, P., J.L. Slack, R. Davis, D.P. Cerretti, C.J. Kozlosky, R.A. Blanton, D. Shows, J.J. Peschon, and R.A. Black. 2000. Functional analysis of the domain structure of tumor necrosis factor-α converting enzyme. J. Biol. Chem. 275:14608–14614. [DOI] [PubMed] [Google Scholar]
- Shi, W., H. Fan, L. Shum, and R. Derynck. 2000. The tetraspanin CD9 associates with transmembrane TGF-α and regulates TGF-α–induced EGF receptor activation and cell proliferation. J. Cell Biol. 148:591–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternlicht, M.D., A. Lochter, C.J. Sympson, B. Huey, J.P. Rougier, J.W. Gray, D. Pinkel, M.J. Bissell, and Z. Werb. 1999. The stromal proteinase MMP3/stromelysin-1 promotes mammary carcinogenesis. Cell. 98:137–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werb, Z. 1997. ECM and cell surface proteolysis: regulating cellular ecology. Cell. 91:439–442. [DOI] [PubMed] [Google Scholar]
- Werb, Z., and Y. Yan. 1998. A cellular striptease act. Science. 282:1279–1280. [DOI] [PubMed] [Google Scholar]