

Abstract
The Saccharomyces cerevisiae mitotic exit network (MEN) is a conserved signaling network that coordinates events associated with the M to G1 transition. We investigated the function of two S. cerevisiae proteins related to the MEN proteins Mob1p and Dbf2p kinase. Previous work indicates that cells lacking the Dbf2p-related protein Cbk1p fail to sustain polarized growth during early bud morphogenesis and mating projection formation (Bidlingmaier, S., E.L. Weiss, C. Seidel, D.G. Drubin, and M. Snyder. 2001. Mol. Cell. Biol. 21:2449–2462). Cbk1p is also required for Ace2p-dependent transcription of genes involved in mother/daughter separation after cytokinesis. Here we show that the Mob1p-related protein Mob2p physically associates with Cbk1p kinase throughout the cell cycle and is required for full Cbk1p kinase activity, which is periodically activated during polarized growth and mitosis. Both Mob2p and Cbk1p localize interdependently to the bud cortex during polarized growth and to the bud neck and daughter cell nucleus during late mitosis. We found that Ace2p is restricted to daughter cell nuclei via a novel mechanism requiring Mob2p, Cbk1p, and a functional nuclear export pathway. Furthermore, nuclear localization of Mob2p and Ace2p does not occur in mob1–77 or cdc14–1 mutants, which are defective in MEN signaling, even when cell cycle arrest is bypassed. Collectively, these data indicate that Mob2p–Cbk1p functions to (a) maintain polarized cell growth, (b) prevent the nuclear export of Ace2p from the daughter cell nucleus after mitotic exit, and (c) coordinate Ace2p-dependent transcription with MEN activation. These findings may implicate related proteins in linking the regulation of cell morphology and cell cycle transitions with cell fate determination and development.
Keywords: cell cycle; polarized growth; cell separation; mitotic exit network; nuclear export
Introduction
Eukaryotic cell division requires spatial and temporal coordination of cell growth, chromosome replication and segregation, and cytokinesis. The intimacy of this coordination is dramatically apparent as cells pass from mitosis to G1, when the processes of cyclin-dependent kinase (CDK)* inactivation, mitotic spindle disassembly, chromatin decondensation, cytokinesis, and activation of early G1 gene transcription are coupled to each other. Many of these processes involve cytoskeleton reorganization. For example, in animal cells the cortical cytoskeleton is reorganized to produce a cytokinetic apparatus aligned with the mitotic spindle midzone (Rappaport, 1996). The onset of cytokinesis, which involves actomyosin ring contraction and delivery of new plasma membrane to an ingressing division furrow, is closely synchronized with the completion of chromosome segregation. The regulatory mechanisms responsible for organizing these diverse cell cycle events in animal cells have not been fully elucidated.
Mitotic exit in the budding yeast Saccharomyces cerevisiae is analogous to that of animal cells and involves close coupling of chromosome segregation with actomyosin ring contraction and septum deposition at the mother–bud junction. In S. cerevisiae, a regulatory cascade called the mitotic exit network (MEN) appears to play a critical role in coordinating several processes associated with mitotic exit. The MEN is comprised of highly conserved proteins that include the protein kinases Cdc5p, Cdc15p, Dbf2p, and Dbf20p, the phosphatase Cdc14p, the GTPase Tem1p and its GEF Lte1p, and Mob1p, a novel protein that binds Dbf2p and Dbf20p (McCollum and Gould, 2001). This regulatory network inactivates mitotic CDK by turning on a cyclin destruction pathway and activating the CDK inhibitor Sic1p, and promoting the action of Swi5p, a transcription factor necessary for G1 transcription (Visintin et al., 1998; Jaspersen et al., 1999). The MEN also controls the onset of cytokinesis: actomyosin ring contraction and septum deposition do not occur when MEN function is abrogated, even when progress from mitosis to G1 is allowed by artificial suppression of mitotic CDK activity (Lippincott et al., 2001; Luca et al., 2001).
The association of Mob1p with Dbf2p is critical for MEN function and likely permits Dbf2p activation by Cdc15p (Komarnitsky et al., 1998; Lee et al., 2001; Mah et al., 2001). Mob1p and Dbf2p are necessary for late mitotic changes in Cdc14p localization that are critical for its regulatory action (Visintin et al., 1998; Shou et al., 1999; Stegmeier et al., 2002). Other Mob1p- and Dbf2p-related proteins are present in S. cerevisiae, raising the possibility that related or parallel pathways control important aspects of cell division. Mob1p is closely related to the nonessential protein Mob2p (Luca and Winey, 1998), and Dbf2p is very similar to the Cbk1p protein kinase. Intriguingly, two-hybrid analysis suggests that Mob2p associates with Cbk1p (Racki et al., 2000). Cbk1p is important for polarized cell growth and required for final separation of mother and daughter cells, an event that follows cytokinesis and involves degradation of the chitin-rich septum deposited between mother and daughter cells (Racki et al., 2000; Bidlingmaier et al., 2001). Cbk1p was shown to localize to the periphery of the growing bud and to the bud neck during late mitosis. Moreover, Cbk1p was demonstrated to be essential for M/G1 transcription of genes that are regulated by the Swi5-related transcription factor Ace2p, which encode proteins that are required for septum degradation and cell separation, such as Cts1p and Scw11p (Dohrmann et al., 1992; Racki et al., 2000; Bidlingmaier et al., 2001; Doolin et al., 2001). It is therefore possible that Mob2p and Cbk1p function analogously to Mob1p and Dbf2p and are part of a MEN-like signaling cascade that controls morphogenesis and transcription at the end of mitosis.
The transcription factor Ace2p is a likely downstream target for Cbk1p. Ace2p drives the expression of cell separation genes at the end of mitosis, and thus its activity must be coordinated with mitotic exit. Ace2p nuclear import is inhibited by CDK phosphorylation (O'Conallain et al., 1999), as is the related Swi5p transcription factor (Moll et al., 1991); its nuclear export is mediated by Xpo1p, a Crm1-like nuclear exportin (Jensen et al., 2000). Recently, and in parallel to work presented here, Ace2p was reported to exhibit Mob2- and Cbk1p-dependent localization to the daughter cell nucleus at the end of mitosis and to control daughter cell–specific transcription of genes that are necessary for septum degradation (Colman-Lerner et al., 2001). The mechanism by which Ace2p is restricted to the daughter cell nucleus is unknown. It is also not known how the nuclear localization or function of Ace2p is coordinated with mitotic exit. Here, we present findings that implicate complexed Mob2p–Cbk1p in the coordination of cell morphogenesis, mitotic exit, and nuclear export–mediated daughter cell localization of active Ace2p.
Results
MOB2 is required for mother/daughter separation after cytokinesis
Since Mob2p is similar to the Dbf2p-binding protein Mob1p but exhibits two-hybrid interaction with Cbk1p (Luca and Winey, 1998; Racki et al., 2000), we asked if mob2Δ cells exhibited defects in mitotic exit, cytokinesis, septum degradation, or cell polarity. We found that mob2Δ strains grow as clumps of round cells joined at regions that stained brightly with calcofluor, a chitin-binding dye (Fig. 1 A). This phenotype is consistent with defective cytokinesis or failure of septum degradation; it is thus essential to distinguish between these possibilities. To do so, we digested the cell wall of formaldehyde-fixed mob2Δ cells. Cell wall degradation is expected to dissociate cells that fail to undergo septum degradation but not cells defective for cytokinesis. Cell wall digestion largely dispersed aggregated mob2Δ cells, whereas similar treatment did not break the cytoplasmic mother/daughter connections of nocodazole-treated wild-type cells, which arrest before cytokinesis (Fig. 1 B). These data indicate that mob2Δ cells fail to complete septum degradation as shown previously for cbk1Δ cells (Racki et al., 2000; Bidlingmaier et al., 2001). To determine if mob2Δ cells exhibit defective actomyosin ring contraction, as demonstrated for mob1 mutants (Luca et al., 2001), we examined the localization of GFP-tagged Myo1p, a contractile ring component. In mob2Δ cells, GFP–Myo1p localized to the mother–daughter cell junction during bud emergence and growth and rapidly contracted and disappeared at the end of mitosis (Fig. 1 C) as described for wild-type cells (Bi et al., 1998). Thus, loss of Mob2p function results in a phenotype equivalent to loss of Cbk1p function. In support of a shared function for Mob2p and Cbk1p, we found that double mutant mob2Δ cbk1Δ cells were phenotypically indistinguishable from mob2Δ and cbk1Δ single mutants (unpublished data).
Figure 1.

Characterization of cell separation defects in mob2 Δ and cbk1 Δ cells. (A) Chitin staining of wild-type (left; FLY93), cbk1Δ (middle; FLY757), and mob2Δ (right; FLY168) cells stained with calcofluor, highlighting mother/daughter junctions and cell shape. (B) Dissociation of attached cells after cell wall digestion. In the control graph on the left, asynchronously growing and nocodazole-arrested wild-type cells were fixed and digested, and cells with one or more buds were scored as associated cells. In the right graph, digested cbk1Δ and mob2Δ cells with more than two associated cell bodies were scored as associated. In both graphs, white bars represent the fraction of associated cells before digestion, and black bars represent the fraction of associated cells after digestion. (C) Time lapse of Myo1–GFP fluorescence in a live mob2Δ cell (FLY770). Note shrinkage and disappearance of Myo1–GFP signal, corresponding with maximal enlargement of daughter bud.
MOB2 is required for maintenance of polarized cell growth
In addition to cell separation defects, mob2Δ cells were rounder than wild-type cells (Fig. 1 A). Similar cell morphology in cbk1Δ cells stems from inefficient apical growth during early bud morphogenesis (Bidlingmaier et al., 2001). To determine if mob2Δ cells fail to sustain apical bud growth, we monitored the pattern of new cell wall deposition. Cells were grown in the presence of the fluorescent lectin FITC-ConA to label their cell walls, and then transferred into FITC-ConA–free medium for 15 min before fixation. In cells so treated, regions of new cell wall deposition are devoid of staining. Buds that undergo highly focused apical growth after removal of FITC-ConA lack staining at the bud tip, whereas buds that grow isotropically exhibit more uniform staining that extends to the bud tip (Tkacz and Lampen, 1972; Bidlingmaier et al., 2001). In wild-type cultures, 39% of buds lacked FITC-ConA staining at tips, indicative of apical growth. In contrast, only 2% of mob2Δ buds and 3% of cbk1Δ buds exhibited this staining pattern (Fig. 2 A). Therefore, buds of mob2Δ cells, like those of cbk1Δ cells, exhibit reduced apical growth.
Figure 2.

Polarized growth and actin organization in mob2 Δ cells. (A) Polarized cell wall deposition in wild-type, mob2Δ, and cbk1Δ strains (FLY93, FLY168, and FLY757). After FITC-ConA treatment and washout, buds were scored for apical exclusion of staining (black bars; example noted by ♦) and staining extending to the bud tip (hatched bars; example noted by ★). n = 100 for all strains. (B) Rhodamine-phalloidin staining of F-actin in asynchronous mob2Δ (left) and wild-type (right) cells. (C) Chitin (calcofluor, left) and F-actin (rhodamine-phalloidin, right) in mating pheromone- (α factor) treated wild-type (first row), ace2Δ (second row; FLY880), mob2Δ (third row), and cbk1Δ (fourth row). Cells in which small mating projections defined by the pattern of chitin deposition do not correspond with F-actin polarization indicated by ★.
Proper actin cytoskeleton architecture is critical for polarized cell growth. Defects in proteins that organize F-actin can profoundly disrupt cell morphogenesis (Pruyne and Bretscher, 2000). When mob2Δ cells were stained with rhodamine-phalloidin, F-actin organization was found to be essentially normal. F-actin patches were restricted to growing buds, and F-actin cables oriented toward the bud (Fig. 2 B). Large budded cells were capable of polarizing F-actin to the bud neck (Fig. 2 B, inset), and unbudded cells polarized F-actin to the presumptive bud site (unpublished data). These findings do not rule out subtle defects in recruitment or persistence of F-actin at sites of polarized growth in mob2Δ cells.
Yeast cells exposed to cognate mating pheromone arrest in G1 and form mating projections. Mating projection formation requires sustained polarized growth and localized wall remodeling, with secretion and F-actin focused to a small region of the cell cortex (Pruyne and Bretscher, 2000). Chitin is deposited in the cell wall at the base of the mating projection. We found wild-type and ace2Δ cells capable of producing obvious mating projections, with F-actin focused to a region of polarized growth flanked by chitin deposition (Fig. 2 C). In contrast, neither cbk1Δ nor mob2Δ cells sustained actin polarization or formed pronounced mating projections when treated with pheromone (Fig. 2 C). Small chitin-rich cell wall deformations were evident in many cbk1Δ and mob2Δ cells, but these generally did not correspond with F-actin polarization (Fig. 2 C, ★). Polarized F-actin organization was not completely absent in pheromone-treated cbk1Δ and mob2Δ cultures: after 1.5 h of treatment, ∼30% of mob2Δ and ∼35% of cbk1Δ cells exhibited polarized F-actin compared with ∼80% of wild-type cells. With extended exposure (3 h), this number dropped to ∼10 and ∼15% in cbk1Δ and mob2Δ cultures, respectively, whereas ∼85% of wild-type cells remained polarized. Expression of a pheromone-induced fus1-lacZ reporter (Trueheart et al., 1987), measured as β-galactosidase activity, was similar in pheromone-treated mob2Δ cells, cbk1Δ cells, and wild-type cells (unpublished data). Furthermore, neither budding nor nuclear division occurred with elevated frequency in pheromone-treated mob2Δ and cbk1Δ strains relative to wild-type cells (unpublished data). Together, these findings indicate that Mob2p and Cbk1p have a shared function in sustaining polarized growth of the mating projection and that the proteins are not required for cell cycle arrest or transcriptional response upon pheromone treatment.
Mob2p associates with Cbk1p
Two-hybrid experiments have suggested that Mob2p and Cbk1p can interact physically (Racki et al., 2000). To test whether functional Mob2p and Cbk1p interact under more physiologically relevant conditions, we performed coimmunoprecipitation experiments using strains expressing epitope-tagged Cbk1p and Mob2p. CBK1-HA, MOB2-myc, and CBK1-myc, MOB2-HA strains, which express these epitope-tagged proteins instead of native Cbk1p and Mob2p, were phenotypically wild type (unpublished data). We immunoprecipitated tagged Mob2p and Cbk1p from extracts of exponentially growing cells and found that Cbk1p coimmunoprecipitated with Mob2p and vice versa, regardless of affinity tag (Fig. 3 A). Thus, Mob2p and Cbk1p associate as a complex.
Figure 3.
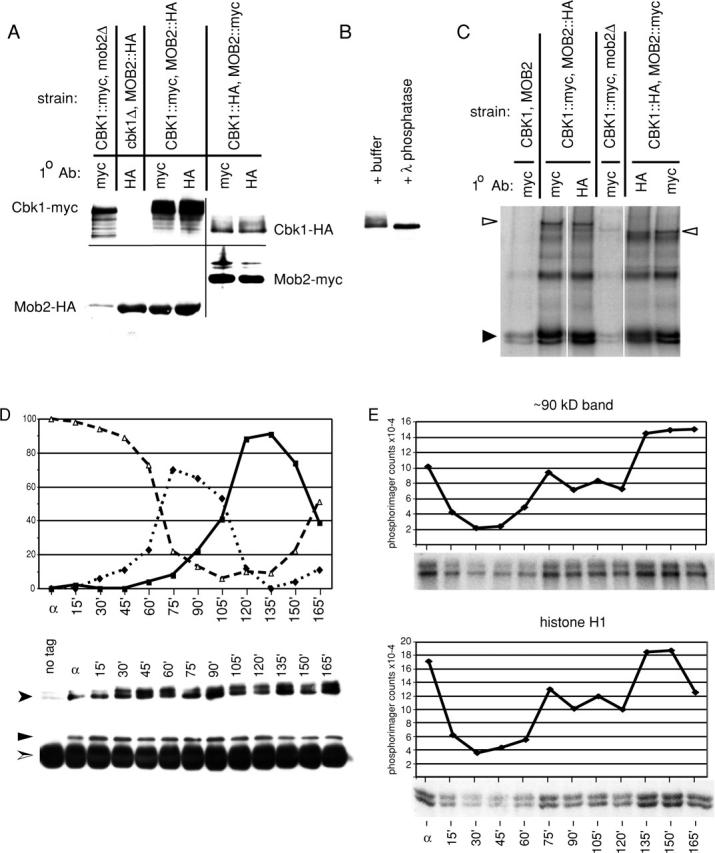
Mob2p physically associates with Cbk1p and is required for Cbk1p kinase activity and hyperphosphorylation. (A) Coimmunoprecipitation of Mob2p and Cbk1p (FLY954 and FLY960), immunoprecipitation of Mob2p from cbk1Δ cells (FLY1005), and immunoprecipitation of Cbk1p from mob2Δ cells (FLY906). The faint HA signal in lane 1 is due to spill over from the neighboring lane. (B) Treatment of immunoprecipitated Cbk1p-HA with λ phosphatase, with λ phosphatase reaction buffer treatment as negative control. (C) Kinase activity present in immunoprecipitates of epitope-tagged Mob2p and Cbk1p. These kinase reactions were performed using the immunoprecipitates characterized by immunoblot shown in A. (D and E) Cells expressing Cbk1p-HA and Mob2-myc (FLY954) were synchronized in G1 (as described in Materials and methods). Anti-HA immunoprecipitations were conducted on extracts of these cells after normalization of protein concentrations. (D, top) Budding morphology at arrest (α) and after release from block. Percentages of unbudded cells are indicated by ▵, small budded cells by ♦, and large budded cells by ▪. (D, bottom) Anti-HA and corresponding anti-myc immunoblots of immunoprecipitated protein. Cbk1p-HA, shown by anti-HA immunoblot, is indicated by a filled arrowhead. Mob2-myc, shown by anti-myc immunoblot, is indicated by ▴. Immunoglobulin heavy chain is indicated by a split arrowhead. (E) Kinase activity present in immunoprecipitated Cbk1p-HA. The top two panels show phosphorylation of a ∼90-kD band, likely Cbk1p autophosphorylation, and the bottom panels show phosphorylation of histone H1. These kinase reactions were performed using the immunoprecipitates characterized by immunoblot (D).
Immunoprecipitated Cbk1p from MOB2 cells migrated as multiple bands on SDS-PAGE. In contrast, Cbk1p isolated from mob2Δ cells did not exhibit the same migration pattern, suggesting that Cbk1p is posttranslationally modified in a MOB2-dependent manner (Fig. 3 A). We found no obvious differences between the migration patterns of Cbk1p isolated directly or through coprecipitation with Mob2p. Slower migrating forms of Cbk1p were eliminated upon treatment of immunoprecipitated protein with λ-phosphatase (Fig. 3 B). Therefore, the slower migration of immunoprecipitated Cbk1p likely reflects Mob2p-dependent hyperphosphorylation.
Cbk1p kinase activity is Mob2p dependent and varies over the cell cycle
In vitro analysis suggests that Dbf2p protein kinase activity requires association with Mob1p (Lee et al., 2001; Mah et al., 2001). To determine if the same is true for Cbk1p and Mob2p, we characterized the in vitro kinase activity of HA- and myc-tagged Cbk1p isolated through both direct immunoprecipitation and in association with HA and myc-tagged Mob2p. Immunoprecipitated Cbk1p phosphorylated the exogenous substrate histone H1 in vitro (Fig. 3 C, filled arrowhead). In addition, a protein of ∼90 kD was phosphorylated by immunoprecipitated Cbk1p-HA, and a ∼120-kD species was phosphorylated by immunoprecipitated Cbk1p-myc (Fig. 3 C, open arrowheads). The difference in apparent molecular weight of these species corresponds to the difference between the sizes of the HA and myc epitope tags, suggesting that these bands represent autophosphorylated Cbk1p. We isolated similar kinase activity in association with Mob2p as predicted by coimmunoprecipitation of Mob2p and Cbk1p. Interestingly, Cbk1p immunoprecipitated from mob2Δ cells had very low in vitro activity (Fig. 3 C). Corresponding immunoblots indicate that similar amounts of Cbk1p were immunoprecipitated from each strain (Fig. 3 A). These findings establish that Mob2p is necessary for Cbk1p activity.
We asked if Mob2p–Cbk1p interaction or Cbk1p kinase activity varied across the cell cycle. We immunoprecipitated Cbk1p-myc from extracts of CBK1-myc, MOB2-HA cells synchronized in G1 by exposure to mating pheromone and examined Cbk1-myc–Mob2-HA coimmunoprecipitation and Cbk1p protein kinase activity. Immunoblots revealed no dramatic variation in protein levels of Cbk1p or associated Mob2p (Fig. 3 D). However, the protein kinase activity of immunoprecipitated Cbk1p varied significantly over the cell cycle (Fig. 3 E). In G1-arrested cells (time zero), Cbk1p kinase activity was relatively high. Given that the kinase is required for mating projection formation, this is not unexpected. Cbk1p activity dropped after release from G1 and rose as cells proceeded into the budding cycle. Late in the cell division cycle, when the majority of cells in culture were large budded, Cbk1p kinase activity was maximal. This peak of activity is consistent with Cbk1p control of cell separation, which is among final events of the cell cycle.
Cbk1p kinase activity is required for cell separation and sustained polarized growth
To evaluate the importance of Cbk1p's protein kinase activity in cell separation and polarized growth, we introduced point mutations predicted to affect catalytic activity. A cbk1 D475A allele, in which an aspartic acid shown to be essential for catalytic activity was changed to alanine (Hanks and Hunter, 1995), failed to complement the cbk1Δ phenotype for both cell separation and mating projection formation (Fig. 4 A). To allow for a conditional block of Cbk1p kinase activity, we replaced methionine 429 with alanine, a substitution corresponding to −as2 alleles that render other protein kinases inhibitable by cell permeable compounds that do not effectively inhibit wild-type kinases (Bishop et al., 2000; Weiss et al., 2000). We found that the cbk1-as2 allele complemented cbk1Δ for cell separation and mating projection formation, although a small number of aggregated cells were present. We then allowed cbk1-as2 and wild-type cells to grow for 3 h in the presence of varying concentrations of the cbk1-as2 inhibitor 1NA-PP1. At 1NA-PP1 concentrations ≥1 μM in cbk1-as2 cells, mother/daughter separation was blocked. Cell separation was unaffected in similarly treated wild-type cells (Fig. 4 B). As with cbk1 D475A cells, inhibitor-treated cbk1-as2 cells were rounder than wild-type cells (Fig. 4 B compared with Fig. 1 A) and formed mating projections poorly (unpublished data). Thus, Cbk1p activity is required for cell separation and polarized growth.
Figure 4.

Inhibition of Cbk1p kinase activity affects cell separation and polarized growth. (A) Chitin and F-actin organization in cells carrying cbk1 D475A (FLY1007). (Top) Chitin staining of asynchronous cells, showing failure of mother/daughter separation and rounded cell shape. (Bottom) Chitin and F-actin organization in cbk1 D475A cells treated with pheromone for 120 min, demonstrating failure of mating projection formation. (B) Cell separation defects caused by 1NA-PP1 treatment of cbk1-as2 cells (FLY1008). The top panel shows dispersed cbk1-as2 cells treated with 1 μM 1NA-PP1 for 2 h; cells counted as instances of mother/daughter separation failure indicated by an asterisk. Graph indicates percentage of cells failing to separate after a 2-h treatment with varying concentration of 1NA-PP1. □, wild-type cells; ♦, cbk1-as2 cells. (C) Maintenance of mating projection growth after inhibition of Cbk1p. The top panels show mating projection formation by cbk1-as2 cells treated with mating pheromone (α-factor; note correspondence of F-actin polarization and chitin deposition) ∼80% of cells exhibited similar morphology (n = 100). The middle panels show a 40-min treatment of these cells with 20 μM 1NA-PP1 in continued presence of pheromone; ∼55% of cells exhibited a depolarized phenotype, ∼30% remained polarized, and ∼15% had not obviously formed mating projections. The bottom panels show control 40-min DMSO treatment of cells shown in top panels; no depolarization was evident.
To determine if Cbk1p kinase activity is required for sustained polarized growth of mating projections subsequent to their initial formation, we added 20 μM 1NA-PP1 to wild-type and cbk1-as2 cells previously induced to form mating projections by pheromone treatment. Prior to 1NA-PP1 addition, ∼80% of both cell types had formed mating projections with corresponding F-actin polarization. After an additional 90-min pheromone treatment in the presence of 1NA-PP1, F-actin was no longer polarized in ∼68% of cbk1-as2 cells that had mating projections identifiable by chitin staining (Fig. 4 C). In contrast, F-actin remained polarized to mating projections in similarly treated wild-type cells and cbk1-as2 cells treated with pheromone and not 1NA-PP1 (Fig. 4 C, bottom). These results further support an important role for Mob2p-dependent Cbk1p kinase activity in maintenance of cell polarization once it has been established.
Mob2p and Cbk1p colocalize at sites of cell growth, the bud neck, and the daughter cell nucleus
Cbk1p was shown previously to localize to the cell cortex during bud growth and to the bud neck at the end of mitosis (Racki et al., 2000; Bidlingmaier et al., 2001). To investigate the subcellular distribution of Mob2p and its relationship to Cbk1p localization, we expressed COOH-terminally tagged Mob2–GFP and Cbk1–GFP in live cells. Both Mob2–GFP and Cbk1–GFP were expressed under the control of their endogenous promoters as the only forms of Mob2p or Cbk1p in these cells. Mob2–GFP and Cbk1–GFP strains exhibited no obvious growth defects (unpublished data). In pheromone-treated cells, both Mob2–GFP and Cbk1–GFP were clearly visible at the tips of mating projections, consistent with the proteins' role in morphogenesis (Fig. 5 A, right). Early in the budding cycle, both Mob2–GFP and Cbk1–GFP localized to incipient bud sites and the cortex of small and medium sized buds (Fig. 5 A, arrowheads). In large budded cells, Mob2–GFP and Cbk1–GFP strongly localized to a bright band at the bud neck with no apparent bias toward the mother or daughter side.
Figure 5.
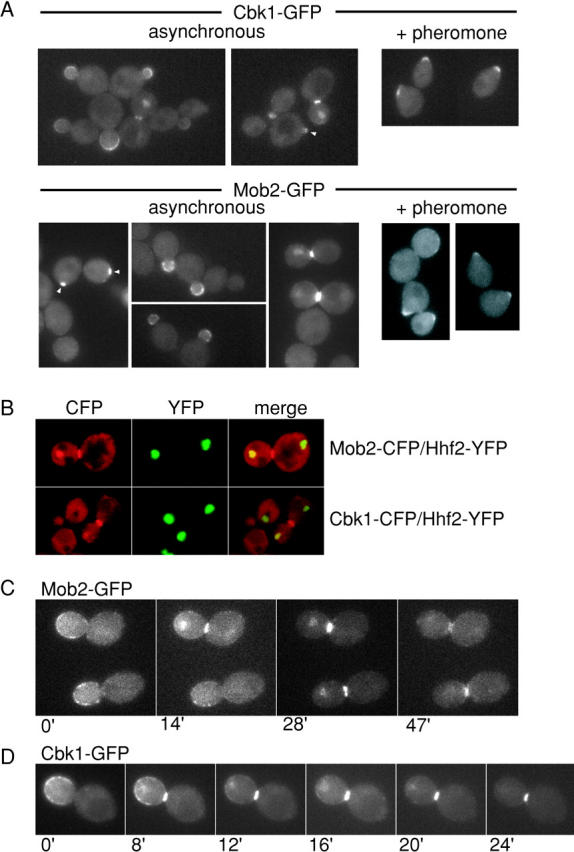
Colocalization of Mob2p and Cbk1p at the cell cortex, bud neck, and daughter nucleus. (A) Localization of Cbk1–GFP (top) and Mob2–GFP (bottom) in asynchronous cells and cells treated with mating pheromone (α-factor) for 90 min. (B) Coexpression of YFP-tagged histone H4 (Hhf2-YFP), a nuclear marker, and CFP-tagged Mob2p (FLY924) or Cbk1p (FLY896). Left panels show CFP fluorescence, center panels show YFP fluorescence, and right panels show overlay. To examine relative timing of localization, we collected fluorescence images of Mob2–GFP localization (FLY893) (C) and Cbk1–GFP localization (FLY895) (D) in time-lapse series. In both cases, daughter nucleus localization of Mob2–GFP and Cbk1–GFP was concomitant with appearance of bud neck localized protein.
In 70–75% of cells with Mob2–GFP concentrated at the bud neck, the protein also exhibited localization consistent with the daughter cell nucleus (Fig. 5 A). Cbk1–GFP showed a similar localization pattern. Using strains expressing CFP-tagged Mob2p or Cbk1p (Mob2–CFP or Cbk1–CFP) and YFP-tagged histone H4 (Hhf2p–YFP), we verified that this indeed reflects daughter cell nuclear localization (Fig. 5 B). Using time-lapse microscopy of Mob2–GFP and Cbk1–GFP, we found that both proteins disappeared from the bud cortex and simultaneously accumulated at the bud neck and daughter cell nucleus. At the end of mitosis or in early G1, Mob2p and Cbk1p disappeared from the daughter nucleus before disappearing from the bud neck (Fig. 5, C and D). We did not find Mob2p or Cbk1p in the mother cell nucleus or in nuclei of G1-arrested cells.
As our report was in preparation, it was reported that Mob2p and Cbk1p localize to bud cortex, bud neck, and daughter cell nucleus in budded cells (Colman-Lerner et al., 2001). However, several of the published images from Colman-Lerner et al. (2001) are ambiguous, particularly with respect to nuclear localization of Mob2p and Cbk1p. It is not clear if this ambiguity is due to poor preservation after fixation, limitations in signal detection, or poor image reproduction. For these reasons, we therefore present our independent analysis of Mob2p and Cbk1p localization in live cells.
Mob2p and Cbk1p are interdependent for localization
To assess the possibility that Cbk1p and Mob2p mediate one another's localization, we examined Mob2–GFP localization in cbk1Δ cells and Cbk1–GFP localization in mob2Δ cells. We found that Mob2–GFP fluorescence was uniformly cytoplasmic in cbk1Δ cells and exhibited no accumulation in nuclei, the cell cortex, or the bud neck (Fig. 6 A), indicating that Mob2p localization is Cbk1p dependent. Similarly, Cbk1p localization was Mob2p dependent. Cbk1–GFP was absent from the cell cortex and bud necks in the vast majority of mob2Δ cells (Fig. 6 B). However, we could detect extremely faint Cbk1–GFP at the bud neck in <1% of cells (not depicted). Thus, our data indicate that Mob2p and Cbk1p are interdependent for cortical localization and that nuclear localization of Mob2p requires Cbk1p. In contrast, the nuclear localization of Cbk1p is not strictly Mob2p dependent. Mob2p was required for temporal and spatial restriction of Cbk1p to the daughter nucleus in late M/early G1. Colman-Lerner et al. (2001) also examined interdependency of Mob2p–Cbk1p localization. However, they concluded that Mob2p localizes to nuclei in cbk1Δ cells. In our studies of live cells, we never observed this despite examining >500 cells. Moreover, our studies indicate that Cbk1–GFP does not localize normally to bud neck and cortex in mob2Δ cells, in contrast to conclusions drawn by Colman-Lerner et al. (2001).
Figure 6.
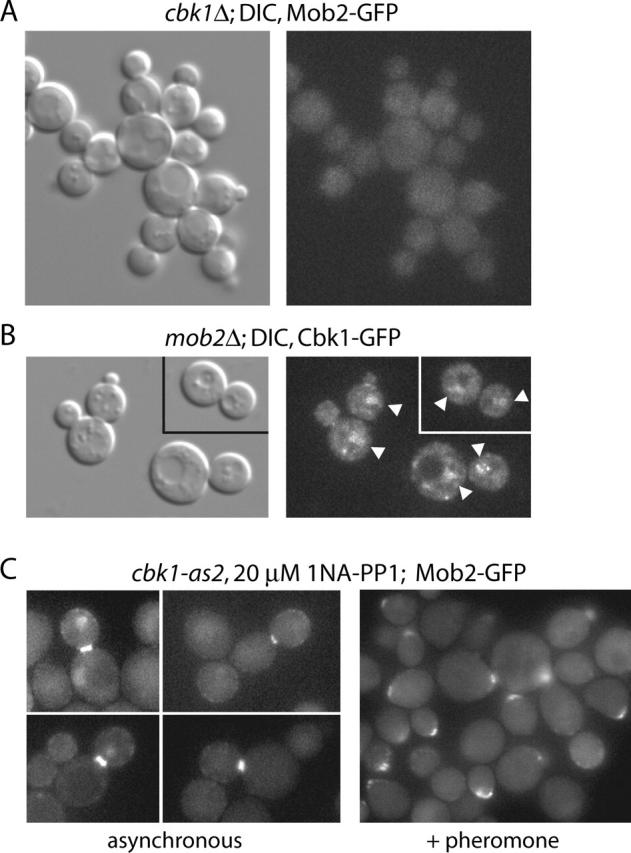
Interdependency of Mob2p and Cbk1p for localization. (A) Mob2–GFP localization in cbk1Δ cells (FLY766); the protein is not found in nuclei or at cortical sites. (B) Cbk1–GFP localization in mob2Δ cells (FLY774). Faint localization of the protein to nuclei was evident in cells with small buds and cells with large buds. (C) Localization of Mob2p upon Cbk1p inhibition in asynchronous (left) and pheromone-treated cells (right; FLY1010).
To determine if Cbk1p kinase activity is required for localization of Mob2p, we examined Mob2–GFP localization in cbk1-as2 cells treated with 20 μM 1NA-PP1. We found Mob2–GFP localized to the daughter cell cortex and bud neck when Cbk1p was inhibited (Fig. 6 C), despite defective polarized growth and cell separation (Fig. 4 B). Similarly, Mob2–GFP localized to a cortical patch in cbk1-as2 cells simultaneously treated with mating pheromone and 1NA-PP1. Nuclear localization of Mob2–GFP was variable. Approximately 20% of cells that had Mob2–GFP present at the bud neck exhibited daughter-specific nuclear localization of Mob2p, whereas ∼20% exhibited very faint Mob2–GFP localization to both mother and daughter nuclei. The remaining ∼60% of cells with Mob2p–GFP at the neck did not have detectable Mob2p nuclear localization. We found essentially identical results using a cbk1Δ MOB2::GFP strain expressing cbk1 D475A, both in exponential growth and mating pheromone exposure (unpublished data). These results indicate that Cbk1p kinase activity is not required for Mob2p localization to cortical sites, including the bud neck, but is required for maximal accumulation of Mob2p in daughter cell nuclei.
Mob2p and Cbk1p restrict Ace2p to daughter cell nuclei
Cbk1p likely controls Ace2p function (Racki et al., 2000; Bidlingmaier et al., 2001). Using overexpression constructs, Ace2p has been found to shuttle between nucleus and cytoplasm in mother and daughter cells (O'Conallain et al., 1999; Jensen et al., 2000). Using Ace2–GFP integrated at the endogenous locus, we found that Ace2p localized specifically to daughter cell nuclei of large budded cells (Fig. 7 A). To assess the cell cycle kinetics of Ace2–GFP localization, we synchronized Ace2–GFP–expressing cells in S phase using hydroxyurea. Ace2–GFP was not readily detectable during and immediately after release from arrest. As cells proceeded into nuclear division, Ace2–GFP initially became visible in both mother and daughter nuclei. As the population progressed into late stages of division, Ace2–GFP accumulated strongly in daughter nuclei and was not apparent in mother cell nuclei (Fig. 7 B). These findings are in agreement with those of Colman-Lerner et al. (2001), who used strains expressing GFP-tagged Ace2p from low copy plasmids.
Figure 7.

Mob2p–Cbk1p–dependent localization of Ace2p to daughter nuclei. (A) Ace2–GFP is localized to daughter cell nuclei in a fraction of large budded cells (FLY811). (B) Localization of Ace2p after release from hydroxyurea arrest. (C) Localization of Ace2p in cbk1Δ cells (top; FLY856) and mob2Δ cells (bottom; FLY850). (D) Localization of Ace2p in hydroxyurea-synchronized cbk1-as2 cells (FLY1009) untreated (top) and treated with 20 mM 1NA-PP1 (bottom). See Results for percentages of symmetric and asymmetric localization. (E) Localization of Cbk1–GFP (top) and Mob2–GFP (bottom) in ace2Δ cells (FLY970 and FLY934).
We found that in cbk1Δ and mob2Δ strains Ace2–GFP localizes to nuclei of both mother and daughter cells (Fig. 7 C), indicating that Mob2p and Cbk1p are required for daughter-specific nuclear localization of Ace2p. We examined the effect of Cbk1p inhibition on Ace2–GFP distribution in hydroxyurea-synchronized cells. In 1NA-PP1–treated cbk1-as2 cells, Ace2–GFP localized faintly to both mother and daughter nuclei of most cells in which the protein was detectable (38/50 cells with visible GFP) (Fig. 7 D). A significant minority of cells (12/50) showed a clear bias in Ace2–GFP localization to the daughter cell nucleus. We also asked if Mob2–GFP and Cbk1–GFP localization requires Ace2p. In ace2Δ cells, Cbk1–GFP and Mob2–GFP localized to cortical sites, but neither protein localized to nuclei (Fig. 7 E). Thus, Cbk1p and Mob2p are required for asymmetric localization of Ace2p to daughter cells. Furthermore, Cbk1p and Mob2p nuclear localizations are Ace2p dependent, consistent with cotransport of the proteins into nuclei. These findings are similar to those reported elsewhere (Colman-Lerner et al., 2001). Importantly, however, we found that high levels of Ace2p accumulation in nuclei required Cbk1p kinase activity.
Mob2p asymmetry is generated by a novel mechanism
The transcription factor Ash1p accumulates in daughter cell nuclei late in cell division and is absent from mother cell nuclei, a localization pattern similar to that of Mob2p, Ace2p, and Cbk1p (Bobola et al., 1996; Sil and Herskowitz, 1996). To determine if Ash1p localization requires MOB2 function, we examined localization of Ash1p–GFP in asynchronous mob2Δ cells. Ash1–GFP localized specifically to daughter cell nuclei in mob2Δ cells (Fig. 8 A). Therefore, Ash1p asymmetry is Mob2p independent. Asymmetric localization of Ash1p was shown to require Myo4p– and Bni1p–dependent localization of ASH1 mRNA to the daughter cell cortex. We found Mob2–GFP localization unaffected in myo4Δ or bni1Δ cells (Fig. 8 B). Ace2–GFP was also unaffected in myo4Δ cells (C. Herbert, personal communication). Thus, asymmetric nuclear localization of Mob2p and Ace2p represents a novel mechanism for asymmetric segregation of proteins that ultimately control gene expression.
Figure 8.

Mob2p-dependent daughter nucleus localization is distinct from Ash1p segregation and requires nuclear export. (A) Nuclear localization of Ash1–GFP in asynchronous mob2Δ cells (FLY841). (B) Daughter nucleus localization of Mob2–GFP in asynchronous myo4Δ cells (top; FLY827) and bni1Δ cells (bottom; FLY823). (C) Localization of Ace2–GFP in xpo1–1 cells (FLY986) at 22°C (top) and 37°C (bottom) after release from hydroxyurea arrest at 22°C.
Daughter cell–specific nuclear localization of Ace2p requires nuclear export
Ace2p nuclear export requires the CRM1-related nuclear export factor Xpo1p (Jensen et al., 2000). Ace2p nuclear accumulation or exclusion could therefore be controlled through regulation of its nuclear import or export. To determine if nuclear export mediates daughter nucleus localization of Ace2p, we examined Ace2–GFP distribution in cells harboring the xpo1–1 conditional loss-of-function allele. We first synchronized these cells in S phase with hydroxyurea and then monitored the progress of nuclear division after release at permissive temperature (22°C). Once nuclear division occurred, we shifted a portion of the culture to restrictive temperature (37°C). In cells maintained at permissive temperature, Ace2–GFP was generally found exclusively in daughter cell nuclei (∼90% of cells with detectable Ace2–GFP [n = 46]) (Fig. 8 C). In cells shifted to 37°C for 1–2 h, Ace2–GFP localized to both mother and daughter nuclei (∼80% of cells with detectable Ace2–GFP [n = 27]) (Fig. 8 C). The levels of Ace2–GFP fluorescence in mother and daughter nuclei were qualitatively equivalent to that in daughter cell nuclei of permissive temperature cells. These results indicate that XPO1 function, and therefore active nuclear export, is necessary for restriction of Ace2p localization to daughter cell nuclei.
Nuclear localization of Mob2p and Ace2p require Mob1p-mediated MEN signaling
Our findings indicate that Mob2p, Cbk1p, and Ace2p control processes that are executed as cells pass from M to G1, coincident with other events controlled by the MEN (McCollum and Gould, 2001). Mob1p, a Mob2p-related protein, and Cdc14p phosphatase are essential components of the MEN. Cells carrying conditional mob1–77 or cdc14–1 alleles arrest in telophase at 34°C due to loss of MEN function (Visintin et al., 1998; Luca et al., 2001). To determine if Mob2p and Ace2p localization require the MEN, we examined their localizations in mob1–77 and cdc14–1 cells. Mob2–GFP localization was clearly abnormal in mob1–77 cells arrested by a 2–3-h shift to 34°C: 65% (n = 90) arrested with Mob2–GFP localized diffusely in the cytoplasm, whereas ∼10% displayed weak Mob2–GFP cortical localization and 25% exhibited weak Mob2–GFP fluorescence at bud necks (Fig. 9 A). Significantly, neither Mob2–GFP nor Ace2–GFP localized to nuclei in telophase-arrested mob1–77 cells. The same is true in cdc14–1 mutants at restrictive temperature (unpublished data).
Figure 9.
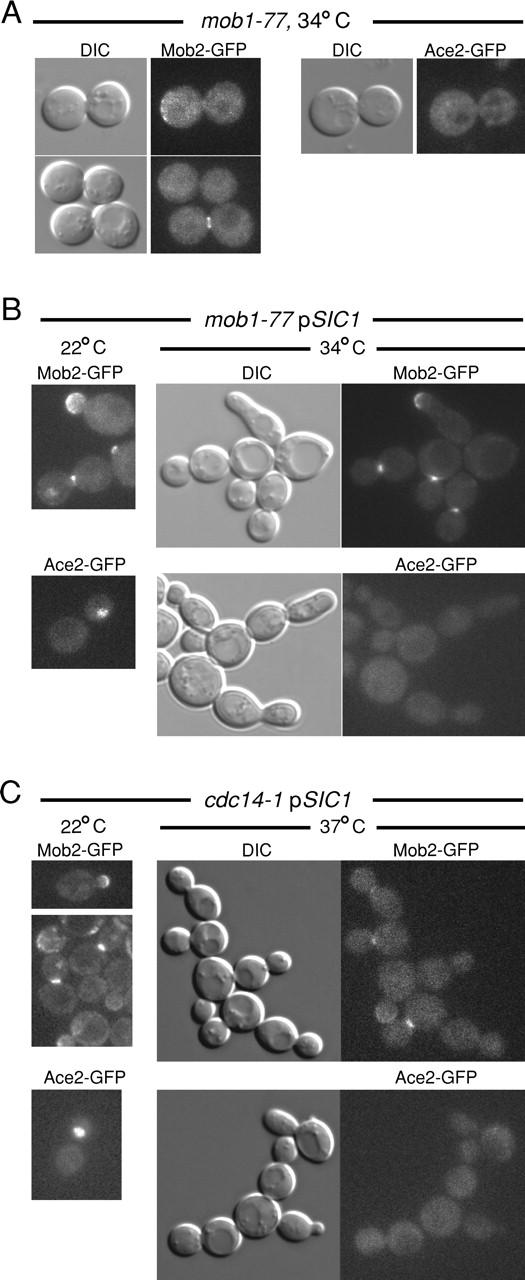
Localization of Mob2p and Ace2p is linked to mitotic exit. (A) Localization of Mob2–GFP and Ace2–GFP in mob1–77 cells (FLY981 and FLY978) at 34°C, which arrest at telophase before mitotic exit. Mob2–GFP was not properly localized to bud necks (as described in Results). Neither Ace2–GFP nor Mob2–GFP was evident in nuclei. (B) Localization of Mob2–GFP and Ace2–GFP in mob1–77 pSIC1 cells at 22 and 34°C (FLY 981 + YEp13-SIC1, FLY978 +YEp13-SIC1). Nuclear division and budding occur in the absence of cytokinesis in these cells at 34°C without nuclear localization of Mob2–GFP (top right) and Ace2–GFP (bottom right) but with cortical and bud neck localization of Mob2p. (C) Localization of Mob2–GFP and Ace2–GFP in cdc14–1 pSIC1 cells at 22 and 37°C (FLY 1011 + YEp13-SIC1and FLY1015 + YEp13-SIC1).
The absence of nuclear Mob2p and Ace2p in arrested mob1–77 and cdc14–1 cells could reflect mitotic exit regulation of Mob2p and Ace2p localization. Alternatively, nuclear accumulation of Mob2p and Ace2p might simply occur at a later point in the cell cycle. To distinguish between these possibilities, we analyzed the localization of Mob2p and Ace2p in mob1–77 and cdc14–1 cells expressing the CDK inhibitor Sic1p from a multicopy plasmid (YEp13-SIC1). This moderate Sic1p overexpression partially bypasses the mitotic exit defect of several MEN mutants, including mob1–77, allowing progression from M to G1 without cytokinesis (Luca et al., 2001). At 22°C, Mob2–GFP and Ace2–GFP localization in mob1–77 YEp13-SIC1 cells were normal (Fig. 9 B). At 34°C, mob1–77 YEp13-SIC1 cells did not arrest in telophase and accumulated as chains of connected cells as described previously. In such cells, Mob2–GFP localized robustly to the bud necks and sites of polarized growth at restrictive temperature but never appeared in nuclei (Fig. 9 B). Similarly, Ace2–GFP also failed to localize to the nuclei of mob1–77 YEp13-SIC1 cells at 34°C (Fig. 9 B). We obtained similar results in cdc14–1 YEp13-SIC1 cells (Fig. 9 C). Therefore, MEN function is essential for nuclear accumulation of Mob2p and Ace2p. In contrast, Mob2p localization to the cell cortex and bud neck is not dependent on MEN activity.
Discussion
Our results indicate that Mob2p and the protein kinase Cbk1p function as a complex that regulates two important and distinct cellular processes: (1) daughter cell–specific subcellular distribution and activation of Ace2p transcription factor, which is necessary for mother–daughter cell separation and (2) maintenance of polarized growth (Fig. 10). In this study, we have demonstrated that Mob2p physically associates with Cbk1p throughout the cell cycle and is essential for both kinase activity and hyperphosphorylation of Cbk1p. We also found that the specific activity of Cbk1p fluctuates as cells bud and divide, suggesting an additional level of regulation. Consistent with the involvement of the physically associated proteins in cell morphogenesis, Cbk1p and Mob2p both localize to actively expanding cell cortex during bud emergence and mating projection formation and later move to the bud neck and daughter cell nucleus during mitotic exit. The latter is in agreement with the findings of a parallel study (Colman-Lerner et al., 2001). Our work further demonstrates that the nuclear localization of Mob2–Cbk1p and Ace2p is achieved by a novel mechanism that requires a functional mitotic exit network and regulated nuclear export. These findings implicate Mob1p/Mob2p-like proteins and their kinase partners in controlling cell morphogenesis, cell cycle, and cell fate.
Figure 10.

Model for Mob2p–Cbk1p function. See Discussion for description.
The role of Mob2p–Cbk1p in cell separation
Budding yeast cytokinesis occurs as cells exit mitosis with actomyosin ring contraction and septum formation accomplishing fission of mother and daughter cell cytoplasms (Bi et al., 1998). After cytokinesis is complete, the septum is destroyed, allowing separation of mother and daughter cells. This event coincides with Ace2p-dependent expression of several genes, including CTS1 and SCW11, which encode proteins required for septum degradation (Spellman et al., 1998; Doolin et al., 2001). Late M/early G1 transcription of these Ace2p-regulated genes is clearly compromised in cells lacking Cbk1p (Bidlingmaier et al., 2001). We found that abrogation of Cbk1p kinase activity, whether caused by cbk1 loss-of-function alleles or absence of Mob2p, has a similar effect. Therefore, we suggest that Ace2p's ability to promote transcription is activated by Cbk1p phosphorylation. The observed increase in Cbk1p activity late in cell division is consistent with this interpretation.
Our results address an interesting additional layer of Mob2p–Cbk1p–dependent regulation of Ace2p. In wild-type cells, Ace2p accumulates specifically in daughter cell nuclei during late mitosis. This localization pattern was also observed recently by others and found to drive expression of Ace2p-dependent genes specifically in the daughter cell (Colman-Lerner et al., 2001). Also in agreement with recently reported results, we found that deletion of either MOB2 or CBK1 permits distribution of Ace2p to both mother and daughter cell nuclei (Colman-Lerner et al., 2001). Interestingly, however, we found that markedly lower amounts of Ace2p accumulate in nuclei when Cbk1p kinase activity is inhibited. This is not due to a simple two-fold dilution of available Ace2p: the transcription factor accumulates to relatively high levels in both nuclei when its nuclear export is blocked. Therefore, the low levels of Ace2p present in nuclei of cells lacking Cbk1p function likely reflect a steady-state level that is reached in the absence of a signal that causes Ace2p nuclear retention.
In agreement with Colman-Lerner et al. (2001), we found that Mob2p and Cbk1p also accumulate in daughter cell nuclei late in cell division and that this localization is ACE2 dependent. Ace2p carries putative nuclear localization sequences (Jensen et al., 2000), whereas Mob2p and Cbk1p do not. Therefore, it is possible that the three proteins form a complex that is cotransported into nuclei. In cells that lack Mob2p, Cbk1p localizes to both mother and daughter nuclei. Colman-Lerner et al. (2001) concluded that Mob2p localizes to mother and daughter nuclei in cbk1Δ cells. Contrary to this conclusion, we found that Mob2p clearly does not accumulate in nuclei of cbk1Δ cells. These findings suggest that Cbk1p and Ace2p are no longer restricted to the daughter bud in mob2Δ cells and can associate and accumulate in both nuclei. On the other hand, Mob2p likely associates with Ace2p through mutual interaction with Cbk1p.
The role of nuclear transport in Ace2p asymmetry
How might Mob2p–Cbk1p promote accumulation of Mob2p, Ace2p, and Cbk1p in daughter cell nuclei, with concomitant disappearance of the transcription factor from mother cell nuclei? Our results indicate that localization of these proteins is separate from Myo4p–Bni1p–mediated sequestration of ASH1 mRNA to the daughter cell cortex, the only previously described pathway for asymmetric partitioning of cell fate determinants in budding yeast (Bobola et al., 1996; Sil and Herskowitz, 1996). Specific localization of Ace2p to daughter cell nuclei could be achieved through regulation of nuclear entry or exit or through degradation of the protein in the mother cell nucleus. Dephosphorylation of three conserved CDK consensus sites on Ace2p permits the protein's nuclear import at the end of mitosis (O'Conallain et al., 1999). Ace2p nuclear export requires Xpo1p, a Crm1-related exportin family protein (Jensen et al., 2000). We found that Ace2p accumulates abundantly in both mother and daughter nuclei when its nuclear export is blocked. Therefore, asymmetric Ace2p accumulation is most likely attributable to suppression of its nuclear export in the daughter cell, rather than differential nuclear import or stability. We propose that Mob2p and Cbk1p mediate suppression of Ace2p nuclear export. Furthermore, we suggest that sequestration of active Mob2p–Cbk1p to the growing daughter cell is ultimately responsible for biased accumulation of Ace2p in daughter nuclei.
Mob2p–Cbk1p may suppress Ace2p export from daughter cell nuclei by various mechanisms. It is unlikely that Mob2p–Cbk1p kinase inhibits Ace2p nuclear export by direct inhibition of Xpo1p, since Xpo1p is an essential protein required for a wide range of nuclear export (Stade et al., 1997). Instead, we favor the idea that Mob2p–Cbk1p–mediated phosphorylation of Ace2p inhibits Ace2p nuclear export, perhaps by making Ace2p unable to interact with nuclear export machinery. Alternately, Mob2p–Cbk1p–mediated phosphorylation might promote anchoring of Ace2p to fixed objects within the nucleus. We found that Ace2p accumulation in nuclei is much lower when Cbk1p kinase activity is inhibited, suggesting that mere association of Mob2–Cbk1p with Ace2p does not block Ace2p nuclear export. Ace2p localization is analogous to that of the transcription factor prospero, which determines ganglion mother cell fate during Drosophila nervous system development (Spana and Doe, 1995). Interestingly, prospero contains a domain that antagonizes the protein's nuclear export in cis, possibly through nuclear export sequence masking (Demidenko et al., 2001). Evaluation of Cbk1p phosphorylation of Ace2p will be an important step toward determining if Ace2p nuclear traffic is controlled in an analogous manner.
Mitotic exit network coordination of Mob2p and Ace2p localization
Septum destruction occurs in earnest only after completion of cytokinesis, reflecting an important temporal coordination of events that occur as cells pass from mitosis into G1. The final events of cell division are controlled by the MEN, a conserved signaling pathway that links initiation of cytokinesis with the completion of nuclear division (McCollum and Gould, 2001). We have shown that Mob2p and Ace2p do not localize to the nucleus when MEN signaling is blocked by mob1–77 or cdc14–1 conditional loss-of-function alleles. More significantly, a partial bypass of mitotic exit block that allows progression into G1 without cytokinesis does not restore nuclear localization of these proteins. Therefore, MEN signaling may directly control Mob2p–Cbk1p's ability to promote nuclear localization and activity of Ace2p. Alternatively, since MEN signaling is required for initiation of cytokinesis, Mob2p–Cbk1p activation may be linked to the actual progress of actomyosin ring contraction and septum deposition. Importantly, cortical and bud neck localization of Mob2p are not strictly dependent on MEN signaling but rather are promoted by progression into subsequent cell cycle stages. These findings further support distinct and separable roles for Mob2p–Cbk1p complex in cortical remodeling and transcriptional control.
The role of Mob2p–Cbk1p in cell surface remodeling
Mob2p–Cbk1p has an important Ace2p-independent role in cell cortex remodeling and polarized growth. Cells lacking Mob2p–Cbk1p function do not sustain polarized growth during budding or mating projection formation, although polarity establishment is not abrogated. The presence of Mob2p and Cbk1p at sites of active cell growth, such as the tips of buds and mating projections, suggest that the proteins perform some function critical for ongoing cortical expansion at these sites. Bud neck localization of Mob2p–Cbk1p late in cell division is consistent with a function in cortical remodeling; this is a site of active cell wall reorganization as cells undergo cytokinesis and cell separation. In contrast to conclusions drawn elsewhere (Colman-Lerner et al., 2001), we found that Mob2p and Cbk1p are clearly interdependent for proper localization to cortical sites. Interestingly, although Cbk1p's kinase activity is required both for maintenance of polarized growth and septum destruction, suppression of Cbk1p kinase activity does not disrupt Mob2p localization to the bud cortex and bud neck. Therefore, recruitment of Mob2p–Cbk1p to sites of cell wall remodeling and polarized growth is likely independent of Cbk1p kinase activity.
The relevant targets of Mob2p–Cbk1p at the cell cortex are not known presently. However, Mob2p–Cbk1p localizes similarly to other components of cell growth machinery, such as the exocyst complex, which directs the targeting of secretory vesicles to the cell cortex (TerBush et al., 1996; Finger and Novick, 1998), and the β-1,3-glucan synthase complex, which is important in cell wall construction (Qadota et al., 1996). These complexes are important for polarized cell morphogenesis and are therefore candidates for regulation by Mob2p–Cbk1p.
Our results indicate that Cbk1p itself may be regulated by phosphorylation and that this control requires Mob2p. Interestingly, the Mob2p-related protein Mob1p is required for regulatory phosphorylation of the Cbk1p-related kinase Dbf2p, most likely by the kinase Cdc15p (Mah et al., 2001). It is not presently clear how Mob2p-dependent phosphorylation of Cbk1p influences the kinase's function. It is plausible that Mob2p–Cbk1p acts within a regulatory pathway organized similarly to the MEN that coordinates events at the cell cortex with cell cycle progression and transcriptional control. Given the conservation of Mob1p-like proteins and Dbf2/Cbk1p kinases, our findings may anticipate a role for related proteins in linking the regulation of cell morphology and cell cycle transitions with cell fate determination and development.
Materials and methods
Strains, plasmids, and growth conditions
Yeast strains (Table I) were derived from S288C and cultured as described previously (Guthrie and Fink, 1991). COOH-terminal GFP, YFP, CFP, 13myc, and HA tags were introduced by homologous recombination using PCR products (oligonucleotides shown on Table II) generated from pFA6a-13xMyc-kanMX6, pFA6a-3xHA-HIS5MX6, pFA6a–GFP (S65T)-kanMX6, pDH5 (YFP), and pDH3 (CFP) (Longtine et al., 1998). Using a pRS316-CBK1 plasmid (a gift from S. Bidlingmaier, Yale University, New Haven, CT), cbk1 D475A and cbk1-as2 alleles were generated by oligonucleotide-directed mutagenesis (QuickChange kit, Stratagene) (Table II).
Table I. List of strains.
| Strain name | Relevant genotype |
|---|---|
| FLY93 | MATα ura3-52 leu2-3,112 trp1Δ1 his3Δ200 (S288C genetic background) |
| FLY95 | MATα ura3-52 leu2-3,112 trp1Δ1 his3Δ200 (S288C genetic background) |
| FLY168 | MATα mob2Δ::HIS3 |
| FLY757 | MATα cbk1Δ::kanMX |
| FLY782 | MATα cbk1Δ::kanMX mob2Δ::HIS3 |
| FLY766 | MATα MOB2-GFP::HIS3 cbk1Δ::kanMX |
| FLY770 | MATα MYO1-GFP::kanMX mob2Δ::HIS3 |
| FLY774 | MATα CBK1-GFP::kanMX mob2Δ::HIS3 |
| FLY811 | MATα ACE2-GFP::kanMX |
| FLY823 | MATα MOB2-GFP::HIS3 bni1Δ::kanMX |
| FLY827 | MATα MOB2-GFP::HIS3 myo4Δ::kanMX |
| FLY841 | MATα ASH1-GFP::kanMX mob2Δ::HIS3 |
| FLY850 | MATα ACE2-GFP::kanMX mob2Δ::HIS3 |
| FLY856 | MATα ACE2-GFP::kanMX cbk1Δ::HIS3 |
| FLY880 | MATα ace2Δ::kanMX |
| FLY893 | MATα/MATα MOB2-GFP::kanMX/ MOB2-GFP::HIS3 |
| FLY895 | MATα/MATα CBK1-GFP::kanMX/CBK1-GFP::kanMX |
| FLY896 | MATα/MATα CBK1-CFP::kanMX/CBK1-CFP::kanMX HHF2-YFP::HIS3/HHF2-YFP::HIS3 |
| FLY906 | MATα CBK1-6xMyc::kanMX mob2Δ::HIS3 |
| FLY924 | MATα/MATα MOB2-CFP::kanMX/MOB2-CFP::kanMX HHF2-YFP::HIS3/HHF2-YFP::HIS3 |
| FLY934 | MATα MOB2-GFP::HIS3 ace2Δ::kanMX |
| FLY954 | MATα CBK1-3xHA::HIS3 MOB2-6xMyc::kanMX |
| FLY960 | MATα CBK1-6xMyc::kanMX MOB2-3xHA::HIS3 |
| FLY970 | MATα CBK1-GFP::kanMX ace2Δ::kanMX |
| FLY978 | MATα ACE2-GFP::kanMX mob1-7 |
| FLY981 | MATα MOB2-GFP:kanMX mob1-77 |
| FLY985* | MATα ACE2-GFP::kanMX xpo1Δ::LEU2 [pKW440-XPO1-HIS3] |
| FLY986* | MATα ACE2-GFP::kanMX xpo1Δ::LEU2 [pKW457-xpo1-1-HIS3] |
| FLY1005 | MATα MOB2-3xHA::HIS3 cbk1Δ::kanMX |
| FLY1007 | MATα cbk1Δ::kanMX [pRS316-cbk1D475A] |
| FLY1008 | MATα cbk1Δ::kanMX [pRS316-cbk1-as2] |
| FLY1009 | MATα ACE2-GFP::kanMX cbk1Δ::HIS3 [pRS316-cbk1-as2] |
| FLY1010 | MATα MOB2-GFP::kanMX cbk1Δ::HIS3 [pRS316-cbk1-as2] |
| FLY1011 | MATα MOB2-GFP::HIS3 cdc14-1 |
| FLY1015 | MATα ACE2-GFP::HIS3 cdc14-1 |
| KWY120 | MATα xpo1Δ::LEU2 ade2-1 ura3-1 his3-11,15 trp1-1 leu2-3,112 can1-100 [pKW440-XPO1-HIS3] |
| KWY121 | MATα xpo1Δ::URA3 ade2-1 ura3-1 his3-11,15 trp1-1 leu2-3,112 can1-100 [pKW457-xpo1-1-HIS3] |
All strains are derived from S288C isolates FLY93 and FLY95, except those noted by *, which are derived from KWY120 and KWY121 (provided by K. Weis, University of California, Berkeley). Plasmids are bracketed.
Table II. List of oligonucleotides.
| MOB2 tagging and deletion | |
|---|---|
| MOB2-F2 | CCGCTGCTACCGTTAATTGAAAGCTTTGAAAAACAGGGCAAAATTATTTATAATGGTGGTCCCGGTG GTCGGATCCCCGGGTTAATTAA |
| MOB2-R1 | GTGTCTAAAATAGGAGTGACCCATGGTAACTAATAATAATAATTGTGTTCACCTGAATTCGAGCTCGT TTAAAC |
| Deletion | See Luca and Winey, 1998 |
| CBK1 tagging and deletion | |
| CBK1-F1 | GGTACACGTCTGCGCTGATCCTTCTGTAATCCTCTGCAGTTTTCGCGGATCCCCGGGTTAATTAA |
| CBK1-F2 | GGCTACACTTACTCCAGATTTGACTATTTGACAAGAAAAAATGCGTTGGGTGGTCCCGGTGGTCGGA TCCCCGGGTTAATTAA |
| CBK1-R1 | CCATAGATAAATACTTGAATAAAGAGGAATGTCCTTAACGCGTCCGAATTCGAGCTCGTTTAAAC |
| CBK1-F | CCAGAGAAGCAGTTGCACTCC |
| CBK1 site-directed mutagenesis | |
| D475A-F | CATAAATTAGGATTCATTCACAGGGCCATTAAACCAGATAATATTTTG |
| D475A-R | CAAAATATTATCTGGTTTAATGGCCCTGTGAATGAATCCTAATTTATG |
| as2-F | GCTCAATACTTATATTTAATCGCCGAATTCTTGCCCGGTGGTGATTTGATG |
| as2-R | CATCAAATCACCACCGGGCAAGAATTCGGCGATTAAATATAAGTATTGAGC |
| HHF2 tagging | |
| HHF2-F2 | TATGCTTTGAAGAGACAAGGTAGAACCTTATATGGTTTCGGTGGTGGTGGTCCCGGTGGTCGGATC CCCGGGTTAATTAA |
| HHF2-R1 | GGCATGAAAATAATTTCAAACACCGATTGTTTAACCACCGATTGTGAATTCGAGCTCGTTTAAAC |
| ACE2 tagging and deletion | |
| ACE2-F1 | ATAACTAAAGAAATCTATAGGACCAAAAACGGTGTTAATACAATCCGGATCCCCGGGTTAATTAA |
| ACE2-F2 | GAGCAAAACTCGAACCGCACCCTTTCAAACGAAACTGATGCTCTCGGTGGTCCCGGTGGTCGGATC CCCGGGTTAATTAA |
| ACE2-R1 | TTACTATTATTTATTATGTTAATATCATGCATAGATAAATGTTCGGAATTCGAGCTCGTTTAAAC |
| ACE2-F | ACATTGGCACTTTGGGAA |
| ASH1 tagging | |
| ASH1-F2 | TACCGTTGCTTATTTTGTAATTACATAACTGAGACAGTAGAGAATGGTGGTCCCGGTGGTCGGATCC CCGGGTTAATTAA |
| ASH1-R1 | TGATAATGTCTCTTATTAGTTGAAAGAGATTCAGTTATCCATGTAGAATTCGAGCTCGTTTAAAC |
| MYO4 deletion | |
| myo4-F1 | ATTTGAAGTAGGAACTAAGTGTTGGTACCCTCACAAAGAACAAGGCGGATCCCCGGGTTAATTAA |
| myo4-R1 | GTATATATACATATATACATATATGGGCGTATATTTACTTTGTTCGAATTCGAGCTCGTTTAAAC |
| myo4-C | GTTTAGCAGTAGTAGATG |
| BNI1 deletion | |
| bni1-F1 | CCTTTTCAACAAACGAGAAGCAAGAAAGGAAGAGAAGGAAAGGAACGGATCCCCGGGTTAATTAA |
| bni1-R1 | GTTTGTTTTGGTATTACTGTTGTCATAATTTTTTGGTTTAATATTGAATTCGAGCTCGTTTAAAC |
| bni1-C | AAGAACAATAGCACGGAC |
Cell synchronization
S phase arrest was induced by growth in medium containing 0.1 M hydroxyurea (Sigma-Aldrich) until ∼90% of cells had large buds. G1 arrest was induced by growth in medium containing 10 μg/ml α-factor (a gift from D. King, University of California, Berkeley) until ∼80% of cells had visible mating projections. Arrested cells were harvested by filtration and released by resuspension in fresh medium. To enrich for G2/M phase cells after release from S phase arrest, we followed nuclear division by staining a sample of cells from the synchronized culture with DAPI.
Protein and immunological techniques
Yeast lysis buffers, wash buffers, and kinase assay conditions were as described previously (Mah et al., 2001) with the addition of 10% glycerol to lysis and wash buffers. To prepare lysates for immunoprecipitations, ∼20 OD600 equivalents of log phase cells were resuspended in 2.5 ml lysis buffer plus 5 ml glass beads in 15 ml conical tubes and vortexed on ice eight times for 30 s. Lysates were clarified by centrifugation, assayed for protein concentration using the Bradford method (Bradford, 1976), adjusted to 8 mg/ml total protein, and preincubated 30 min with 100 μl protein A beads. 25 μg of either anti-HA (14b11; Covance) or anti-myc (9E10) monoclonal antibody was added to 1 ml lysate and incubated on ice, then added to 100 μl of protein A beads (Amersham Biosciences) and rotated 1 h at 4°C. Washing of beads, preparation for kinase assays, SDS-PAGE, and immunoblotting were performed as described previously (Mah et al., 2001). Immunoblots were probed with appropriate primary antibody and detected by sheep anti–mouse peroxidase-conjugated secondary antibody and ECL (Amersham Biosciences). Nonreducing sample buffer was used for Western blot analysis of Mob2p-myc. Kinase assays were quantified using a PhosphorImager (Molecular Dynamics).
Cytology, microscopy, and image analysis
Staining of F-actin with rhodamine-phalloidin, staining of chitin with calcofluor, and FITC-ConA pulse labeling were performed as described (Tkacz and Lampen, 1972; Pringle, 1991). Fluorescence/DIC microscopy and image acquisition were either performed as described previously (Luca et al., 2001) or using a Nikon Eclipse TE300 fluorescence microscope equipped with a Hamamatsu ORCA CCD camera. Cell wall digestion was performed on formaldehyde-fixed cells by treatment with 0.1 mg/ml zymolyase (ICN Biomedicals) in PBS. In scoring cell separation defects upon inhibitor treatment, cells sonicated for 2 s at low power were allowed to grow in indicated conditions, fixed in 5% formaldehyde, and calcofluor stained. Each cluster of more than two attached cell bodies was counted as cell separation failure.
Acknowledgments
We thank Waldi Racki and Chris Herbert for generously sharing unpublished results, Scott Bidlingmaier and Michael Snyder for pRS316-CBK1 plasmid, and Karsten Weis for xpo1-1 strains and advice. We also thank Erfei Bi, David Nix, and Daria Siekhaus for critical reading of this article, and Keith Kozminski, Michael Eisen, and Jeremy Thorner for helpful discussions.
This work was supported by National Institutes of Health grant RO1 GM60575 to F.C. Luca., National Institutes of Health grant RO1 GM50399 to D.G. Drubin, and National Institutes of Health grant RO1 AI44009 to K. Shokat. E.L. Weiss is supported as a Research Special Fellow of the Leukemia and Lymphoma Society.
E.L. Weiss and C. Kurischko contributed equally to this work.
Footnotes
Abbreviations used in this paper: CDK, cyclin-dependent kinase; MEN, mitotic exit network.
References
- Bi, E., P. Maddox, D.J. Lew, E.D. Salmon, J.N. McMillan, E. Yeh, and J.R. Pringle. 1998. Involvement of an actomyosin contractile ring in Saccharomyces cerevisiae cytokinesis. J. Cell Biol. 142:1301–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bidlingmaier, S., E.L. Weiss, C. Seidel, D.G. Drubin, and M. Snyder. 2001. The Cbk1p pathway is important for polarized cell growth and cell separation in Saccharomyces cerevisiae. Mol. Cell. Biol. 21:2449–2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop, A.C., J.A. Ubersax, D.T. Petsch, D.P. Matheos, N.S. Gray, J. Blethrow, E. Shimizu, J.Z. Tsien, P.G. Schultz, M.D. Rose, et al. 2000. A chemical switch for inhibitor-sensitive alleles of any protein kinase. Nature. 407:395–401. [DOI] [PubMed] [Google Scholar]
- Bobola, N., R.P. Jansen, T.H. Shin, and K. Nasmyth. 1996. Asymmetric accumulation of Ash1p in postanaphase nuclei depends on a myosin and restricts yeast mating-type switching to mother cells. Cell. 84:699–709. [DOI] [PubMed] [Google Scholar]
- Bradford, M.M. 1976. A rapid and sensitive method for the quatitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248–254. [DOI] [PubMed] [Google Scholar]
- Colman-Lerner, A., T.E. Chin, and R. Brent. 2001. Yeast Cbk1 and Mob2 activate daughter-specific genetic programs to induce asymmetric cell fates. Cell. 107:739–750. [DOI] [PubMed] [Google Scholar]
- Demidenko, Z., P. Badenhorst, T. Jones, X. Bi, and M.A. Mortin. 2001. Regulated nuclear export of the homeodomain transcription factor Prospero. Development. 128:1359–1367. [DOI] [PubMed] [Google Scholar]
- Dohrmann, P.R., G. Butler, K. Tamai, S. Dorland, J.R. Greene, D.J. Thiele, and D.J. Stillman. 1992. Parallel pathways of gene regulation: homologous regulators SWI5 and ACE2 differentially control transcription of HO and chitinase. Genes Dev. 6:93–104. [DOI] [PubMed] [Google Scholar]
- Doolin, M.T., A.L. Johnson, L.H. Johnston, and G. Butler. 2001. Overlapping and distinct roles of the duplicated yeast transcription factors Ace2p and Swi5p. Mol. Microbiol. 40:422–432. [DOI] [PubMed] [Google Scholar]
- Finger, F.P., and P. Novick. 1998. Spatial regulation of exocytosis: lessons from yeast. J. Cell Biol. 142:609–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guthrie, C., and G.R. Fink. 1991. Guide to yeast genetics and molecular biology. In Methods Enzymol. Vol. 194. 1–933. [PubMed]
- Hanks, S.K., and T. Hunter. 1995. Protein kinases 6. The eukaryotic protein kinase superfamily: kinase (catalytic) domain structure and classification. FASEB J. 9:576–596. [PubMed] [Google Scholar]
- Jaspersen, S.L., J.F. Charles, and D.O. Morgan. 1999. Inhibitory phosphorylation of the APC regulator Hct1 is controlled by the kinase Cdc28 and the phosphatase Cdc14. Curr. Biol. 9:227–236. [DOI] [PubMed] [Google Scholar]
- Jensen, T.H., M. Neville, J.C. Rain, T. McCarthy, P. Legrain, and M. Rosbash. 2000. Identification of novel Saccharomyces cerevisiae proteins with nuclear export activity: cell cycle-regulated transcription factor ace2p shows cell cycle-independent nucleocytoplasmic shuttling. Mol. Cell. Biol. 20:8047–8058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komarnitsky, S.I., Y.C. Chiang, F.C. Luca, J. Chen, J.H. Toyn, M. Winey, L.H. Johnston, and C.L. Denis. 1998. DBF2 protein kinase binds to and acts through the cell cycle-regulated MOB1 protein. Mol. Cell. Biol. 18:2100–2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, S.E., L.M. Frenz, N.J. Wells, A.L. Johnson, and L.H. Johnston. 2001. Order of function of the budding-yeast mitotic exit-network proteins Tem1, Cdc15, Mob1, Dbf2, and Cdc5. Curr. Biol. 11:784–788. [DOI] [PubMed] [Google Scholar]
- Lippincott, J., K.B. Shannon, W. Shou, R.J. Deshaies, and R. Li. 2001. The Tem1 small GTPase controls actomyosin and septin dynamics during cytokinesis. J. Cell Sci. 114:1379–1386. [DOI] [PubMed] [Google Scholar]
- Longtine, M.S., A. McKenzie, III, D.J. Demarini, N.G. Shah, A. Wach, A. Brachat, P. Philippsen, and J.R. Pringle. 1998. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast. 14:953–961. [DOI] [PubMed] [Google Scholar]
- Luca, F.C., and M. Winey. 1998. MOB1, an essential yeast gene required for completion of mitosis and maintenance of ploidy. Mol. Biol. Cell. 9:29–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luca, F.C., M. Mody, C. Kurischko, D.M. Roof, T.H. Giddings, and M. Winey. 2001. Saccharomyces cerevisiae Mob1p is required for cytokinesis and mitotic exit. Mol. Cell. Biol. 21:6972–6983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mah, A.S., J. Jang, and R.J. Deshaies. 2001. Protein kinase Cdc15 activates the Dbf2-Mob1 kinase complex. Proc. Natl. Acad. Sci. USA. 98:7325–7330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCollum, D., and K.L. Gould. 2001. Timing is everything: regulation of mitotic exit and cytokinesis by the MEN and SIN. Trends Cell Biol. 11:89–95. [DOI] [PubMed] [Google Scholar]
- Moll, T., G. Tebb, U. Surana, H. Robitsch, and K. Nasmyth. 1991. The role of phosphorylation and the CDC28 protein kinase in cell cycle-regulated nuclear import of the S. cerevisiae transcription factor SWI5. Cell. 66:743–758. [DOI] [PubMed] [Google Scholar]
- O'Conallain, C., M.T. Doolin, C. Taggart, F. Thornton, and G. Butler. 1999. Regulated nuclear localisation of the yeast transcription factor Ace2p controls expression of chitinase (CTS1) in Saccharomyces cerevisiae. Mol. Gen. Genet. 262:275–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pringle, J.R. 1991. Staining of bud scars and other cell wall chitin with calcofluor. Methods Enzymol. 194:732–735. [DOI] [PubMed] [Google Scholar]
- Pruyne, D., and A. Bretscher. 2000. Polarization of cell growth in yeast. I. Establishment and maintenance of polarity states. J. Cell Sci. 113(Pt 3):365–375. [DOI] [PubMed]
- Qadota, H., C.P. Python, S.B. Inoue, M. Arisawa, Y. Anraku, Y. Zheng, T. Watanabe, D.E. Levin, and Y. Ohya. 1996. Identification of yeast Rho1p GTPase as a regulatory subunit of 1,3-beta-glucan synthase. Science. 272:279–281. [DOI] [PubMed] [Google Scholar]
- Racki, W.J., A.M. Becam, F. Nasr, and C.J. Herbert. 2000. Cbk1p, a protein similar to the human myotonic dystrophy kinase, is essential for normal morphogenesis in Saccharomyces cerevisiae. EMBO J. 19:4524–4532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappaport, R. 1996. Cytokinesis in Animal Cells. Cambridge University Press, Cambridge, U.K. 386 pp.
- Shou, W., J.H. Seol, A. Shevchenko, C. Baskerville, D. Moazed, Z.W. Chen, J. Jang, H. Charbonneau, and R.J. Deshaies. 1999. Exit from mitosis is triggered by Tem1-dependent release of the protein phosphatase Cdc14 from nucleolar RENT complex. Cell. 97:233–244. [DOI] [PubMed] [Google Scholar]
- Sil, A., and I. Herskowitz. 1996. Identification of asymmetrically localized determinant, Ash1p, required for lineage-specific transcription of the yeast HO gene. Cell. 84:711–722. [DOI] [PubMed] [Google Scholar]
- Spana, E.P., and C.Q. Doe. 1995. The prospero transcription factor is asymmetrically localized to the cell cortex during neuroblast mitosis in Drosophila. Development. 121:3187–3195. [DOI] [PubMed] [Google Scholar]
- Spellman, P.T., G. Sherlock, M.Q. Zhang, V.R. Iyer, K. Anders, M.B. Eisen, P.O. Brown, D. Botstein, and B. Futcher. 1998. Comprehensive identification of cell cycle-regulated genes of the yeast Saccharomyces cerevisiae by microarray hybridization. Mol. Biol. Cell. 9:3273–3297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stade, K., C.S. Ford, C. Guthrie, and K. Weis. 1997. Exportin 1 (Crm1p) is an essential nuclear export factor. Cell. 90:1041–1050. [DOI] [PubMed] [Google Scholar]
- Stegmeier, F., R. Visintin, and A. Amon. 2002. Separase, polo kinase, the kinetochore protein Slk19, and Spo12 function in a network that controls Cdc14 localization during early anaphase. Cell. 108:207–220. [DOI] [PubMed] [Google Scholar]
- TerBush, D.R., T. Maurice, D. Roth, and P. Novick. 1996. The Exocyst is a multiprotein complex required for exocytosis in Saccharomyces cerevisiae. EMBO J. 15:6483–6494. [PMC free article] [PubMed] [Google Scholar]
- Tkacz, J.S., and J.O. Lampen. 1972. Wall replication in Saccharomyces species: use of fluorescein-conjugated concanavalin A to reveal the site of mannan insertion. J. Gen. Microbiol. 72:243–247. [DOI] [PubMed] [Google Scholar]
- Trueheart, J., J.D. Boeke, and G.R. Fink. 1987. Two genes required for cell fusion during yeast conjugation: evidence for a pheromone-induced surface protein. Mol. Cell. Biol. 7:2316–2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visintin, R., K. Craig, E.S. Hwang, S. Prinz, M. Tyers, and A. Amon. 1998. The phosphatase Cdc14 triggers mitotic exit by reversal of Cdk-dependent phosphorylation. Mol. Cell. 2:709–718. [DOI] [PubMed] [Google Scholar]
- Weiss, E.L., A.C. Bishop, K.M. Shokat, and D.G. Drubin. 2000. Chemical genetic analysis of the budding-yeast p21-activated kinase Cla4p. Nat. Cell Biol. 2:677–685. [DOI] [PubMed] [Google Scholar]