

Abstract
N-cadherin, a member of the Ca2+-dependent cell–cell adhesion molecule family, plays an essential role in skeletal muscle cell differentiation. We show that inhibition of N-cadherin–dependent adhesion impairs the upregulation of the two cyclin-dependent kinase inhibitors p21 and p27, the expression of the muscle-specific genes myogenin and troponin T, and C2C12 myoblast fusion. To determine the nature of N-cadherin–mediated signals involved in myogenesis, we investigated whether N-cadherin–dependent adhesion regulates the activity of Rac1, Cdc42Hs, and RhoA. N-cadherin–dependent adhesion decreases Rac1 and Cdc42Hs activity, and as a consequence, c-jun NH2-terminal kinase (JNK) MAPK activity but not that of the p38 MAPK pathway. On the other hand, N-cadherin–mediated adhesion increases RhoA activity and activates three skeletal muscle-specific promoters. Furthermore, RhoA activity is required for β-catenin accumulation at cell–cell contact sites. We propose that cell–cell contacts formed via N-cadherin trigger signaling events that promote the commitment to myogenesis through the positive regulation of RhoA and negative regulation of Rac1, Cdc42Hs, and JNK activities.
Keywords: N-cadherin; Rho GTPases; JNK; β-catenin, myogenesis; PL, polylysine
Introduction
Skeletal myogenesis is a multistep process that is temporally and spatially regulated in the embryo. In addition to diffusible molecules, such as the growth and differentiation factors IGF, FGF, Wnt, and sonic hedgehog, cell–extracellular matrix and cell–cell adhesion are also involved in the regulation of myogenesis (Cossu et al., 1995; Buckingham, 2001). Cadherins are transmembrane glycoproteins that promote calcium-dependent homophilic cell–cell adhesion. Cadherin cytoplasmic domains provide F-actin cytoskeleton attachment points through association of catenins and other cytoskeletal-associated proteins (Kemler, 1993). Developing skeletal muscle expresses N-, M-, R-, and 11-cadherins (Knudsen, 1990). Cadherin-dependent adhesion is required for a variety of cellular events during skeletal myogenesis. M-cadherin has been implicated in terminal myoblast differentiation and particularly in myoblast fusion (Zeschnigk et al., 1995). In early embryogenesis, N-cadherin is expressed in somites, myotomes, and embryonic muscles (Duband et al., 1987; Hatta et al., 1987; Linask et al., 1998). Several studies suggest that N-cadherin–mediated cell–cell adhesion plays an important role in skeletal myogenesis. Antibodies that perturb the function of N-cadherin inhibit myotube formation (Knudsen et al., 1990; Mege et al., 1992) and inhibit myosin accumulation and decrease MyoD expression in primitive streak stage epiblast cells (George-Weinstein et al., 1997). Expression of N-cadherin in baby hamster kidney cells stimulates expression of MyoD and sarcomeric myosin (Redfield et al., 1997). Furthermore, N-cadherin–coated beads induce myogenesis in cultured myoblasts, suggesting a role of N-cadherin–mediated adhesion in myogenic induction (Goichberg and Geiger, 1998). Whereas cadherins have been long considered to principally have a structural role providing a link for the F-actin cytoskeleton, these data show that cadherin-dependent adhesion activates intracellular signaling pathways, still unidentified, which promote myogenic induction. In skeletal muscle cells, the Rho GTPases RhoA, Rac1, and Cdc42Hs differentially regulate the commitment to myogenesis. RhoA positively regulates MyoD expression and skeletal muscle cell differentiation (Carnac et al., 1998; Takano et al., 1998; Wei et al., 1998), whereas Rac1 and Cdc42Hs play a dual role in myogenesis. On one hand, inhibition of Rac1 and Cdc42Hs, and a corresponding decrease in p38 MAPK activity, interferes with myogenesis (Meriane et al., 2000). On the other hand, expression of constitutively active forms of Rac1 and Cdc42Hs inhibits myogenesis through activation of the c-jun NH2-terminal kinase (JNK)* cascade (Gallo et al., 1999; Meriane et al., 2000). These data suggest that myogenesis induction involves precise regulation of Rho GTPases, JNK, and p38 MAPK activity.
In the present study, we have investigated the influence of N-cadherin–dependent cell–cell contacts on mouse C2C12 myoblasts myogenesis and on Rho GTPases and MAPK activity. Using antibodies that specifically recognize the extracellular domain of N-cadherin or N-cadherin ligand, allowing us to mimic N-cadherin–mediated adhesion, we demonstrate that N-cadherin–dependent cell–cell adhesion negatively regulates the activity of Rac1, Cdc42Hs, and JNK but increases RhoA activity and induces three skeletal muscle-specific promoters. We also observed RhoA-dependent β-catenin accumulation at the cell–cell contact sites. Together, these results suggest a crucial role for N-cadherin–dependent cell–cell contacts in the regulation of RhoA, Rac1, Cdc42Hs, and JNK activity during myogenesis.
Results
Inhibition of N-cadherin–dependent cell–cell contacts impairs skeletal muscle differentiation
To be able to specifically interfere with N-cadherin–dependent cell–cell contacts, we raised three antisera against the extracellular domain of the N-cadherin molecule. All of these antisera were extensively characterized and shown to specifically recognize N-cadherin in mouse C2C12 myoblasts (unpublished data). To determine whether the blockade of cell to cell adhesion mediated by N-cadherin affects the engagement in skeletal muscle differentiation of mouse C2C12 myoblasts, we then treated C2C12 myoblasts with either preimmune serum or with these anti–N-cadherin antibodies and followed myoblast differentiation by time-lapse imaging and analysis of the expression of the muscle-specific proteins myogenin, troponin T, and that of the cyclin-dependent kinase inhibitors p21 and p27, proteins normally upregulated during myogenesis (Lassar et al., 1994; Walsh, 1997). Table I summarizes the data obtained, showing that addition of anti–N-cadherin antibodies strongly affected the expression of all four proteins and myotube formation.
Table I. Quantitative analysis of the cyclin-dependent kinase inhibitors p21 and p27 and muscle-specific proteins expression and myotubes formation in control and anti–N-cadherin antibodies-treated cells.
| % of p21 expression | % of p27 expression | % of myogenin | % of troponin T | % of myotubes | |||||
|---|---|---|---|---|---|---|---|---|---|
| D4 | D4 | D3 | D5 | D3 | D5 | D3 | D5 | D7 | |
| Control cells (preimmune serum) | 100 | 100 | 68 | 89 | 52 | 85 | 21 | 53 | 61 |
| Anti–N-cadherin antibody (#1) | ND | ND | 1 | 3 | 2 | 5 | 0 | 2 | 6 |
| Anti–N-cadherin antibody (#2) | 20 | 25 | 2 | 3 | 1 | 2 | 0 | 3 | 5 |
| Anti–N-cadherin antibody (#3) | 18 | 21 | 0 | 1 | 0 | 2 | 0 | 0 | 0 |
Results are means of several experiments (n = 5) and are the percentage of muscle-specific proteins expression and myotubes formation. All SEMs were <5%. C2 myoblasts were treated with preimmune serum or anti–N-cadherin antibodies (three differents antisera were used #1, #2, and #3), then placed in differentiation medium, fixed, and analyzed for myogenin and troponin T expression and myotube formation at the indicated times. For p21 and p27 detection, equal amount of protein extract from C2C12 myoblasts treated as described above were analyzed by immunoblotting and quantified as described in Materials and methods. D, time in culture in days.
These data indicate that our antibodies directed against the extracellular domain of the N-cadherin inhibit the early stages of myogenic differentiation and are thus suitable tools to specifically interfere with N-cadherin–dependent cell–cell adhesion.
N-cadherin–dependent cell–cell contact decreases Rac1 and Cdc42Hs activities
Since active Rac1 and Cdc42Hs inhibit myogenesis (Meriane et al., 2000), we examined whether N-cadherin–dependent cell–cell contacts control Rac1 and Cdc42Hs activity. Cadherins require Ca2+ to form homophilic cell–cell adhesions; thus Ca2+ chelation with EGTA, followed by Ca2+ readdition, offers a simple method to study the adhesive properties of these surface molecules (Volberg et al., 1986). Whereas control cells present a typical pattern of N-cadherin and β-catenin immunostaining at the level of cell–cell contacts (Fig. 1, a and f), in EGTA-treated cells, N-cadherin and β-catenin are lost from the cell–cell contact sites (Fig. 1, b and g). Upon restoration of extracellular Ca2+, cell–cell contacts were rapidly reformed as shown by both N-cadherin and β-catenin staining (Fig. 1, c and h). In contrast, the presence of anti–N-cadherin antibodies blocked calcium-restored N-cadherin or β-catenin accumulation at the cell–cell contact sites (Fig. 1, d and i) as did addition of anti–N-cadherin antibodies during contact establishment (Fig. 1, e and j). Higher magnification and deconvolved views of N-cadherin and β-catenin at the contact sites are shown in Fig. 1 (k to y).
Figure 1.
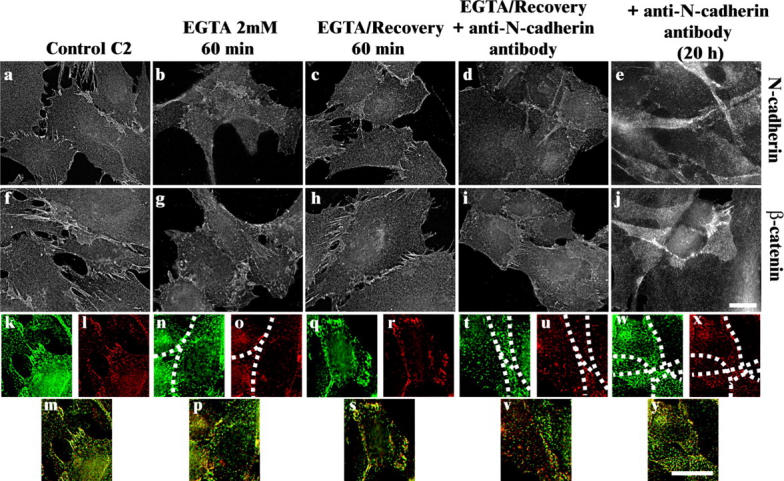
N-cadherin and β-catenin accumulation in cell–cell junctions is dependent on calcium and N-cadherin activity. C2C12 cells were left untreated (a, f, and k–m) or treated with 2 mM EGTA for 60 min (b, g, and n–p). The EGTA-containing medium was then replaced with calcium containing medium alone for 60 min (c, h, q, r, and s) or in the presence of anti–N-cadherin antibodies (d, i, and t–v). Alternatively, anti–N-cadherin antibody was added for 20 h during cell–cell contact establishment (e, j, w, x, and y). Enlarged views of the cell–cell contact sites (k–y). Cells were stained for N-cadherin (a–e, k, n, q, t, and w) or for β-catenin (f–j, l, o, r, u, and x). Dotted lines represent the cell–cell contact region. Bars, 10 μm.
We next examine the effect of N-cadherin engagement on Rac1 and Cdc42Hs activity using pull-down assays. Isolated myoblasts showed a fourfold increase in the level of activated Rac1 and Cdc42Hs relative to cells in contact (Fig. 2, A and B, lanes 1 and 2). 60 min after EGTA addition to contacting cells, the level of Rac1 and Cdc42Hs activity is almost similar to that measured in isolated cells (Fig. 2, A and B, lane 3). Addition of extracellular Ca2+ rapidly diminished Rac1 and Cdc42Hs activity to that measured in contacting cells (Fig. 2, A and B, lane 4), although this diminution was not seen when the anti–N-cadherin antibody was added during recovery (Fig. 2, A and B, lane 5). Moreover, addition of inhibitory anti–N-cadherin antibody during contact establishment clearly impaired the decrease of Rac1 and Cdc42Hs activity observed in contacting cells whereas addition of preimmune serum had no effect (Fig. 2, A and B, lane 6 compared with 7). To rule out the possibility that the calcium switch procedure affects Rac1 and Cdc42Hs activity, we measured their activities after EGTA treatment of isolated cells. No significant modification of the GTP loading of Rac1 and Cdc42H is detectable under these conditions (Fig. 2 C).
Figure 2.
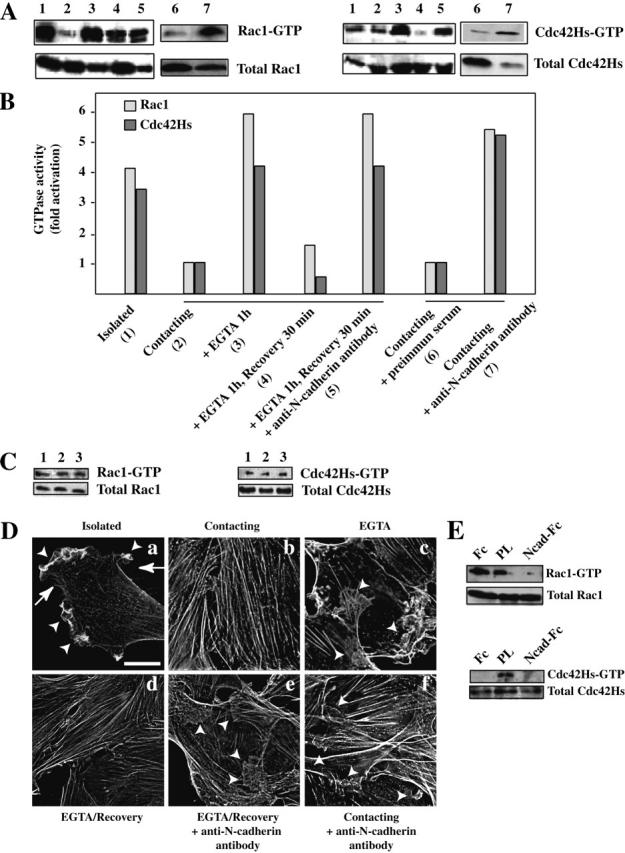
N-cadherin–dependent cell–cell contact formation decreases Rac1 and Cdc42Hs activity. (A) The level of GTP-bound Rac1 and Cdc42Hs was measured using GST–Pak CRIB in lysates obtained from cells treated as shown below the bars in B. Rac1 and Cdc42Hs molecules were detected by immunoblotting. (B) The results in A were analyzed by densitometry as described in Materials and methods. The histogram represents the GTPase activity normalized for the amount of total protein. Data are representative of more than three independent experiments. (C) The level of GTP-bound Rac1 and Cdc42Hs was measured in lysates obtained from isolated C2C12 myoblasts left untreated (1), treated with EGTA (2), and treated with EGTA before Ca2+ restoration (3). Data are representative of three independent experiments. (D) C2C12 myoblasts treated as in A were stained for F-actin distribution with rhodamine-labeled phalloidin. Shown are deconvolved images of F-actin staining. Filopodia (arrows); lamellipodia (arrowheads). Cells shown are representative of >100 observed cells. Bar, 10 μm. (E) The level of GTP-bound Rac1 and Cdc42Hs was measured in lysates obtained 2 h after plating of C2C12 on surface coated with either Fc fragment, PL, or Ncad-Fc. RhoA molecules were detected by Immunoblotting. Data are representative of three independent experiments.
To further demonstrate that cell–cell contact decreased Rac1 and Cdc42Hs activity in C2C12 myoblasts, we used the organization of the F-actin cytoskeleton as functional read-out. Rac1 mediates local actin polymerization, leading to the formation of ruffles and lamellipodia, whereas Cdc42Hs induces the formation of peripheral actin microspikes, filopodia, and dorsal microvilli (Ridley et al., 1992; Meriane et al., 2000). Typical lamellipodia and filopodia were detected in isolated C2C12 myoblasts (Fig. 2 D, a, arrowhead, lamellipodia, and arrow, filopodia). In contrast, all these structures were virtually undetectable in cells in contact (Fig. 2 D, b). Lamellipodia are clearly visible in EGTA-treated and anti–N-cadherin–treated cells even though the cells are in contact (Fig. 2 D, c, e, and f), whereas no such F-actin–rich membrane structures are seen after Ca2+ addition to EGTA-treated cells (Fig. 2 D, d).
To address the question of whether cadherins themselves signal directly to Rac1 and Cdc42Hs, we examined N-cadherin engagement in the absence of cell–cell adhesion. Isolated C2C12 were plated on dishes coated with either Fc fragment (Fc), polylysine (PL), or Ncad-Fc ligand, which allowed us to mimic N-cadherin–mediated adhesion. We verifited that adhesion to Ncad-Fc–coated surfaces is dependent on calcium (unpublished data). 2 h after plating, we observed a marked Rac1 activation on Fc and PL and Cdc42Hs activity on PL (Fig. 2 E). No such Rac1 and Cdc42Hs activity was detected when C2C12 myoblasts were plated on surface coated with Ncad-Fc. These data demonstrate that N-cadherin engagement strongly inhibited Rac1 and Cdc42Hs activity.
N-cadherin–dependent cell–cell contact decreases JNK activity without modification of p38 activity
Activation of the JNK pathway by Rac1 and Cdc42Hs GTPases inhibits myogenesis (Meriane et al., 2000). Therefore we examined whether N-cadherin–dependent cell–cell contact regulates JNK activity (Fig. 3 A). In cells treated with EGTA, JNK activity was similar to that in isolated cells. Addition of anti–N-cadherin antibody blocked the calcium-dependent decrease in JNK activity. To assess whether the control of JNK activity by cell–cell contacts depends on Rac1 and Cdc42Hs activity, we transfected C2C12 myoblasts with constructs expressing the dominant negative T17N mutants of Rac1 and Cdc42Hs (Fig. 3 B). The level of JNK activity in isolated cells is decreased by expression of Rac1N17 or Cdc42HsN17. In addition, Rac1N17 or Cdc42HsN17 expression impaired JNK activation by EGTA treatment of cells in contact.
Figure 3.
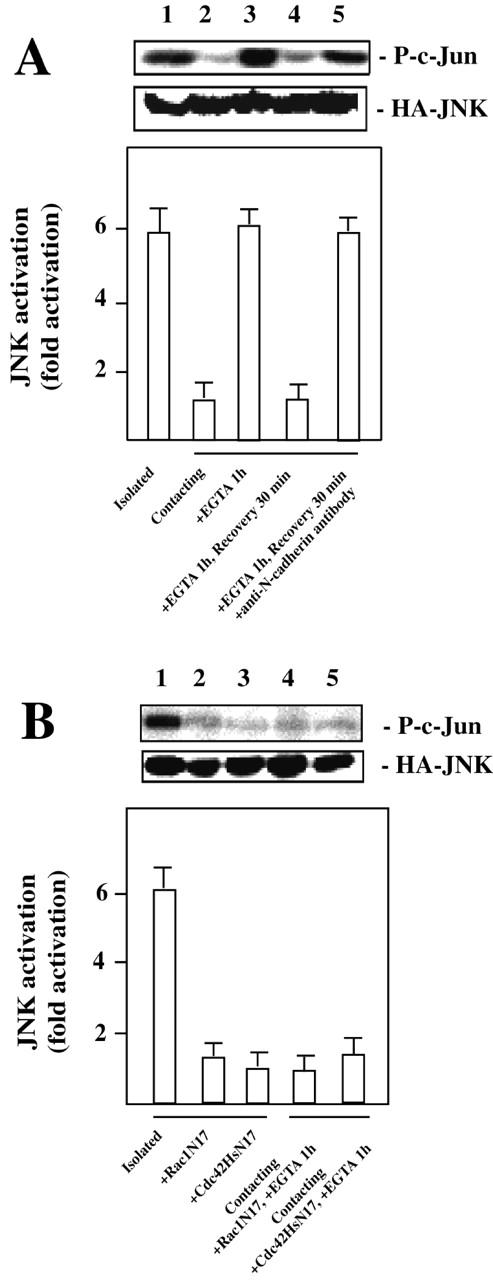
Activation of JNK is inhibited by cell confluency in a N-cadherin– and Rac1-Cdc42Hs–dependent manner. C2C12 myoblasts were transfected with HA-tagged JNK alone (A) or together with GFP-tagged Rac1N17 or Cdc42HsN17 (B). JNK activities in lysates of cells treated as indicated were measured by immunocomplex kinase assays using GST–c-Jun as the substrate. The results are presented as the mean of five independent experiments. Typical data (phosphorylated c-Jun and JNK expression) from one experiment are shown.
Rac1 and Cdc42Hs GTPases also regulate the p38 MAPK pathway (Coso et al., 1995; Minden et al., 1995). We analyzed whether p38 activity was affected by cell–cell contact in C2C12 myoblasts (Fig. 4). Almost no modification in the level of p38 activity was detected whatever the confluency status, whereas addition of differentiation medium led to a threefold increase in p38 activity (Meriane et al., 2000). These data show that JNK activity is negatively controlled by N-cadherin–dependent cell–cell contact in a Rac1- and Cdc42Hs-dependent manner, whereas p38 activity is insensitive to confluency.
Figure 4.
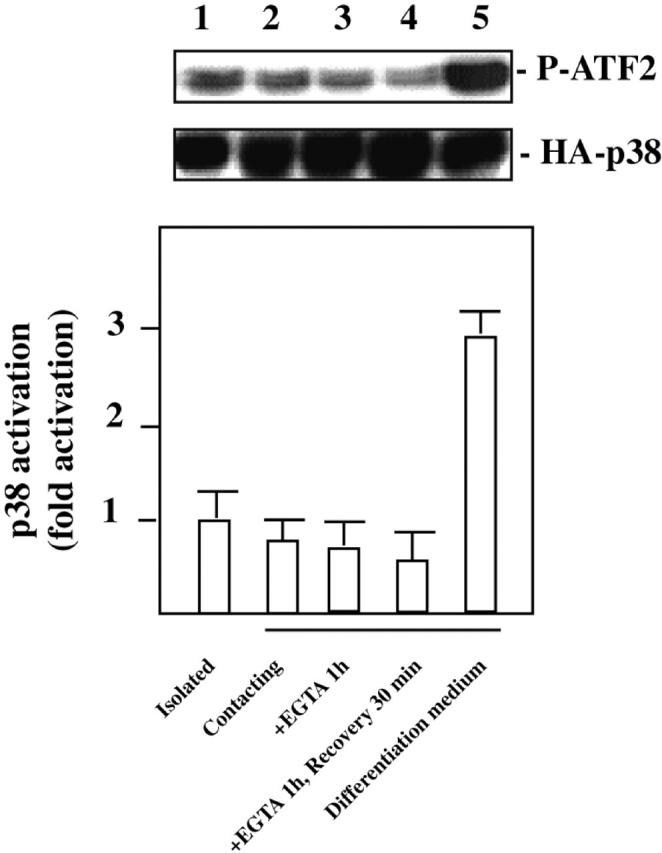
p38 activity is independent of the cell confluency status. C2C12 myoblasts were transfected with HA-tagged p38. p38 activity in lysates of cells treated as indicated was measured by immunocomplex kinase assays using GST–ATF2 as the substrate. The results are presented as mean of five independent experiments. Typical data (phosphorylated ATF2 and p38 expression) from one experiment are shown.
N-cadherin–dependent cell–cell contact activates RhoA in C2C12 myogenic cells
RhoA is an integral part of the skeletal muscle differentiation pathway (Carnac et al., 1998; Wei et al., 1998; Meriane et al., 2000). To find out whether N-cadherin–dependent cell–cell contact activates endogenous RhoA in myogenic C2C12 cells, we measured its activity using pull-down assays. Cell–cell contact induces a sixfold increase in the level of RhoA activity (Fig. 5, A and B, lane 1 compared with 2). Addition of EGTA led to a marked decrease in RhoA activity (Fig. 5, A and B, lane 3), to the level measured in isolated cells. Addition of extracellular Ca2+ restored RhoA activity (Fig. 5, A and B, lane 4), an effect blocked by the addition of anti–N-cadherin antibodies (Fig. 5, A and B, lane 5). Addition of anti–N-cadherin antibody during cell–cell contact establishment led also to a decrease in RhoA activity (Fig. 5, A and B, lane 7), whereas control preimmune serum had no effect (Fig. 5, A and B, lane 6). RhoA activity is not modified by the Ca2+ switch procedure performed on isolated C2C12 myoblasts (Fig. 5 C), ruling out an effect of EGTA on GTP loading of RhoA.
Figure 5.
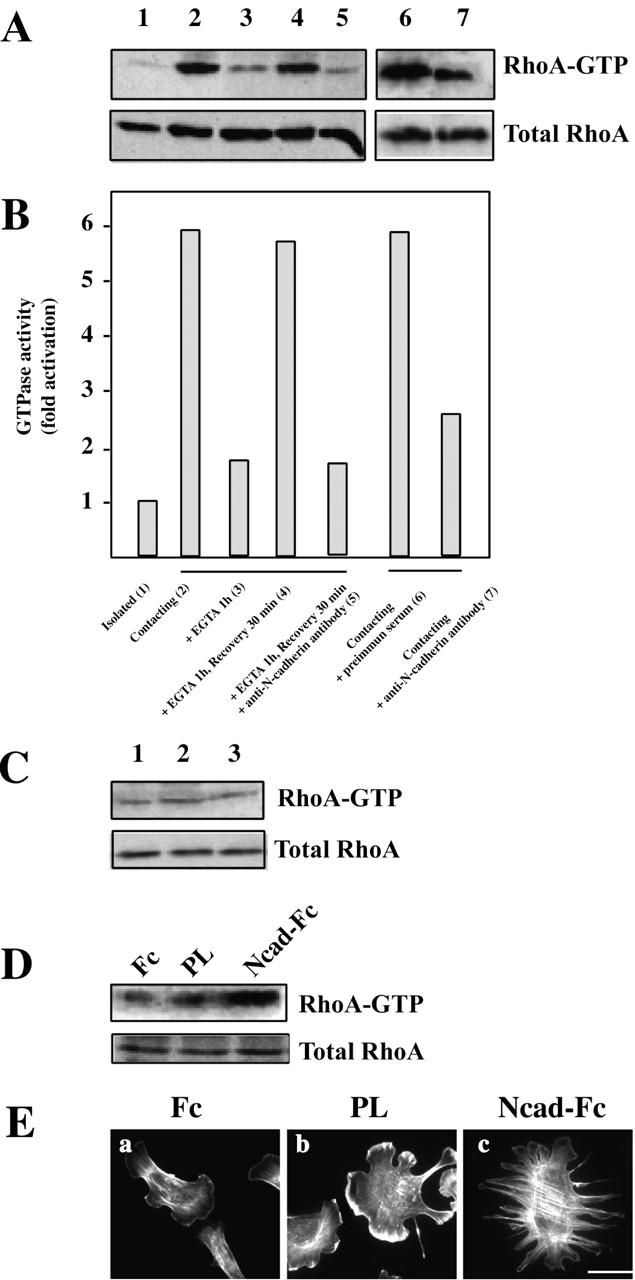
N-cadherin–dependent cell–cell contact formation activates RhoA GTPase. (A) The level of GTP-bound RhoA was measured using GST fused to the Rho-binding domain of the RhoA effector Rhotekin (GST-TRBD) in lysates obtained from cells treated as shown below the bars in B. RhoA molecules were detected by immunoblotting. (B) The results in A were analyzed by densitometry as described in Materials and methods. The histogram represents the GTPase activity normalized for the amount of total protein. Data are representative of more than three independent experiments. (C) The level of GTP-bound RhoA was measured in lysates obtained from isolated C2C12 myoblasts left untreated (1), treated with EGTA (2), and treated with EGTA before Ca2+ restoration (3). Data are representative of three independent experiments. (D) The level of GTP-bound RhoA was measured in lysates obtained 2 h after plating of C2C12 on surface coated with either Fc fragment, PL, or Ncad-Fc. Total and bound RhoA molecules are shown. Data are representative of three independent experiments. (E) C2C12 myoblasts plated onto surface coated with Fc fragment (a), PL (b), or Ncad-Fc (c) were stained for F-actin distribution with rhodamine-labeled phalloidin. Bar, 10 μm.
To determine whether cadherins themselves signal directly to RhoA, isolated C2C12 were plated on dishes coated with an Ncad-Fc ligand (Fig. 5 D). 2 h after plating on Ncad-Fc–coated dishes, a marked RhoA activity was detected in contrast to Fc fragment alone or PL-coated surfaces. This RhoA activation was further confirmed using F-actin cytoskeleton staining (Fig. 5 E). Although cells plated on Fc- or PL-coated surface show peripheral lamellipodia formation (Fig. 5 E, a and b), plating on Ncad-Fc–coated surface induces stress fiber formation (Fig. 5 E, c), a hallmark of RhoA activation. These data demonstrate that N-cadherin engagement activated RhoA GTPase.
N-cadherin–dependent cell–cell contact activates muscle-specific promoters
We then determined whether the effect of N-cadherin–dependent cell–cell contact extends to muscle-specific promoters shown previously to be controlled by RhoA, the skeletal α-actin (SkA), and the myosin light chain (MLC)1A promoters (Fig. 6, A and B) (Carnac et al., 1998; Wei et al., 1998). Both reporter constructs are significantly more active in cells in contact, whereas the effect is weak with the SkA promoter. We also manipulated cadherin-mediated junction formation using the calcium switch procedure. EGTA addition reduced the promoter activity of both constructs to that measured in isolated cells. After Ca2+ addition, SkA and MLC1A promoter activities increased, although to different extents. This increase was partially (MLC1A) or fully (SkA) sensitive to the addition of anti–N-cadherin antibodies. Controls were performed with serum addition and expression of constitutively active RhoA. Serum addition, which as contact formation, activates endogenous pathway led to a twofold increased in the SkA promoter, whereas expression of constitutively active RhoA strongly activates the MLC1A promoter.
Figure 6.

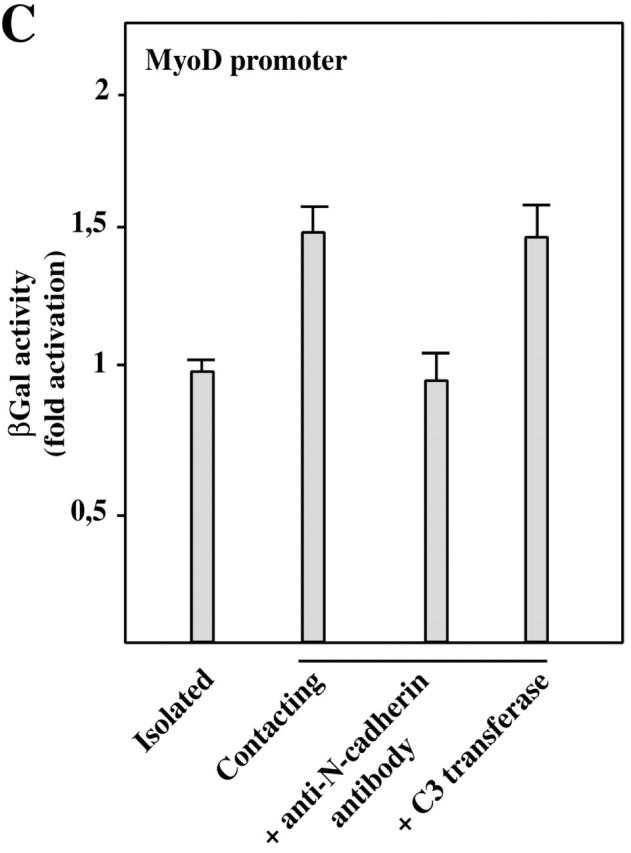
N-cadherin cell contact formation activates the SkA, MLC1A, and MyoD promoters in C2C12 myoblasts. (A) C2C12 myoblasts were cotransfected with plasmids containing the luciferase reporter gene driven by the SkA promoter and a renilla luciferase reporter gene driven by the CMV promoter. After the different treatments indicated on the graph, firefly luciferase activity was measured and corrected with respect to renilla luciferase activity. Data are the means ± SEM of three independent experiments. (B) C2C12 myoblasts were cotransfected with a plasmid containing the first 630 bp of the mouse MLC1A promoter linked to the CAT reporter gene and a β-gal reporter gene driven by the CMV promoter. After the different treatments indicated on the graph, CAT activity was measured and corrected with respect to β-gal activity. Data are the means ± SEM of at least three independent experiments. (C) C2C12 cells stably transfected with MyoD promoter (DRR and PRR regions driving a β-gal reporter gene) were grown in isolated (lane 1) or confluent conditions alone (lane 2), in the presence of anti– N-cadherin antibodies (lane 3), or with C3 transferase (lane 4). β-gal activity is shown, normalized to total protein content. β-gal values are expressed relative to that of isolated myoblasts taken as 1. Data are the means ± SEM of three independent experiments.
RhoA GTPase also controls MyoD expression during myoblast differentiation (Carnac et al., 1998). Thus, we tested if N-cadherin–dependent cell–cell contact regulates MyoD promoter activity via RhoA (Fig. 6 C). Culture conditions favoring cell–cell contacts led to a twofold increased in MyoD promoter activity compared with isolated conditions. Preventing N-cadherin–dependent cell–cell contacts by addition of anti–N-cadherin antibody during establishment of cell confluency reduced MyoD promoter activity to the isolated cell level. Inactivating Rho GTPases by addition of C3 transferase for 24 h led to a similar decrease in MyoD promoter activity. In conclusion, N-cadherin–dependent cell–cell contact controls Ska, MLC1A, and MyoD promoter activity most likely through Rho GTPase activity.
RhoA activity is required for β-catenin recruitment to intercellular adhesions sites
Recently, β-catenin was shown to be translocated and accumulated to adherens junctions in differentiating myoblasts (Goichberg et al., 2001). Moreover, Rho GTPases have been shown to regulate adherens junction assembly. We wondered whether the N-cadherin–dependent RhoA increase we observed might regulate β-catenin translocation to cell–cell contact sites. We first analyzed RhoA and β-catenin localization (Fig. 7 A). Endogenous RhoA (Fig. 7 A, a) and β-catenin (Fig. 7 A, b) were closely associated at cell–cell junctions (Fig. 7 A, c, merge). We next examined RhoA localization during the calcium switch procedure by time-lapse microscopy. C2C12 cells were transfected with GFP–RhoA. Fig. 7 B (videos 1–5 available at http://www.jcb.org/cgi/content/full/jcb.200202034/DC1) shows that under normal calcium conditions, GFP–RhoAWT, is both cytoplasmic and found at cell–cell contact sites (Fig. 7 B, a and d; videos 1 and 4). In cells treated with EGTA, GFP–RhoAWT is cytoplasmic but barely detectable at the cell–cell contact sites (Fig. 7 B, b; video 2). Upon addition of extracellular Ca2+, GFP–RhoAWT was rapidly revisualized at cell–cell contact sites (Fig. 7 B, c; video 3), except when anti–N-cadherin antibodies were added (Fig. 7 B, e; video 5).
Figure 7.

RhoA activity controls β-catenin localization to cell–cell contact sites. (A) C2C12 myoblasts were stained for RhoA (a) and β-catenin proteins (b). Bar, 10 μm. (B) Images of GFP–RhoAWT–expressing cells were captured every 10 s. Inverted contrast images are displayed from control GFP–RhoAWT–expressing cells untreated (a and d), treated with EGTA (b), or treated with EGTA and allowed to recover in Ca2+-containing medium alone (c) or together with anti–N-cadherin antibody (e). Bar, 10 μm. (C) C2C12 myoblasts transfected with GFP-tagged RhoAV14 (a) or RhoAN19 (c) were stained for β-catenin (b and d). Bar, 10 μm. See also videos available at http://www.jcb.org/cgi/content/full/jcb.200202034/DC1.
Finally, we analyzed whether expression of GFP-tagged activated or dominant negative forms of RhoA (GFP–RhoAV14 and GFP–RhoAN19, respectively) modify β-catenin accumulation at adherens junctions (Fig. 7 C). C2C12 expressing GFP–RhoAV14 (Fig. 7 C, a) presented a more prominent staining of β-catenin at the cell–cell junctions (Fig. 7 C, b). In contrast, in cells expressing GFP–RhoAN19 (Fig. 7 C, c), β-catenin was excluded from the junctions (Fig. 7 C, d). Similarly, expression of activated or dominant negative forms of RhoA modified p120 localization (unpublished data). Although Rac1 and Cdc42Hs GTPases also localize to the cell–cell contact sites, expression of dominant negative forms of either of these two GTPases did not impair β-catenin accumulation at the adherens junction (unpublished data). Together, these results indicate that RhoA can be recruited at the cell–cell adhesion sites after N-cadherin–dependent signaling and that RhoA activity is required for β-catenin accumulation at these adhesion sites.
Discussion
Cellular interactions mediated by members of the cadherin family of Ca2+-dependent cell–cell adhesion molecules have been shown to play important roles throughout myogenesis. Although N-cadherin was initially proposed to be involved in myoblast fusion, this process still occurs in myoblasts lacking N-cadherin, probably due to other cadherin molecules acting in place of the targeted one (Knudsen et al., 1990; Mege et al., 1992; Charlton et al., 1997). Indeed, inhibition of N-cadherin–dependent adhesion impairs myogenesis and perturbs the expression of skeletal muscle differentiation markers expression (Holt et al., 1994; George-Weinstein et al., 1997). The data we present, using three antisera that disrupt N-cadherin function, indicate that N-cadherin–dependent intercellular adhesion has a major role in cell cycle exit and in the skeletal muscle differentiation program. In support of this, addition of N-cadherin–coated beads to isolated myoblasts triggers myogenesis (Goichberg and Geiger, 1998), and expression of N-cadherin in BHK cells, which express MyoD but fail to differentiate, induces skeletal myogenesis by upregulating myogenin (Seghatoleslami et al., 2000). The important question now is to determine the nature of the adherens junction-mediated signals involved in myogenesis.
To address these signaling pathways, we investigated whether Rho GTPase activities might be affected by N-cadherin–mediated cell–cell adhesion. We found that N-cadherin–dependent adhesion decreases the activity of Rac1 and Cdc42Hs, two GTPases shown previously to have an inhibitory role in myogenesis (Gallo et al., 1999; Meriane et al., 2000). Two mechanisms have been proposed to explain their inhibitory effect. The first one involves JNK activation, which inhibits Myf5 nuclear localization (Meriane et al., 2000). We found here that JNK activity is diminished after N-cadherin–dependent cell–cell adhesion. This is in agreement with previous results showing that in fibroblasts cell density decreases JNK activity (Lallemand et al., 1998). The second one consists of active Rac1 and Cdc42Hs inhibiting cell cycle withdrawal in myoblasts (Heller et al., 2001; Meriane et al., 2002), a prerequisite for differentiation, and activation of muscle-specific gene expression (Lassar et al., 1994). Interestingly, cadherin-dependent adhesion is important for contact-dependent growth inhibition in CHO cells and regulates the level of the cyclin-dependent kinase inhibitor p27 (St Croix et al., 1998; Levenberg et al., 1999). We demonstrate here that the inhibition of N-cadherin–dependent adhesion impairs the expression of the two cyclin-dependent kinase inhibitors p21 and p27, which are normally upregulated during myogenesis (Guo et al., 1995; Ragolia et al., 2000). The N-cadherin–dependent decrease of Rac1 and Cdc42Hs activity that we observed might thus contribute to both cell cycle withdrawal and extinction of a myogenic inhibitory signal, such as JNK activity (Fig. 8). Notably, the decreased level of Rac1 and Cdc42Hs activity upon N-cadherin–dependent cell–cell contact formation remains sufficient to allow p38 activity (Cuenda and Cohen, 1999; Zetser et al., 1999; Meriane et al., 2000).
Figure 8.
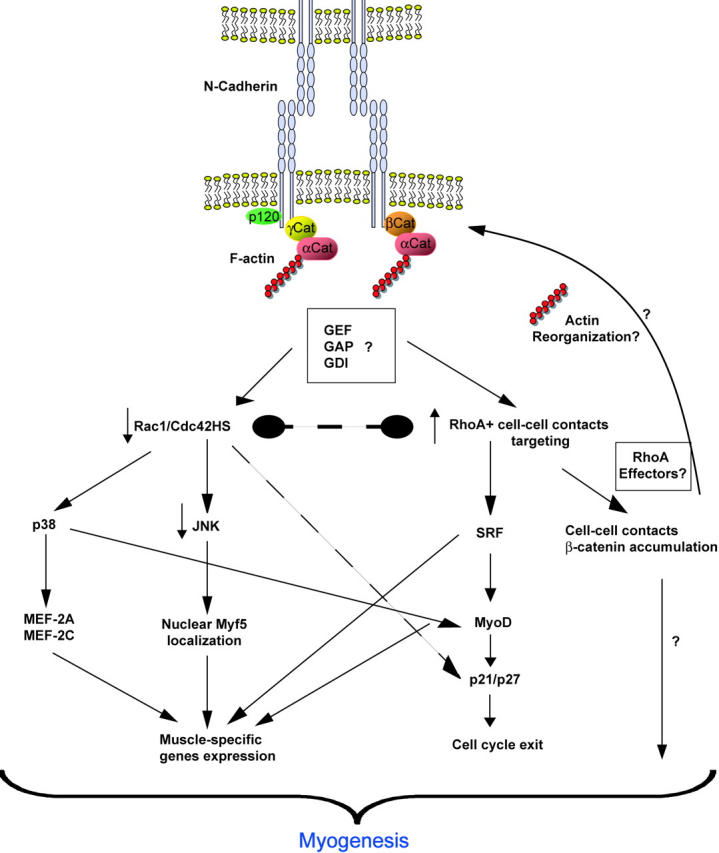
N-cadherin–mediated adhesion and myogenesis. N-cadherin activation through cell–cell contact formation affects the RhoA, Rac1, and Cdc42Hs level activation via an unidentified mechanism. The decrease of Rac1 and Cdc42Hs activity leads to a decrease in the JNK level, which allows Myf5 nuclear localization (Meriane et al., 2000) and also favors cell cycle exit (Heller et al., 2001; Meriane et al., 2002). The decreased level of Rac1 and Cdc42Hs activity remains sufficient to allow p38 activity, which is required for MyoD- and MEF-2–responsive genes expression (Zetser et al., 1999; Wu et al., 2000). Elevation of RhoA level activates SRF (Carnac et al., 1998; Wei et al., 1998) and leads to MyoD expression (Gauthier-Rouviere et al., 1996), which allowed p21 and p27-dependent cell cycle exit (Walsh, 1997) and muscle-specific genes expression (Tapscott et al., 1988). RhoA is required for β-catenin localization at the cell–cell contacts sites, another event involved in myogenic induction (Goichberg et al., 2001).
Our data also demonstrate that N-cadherin–dependent cell–cell contact formation stimulates RhoA GTPase activity. This GTPase is crucial for myogenic differentiation, since RhoA controls both the expression and activity of SRF, a transcription factor which binds to CArG motifs present in the promoter regions of many muscle-specific genes (Gauthier-Rouviere et al., 1996; Wei et al., 1998). In addition, SRF controls the expression of the muscle-determining factor MyoD, which can convert mesenchymal cells into skeletal myoblasts (Lassar et al., 1986; Tapscott et al., 1988; Choi et al., 1990). MyoD binds to promoter elements containing the E-box and activates genes that encode skeletal muscle-specific proteins. We clearly demonstrate that the MyoD promoter and two skeletal muscle genes are stimulated by N-cadherin–dependent cell–cell contact. Therefore, our data identify N-cadherin–dependent adhesion as an event upstream of the RhoA/SRF pathway (Fig. 8).
Not only was RhoA activated in our system, but it was also recruited to sites of N-cadherin–mediated cell–cell adhesion. The localization and activity of Rho GTPases is controlled by three classes of regulators: Rho GDP dissociation inhibitors, guanine nucleotide exchange factors (GEFs), and GTPase-activating proteins. All of these regulators are potential interesting candidates to link N-cadherin–dependent adhesion to Rho GTPase activity. Regulators of Rho GDP dissociation inhibitor activity (ezrin/radixin/moesin proteins) and GEF family members were shown to localize or interact with the cadherin–catenins complexes (Takahashi et al., 1998; Hiscox and Jiang, 1999; Sander et al., 1999). Recently, it has been shown that free p120 catenin, which normally binds to the cytoplasmic domain of the cadherins in the juxtamembrane region interacts with the GEF Vav2 and modulates the activity of Rho GTPases, suggesting that regulating p120 localization might also control Rho GTPases activity (Noren et al., 2000). Whether the activity of these Rho GTPases regulators is directly controlled by cadherin activation or results from a functional interaction between activated cadherin and classical growth receptors remains to be determined. Recently, N-cadherin has been shown to interact with the FGF receptor in neuronal cells (Williams et al., 2001). A balance between Rac1-Cdc42Hs and Rho activities determines the cellular phenotype and biological behavior in various cell systems: actin cytoskeleton organization (Sander et al., 1999; Vignal et al., 2001), formation of focal adhesions (Rottner et al., 1999), neurite extension (Kozma et al., 1997; Leeuwen et al., 1997), and myogenesis (Meriane et al., 2000). This was clearly demonstrated in NIH3T3, where Rac1 signaling is able to antagonize Rho activity directly at the GTPase level (Sander et al., 1999). Various other studies reported a similar antagonism, but the precise molecular mechanism remains a debated issue. Nevertheless, expression of activated form of RhoA in C2C12 myoblasts decreases Rac1 and Cdc42Hs activities, suggesting that N-cadherin signaling leading to RhoA activation might in turn downregulate Rac1 and Cdc42Hs activities (unpublished data). The identification of the missing links between N-cadherin and Rho GTPases will shed light on this process.
RhoA activity is clearly required for β-catenin localization at the contact sites, an event involved in myogenic induction (Goichberg et al., 2001). RhoA GTPase activity is mediated by downstream effectors, such as Rho kinase family of serine/threonine kinases and the proline-rich formin homology domain–containing proteins mDia1 and 2, which regulate actin stress fiber organization and alignment of actin bundles and microtubules (Bishop and Hall, 2000). Whether these RhoA effectors participate in the regulation of β-catenin localization through signaling toward actin filaments remains to be determined.
Previous studies examining the regulation of the Rho family GTPases by the formation of adherens junctions have concentrated on E-cadherin–dependent events. Interestingly, the results are the inverse of those obtained in myoblasts, since they found a stimulation of Rac1 and Cdc42Hs and a decrease of RhoA activities (Fukata and Kaibuchi, 2001). The differences between E- and N-cadherin–dependent intracellular signaling might be due to the cadherin molecule itself and/or to the different cell types in which these cadherins are expressed. Further studies are required to address this issue.
In conclusion, our data show that N-cadherin–dependent adhesion controls different crucial steps of the myogenic differentiation program. In the developing embryo, N-cadherin–mediated RhoA activation might play an important role during somitogenesis, myotome formation, and also terminal skeletal myogenesis, which lead to formation of the different body muscles. This would thus constitute an integral part of the community effect process which governs myogenesis. Further studies will be necessary to further correlate the activity of Rho GTPases with the skeletal myogenesis during development.
Materials and methods
Cell culture
C2C12 mouse myoblasts were grown in DME/HAM F-12 (1:1) supplemented with 10% FCS (HyClone). To induce differentiation, growth medium was replaced with differentiation medium consisting of DME/HAM F-12 supplemented with 2% FCS. EGTA was used at 2 mM for 30 to 60 min. C2.7 myoblasts stably transfected with MyoD promoter (DRR and PRR regions driving by β-gal reporter gene [Carnac et al., 1998]) were cultured as described above. Botilium C3 transferase (a gift from P. Bocquet, Institut National de la Santé et de la Recherche Médicale, U452, Nice, France) was used at 15 μg/ml for 24 h.
For density experiments, C2C12 cells were trypsinized and extensively homogenized to generate a dispersed cell suspension. Isolated cells were plated at a density of 1,250 cells/cm2, and confluent cells were plated at a density of 6,250 cells/cm2. Cells were allowed to recover for 20 h before measurement of Rac1, Cdc42Hs, RhoA, JNK, and p38 activities as described later. Quantification was performed by densitometric analysis of Western blots using Aida/2D densitometry software. The relative amount of active protein was determined by measuring the amount of protein sedimented or immunoprecipitated relative to the amount in whole cell lysates.
Cell–cell contact manipulation
Isolated C2C12 myoblasts were either left untreated, treated with EGTA for 60 min, or treated with EGTA for 60 min and allowed to recover in fresh medium. Confluent C2C12 myoblasts were either left untreated, treated with EGTA for 60 min, treated with EGTA for 60 min, and allowed to recover in fresh medium in the presence or not of anti–N-cadherin antibodies for 30 min. Confluent C2C12 myoblasts were allowed to establish cell–cell contacts with preimmune serum or anti–N-cadherin antibody. Alternatively, isolated cells were settle onto Ncad-Fc–coated Petri dishes.
Polyclonal anti–N-cadherin antibody production
The EcoRV fragment of N-cadherin (NM 007664) corresponding to amino acids 75–706 was cloned in the pGEX5X vector. GST–N-cadherin fragment was produced as described (Mary et al., 2002).
Three rabbits were injected with 80–100 μg of GST–N-cadherin protein. Antisera were tested by immunoblotting and immunofluorescence. Affinity purification was performed by incubation with the GST–N-cadherin fragment spotted onto nitrocellulose.
Differentiation inhibition assays
C2C12 cells were plated in 35-mm dish in DME/HAM F-12 supplemented with 10% FCS (HyClone). 2 h after plating, anti–N-cadherin antibody was added. To induce differentiation, growth medium was replaced with differentiation medium (DME/HAM F-12 supplemented with 2% FCS) containing anti–N-cadherin antibody. The medium was replaced every 24 h for 7 d. The differentiation of control or anti–N-cadherin–treated cells was followed by time-lapse imaging, analyzing the expression of three markers for myoblast differentiation (myogenin, Troponin T and MHC) on fixed cells, or measuring the levels of cyclin-dependent kinase inhibitors p21 and p27 by immunoblotting.
Transfection and kinase assay
C2C12 myoblasts cultured in 60-mm dishes were transfected or cotransfected with 2 μg HA-JNK, HA-p38 (provided by B. Dérijard, Centre National de la Recherche Scientifique, UMR 6548, Nice, France), GFP–Rac1N17, GFP–Cdc42HsN17 plasmids as described by the supplier (Life Technologies). 4 h after the transfection, the medium was replaced with DME/HAM F12 supplemented with 10% FCS. 24 h after transfection, cell–cell contacts were manipulated, and cells were lysed and processed as described (Meriane et al., 2000). Scanned autoradiographs were quantified using Aida/2D densitometry software and normalized as a function of the expression of the various proteins.
Preparation of Ncad-Fc–coated dishes
The Ncad-Fc chimera (chicken N-cadherin ectodomain fused to the Fc fragment of mouse IgG2b) was produced in eukaryotic cells as described (Lambert et al., 2000). The production media was collected every 2–3 d and analyzed for the presence of the fusion protein by Western blot analysis. 100-mm Petri dishes were first coated with goat anti–mouse Fcγ antibody (Jackson ImmunoResearch Laboratories) at 1 μg/cm2 in 0.1 borate buffer (pH 8.5) for 18 h at 4°C. Dishes were washed with borate buffer and incubated with Ncad-Fc medium (375 μg to obtain a theoretical coating value of 7 μg/cm2). Dishes were washed in PBS and used immediately. Coating efficiency was controlled by Western blot analysis of the coated material in a dish. Petri dishes were also coated with poly-l-lysine (0.001% in PBS; Sigma-Aldrich) for 10 min. RhoA, Rac1, and Cdc42Hs activity was measured 2 h after plating.
GTPase activity assays
After cell–cell contact manipulation, cells were processed as described to measure Rac1 and Cdc42Hs activity (Meriane et al., 2002). For RhoA activity assay, C2C12 cells were lysed in 50 mM Tris, pH 7.2, 1% Triton X-100, 0.5% sodium deoxycholate, 500 mM NaCl, 10 mM MgCl2, 1 mM PMSF, and cocktail protease inhibitors (Sigma-Aldrich). Cleared lysate was incubated with 25 μg of GST fusion protein containing the RhoA-binding domain of Rhotekin (GST-TRBD) beads for 40 min at 4°C, and then the beads were washed four times in Tris buffer containing 1% Triton X-100, 150 mM NaCl, 10 mM MgCl2, 0.1 mM PMSF and cocktail protease inhibitors before addition of SDS sample buffer containing DTT. Rho A fractions were analyzed by Western blotting with RhoA antibody (Santa Cruz Biotechnology, Inc.). Scanned autoradiographs were quantified using Aida/2D densitometry software and normalized as a function of the expression of the various proteins.
Luciferase assay
C2C12 cells plated in 35-mm dishes (20,000 cells for subconfluent condition and 50,000 cells for confluent condition) were cotransfected using the lipofectamine method with 0.7 μg of chimeric construct containing the −394/+24 skeletal actin (SkA) promoter fused to the luciferase gene (MacLellan et al., 1994), 0.7 μg of pEGFP empty vector (CLONETECH Laboratories, Inc.), and 40 ng of pRL cytomegalovirus (CMV) vector (renilla luciferase CMV). 48 h after transfection, cell–cell junctions were manipulated and luciferase activity was measured using the Dual-Luciferase Reporter Assay system (Promega).
Chloramphenicol acetyltransferase assay
C2C12 cells plated in 60-mm dishes (40,000 cells for subconfluent condition and 100,000 cells for confluent condition) were cotransfected using Lipofectamine with 0.8 μg of chimeric construct containing the first 630 bp of the mouse MLC1A promoter driving chloramphenicol acetyltransferase (CAT) gene expression (Catala et al., 1995) (provided by M. Buckingham, Institut Pasteur, Paris, France) with either 0.8 μg of pEGFP empty vector or GFP-tagged RhoA V14 and 0.4 μg of CMV driving β-galactosidase expression vector (CMV–β-gal). 48 h after transfection, cells were submitted to different treatments, and CAT (CAT Elisa; Roche Molecular Biology) and β-gal (β-gal reporter gene assay, chemiluminescent; Roche Molecular Biology) activities were determined as described by the supplier. CAT activity was corrected with respect to β-gal activity.
β-Gal assay
C2–7 stably expressing MyoD promoter fused to β-gal reporter gene (Carnac et al., 1998) were analyzed for β-gal activity in isolated or contacting culture conditions as described by the supplier. Alternatively, cells were allowed to form contacts in the presence of anti–N-cadherin antibodies or C3 transferase for 24 h. Protein determination (BCA assay) was determined to normalize results.
Immunofluorescence
Cells growing onto 35-mm dishes were fixed in 3.7% formaldehyde in PBS followed by a 5-min permeabilization in 0.1% Triton X-100 in PBS and incubated in PBS containing 0.1% BSA. Myogenin and Troponin T expression was detected as described (Meriane et al., 2000). Anti–β-catenin and anti–N-cadherin antibodies are from Transduction Laboratories (Interchim). All of these mouse antibodies were revealed with either an Alexa Fluor 546– or an Alexa Fluor 488–conjugated goat anti–mouse antibody (Molecular Probes and Interchim). Anti-Rho antibody is from Upstate Biotechnology (Euromedex), revealed by a either an Alexa Fluor 546– or an Alexa Fluor 488–conjugated goat anti–rabbit antibody (Molecular Probes and Interchim). Cells were stained for F-actin using TRITC-conjugated phalloidin (Sigma-Aldrich). Cells were prepared and observed as described (Mary et al., 2002). Fluorescent images were deconvolved using the maximum likelihood estimation algorithm (Huygens, Scientific Volume Imaging). The restored images were saved as Tif files that were mounted using Adobe Photoshop® and Adobe Illustrator®.
Gel electrophoresis and immunoblotting
Cells cultured in 60-mm dishes were rinsed in cold PBS and lysed in 50 mM Tris-HCl, pH 7.5, 120 mM NaCl, 20 mM NaF, 1 mM EDTA, 6 mM EGTA, 15 mM NaPPi, 15 mMpNPP, 1 mM benzamidine, 0.1 mM phenylmethylsulfonyl fluoride, 0.5 mM vanadate, and 1% Nonidet P-40. 15 μg of proteins were loaded on a 12.5% polyacrylamide gel and then transferred onto nitrocellulose. After saturation in 5% milk in PBS, membranes were incubated with rabbit anti-p21 and anti-p27 antibodies (Santa Cruz Biotechnology, Inc.). After washing, membranes were processed as described (Mary et al., 2002).
The relative amounts of p21 and p27 in each sample were calculated by densitometric analysis of Western blots using Aida/2D densitometry software. A 100% value was arbitrary affected to the control condition.
Time-lapse imaging
Time-lapse epifluorescence microscopy was performed as described previously (Mary et al., 2002). Fluorescent images were deconvolved using the maximum likelihood estimation algorithm (Huygens, Scientific Volume Imaging). The restored images were saved as Tif files that were edited with NIH Image and compiled into QuickTime movies or directly mounted using Adobe Photoshop® and Illustrator®.
Online supplemental material
Videos 1–5, available at http://www.jcb.org/cgi/content/full/jcb.200202034/ DC1, correspond to Fig. 7. C2C12 cells were transfected with GFP–RhoAWT cDNA, and images of GFP fluorescence were recorded 18 h after transfection. Note the accumulation of GFP–RhoA at the cell–cell contact sites in control cells (videos 1 and 2), whereas no such GFP–RhoA accumulation is detectable in EGTA- (video 3) or anti–N-cadherin–treated cells (video 4). GFP–RhoA is again found at the cell–cell contact sites upon restoration of extracellular Ca2+ (video 5).
Supplemental Material
Acknowledgments
We thank P. Fort and G. Carnac for fruitful discussions. We thank M. Buckingham (Institut Pasteur, Paris, France), R. Schwartz (Baylor College of Medicine, Houston, Texas) for MLC1A and Ska constructs, R.M. Mege and M. Lambert for Ncad-Fc construct, and Bob Hipskind for critical reading of the manuscript.
This work was supported by contracts for the Ligue Nationale contre le Cancer (“Equipe labelisee”), the Association pour la Recherche contre le Cancer (contract no. 5668), the Association Francaise contre les myopathies, and institutional grants from Centre National de la Resherche. During this work S. Charrasse was supported by a fellowship from the Association pour la Recherche sur le Cancer.
The online version of this article contains supplemental material.
Footnotes
Abbreviations used in this paper: β-gal, β-galactosidase; CAT, chloramphenicol acetyltransferase; CMV, cytomegalovirus; GEF, guanine nucleotide exchange factor; JNK, c-jun NH2-terminal kinase; MLC, myosin light chain; SkA, skeletal α-actin.
References
- Bishop, A.L., and A. Hall. 2000. Rho GTPases and their effector proteins. Biochem. J. 348(Pt 2):241–255. [PMC free article] [PubMed]
- Buckingham, M. 2001. Skeletal muscle formation in vertebrates. Curr. Opin. Genet. Dev. 11:440–448. [DOI] [PubMed] [Google Scholar]
- Carnac, G., M. Primig, M. Kitzmann, P. Chafey, D. Tuil, N. Lamb, and A. Fernandez. 1998. RhoA GTPase and serum response factor control selectively the expression of MyoD without affecting myf5 in mouse myoblasts. Mol. Biol. Cell. 9:1891–1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catala, F., R. Wanner, P. Barton, A. Cohen, W. Wright, and M. Buckingham. 1995. A skeletal muscle-specific enhancer regulated by factors binding to E and CArG boxes is present in the promoter of the mouse myosin light-chain 1A gene. Mol. Cell. Biol. 15:4585–4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlton, C.A., W.A. Mohler, G.L. Radice, R.O. Hynes, and H.M. Blau. 1997. Fusion competence of myoblasts rendered genetically null for N-cadherin in culture. J. Cell Biol. 138:331–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi, J., M.L. Costa, C.S. Mermelstein, C. Chagas, S. Holtzer, and H. Holtzer. 1990. MyoD converts primary dermal fibroblasts, chondroblasts, smooth muscle, and retinal pigmented epithelial cells into striated mononucleated myoblasts and multinucleated myotubes. Proc. Natl. Acad. Sci. USA. 87:7988–7992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coso, O.A., M. Chiariello, J.C. Yu, H. Teramoto, P. Crespo, N. Xu, T. Miki, and J.S. Gutkind. 1995. The small GTP-binding proteins Rac1 and Cdc42 regulate the activity of the JNK/SAPK signaling pathway. Cell. 81:1137–1146. [DOI] [PubMed] [Google Scholar]
- Cossu, G., R. Kelly, S. Di Donna, E. Vivarelli, and M. Buckingham. 1995. Myoblast differentiation during mammalian somitogenesis is dependent upon a community effect. Proc. Natl. Acad. Sci. USA. 92:2254–2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuenda, A., and P. Cohen. 1999. Stress-activated protein kinase-2/p38 and a rapamycin-sensitive pathway are required for C2C12 myogenesis. J. Biol. Chem. 274:4341–4346. [DOI] [PubMed] [Google Scholar]
- Duband, J.L., S. Dufour, K. Hatta, M. Takeichi, G.M. Edelman, and J.P. Theiry. 1987. Adhesion molecules during somitogenesis in the avian embryo. J. Cell Biol. 104:1361–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukata, M., and K. Kaibuchi. 2001. Rho-family GTPases in cadherin-mediated cell-cell adhesion. Nat. Rev. Mol. Cell Biol. 2:887–897. [DOI] [PubMed] [Google Scholar]
- Gallo, R., M. Serafini, L. Castellani, G. Falcone, and S. Alema. 1999. Distinct effects of rac1 on differentiation of primary avian myoblasts. Mol. Biol. Cell. 10:3137–3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauthier-Rouviere, C., M. Vandromme, D. Tuil, N. Lautredou, M. Morris, M. Soulez, A. Kahn, A. Fernandez, and N. Lamb. 1996. Expression and activity of serum response factor is required for expression of the muscle-determining factor MyoD in both dividing and differentiating mouse C2C12 myoblasts. Mol. Biol. Cell. 7:719–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George-Weinstein, M., J. Gerhart, J. Blitz, E. Simak, and K.A. Knudsen. 1997. N-cadherin promotes the commitment and differentiation of skeletal muscle precursor cells. Dev. Biol. 185:14–24. [DOI] [PubMed] [Google Scholar]
- Goichberg, P., and B. Geiger. 1998. Direct involvement of N-cadherin-mediated signaling in muscle differentiation. Mol. Biol. Cell. 9:3119–3131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goichberg, P., M. Shtutman, A. Ben-Ze'ev, and B. Geiger. 2001. Recruitment of beta-catenin to cadherin-mediated intercellular adhesions is involved in myogenic induction. J. Cell Sci. 114:1309–1319. [DOI] [PubMed] [Google Scholar]
- Guo, K., J. Wang, V. Andres, R.C. Smith, and K. Walsh. 1995. MyoD-induced expression of p21 inhibits cyclin-dependent kinase activity upon myocyte terminal differentiation. Mol. Cell. Biol. 15:3823–3829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatta, K., S. Takagi, H. Fujisawa, and M. Takeichi. 1987. Spatial and temporal expression pattern of N-cadherin cell adhesion molecules correlated with morphogenetic processes of chicken embryo. Dev. Biol. 120:215–227. [DOI] [PubMed] [Google Scholar]
- Heller, H., E. Gredinger, and E. Bengal. 2001. Rac1 inhibits myogenic differentiation by preventing the complete withdrawal of myoblasts from the cell cycle. J. Biol. Chem. 276:37307–37316. [DOI] [PubMed] [Google Scholar]
- Hiscox, S., and W.G. Jiang. 1999. Ezrin regulates cell-cell and cell-matrix adhesion, a possible role with E-cadherin/beta-catenin. J. Cell Sci. 112(Pt 18):3081–3090. [DOI] [PubMed]
- Holt, C.E., P. Lemaire, and J.B. Gurdon. 1994. Cadherin-mediated cell interactions are necessary for the activation of MyoD in Xenopus mesoderm. Proc. Natl. Acad. Sci. USA. 91:10844–10848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemler, R. 1993. From cadherins to catenins: cytoplasmic protein interactions and regulation of cell adhesion. Trends Genet. 9:317–321. [DOI] [PubMed] [Google Scholar]
- Knudsen, K.A. 1990. Cell adhesion molecules in myogenesis. Curr. Opin. Cell Biol. 2:902–906. [DOI] [PubMed] [Google Scholar]
- Knudsen, K.A., L. Myers, and S.A. McElwee. 1990. A role for the Ca2(+)-dependent adhesion molecule, N-cadherin, in myoblast interaction during myogenesis. Exp. Cell Res. 188:175–184. [DOI] [PubMed] [Google Scholar]
- Kozma, R., S. Sarner, S. Ahmed, and L. Lim. 1997. Rho family GTPases and neuronal growth cone remodelling: relationship between increased complexity induced by Cdc42Hs, Rac1, and acetylcholine and collapse induced by RhoA and lysophosphatidic acid. Mol. Cell. Biol. 17:1201–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lallemand, D., J. Ham, S. Garbay, L. Bakiri, F. Traincard, O. Jeannequin, C.M. Pfarr, and M. Yaniv. 1998. Stress-activated protein kinases are negatively regulated by cell density. EMBO J. 17:5615–5626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert, M., F. Padilla, and R.M. Mege. 2000. Immobilized dimers of N-cadherin-Fc chimera mimic cadherin-mediated cell contact formation: contribution of both outside-in and inside-out signals. J. Cell Sci. 113:2207–2219. [DOI] [PubMed] [Google Scholar]
- Lassar, A.B., B.M. Paterson, and H. Weintraub. 1986. Transfection of a DNA locus that mediates the conversion of 10T1/2 fibroblasts to myoblasts. Cell. 47:649–656. [DOI] [PubMed] [Google Scholar]
- Lassar, A.B., S.X. Skapek, and B. Novitch. 1994. Regulatory mechanisms that coordinate skeletal muscle differentiation and cell cycle withdrawal. Curr. Opin. Cell Biol. 6:788–794. [DOI] [PubMed] [Google Scholar]
- Leeuwen, F.N., H.E. Kain, R.A. Kammen, F. Michiels, O.W. Kranenburg, and J.G. Collard. 1997. The guanine nucleotide exchange factor Tiam1 affects neuronal morphology; opposing roles for the small GTPases Rac and Rho. J. Cell Biol. 139:797–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levenberg, S., A. Yarden, Z. Kam, and B. Geiger. 1999. p27 is involved in N-cadherin-mediated contact inhibition of cell growth and S-phase entry. Oncogene. 18:869–876. [DOI] [PubMed] [Google Scholar]
- Linask, K.K., C. Ludwig, M.D. Han, X. Liu, G.L. Radice, and K.A. Knudsen. 1998. N-cadherin/catenin-mediated morphoregulation of somite formation. Dev. Biol. 202:85–102. [DOI] [PubMed] [Google Scholar]
- MacLellan, W.R., T.C. Lee, R.J. Schwartz, and M.D. Schneider. 1994. Transforming growth factor-beta response elements of the skeletal alpha-actin gene. Combinatorial action of serum response factor, YY1, and the SV40 enhancer-binding protein, TEF-1. J. Biol. Chem. 269:16754–16760. [PubMed] [Google Scholar]
- Mary, S., S. Charrasse, M. Meriane, F. Comunale, P. Travo, A. Blangy, and C. Gauthier-Rouviere. 2002. Biogenesis of N-cadherin-dependent cell-cell contacts in living fibroblasts is a microtubule-dependent kinesin-driven mechanism. Mol. Biol. Cell. 13:285–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mege, R.M., D. Goudou, C. Diaz, M. Nicolet, L. Garcia, G. Geraud, and F. Rieger. 1992. N-cadherin and N-CAM in myoblast fusion: compared localisation and effect of blockade by peptides and antibodies. J. Cell Sci. 103:897–906. [DOI] [PubMed] [Google Scholar]
- Meriane, M., P. Roux, M. Primig, P. Fort, and C. Gauthier-Rouviere. 2000. Critical activities of Rac1 and Cdc42Hs in skeletal myogenesis: antagonistic effects of JNK and p38 pathways. Mol. Biol. Cell. 11:2513–2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meriane, M., S. Charrasse, F. Comunale, A. Mery, P. Fort, P. Roux, and C. Gauthier-Rouviere. 2002. Participation of small GTPases Rac1 and Cdc42Hs in myoblast transformation. Oncogene. 21:2901–2907. [DOI] [PubMed] [Google Scholar]
- Minden, A., A. Lin, F.X. Claret, A. Abo, and M. Karin. 1995. Selective activation of the JNK signaling cascade and c-Jun transcriptional activity by the small GTPases Rac and Cdc42Hs. Cell. 81:1147–1157. [DOI] [PubMed] [Google Scholar]
- Noren, N.K., B.P. Liu, K. Burridge, and B. Kreft. 2000. p120 catenin regulates the actin cytoskeleton via Rho family GTPases. J. Cell Biol. 150:567–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragolia, L., Q. Zuo, and N. Begum. 2000. Inhibition of myogenesis by depletion of the glycogen-associated regulatory subunit of protein phosphatase-1 in rat skeletal muscle cells. J. Biol. Chem. 275:26102–26108. [DOI] [PubMed] [Google Scholar]
- Redfield, A., M.T. Nieman, and K.A. Knudsen. 1997. Cadherins promote skeletal muscle differentiation in three-dimensional cultures. J. Cell Biol. 138:1323–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridley, A.J., H.F. Paterson, C.L. Johnston, D. Diekmann, and A. Hall. 1992. The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell. 70:401–410. [DOI] [PubMed] [Google Scholar]
- Rottner, K., A. Hall, and J.V. Small. 1999. Interplay between Rac and Rho in the control of substrate contact dynamics. Curr. Biol. 9:640–648. [DOI] [PubMed] [Google Scholar]
- Sander, E.E., J.P. ten Klooster, S. van Delft, R.A. van der Kammen, and J.G. Collard. 1999. Rac downregulates Rho activity: reciprocal balance between both GTPases determines cellular morphology and migratory behavior. J. Cell Biol. 147:1009–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seghatoleslami, M.R., L. Myers, and K.A. Knudsen. 2000. Upregulation of myogenin by N-cadherin adhesion in three-dimensional cultures of skeletal myogenic BHK cells. J. Cell. Biochem. 77:252–264. [DOI] [PubMed] [Google Scholar]
- St Croix, B., C. Sheehan, J.W. Rak, V.A. Florenes, J.M. Slingerland, and R.S. Kerbel. 1998. E-cadherin-dependent growth suppression is mediated by the cyclin-dependent kinase inhibitor p27(KIP1). J. Cell Biol. 142:557–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi, K., T. Sasaki, A. Mammoto, I. Hotta, K. Takaishi, H. Imamura, K. Nakano, A. Kodama, and Y. Takai. 1998. Interaction of radixin with Rho small G protein GDP/GTP exchange protein Dbl. Oncogene. 16:3279–3284. [DOI] [PubMed] [Google Scholar]
- Takano, H., I. Komuro, T. Oka, I. Shiojima, Y. Hiroi, T. Mizuno, and Y. Yazaki. 1998. The Rho family G proteins play a critical role in muscle differentiation. Mol. Cell. Biol. 18:1580–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapscott, S.J., R.L. Davis, M.J. Thayer, P.F. Cheng, H. Weintraub, and A.B. Lassar. 1988. MyoD1: a nuclear phosphoprotein requiring a Myc homology region to convert fibroblasts to myoblasts. Science. 242:405–411. [DOI] [PubMed] [Google Scholar]
- Vignal, E., A. Blangy, M. Martin, C. Gauthier-Rouviere, and P. Fort. 2001. Kinectin is a key effector of RhoG microtubule-dependent cellular activity. Mol. Cell. Biol. 21:8022–8034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volberg, T., B. Geiger, J. Kartenbeck, and W.W. Franke. 1986. Changes in membrane-microfilament interaction in intercellular adherens junctions upon removal of extracellular Ca2+ ions. J. Cell Biol. 102:1832–1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh, K. 1997. Coordinate regulation of cell cycle and apoptosis during myogenesis. Prog. Cell Cycle Res. 3:53–58. [DOI] [PubMed] [Google Scholar]
- Wei, L., W. Zhou, J.D. Croissant, F.E. Johansen, R. Prywes, A. Balasubramanyam, and R.J. Schwartz. 1998. RhoA signaling via serum response factor plays an obligatory role in myogenic differentiation. J. Biol. Chem. 273:30287–30294. [DOI] [PubMed] [Google Scholar]
- Williams, E.J., G. Williams, F.V. Howell, S.D. Skaper, F.S. Walsh, and P. Doherty. 2001. Identification of an N-cadherin motif that can interact with the fibroblast growth factor receptor and is required for axonal growth. J. Biol. Chem. 276:43879–43886. [DOI] [PubMed] [Google Scholar]
- Wu, Z., P.J. Woodring, K.S. Bhakta, K. Tamura, F. Wen, J.R. Feramisco, M. Karin, J.Y. Wang, and P.L. Puri. 2000. p38 and extracellular signal-regulated kinases regulate the myogenic program at multiple steps. Mol. Cell. Biol. 20:3951–3964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeschnigk, M., D. Kozian, C. Kuch, M. Schmoll, and A. Starzinski-Powitz. 1995. Involvement of M-cadherin in terminal differentiation of skeletal muscle cells. J. Cell Sci. 108:2973–2981. [DOI] [PubMed] [Google Scholar]
- Zetser, A., E. Gredinger, and E. Bengal. 1999. p38 mitogen-activated protein kinase pathway promotes skeletal muscle differentiation. Participation of the Mef2c transcription factor. J. Biol. Chem. 274:5193–5200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.