

Abstract
Tight junctions (TJs) play a crucial role in the establishment of cell polarity and regulation of paracellular permeability in epithelia. Here, we show that upon calcium-induced junction biogenesis in Madin-Darby canine kidney cells, ABαC, a major protein phosphatase (PP)2A holoenzyme, is recruited to the apical membrane where it interacts with the TJ complex. Enhanced PP2A activity induces dephosphorylation of the TJ proteins, ZO-1, occludin, and claudin-1, and is associated with increased paracellular permeability. Expression of PP2A catalytic subunit severely prevents TJ assembly. Conversely, inhibition of PP2A by okadaic acid promotes the phosphorylation and recruitment of ZO-1, occludin, and claudin-1 to the TJ during junctional biogenesis. PP2A negatively regulates TJ assembly without appreciably affecting the organization of F-actin and E-cadherin. Significantly, inhibition of atypical PKC (aPKC) blocks the calcium- and serum-independent membrane redistribution of TJ proteins induced by okadaic acid. Indeed, PP2A associates with and critically regulates the activity and distribution of aPKC during TJ formation. Thus, we provide the first evidence for calcium-dependent targeting of PP2A in epithelial cells, we identify PP2A as the first serine/threonine phosphatase associated with the multiprotein TJ complex, and we unveil a novel role for PP2A in the regulation of epithelial aPKC and TJ assembly and function.
Keywords: PP2A; aPKC; ZO-1; occludin; claudin
Introduction
Tight junctions (TJs)* serve as occluding barriers, maintain polarity and homeostasis, and regulate the permeability characteristics of the paracellular space in epithelia (Mitic et al., 2000). ZO-1, a member of the MAGUK family of proteins, acts as a scaffold to organize transmembrane TJ proteins and recruit various signaling molecules and the actin cytoskeleton to TJs (Gonzalez-Mariscal et al., 2000). Occludin binds to ZO-1 and regulates TJ paracellular permeability (Furuse et al., 1993; Wong and Gumbiner, 1997). Several lines of evidence point to proteins of the claudin family as key components of TJ fibrils and the primary seal-forming proteins responsible for mediating TJ's physiological barrier (Tsukita and Furuse, 2000). Although TJs serve as organizing centers for numerous other proteins involved in trafficking and signaling (Mitic et al., 2000), little is known about the molecular mechanisms involved in the dynamic regulation of these multiprotein complexes. Translocation of occludin and ZO-1 from the cytoplasm to the membrane during Ca2+-induced TJ biogenesis is accompanied by their phosphorylation (Stuart and Nigam, 1995; Wong, 1997; Sakakibara et al., 1997; Farshori and Kachar, 1999). However, so far, how dephosphorylation events are involved in the structural/functional regulation of TJs remains obscure.
Protein phosphatase (PP)2A enzymes are major Ser/Thr protein phosphatases. The core enzyme is a dimer containing a catalytic subunit (C) and a regulatory subunit (A), which can associate to a regulatory subunit (B). Several families of B subunits have been identified and modulate PP2A catalytic activity and substrate specificity (Sontag, 2001). Distinct B subunits contribute to targeting PP2A to defined intracellular domains and recruiting PP2A to signaling complexes, thereby ensuring its functional specificity (Sim and Scott, 1999; Sontag, 2001). Notably, the holoenzyme containing the Bα subunit is a major PP2A isoform involved in cell growth and cytoskeletal regulation in numerous cell types (Sontag, 2001). Here, we chose to undertake a detailed analysis of ABαC behavior in epithelial cells, a cell type for which PP2A properties and functions are poorly documented. We show that ABαC is targeted to the TJ complex and identify a novel role for PP2A in TJ regulation.
Results
Ca2+-dependent recruitment of ABαC to regions of cell–cell contact
In Madin-Darby canine kidney (MDCK) cells, Ca2+ depletion from the culture medium results in disruption of intercellular junctions and cell rounding; conversely, the formation of functional junctional complexes can be triggered upon transferring cells cultured in low Ca2+ (LC) medium to normal Ca2+ (NC) medium (Gonzalez-Mariscal et al., 1990; Cereijido et al., 2000). Because the Bα subunit is always complexed to the AC core enzyme (Sontag, 2001), immunofluorescent microscopy and immunoblotting with anti-Bα antibodies were used to assess the behavior of ABαC during Ca2+ switch experiments in confluent MDCK cells. Ca2+ deprivation of cells resulted in progressive cell retraction and disappearance of the peripheral membrane staining for ABαC (Fig. 1 A). When cells were Ca2+ starved overnight and then switched to NC medium to induce junction biogenesis, a pool of ABαC gradually reconcentrated at cell–cell contact sites (Fig. 1 B). Significant amounts of ABαC copurified with the membrane fraction from cells cultured in NC medium, whereas most of the holoenzyme was present in the cytosolic fraction from cells transferred to LC medium (Fig. 1 C). The proportion of membrane-bound ABαC increased considerably during junction biogenesis and remained unchanged 24 h after the Ca2+ switch, at which time TJs were completely reformed (Fig. 1 D).
Figure 1.
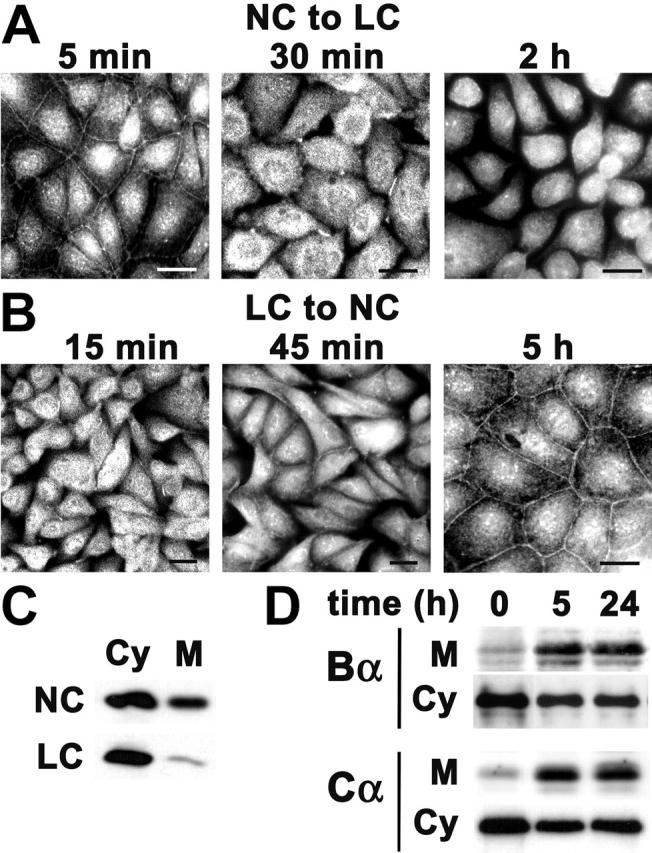
Ca 2+ -dependent membrane localization of ABαC in MDCK cells. (A) Confluent MDCK cells grown on glass coverslips in NC medium were switched for 5 min, 30 min, or 2 h to LC medium containing 1 mM EGTA to induce rapid TJ disassembly (NC to LC), and then processed for indirect immunofluorescence with rabbit anti-Bα Δ237 antibody. (B) MDCK cells were Ca2+ starved overnight then switched to NC medium to induce TJ biogenesis (LC to NC). Cells were fixed 15 min, 45 min, and 5 h after initiation of cell–cell contact and analyzed as described in A. Bars, 10 μm. (C) MDCK cells cultured in NC medium (NC) were switched overnight to LC medium (LC). Equivalent amounts of proteins from cytosolic (Cy) and membrane (M) fractions were analyzed by immunoblotting with anti-Bα antibodies. (D) MDCK cells were switched from LC to NC medium for 0, 5, or 24 h. Equivalent amounts of proteins from cytosolic (Cy) and membrane (M) fractions were analyzed by immunoblotting for the presence of PP2A Bα and Cα subunits.
Ca2+-dependent interaction of ABαC with the TJ complex
The dependency of the membrane localization of ABαC on the presence of both Ca2+ and cell–cell contacts suggests that ABαC associates with junctional complexes. Thus, we compared by confocal microscopy the distribution of ABαC with that of known junctional proteins in well-polarized MDCK cells. Immunolabeling with anti-Bα antibodies indicated that endogenous ABαC was present at areas of intercellular contact (Fig. 2 A). A similar belt-like pattern of membrane immunostaining was observed with anti-HA antibody in MDCK cells stably expressing HA-tagged Bα (MDCK-Bα), confirming the specificity of the signal obtained with anti-Bα antibodies. Images of x-z sections performed across the cell monolayers showed that endogenous or expressed Bα subunits were concentrated at both the apical membrane and apex of lateral cell borders, but essentially absent from basolateral domains. By comparison, the distribution of ZO-1, occludin, and claudin-1 was characteristically restricted to the apex of the lateral cell borders, whereas E-cadherin, a marker of adherens junctions (AJs), was primarily enriched along the lateral membrane (Fig. 2 B). Double labeling of cells further confirmed that ABαC partially colocalized with ZO-1 at the apical membrane, but not with E-cadherin in the basolateral borders (Fig. 2 C). Immunofluorescent results indicate that a portion of membrane-associated ABαC is in proximity to TJs in MDCK cells. Because the apical junctional complex is better defined in intestinal cells, immunogold–electron microscopy with anti-Bα antibodies was used to examine ABαC localization in human colon, and revealed a pool of TJ-associated PP2A (Fig. 2 D). However, in contrast to MDCK cells, ABαC was not present at the apical membrane of intestinal cells, suggesting a cell type–specific distribution of the phosphatase.
Figure 2.
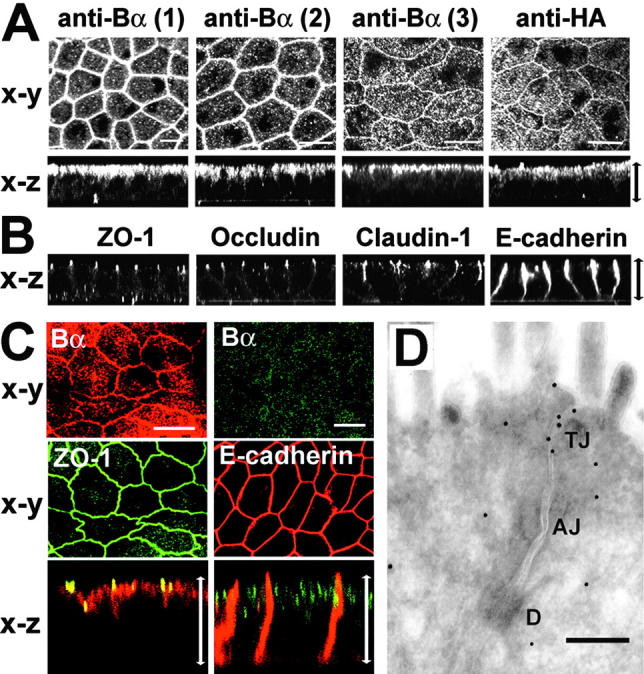
Analysis of the distribution of ABαC at junctional complexes in polarized MDCK cells and in human colon. (A–C) Analysis of polarized cells by confocal microscopy. Arrows in x-z images indicate the thickness of the monolayers. Bars, 10 μm. (A) Representative apical x-y sections and transversal x-z views obtained in MDCK cells labeled with either Δ237 (1), P9 (2), or 2G9 (3) anti-Bα antibodies, and in MDCK-Bα cells labeled with anti-HA antibody (anti-HA). (B) Representative x-z sections of ZO-1, occludin, claudin-1, or E-cadherin staining in MDCK cells. (C) MDCK cells were double labeled with rabbit anti-Bα and either rat anti–ZO-1 or mouse anti–E-cadherin antibodies. Representative apical (for Bα/ZO-1) and basolateral (for Bα/E-cadherin) x-y sections, and corresponding x-z views are shown. (D) Immunogold labeling was performed with the anti-B Δ237 antibody (diluted 1:80) on frozen ultrathin sections obtained from a normal human adult colon tissue biopsy. Note the concentration of gold particles in the TJ region beneath the lumen of the microvilli. Similar results were obtained with the P9 antibody; no TJ labeling was obtained with corresponding preimmune sera (not depicted). Bar, 150 nm. D, desmosome.
These observations prompted us to use immunoprecipitation assays to investigate whether PP2A could interact with TJ proteins. Because most antisubunit antibodies do not quantitatively immunoprecipitate PP2A heterotrimers, cell extracts were prepared from MDCK-Bα cells and MDCK cells stably expressing HA-tagged wild-type C (MDCK-Wt C), and immunoprecipitated using anti-HA antibody (Ogris et al., 1997; Goedert et al., 2000). Large amounts of ZO-1, occludin, and claudin-1 were detected in the immunoprecipitates prepared from MDCK-Wt C and MDCK-Bα, but not from control cells (Fig. 3 A). TJ proteins were enriched, whereas E-cadherin could not be detected in the HA-Bα immunoprecipitates, strengthening the hypothesis that ABαC preferentially interacts with TJ proteins. Detergent insolubility is commonly used as an indicator of the cytoskeletal association and incorporation of proteins into large junctional complexes (Stuart and Nigam, 1995; Wong, 1997). Substantial amounts of ABαC were detected in ZO-1, occludin, or claudin-1, but not in E-cadherin immunoprecipitates prepared from detergent-insoluble MDCK cell fractions (Fig. 3 B). Other Ser/Thr phosphatases, including PP1α or PP2A holoenzymes containing the B56γ subunit, were not found in the immunoprecipitates, in support of a specific association of ABαC with the TJ complex. ABαC was also present in occludin immunoprecipitates prepared from rat colon or kidney tissue homogenates (Fig. 3 C). Comparative analysis of immunoprecipitates from cytosolic and membrane fractions prepared from MDCK cells cultured in NC medium or transferred to LC medium demonstrated that the interaction between PP2A and TJ proteins was Ca2+-dependent and primarily occurred in the membrane fraction (Fig. 3 D).
Figure 3.

Ca 2+ -dependent association of ABαC with membrane-associated TJ proteins. (A) Immunoblots of total cell lysates prepared from MDCK, MDCK-Wt C, or MDCK-Bα cells and immunoprecipitated with anti–HA-coupled affinity matrix. (B) Immunoblots of detergent-insoluble fractions prepared from MDCK cells and immunoprecipitated with either anti–ZO-1, –occludin, –claudin-1, or –E-cadherin antibodies (+), or no antibody (−). (C) Immunoblots of homogenates prepared from rat colon or kidney tissue and immunoprecipitated with (+) or without (−) anti-occludin antibody. (D) Immunoblots of cytosolic (Cy) and membrane (M) fractions prepared from MDCK, MDCK-Bα, and MDCK-Wt C cells cultured in NC medium (NC) then switched overnight to LC medium (LC), and immunoprecipitated with either anti–ZO-1 or anti–HA antibodies.
PP2A can dephosphorylate TJ proteins at the membrane, resulting in enhanced TJ permeability
Because PP2A is appropriately localized to dephosphorylate TJ-associated proteins, we next examined how changes in PP2A activity influence TJ protein regulation and TJ barrier function. Potent inhibition of endogenous PP2A activity was achieved by treating MDCK cells with the toxin okadaic acid (OA) (Haystead et al., 1989). Increased levels of endogenous PP2A activity were obtained after expression of Wt C subunit in MDCK cells. Analysis of stable polarized MDCK-Wt C cells by confocal microscopy indicated that HA-tagged Wt C subunits were homogenously expressed and localized at the apical membrane (Fig. 4 A), as observed earlier for Bα. First, to determine whether changes in PP2A activity affect the phosphorylation state of TJ proteins at mature TJs, ZO-1, occludin, and claudin-1 were immunoprecipitated from membrane fractions prepared from polarized MDCK, OA-treated MDCK or MDCK-Wt C cells, and analyzed by immunoblotting with anti-phosphoserine and anti-TJ protein antibodies (Fig. 4 B). Cells were examined before and after incubation with sodium butyrate, which significantly enhances the expression of transfected proteins. After treatment with sodium butyrate, endogenous PP2A activity levels were increased by ∼30% in MDCK-Wt C relative to control cells (unpublished data). Cell incubation with OA resulted in ∼70% PP2A inhibition. As expected from previous studies showing that TJ-associated ZO-1 is a Ser phosphoprotein (Anderson et al., 1988), ZO-1 was immunoreactive with the anti-phosphoserine antibody in MDCK cell membrane fractions. OA-mediated inhibition of PP2A activity induced an increase in the amounts of phosphorylated ZO-1 that correlated with an upward shift in its electrophoretic mobility. Expression of Wt C resulted in marked dephosphorylation and increased electrophoretic mobility of ZO-1. TJ-associated occludin migrates as multiple bands encompassing slow-migrating, high molecular weight occludin forms that are primarily phosphorylated on Ser residues, and fast-migrating, low molecular weight, dephosphorylated occludin species (Wong, 1997). Accordingly, only slow-migrating occludin bands were immunoreactive with the anti-phosphoserine antibody in MDCK cells. OA induced a significant increase in the phosphorylation levels of occludin that was accompanied by changes in its banding pattern. Expressed Wt C dephosphorylated occludin, as judged by the complete disappearance of the phosphorylated, upper occludin band. The amounts of slow-migrating, phosphorylated claudin-1 were increased after incubation of MDCK cells with OA, whereas expressed Wt C caused the accumulation of a fast-migrating, dephosphorylated claudin-1 species.
Figure 4.

Effects of deregulating PP2A activity on the phosphorylation state of TJ proteins and TJ barrier properties of MDCK cells cultured in NC medium. (A–E) Confluent cells were grown in NC medium. (A) Polarized MDCK-Wt C cells were analyzed by confocal microscopy with anti-HA antibody for the distribution of expressed Wt C subunit. Representative apical x-y and x-z views are shown. Bar, 10 μm; arrow, thickness of the cell monolayer. (B) ZO-1, occludin, and claudin-1 were immunoprecipitated from membrane fractions prepared from control MDCK cells (lanes 1 and 4), MDCK cells incubated for 1 h with 200 nM OA before harvesting (lanes 2 and 5), or stable MDCK-Wt C cells (lanes 3 and 6), before (lanes 1–3) or after (lanes 4–6) preincubation with sodium butyrate. The samples were analyzed by SDS-PAGE and immunoblotting with anti-phosphoserine antibody. The blots were reprobed with anti–ZO-1, occludin, or claudin-1 antibodies. In parallel, aliquots of membrane fractions from OA-treated cells were left untreated (lane 7) or treated (lane 8) with alkaline phosphatase, and analyzed by immunoblotting with anti–ZO-1, occludin, or claudin-1 antibodies. Note that the upward band shift caused by OA results from phosphorylation, as it can be abolished after alkaline phosphatase treament. Arrows and brackets (]*) indicate the position of dephosphorylated and phosphorylated TJ protein species, respectively. (C) TER was measured in control MDCK (1–2) and MDCK-Wt C cells expressing distinct levels of transfected Wt C subunit (3–5), before and after preincubation with sodium butyrate. Subsets of cells were incubated for 1 h with (+ OA) or without (− OA) 100 nM OA. Results are expressed as the mean TER ± SD of duplicate determinations performed in four separate experiments with monolayers of equivalent cell density. The representative blot in the bottom panel shows the relative amounts of expressed Wt C subunits detected with anti-HA antibody in total extracts prepared from the cell lines analyzed for TER. (D) Paracellular diffusion of [3H]-mannitol and [3H]-inulin was determined in identical subsets of MDCK-Wt C monolayers treated with or without 100 nM OA, before and after preincubation with sodium butyrate. Results are expressed as percent of the relative tracer flux measured in untreated cells before addition of sodium butyrate, and are the mean ± SD of triplicate determinations performed in four distinct stable MDCK-Wt C populations. (E) Paracellular diffusion of [3H]-mannitol and [3H]-inulin was determined in MDCK cells incubated with increasing concentrations of OA. Results are expressed as percent of the relative tracer flux measured in untreated cells, and are the mean ± SD of triplicate determinations from six separate experiments.
Next, we examined how PP2A-mediated changes in the phosphorylation levels of TJ-associated proteins affected TJ barrier properties. TJs not only regulate paracellular ion flow, as measured by transepithelial resistance (TER), but also the diffusion of nonionic molecules through the paracellular space. Increasing levels of expressed Wt C caused a gradual and substantial decrease in the TER of MDCK monolayers (Fig. 4 C). Treatment with 100 nM OA marginally enhanced the TER of control cells, but markedly inhibited the TER decrease induced by expressed Wt C. The paracellular diffusion of radioactive tracers across MDCK-Wt C monolayers increased by up to fourfold after sodium butyrate–induced Wt C expression, and was sensitive to 100 nM OA (Fig. 4 D). 50 nM OA had no effect, whereas incubation of MDCK cells with 100–250 nM OA slightly decreased tracer fluxes (Fig. 4 E). At higher concentrations, OA induced prominent TJ leakiness, but this effect was associated with substantial cell rounding. Thus, changes in PP2A activity can influence the phosphorylation state of ZO-1, occludin, and claudin-1 at mature TJs, and modulate TJ barrier properties.
Inhibition of PP2A promotes TJ protein phosphorylation and TJ assembly
To explore the potential role of PP2A in TJ assembly, the distribution of ZO-1, occludin, and claudin-1 was compared by immunofluorescent microscopy and immunoblotting during Ca2+ switch experiments performed in untreated or OA-treated control MDCK cells, and in MDCK-Wt C cells (Figs. 5 and 6). Cells were Ca2+-starved overnight to induce TJ downregulation, resulting in internalization/redistribution of TJ proteins from the cell periphery to the cytosol, and then transferred to NC medium to induce TJ biogenesis. The Ca2+ switch initiates a rapid sorting of TJ proteins from the cytosol to the membrane; however, complete TJ stabilization and resealing, as measured by TER restoration, is only achieved >20 h after the calcium switch (Stuart and Nigam, 1995; Farshori and Kachar, 1999). 1 h after the Ca2+ switch, a portion of total TJ proteins had already migrated to the cell periphery in control MDCK cells, but this redistribution was largely inhibited after expression of Wt C (Fig. 5 A). The peripheral membrane staining for TJ proteins appeared more continuous after incubation of MDCK cells with OA. The critical role played by PP2A during early TJ biogenesis was even more clearly demonstrated during Ca2+ switch experiments performed in the absence of serum (Fig. 5 B). OA promoted, whereas expression of Wt C severely prevented the accumulation of TJ proteins at cell-cell contact sites. The inhibitory effect of expressed Wt C could be partially reversed by OA, reinforcing the idea that PP2A negatively regulates the initial sorting of TJ proteins to the membrane. 4 h after the Ca2+ switch, a complete junctional distribution of ZO-1 was achieved over the entire circumference of MDCK cells (Fig. 6 A). OA accelerated the formation of the TJ network. Expression of Wt C noticeably delayed the accumulation of TJ proteins at junctional areas, as revealed by the predominant cytosolic concentration and fragmentary staining of ZO-1 at cell–cell boundaries. 24 h after the Ca2+ switch and thereafter, all the cells displayed the typical chicken wire staining pattern of ZO-1. However, cytosolic pools of TJ proteins were still visible at that stage in MDCK-Wt C cells. Significantly, in contrast to TJ proteins, changes in PP2A activity did not affect the redistribution of E-cadherin to areas of cell–cell contact during AJ formation.
Figure 5.
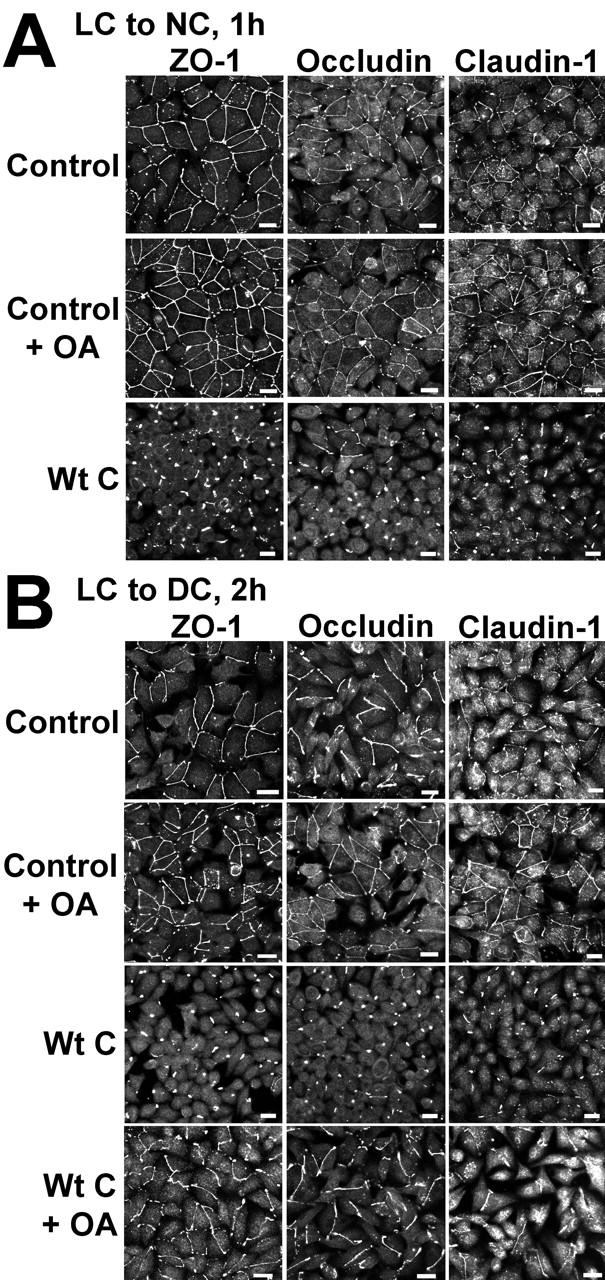
Effects of deregulating PP2A activity on the initial redistribution of TJ proteins during Ca2 + -induced TJ biogenesis. Control MDCK or MDCK-Wt C cell monolayers were incubated overnight in LC medium, then switched for 1 h to NC medium (A) or for 2 h to DC medium (B), in the absence or presence of 100 nM OA. Cells were analyzed by immunofluorescence for the distribution of ZO-1, occludin, and claudin-1. Bars, 10 μM.
Figure 6.
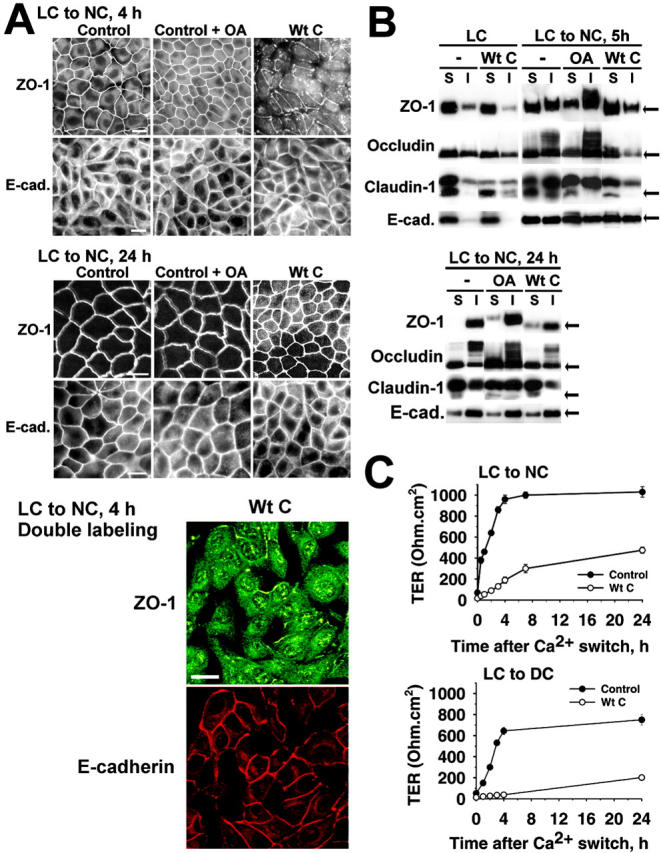
Effects of deregulating PP2A activity on the distribution, detergent solubility, and phosphorylation state of junctional proteins, and development of TER during junctional assembly. (A) Control MDCK or MDCK-Wt C cell monolayers were switched for 4 or 24 h from LC to NC medium, and analyzed by immunofluorescence for the distribution of ZO-1 or E-cadherin. Subsets of control cells were incubated for 1 h with 100 nM OA before staining. The distribution of ZO-1 was comparable to those of occludin and claudin-1 (not depicted). Bars, 10 μM. (B) Control MDCK or MDCK-Wt C cells were switched for 5 or 24 h from LC to NC medium. Control cells were incubated for 2 h with (OA) or without (−) 100 nM OA before harvesting. Equivalent amounts of proteins (∼20 μg) from detergent-soluble (S) and -insoluble (I) fractions were prepared from the cells and simultaneously analyzed by Western blotting for the presence of ZO-1, occludin, claudin-1, or E-cadherin. Arrows indicate the position of dephosphorylated proteins. (C) Polarized control MDCK and MDCK-Wt C cells were incubated for 90 min in LC medium containing 1 mM EGTA to induce TJ opening (t = 0; TER < 30 Ω.cm2), then switched to NC or DC medium for the indicated time. Results are expressed as the mean TER ± SD of duplicate determinations from seven separate experiments performed with seven distinct stable cell populations.
TJ assembly is associated with resistance of newly formed TJ protein complexes to nonionic detergent extraction (Stuart and Nigam, 1995; Wong, 1997; Farshori and Kachar, 1999). To further determine how PP2A influences the phosphorylation state and association of ZO-1, occludin, and claudin-1 with TJs, we compared by immunoblotting their distribution in detergent-soluble/insoluble cell fractions during Ca2+ switch experiments (Fig. 6 B). The experimental conditions were optimized to allow for visualization of changes in the electrophoretic mobility/banding pattern of TJ proteins, which could be linked to changes in their phosphorylation levels (Fig. 4 B). When MDCK monolayers were exposed to prolonged Ca2+ starvation, ZO-1, occludin, and claudin-1 were predominantly observed in the detergent-soluble fraction, as reported previously (Stuart and Nigam, 1995; Wong, 1997; Farshori and Kachar, 1999). Expressed Wt C stimulated the accumulation of detergent-soluble, dephosphorylated TJ proteins. TJ reassembly was induced by switching MDCK cells from LC to NC medium and correlated with the progressive appearance of detergent-insoluble, slow migrating phosphorylated ZO-1 and occludin species (Wong, 1997). In accordance with literature data showing that claudin-1 becomes more resistant to detergent extraction during TJ assembly (Chen et al., 2000), the Ca2+ switch was marked by the buildup of a detergent-insoluble, slow-migrating claudin-1 band. 24 h after the Ca2+switch, most of ZO-1 and occludin were phosphorylated and detergent-insoluble in MDCK cells, as expected from previous studies (Stuart and Nigam, 1995; Sakakibara et al., 1997; Farshori and Kachar, 1999) and from our immunofluorescent pictures showing complete membrane redistribution of ZO-1 (Fig. 6 A). Claudin-1 was more detergent extractable than ZO-1 and occludin (Nishiyama et al., 2001), and slow-migrating claudin-1 species were present in both soluble/insoluble fractions. Expression of Wt C dramatically inhibited the phosphorylation and recruitment of TJ proteins to detergent-insoluble fractions during TJ biogenesis. Even 24 h after the Ca2+ switch, higher levels of detergent-soluble, dephosphorylated TJ proteins were still present in MDCK-Wt C compared to control cells, in agreement with our immunofluorescent data showing residual ZO-1 cytosolic staining in MDCK-Wt C cells (Fig. 6 A). Reciprocally, the relative amounts of detergent-insoluble, phosphorylated TJ proteins were largely increased in OA-treated cells during TJ assembly, supporting the hypothesis that inhibition of PP2A activity promotes the phosphorylation and association of ZO-1, claudin-1, and occludin with TJs. In contrast, changes in PP2A activity did not affect the redistribution of E-cadherin during junctional biogenesis.
TJ assembly also correlates with the development of TER. When MDCK-Wt C cells were switched from LC to NC medium to induce TJ resealing, they developed TER with much slower kinetics than control cells (Fig. 6 C). This delay in TER development was exacerbated when the Ca2+ switch was performed under serum-free conditions (LC to DC), validating our hypothesis that PP2A activity negatively regulates TJ assembly.
Effects of PP2A deregulation on F-actin
The complete reestablishment of the actin cytoskeleton architecture plays a crucial role during Ca2+-mediated junctional biogenesis, and the contraction of perijunctional F-actin critically regulates the TJ permeability barrier (Denker and Nigam, 1998). Thus, we addressed the hypothesis that PP2A regulates TJs via F-actin remodeling. As shown in Fig. 7, we could not observe any significant difference in the organization of F-actin in MDCK cells subjected to a Ca2+ switch, whether they were treated with OA, or transiently or stably expressing HA-tagged Wt C. Notably, subconfluent MDCK cells expressing high levels of transfected Wt C subunits exhibited a flattened, irregular shape with occasional lamellipodia, that was somewhat reminiscent of the morphology of cells expressing dominant-negative mutants of atypical protein kinase C (aPKC) (Suzuki et al., 2001). Overall, OA treatment or Wt C expression did not dramatically affect the appearance of the cortical F-actin bundles (see “apical” sections) and stress fibers (see “basal” sections) present in confluent cells cultured in NC medium. However, now and then, the pattern of stress fibers appeared interwoven or less dense in confluent MDCK-Wt C cells.
Figure 7.
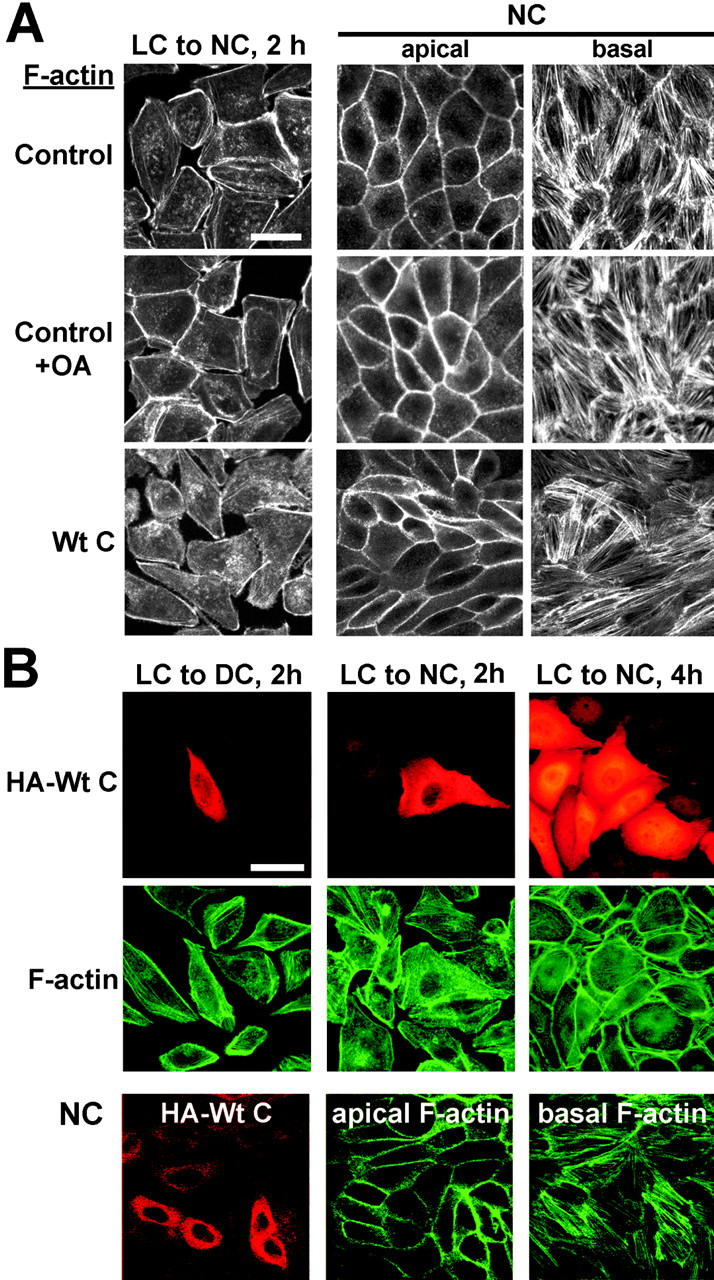
Effects of PP2A deregulation on the organization of F-actin. (A) Immunofluorescent localization of F-actin in control MDCK or MDCK-Wt C cells switched for 2 h from LC to NC medium or cultured in NC medium. Subsets of control cells were incubated for 1 h with 100 nM OA before staining. (B) MDCK cells were transiently transfected with the pcDNA3.1 vector encoding HA-tagged Wt C, and seeded at confluency on coverslips 24 h posttransfection. After plating, cells were either incubated overnight in LC medium then switched to DC or NC medium for the indicated time, or cultured in NC medium. Cells were double stained for the HA epitope and F-actin. For A and B, representative apical and basal x-y sections are shown for cells cultured in NC medium. Bars, 10 μM.
aPKC-dependent regulation of TJ proteins by PP2A
The aPKC isoforms, PKCζ, and PKCλ, are major TJ-associated protein kinases that critically participate in the establishment of epithelial TJ structures (Suzuki et al., 2001). In light of the functional importance of aPKC in TJ biogenesis, we investigated its role in PP2A-dependent TJ protein regulation. First, we found that ABαC was able to dephosphorylate in vitro PKCζ-phosphorylated ZO-1, occludin, and claudin-1 in an OA-sensitive manner (Fig. 8 A). We then examined the effects of inhibiting aPKC on OA-induced redistribution of TJ proteins. aPKC activity was blocked by incubating MDCK cells with the cell-permeable, myristoylated PKCζ/λ pseudosubstrate (ZI), which directly inhibits aPKC autophosphorylation and transactivation (Standaert et al., 1999a). Although OA alone was sufficient to promote the translocation of ZO-1, occludin, and claudin-1 from the cytosol to the membrane in MDCK cells incubated in LC medium, this effect was blocked by preincubating cells with ZI (Fig. 8 B). OA-stimulated accumulation of TJ proteins at junctional areas was also greatly affected by ZI in cells switched from LC to DC medium, as indicated by the disrupted junctional staining pattern (Fig. 8 B) and reduced amounts of detergent-insoluble TJ proteins (Fig. 8 C) in ZI-treated cells.
Figure 8.
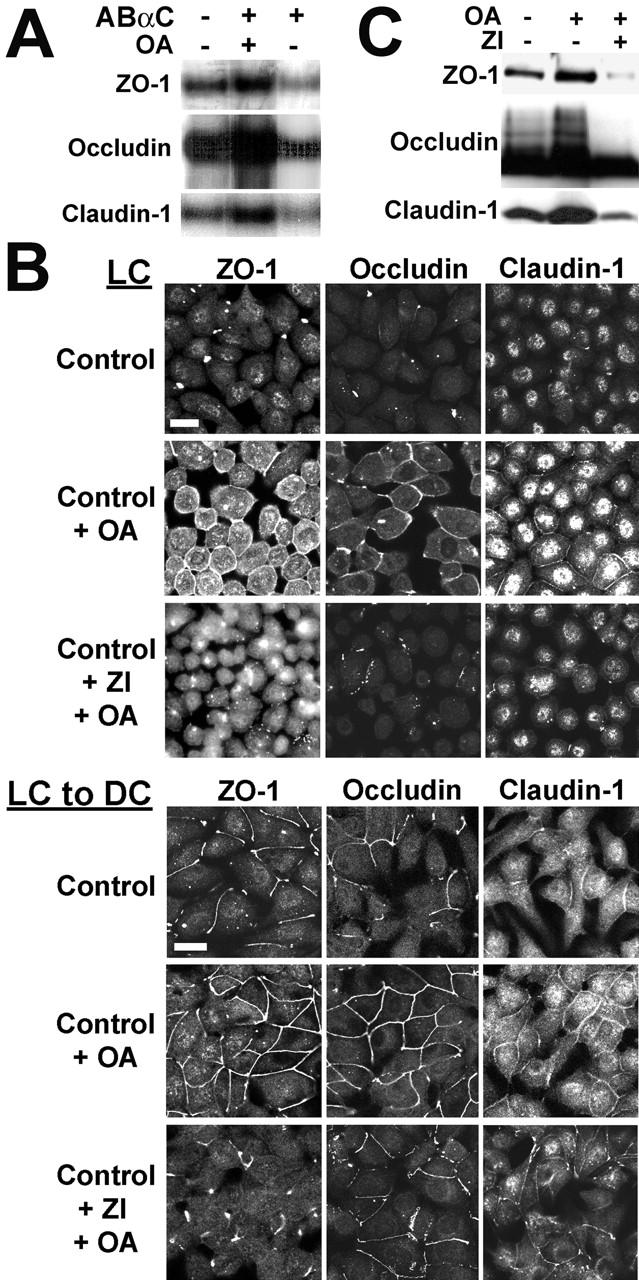
aPKC-dependent regulation of TJ proteins by PP2A. (A) In vitro PKCζ-phosphorylated TJ proteins were incubated with 100 nM ABαC, 100 nM ABαC plus 1 μM OA, or the phosphorylation buffer alone. The samples were resolved by SDS-PAGE and analyzed by autoradiography. Only major radioactive bands corresponding to PKCζ-phosphorylated ZO-1, occludin, and claudin-1 could be detected on the autoradiographs. (B) Immunofluorescent localization of TJ proteins in confluent MDCK cells incubated overnight in LC medium, or switched for 2 h from LC to DC medium. Subsets of cells were preincubated for 1 h in the absence or presence of 20 μM myristoylated PKCζ/λ pseudosubstrate (ZI), then incubated for 1 h with 50 nM OA before staining. Bars, 10 μM. (C) Confluent MDCK cells were switched for 1 h from LC to DC medium containing or not 50 μM ZI, then incubated for 2 h with or without 100 nM OA. Equivalent amounts of proteins from detergent-insoluble fractions were prepared from the cells and analyzed by Western blotting for the presence of TJ proteins.
PP2A interacts with and regulates aPKC
We have previously reported that PP2A is a critical regulator of PKCζ signaling in fibroblasts (Sontag et al., 1997). Moreover, OA activates aPKCζ/λ in adipocytes (Standaert et al., 1999b), raising the possibility that PKCζ is a target for PP2A in epithelial cells. Indeed, ABαC colocalized with PKCζ at the apical membrane of MDCK cells (Fig. 9 A) and associated with both cytosolic and membrane-associated PKCζ during immunoprecipitation assays (Fig. 9 B). OA clearly promoted the membrane translocation of PKCζ in LC medium and its accumulation at junctional areas during TJ assembly performed under serum-free conditions (Fig. 9 C). In contrast, severe defects in the recruitment of PKCζ into junctional complexes were observed in MDCK-Wt C cells during TJ biogenesis. Compared to control cells, the staining pattern of PKCζ at the membrane appeared irregular and discontinuous. Abundant cytoplasmic pools of the kinase remained present in MDCK-Wt C cells, even 24 h after the Ca2+ switch. In support for a direct role of PP2A in the regulation of aPKC during TJ assembly, OA induced a ∼2.7-fold activation of membrane-associated aPKC activity in MDCK cells switched from LC to DC medium, whereas aPKC activity was inhibited by ∼50% in MDCK-Wt C cell membrane fractions, relative to the control (Fig. 9 D). Phosphorylation of PKCζ on Thr410 is essential for intrinsic PKCζ activity and subsequent activation (Standaert et al., 1999a). Accordingly, OA promoted, whereas expressed Wt C prevented the phosphorylation of PKCζ on Thr410 during TJ assembly performed in the absence of serum (Fig. 9 E). As with PKCζ, PP2A also associated with and regulated PKCλ activity and distribution (unpublished data). Thus, PP2A directly participates in the regulation of aPKC in MDCK cells.
Figure 9.
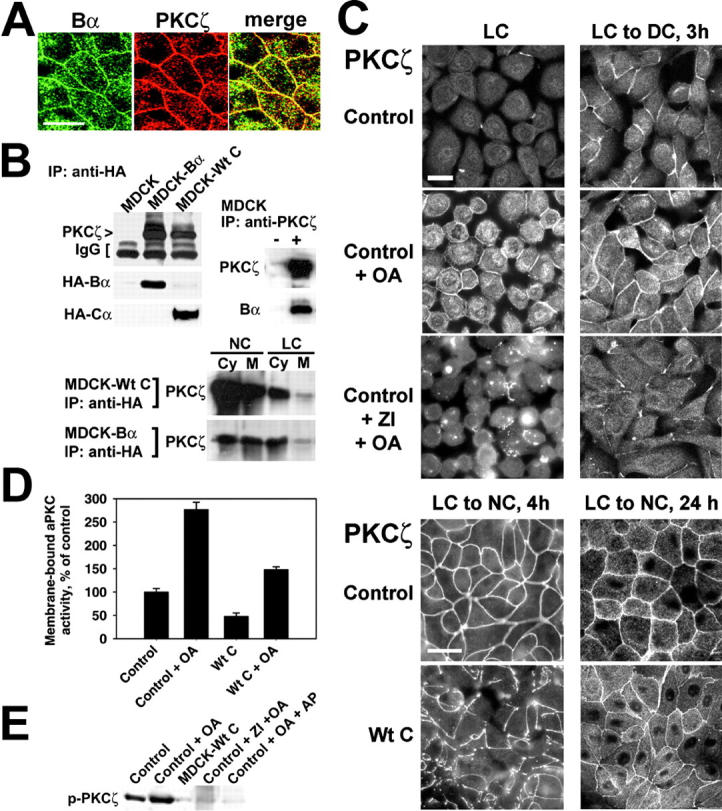
PP2A associates with and regulates aPKC. (A) Representative apical x-y sections of polarized MDCK cells double labeled with rabbit anti-Bα and rat anti-PKCζ antibodies. Bar, 10 μM. (B) Representative immunoblots of MDCK, MDCK-Bα, and MDCK-Wt C cell extracts immunoprecipitated with anti-HA antibody, of MDCK cell detergent-insoluble fractions immunoprecipitated with anti-PKCζ (+) or no (−) antibody, and of cytosolic (Cy) and membrane (M) fractions prepared from MDCK-Wt C or MDCK-Bα cells cultured in NC medium (NC) or LC medium (LC), and immunoprecipitated with anti-HA antibody. IgG, immunoglobulins. (C) Control and MDCK-Wt C cells were incubated overnight in LC medium and switched for the indicated time to DC or NC medium. Subsets of control cells were incubated for 2 h in the absence or presence of 20 μM ZI, then for 1 h with 50 nM OA before fixation and staining with rat anti-PKCζ antibody. Bars, 10 μM. (D) Control and MDCK-Wt C cells were switched from LC to DC medium in the absence or presence of 100 nM OA. PKCζ immunoprecipitates were prepared from membrane fractions 2 h after the Ca2+ switch and assayed for aPKC kinase activity. aPKC activity was completely abolished when kinase assays were performed in the presence of 10 μM ZI (not depicted). For (A–D), similar results were obtained when using either mouse anti-PKCλ antibody or rabbit anti-PKCζ (C-20) antibody, which recognizes both PKCζ and PKCλ (not depicted). (E) Control MDCK or MDCK-Wt C cells were switched for 1 h to DC medium with or without 50 μM ZI, then incubated for another 2 h with or without 100 nM OA. Detergent-insoluble fractions were prepared from the cells and equivalent amounts of proteins (∼100 μg) were analyzed by immunoblotting for the presence of phosphorylated PKCζ using anti–p-nPKCζ (Thr410) antibody. A duplicate aliquot of OA-treated cell fraction was incubated with alkaline phosphatase (+ AP). Note the disappearance of the signal for phosphorylated PKCζ after AP treatment.
Discusssion
We show that a pool of ABαC is recruited to peripheral regions of intercellular contact in a Ca2+-dependent manner. To our knowledge, this is the first example of Ca2+-dependent translocation of ABαC. Although the importance of signal-regulated translocation of protein kinases is well recognized (Sim and Scott, 1999), the concept of signal-dependent redistribution of specific PP2A enzymes is fairly groundbreaking. We show that ABαC is concentrated at the apical membrane of polarized MDCK cells, where it interacts with TJ proteins. Our experimental conditions were aimed at visualizing membrane-associated PP2A; PP2A is likely present in other subcellular compartments not described here. Because aPKC indirectly binds to ZO-1 via the junctional-adhesion molecule (JAM) (Ebnet et al., 2001; Itoh et al., 2001) and may interact with occludin (Nusrat et al., 2000), and ZO-1 binds to both occludin (Furuse et al., 1994) and claudin-1 (Itoh et al., 1999), the precise PP2A binding partner at the TJ has yet to be identified. Nevertheless, our conclusion that ABαC interacts with a multiprotein TJ complex is strengthened by our findings showing the Ca2+ dependency of the association of PP2A with TJ proteins at the membrane, and the presence of PP2A at TJs in human colon. In contrast to other signaling molecules, a direct interaction of specific Ser/Thr phosphatases with TJs has never been described. Thus, it is significant that ABαC associates with the TJ complex; because of its localization, it is a strong candidate for regulating the phosphorylation state of TJ proteins.
So far, the evidence for a role for Ser/Thr phosphatases in TJ regulation is fragmentary, and derives entirely from studies utilizing OA and calyculin A, which inhibit PP2A/PP1 enzymes in vivo in a dose- and time-dependent manner. Interestingly, these inhibitors are naturally occurring toxins that modulate intestinal paracellular permeability and are responsible for diarrheic shellfish poisoning in humans (Tripuraneni et al., 1997; Okada et al., 2000). As reported previously in other epithelial cells (Singer et al., 1994; Tripuraneni et al., 1997; Okada et al., 2000), we observed that high concentrations of OA or prolonged incubation of MDCK cells with this inhibitor induced cell rounding and TJ leakiness. Under these conditions, not only PP2A but also PP1 enzymes became inhibited, and a direct role for PP2A in TJ regulation cannot be meaningfully evaluated. Instead, lower OA concentrations more selective for PP2A did not appreciably alter the organization of the TJ network and the resistance and paracellular permeability of MDCK cells grown in NC medium, in agreement with previous studies (Pasdar et al., 1995; Tripuraneni et al., 1997; Okada et al., 2000). At first sight, this may suggest that PP2A does not regulate TJs once formed. However, enhanced PP2A activity induced dephosphorylation of membrane-associated TJ proteins, leading to decreased TER and increased TJ permeability. Based on previous studies pointing to an important role for occludin dephosphorylation in promoting TJ opening (Farshori and Kachar, 1999; Clarke et al., 2000; Simonovic et al., 2000), reduced levels of TJ-associated, phosphorylated occludin may contribute in part to enhanced TJ leakiness in MDCK-Wt C cells. The fact that OA can inhibit the effects of expressed Wt C suggests that PP2A-dependent changes in the phosphorylation state of TJ-bound proteins modulate the dynamic opening/closing of mature TJs. Many signaling molecules mediate changes in TJ barrier properties via remodeling of the actin cytoskeleton (Denker and Nigam, 1998). Yet, we were unable to demonstrate that changes in PP2A activity induce clear-cut effects on F-actin organization under our experimental conditions. Likewise, OA does not significantly affect F-actin distribution in carcinoma cells (Strnad et al., 2001). However, the effects of OA on F-actin are dose- and time-dependent (Leira et al., 2001); high OA concentrations consistent with PP1 inhibition disrupt F-actin (Fiorentini et al., 1996).
OA promoted the phosphorylation and recruitment of TJ proteins to TJs during junctional assembly. Reciprocally, expressed Wt C induced the accumulation of soluble, dephosphorylated forms of TJ proteins, which correlated with a severe delay in the sorting of TJ proteins from the cytosol to TJs, and retardation in TER development. The observation that OA induces the membrane translocation of TJ proteins in LC medium and reverses the effects of Wt C in the absence of serum is consistent with the hypothesis that inhibition of PP2A promotes TJ assembly. Thus, the phosphorylation state of TJ proteins is controlled by PP2A and is closely linked to their ability to redistribute to the membrane during junctional biogenesis. It has been proposed that the nucleation of AJ assembly by cadherin/catenin complexes controls TJ assembly (Gumbiner et al., 1988). Although PP2A Cα subunit associates with and stabilizes the β-catenin/E-cadherin complex in immature blastocysts (Gotz et al., 2000), we were unable to colocalize or coimmunoprecipitate ABαC and E-cadherin in polarized MDCK cells. The accumulation of E-cadherin at regions of cell–cell contact also proceeded normally despite PP2A deregulation, suggesting that PP2A-dependent defects in TJ assembly do not occur secondary to abnormalities in AJ formation. Together with the observations that AB56C, but not ABαC, regulates β-catenin in the Wnt signaling pathway (Li et al., 2001), and that ABαC, but not AB56C, associates with TJ proteins, our findings suggest that the regulation of TJs by PP2A likely involves an E-cadherin–independent pathway (Stuart and Nigam, 1995) controlled by ABαC. However, the hierarchical regulation of junctional complexes is not absolute but rather multifaceted, as inhibition of cadherin adhesion can exert both negative and positive effects on TJ biogenesis in MDCK cells (Troxell et al., 2000). It is thus possible that distinct PP2A holoenzymes differentially affect separate signaling pathways that converge on TJs.
The results from our inhibitor studies indicate that aPKC participates in the regulation of ZO-1, occludin, and claudin-1 by PP2A. OA induced a 2.7-fold activation of membrane-bound aPKC, in agreement with the finding that membrane-associated PKC activity more than doubles during TJ assembly (Stuart and Nigam, 1995). Our conclusions support those of previous reports showing the importance of aPKC-mediated TJ protein phosphorylation for Ca2+-induced TJ assembly (Stuart and Nigam, 1995; Suzuki et al., 2001). PP2A associated with both soluble and membrane-bound aPKC and modulated aPKC activity and distribution. Thus, PP2A may be part of and directly regulate the aPKC/Par-3 signaling complex that is involved in regulation of TJ formation and cell polarity. The aPKC/Par-3 complex is tethered to TJs via JAM. It is noteworthy that, as with Wt C, overexpression of a JAM mutant disrupts the localization of aPKC and ZO-1 without significantly affecting the localization of E-cadherin (Ebnet et al., 2001).
In conclusion, we demonstrate that ABαC is recruited to and is a novel component and regulator of the TJ signaling complex. The regulation of TJs by PP2A is dependent on functional aPKC. We establish PP2A as a novel regulator of aPKC and identify the TJ proteins, ZO-1, occludin, and claudin-1, as targets of PP2A/aPKC signaling in epithelial cells.
Materials and methods
Plasmids and antibodies
The pcDNA3.1 vector encoding HA-tagged Wt C has been described previously (Ogris et al., 1997; Goedert et al., 2000). The pcDNA3.1 vector (Invitrogen) encoding HA-tagged Bα was prepared from pcDNA1/Amp-HA-Bα (Yu et al., 2001) via NdeI/XhoI restriction sites. Antibodies included: anti-HA (Covance); anti-ZO-1 (Chemicon International, Inc.); anti–claudin-1, -occludin, and -phosphoSer (Zymed Laboratories, Inc.); anti-Cα, anti–E-cadherin, and anti-PKCλ (Transduction Laboratories); anti-PKCζ (Alexis Corporation); anti-PKCζ (C-20), anti–p-nPKCζ (Thr410) (Santa Cruz Biotechnology, Inc.); anti-PP1α (Calbiochem); anti-B56γ subunit (Tehrani et al., 1996); and affinity-purified FITC- and Texas red–coupled secondary antibodies (Jackson ImmunoResearch Laboratories). The rabbit anti-B Δ237 antibody was raised against a fusion protein containing a 6 × His tag fused to amino acids 238–447 of the rat Bα subunit. The rabbit anti-B P9 and monoclonal 2G9 antibodies were raised against a synthetic peptide corresponding to amino acids 398–411 of the rat Bα sequence. Antibodies raised against Bα may crossreact with the β, δ, or γ isoforms of the B subunit family, but expression of Bβ and Bγ is restricted to testis and/or brain, and Bδ is a cytosolic subunit almost undetectable in kidney (for review see Sontag, 2001).
Cell culture and characterization of stable MDCK cell lines
All experiments were performed in the highly polarized MDCK strain II D5 clonal cell line (Brewer and Roth, 1991). Cells were maintained on plastic dishes in DME (Gibco BRL) containing 10% FBS (Hyclone). For most experiments, cells were plated at high density on Transwell filters (Costar) and cultured until complete polarization, as verified by measuring TER. Cells were transfected using Lipofectamine Plus reagent (Gibco BRL). Stable clones were obtained after selection with 800 μg/ml geneticin (Gibco BRL), analyzed for expression of the transfectant, and pooled together to minimize the effects of clonal variations. Seven separate pooled populations of stable transfectants were utilized throughout our studies. The expression level of transfected proteins was constantly monitored by immunofluorescence and immunoblotting. Immunoblotting indicated that HA-tagged Wt C subunit was expressed at ∼30–50% of the levels of endogenous C in stable MDCK cell lines, as previously reported in NIH 3T3 cells (Ogris et al., 1997). HA-tagged Bα subunits were expressed at ∼20% of the levels of endogenous Bα. Stable MDCK cells transfected with the pcDNA3.1 vector were used as controls (control MDCK cells) and behaved like nontransfected cells in our experiments. Sodium butyrate (2.5–5 mM; Sigma-Aldrich) was added in the culture medium for ∼16 h before each experiment to enhance expression of the transfected proteins, and had no detrimental effects on any of the parameters measured in our studies, as reported previously (Brewer and Roth, 1991).
Calcium switch experiments
To induce rapid TJ disassembly, monolayers of MDCK cells grown in normal Ca2+ (NC) medium (DMEM + 10% FBS; 1.8 mM Ca2+) were incubated in LC medium (Ca2+-free S-MEM; Gibco BRL) containing 1 mM EGTA. For prolonged Ca2+ removal, cells were incubated overnight in LC medium containing 1% dialyzed FBS. For the Ca2+ switch, Ca2+-starved cells were transferred to either NC or DC (DMEM + 1% dialyzed FBS) medium. If indicated, OA (Alexis Biochemicals) or the vehicle (100% DMSO) was added to the medium during the Ca2+ switch.
Transepithelial resistance measurements
Cells were plated at confluency and grown on sixwell Transwell filters in NC medium. TER values were measured in duplicate wells using an Endohm™ voltohmmeter (World Precision Instruments). TER values (Ω.cm2) were normalized to the area of the monolayer (filter), and calculated by subtracting the blank values (∼18 Ω.cm2) from the filter and the bathing medium. All cell culture media were supplemented with 25 mM Hepes, pH 7.4, and the integrity and cell density of the monolayers were carefully monitored during TER measurement studies.
Paracellular diffusion measurement
Cells were grown on 6.5-mm Transwell filters until complete polarization. TJ leakiness was assessed by measuring the diffusion of [3H]-inulin (Amersham Biosciences) and [3H]-mannitol (NENTM Life Science Products, Inc.) across the membrane in monolayers of equivalent cell density (Wong and Gumbiner, 1997). The tracer diffusion was examined by replacing the apical compartment medium by fresh NC medium containing the tracer (2.5 μCi/ml), and the basal compartment medium with the same medium without tracer. When indicated, OA or the vehicle (DMSO) was added in both basal and apical bathing media. The compartment media were collected 60 min afterwards, and counted by liquid scintillation.
Confocal microscopy
Cells grown on Transwell filters or on glass coverslips were fixed with methanol for 5 min at −20°C, and labeled sequentially for 1 h with primary and secondary antibodies at a 1:100 dilution (Sontag et al., 1995). For the visualization of F-actin, cells were fixed for 20 min with 4% paraformaldehyde, permeabilized for 5 min with 0.1% Triton X-100, and labeled for 30 min with FITC-labeled phalloidin (Sigma-Aldrich). The samples were mounted using Fluoromount-G (Fisher) and examined on a Leica TCS SP confocal microscope using a 63× objective. Images (16 x-y or x-z sections across cells) were directly captured, saved and transferred to Adobe Photoshop® 5.5 for printing. The specificity of the labeling was verified by omitting first or second antibodies during the staining procedures, and by using pre-immune sera and antibodies that had been preadsorbed with the corresponding antigen.
Electron microscopy
Cryosections (80 nm) from normal human adult colon fixed in 1% glutaraldehyde/4% paraformaldehyde were picked up using a 1.8-mm loop with a droplet of frozen 2.3 M sucrose, thawed, moved onto Formvar-covered and carbon-coated glow-discharged 100 mesh copper grids and stored on buffer. Immunogold labeling of the cryosections was performed by the Tokuyasu method (Tokuyasu, 1986) by which the grids are moved through series of droplets containing blocking agents, antibodies and are ultimately coated with a layer of 0.2% uranyl acetate in 2% methylcellulose. The cryosections were incubated for 30 min each with serial dilutions of the primary antibody in PBS containing 5% milk, and then with protein A gold (10 nm diameter; 1:70 dilution; a gift from G. Posthuma, Utrecht University Medical Center, The Netherlands). The cryosections were examined using a JEOL 1200EX Transmission Electron Microscope operating at 120kV. Electron micrographs were taken using Eastman Kodak SO-163 electron image film.
Cell fractionation and Western blotting
To prepare detergent-soluble and -insoluble fractions, confluent cells were washed in PBS, harvested in 400 μl buffer 1 (25 mM Tris, 150 mM NaCl, 1% NP-40, 4 mM EDTA, 25 mM sodium fluoride, 1 mM sodium orthovanadate, 1 μM OA, pH 7.4) containing a cocktail of protease inhibitors (Roche) and incubated for 30 min at 4°C. The extracts were then centrifuged for 30 min at 4°C in a microfuge to yield a soluble fraction (supernatant), after which the remaining insoluble fraction (pellet) was harvested in the same buffer, sonicated to disrupt protein aggregates, then recentrifuged to eliminate insoluble material. To prepare membrane fractions, cells were washed in PBS and incubated for 5 min at room temperature in buffer S (0.25 M sucrose, 1 mM imidazole, 5 mM MgCl2). The medium was aspirated and cells were harvested in buffer S containing 1 μM OA, 1 mM DTT and a cocktail of protease inhibitors, incubated on ice for 15 min then dounce homogenized. Cells were centrifuged at 800 g at 4°C to pellet nuclei, after which the supernatant was centrifuged at 4°C for 45 min at 100,000 g in a Beckman TL-100.3 rotor. The supernatant (cytosol) was collected and the remaining pellet (membrane fraction) was resuspended in buffer 1 and sonicated. Equivalent aliquots of proteins from the cell fractions determined using Bio-Rad protein assay kit were resolved by SDS-PAGE on 5% (for ZO-1), 8% (for occludin, E-cadherin, and aPKC) and 12% (for PP2A subunits and claudin-1) polyacrylamide gels, and analyzed by immunoblotting for the presence of PP2A subunits or junctional proteins. Immunoreactive proteins were detected using SuperSignal Chemiluminescence substrates (Pierce Chemical Co.).
Immunoprecipitation
Cytosolic/membrane fractions were normalized for protein concentration and volume, after which the buffer was adjusted to 150 mM NaCl and 1% NP-40. After preclearing, total, detergent-soluble/insoluble, or cytosolic/membrane fractions were incubated overnight at 4°C with either the indicated antibodies (∼7 μl antibody/ml cell extract), or no antibody to assess nonspecific binding. The immunoprecipitates were collected using either protein A Sepharose or G PLUS-agarose beads (Santa Cruz Biotechnology), washed extensively in buffer 1, and resuspended in Laemmli sample buffer. In some experiments, the immunoprecipitations were carried out with HA-tagged antibody-coupled affinity matrix (Covance) and control nontransfected cells were used in parallel assays to verify the specificity of the immunoprecipitations (Goedert et al., 2000). Equivalent aliquots of the immunoprecipitates were analyzed by SDS-PAGE on 4–15% gradient Ready gels (Bio-Rad Laboratories) and transferred to nitrocellulose. The blots were cut into strips and simultaneously immunoblotted with antibodies directed against PP2A subunits, PP1α, and junctional proteins to allow for comparative analysis of the relative amounts of immunoprecipitated material in each condition. In other experiments, the colon and kidney from a 10-wk old rat was minced, homogenized in buffer 1 with a tissue grinder and centrifuged at 13,000 g for 15 min to remove insoluble material. Aliquots of the supernatants were immunoprecipitated with mouse anti-occludin antibody and analysed as described above.
Analysis of TJ protein phosphorylation
MDCK cell detergent-insoluble fractions were immunoprecipitated as described above with anti–ZO-1, -occludin, or -claudin-1 antibodies. The immunoprecipitates were washed and resuspended in P buffer (50 mM Tris-HCl, pH 7.4, 5 mM MgCl2, 0.5 mM EGTA, 1 mM DTT, 1 mM sodium fluoride, 100 μM sodium orthovanadate). Phosphorylation of TJ proteins was performed for 1 h at 30°C by adding 10 μg phosphatidylserine, a gift from P. Sternweis (UT Southwestern, Dallas TX), ∼1 μg recombinant human aPKC ζ (Calbiochem), and 100 μM [γ-32P]ATP (5 μCi) per reaction. The tubes containing phosphorylated TJ proteins were transferred on ice and carefully divided into 15-μl aliquots. Either 100 nM purified ABαC (Goedert et al., 2000), 100 nM ABαC preincubated on ice for 20 min with 1 μM OA, or buffer alone, were added into the reaction mixtures. The samples (25 μl) were incubated for another 30 min at 30°C, after which the reactions were terminated by addition of 3× sample buffer for SDS-PAGE. The samples were boiled for 5 min then simultaneously analyzed by SDS-PAGE on 4-20% gradient gels (Bio-Rad Laboratories), followed by autoradiography. In other experiments, ZO-1, occludin, and claudin-1 were immunoprecipitated from membrane fractions, resolved by SDS-PAGE and analysed by immunoblotting using rabbit anti-phosphoserine antibody. The blots were reprobed with anti-ZO-1, occludin, and claudin-1 antibodies. Phosphorylation of PKCζ was examined by immunoblotting using p-nPKCζ (Thr410) antibody (Standaert et al., 1999a). In parallel, aliquots of cell fractions were resuspended in AP buffer (50 mM Tris, pH. 7.4, 50 mM NaCl, 1 mM MgCl2, 1 mM DTT, 0.1% NP-40, and cocktail of protease inhibitors) and incubated for 1 h at 30°C with or without alkaline phosphatase (20 U/sample; Roche) before being analyzed by immunoblotting.
aPKC activity assays
aPKC activity was measured in immunoprecipitates as described previously (Standaert et al., 1999a). After washing in P buffer, aPKC immunoprecipitates were incubated for 15 min at 30°C with 50 μl of P buffer containing 50 μM ATP, 1 μCi [γ-32P]ATP, and 5 μg serine analogue of PKCε-pseudosubstrate (BioSource International), a selective aPKC substrate. The reactions were performed in the presence or absence of 10 μM PKCζ/λ pseudosubstrate (BioSource International), as described previously (Sontag et al., 1997). The reaction mixtures were spotted onto P-81 phosphocellulose paper and washed with 75 mM phosphoric acid. Incorporation of 32P was determined by liquid scintillation counting.
PP2A activity assays
Aliquots of cell extracts were assayed for PP2A activity for 6 min at 30°C using the RRREEE(pT)EEE phosphopeptide (Biosynthesis, Inc.) as a substrate. Dephosphorylation of the peptide was determined by measuring the release of Pi using a quantitative colorimetric assay (Sontag et al., 1999).
Acknowledgments
We thank Dr. M. Roth (Uiversity of Texas Southwestern Medical Center, Dallas, TX) for MDCK cells and technical advice, Dr. D. Pallas (Emory University School of Medicine, Atlanta, GA) for PP2A plasmids, Dr. E. Bigio (University of Texas Southwestern Medical Center) for tissue samples, Dr. C. Kamibayashi (Uiversity of Texas Southwestern Medical Center) for anti-B56 antibodies, and Sivapong Satumtira for technical assistance.
E. Ogris serves as a consultant to Upstate Biotechnology, Inc. This work was supported by National Institutes of Health grant AG18883 to E. Sontag, and a grant from the Austrian Science Foundation (FWF, P13707-MOB) to E. Ogris.
T. Machleidt's present address is RW Johnson Pharmaceutical Research Institute, La Jolla, CA 92121.
Footnotes
Abbreviations used in this paper: AJ, adherens junction; aPKC, atypical PKC; JAM, junctional-adhesion molecule; LC, low Ca2+; MDCK, Madin-Darby canine kidney; NC, normal Ca2+; OA, okadaic acid; PP, protein phosphatase; TER, transepithelial resistance; TJ, tight junction; ZI, PKCζ/λ pseudosubstrate.
References
- Anderson, J.M., B.R. Stevenson, L.A. Jesaitis, D.A. Goodenough, and M.S. Mooseker. 1988. Characterization of ZO-1, a protein component of the tight junction from mouse liver and Madin-Darby canine kidney cells. J. Cell Biol. 106:1141–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer, C.B., and M.G. Roth. 1991. A single amino acid change in the cytoplasmic domain alters the polarized delivery of influenza virus hemagglutinin. J. Cell Biol. 114:413–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cereijido, M., L. Shoshani, and R.G. Contreras. 2000. Molecular physiology and pathophysiology of tight junctions. I. Biogenesis of tight junctions and epithelial polarity. Am. J. Physiol. Gastrointest. Liver Physiol. 279:G477–G482. [DOI] [PubMed] [Google Scholar]
- Chen, Y., Q. Lu, E.E. Schneeberger, and D.A. Goodenough. 2000. Restoration of tight junction structure and barrier function by down-regulation of the mitogen-activated protein kinase pathway in ras-transformed Madin-Darby canine kidney cells. Mol. Biol. Cell. 11:849–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke, H., A.P. Soler, and J.M. Mullin. 2000. Protein kinase C activation leads to dephosphorylation of occludin and tight junction permeability increase in LLC-PK1 epithelial cell sheets. J. Cell Sci. 113:3187–3196. [DOI] [PubMed] [Google Scholar]
- Denker, B.M., and S.K. Nigam. 1998. Molecular structure and assembly of the tight junction. Am. J. Physiol. 274:F1–F9. [DOI] [PubMed] [Google Scholar]
- Ebnet, K., A. Suzuki, Y. Horikoshi, T. Hirose, M.K. Meyer Zu Brickwedde, S. Ohno, and D. Vestweber. 2001. The cell polarity protein ASIP/PAR-3 directly associates with junctional adhesion molecule (JAM). EMBO J. 20:3738–3748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farshori, P., and B. Kachar. 1999. Redistribution and phosphorylation of occludin during opening and resealing of tight junctions in cultured epithelial cells. J. Membr. Biol. 170:147–156. [DOI] [PubMed] [Google Scholar]
- Fiorentini, C., P. Matarrese, A. Fattorossi, and G. Donelli. 1996. Okadaic acid induces changes in the organization of F-actin in intestinal cells. Toxicon. 34:937–945. [DOI] [PubMed] [Google Scholar]
- Furuse, M., T. Hirase, M. Itoh, A. Nagafuchi, S. Yonemura, and S. Tsukita. 1993. Occludin: a novel integral membrane protein localizing at tight junctions. J. Cell Biol. 123:1777–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuse, M., M. Itoh, T. Hirase, A. Nagafuchi, S. Yonemura, S. Tsukita, and S. Tsukita. 1994. Direct association of occludin with ZO-1 and its possible involvement in the localization of occludin at tight junctions. J. Cell Biol. 127:1617–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert, M., S. Satumtira, R. Jakes, M.J. Smith, C. Kamibayashi, C.L. White, and E. Sontag. 2000. Reduced binding of protein phosphatase 2A to tau protein with frontotemporal dementia and parkinsonism linked to chromosome 17 mutations. J. Neurochem. 75:2155–2162. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Mariscal, L., A. Betanzos, and A. Avila-Flores. 2000. MAGUK proteins: structure and role in the tight junction. Semin. Cell Dev. Biol. 11:315–324. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Mariscal, L., R.G. Contreras, J.J. Bolivar, A. Ponce, D.R. Chavez, and M. Cereijido. 1990. Role of calcium in tight junction formation between epithelial cells. Am. J. Physiol. 259:C978–C986. [DOI] [PubMed] [Google Scholar]
- Gotz, J., A. Probst, C. Mistl, R.M. Nitsch, and E. Ehler. 2000. Distinct role of protein phosphatase 2A subunit Calpha in the regulation of E-cadherin and beta-catenin during development. Mech. Dev. 93:83–93. [DOI] [PubMed] [Google Scholar]
- Gumbiner, B., B. Stevenson, and A. Grimaldi. 1988. The role of the cell adhesion molecule uvomorulin in the formation and maintenance of the epithelial junctional complex. J. Cell Biol. 107:1575–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haystead, T.A.J., A.T.R. Sim, D. Carling, R.C. Honnor, Y. Tsukitani, P. Cohen, and D.G. Hardie. 1989. Effects of the tumour promoter okadaic acid on intracellular protein phosphorylation and metabolism. Nature. 337:78–81. [DOI] [PubMed] [Google Scholar]
- Itoh, M., M. Furuse, K. Morita, K. Kubota, M. Saitou, and S. Tsukita. 1999. Direct binding of three tight junction-associated MAGUKs, ZO-1, ZO-2, and ZO-3, with the COOH termini of claudins. J. Cell Biol. 147:1351–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh, M., H. Sasaki, M. Furuse, H. Ozaki, T. Kita, and S. Tsukita. 2001. Junctional adhesion molecule (JAM) binds to PAR-3: a possible mechanism for the recruitment of PAR-3 to tight junctions. J. Cell Biol. 154:491–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leira, F., C. Alvarez, J.M. Vieites, M.R. Vieytes, and L.M. Botana. 2001. Study of cytoskeletal changes induced by okadaic acid in BE(2)-M17 cells by means of a quantitative fluorimetric microplate assay. Toxicol. In Vitro. 15:277–282. [DOI] [PubMed] [Google Scholar]
- Li, X., H.J. Yost, D.M. Virshup, and J.M. Seeling. 2001. Protein phosphatase 2A and its B56 regulatory subunit inhibit Wnt signaling in Xenopus. EMBO J. 20:4122–4131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitic, L.L., C.M. Van Itallie, and J.M. Anderson. 2000. Molecular physiology and pathophysiology of tight junctions I. Tight junction structure and function: lessons from mutant animals and proteins. Am. J. Physiol. Gastrointest. Liver Physiol. 279:G250–G254. [DOI] [PubMed] [Google Scholar]
- Nishiyama, R., T. Sakaguchi, T. Kinugasa, X. Gu, R.P. MacDermott, D.K. Podolsky, and H.C. Reinecker. 2001. IL-2 receptor beta subunit dependent and independent regulation of intestinal epithelial tight junctions. J. Biol. Chem. 276:35571–35580. [DOI] [PubMed] [Google Scholar]
- Nusrat, A., J.A. Chen, C.S. Foley, T.W. Liang, J. Tom, M. Cromwell, C. Quan, and R.J. Mrsny. 2000. The coiled-coil domain of occludin can act to organize structural and functional elements of the epithelial tight junction. J. Biol. Chem. 275:29816–29822. [DOI] [PubMed] [Google Scholar]
- Ogris, E., D.M. Gibson, and D.C. Pallas. 1997. Protein phosphatase 2A subunit assembly: the catalytic subunit carboxy terminus is important for binding cellular B subunit but not polyomavirus middle tumor antigen. Oncogene. 15:911–917. [DOI] [PubMed] [Google Scholar]
- Okada, T., A. Narai, S. Matsunaga, N. Fusetani, and M. Shimizu. 2000. Assessment of the marine toxins by monitoring the integrity of human intestinal Caco-2 cell monolayers. Toxicol. In Vitro. 14:219–226. [DOI] [PubMed] [Google Scholar]
- Pasdar, M., Z. Li, and H. Chan. 1995. Desmosome assembly and disassembly are regulated by reversible protein phosphorylation in cultured epithelial cells. Cell Motil. Cytoskeleton. 30:108–121. [DOI] [PubMed] [Google Scholar]
- Sakakibara, A., M. Furuse, M. Saitou, Y. Ando-Akatsuka, and S. Tsukita. 1997. Possible involvement of phosphorylation of occludin in tight junction formation. J. Cell Biol. 137:1393–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sim, A.T., and J.D. Scott. 1999. Targeting of PKA, PKC and protein phosphatases to cellular microdomains. Cell Calcium. 26:209–217. [DOI] [PubMed] [Google Scholar]
- Simonovic, I., J. Rosenberg, A. Koutsouris, and G. Hecht. 2000. Enteropathogenic Escherichia coli dephosphorylates and dissociates occludin from intestinal epithelial tight junctions. Cell Microbiol. 2:305–315. [DOI] [PubMed] [Google Scholar]
- Singer, K.L., B.R. Stevenson, P.L. Woo, and G.L. Firestone. 1994. Relationship of serine/threonine phosphorylation/dephosphorylation signaling to glucocorticoid regulation of tight junction permeability and ZO-1 distribution in nontransformed mammary epithelial cells. J. Biol. Chem. 269:16108–16115. [PubMed] [Google Scholar]
- Sontag, E. 2001. Protein phosphatase 2A: the Trojan Horse of cellular signaling. Cell. Signal. 13:7–16. [DOI] [PubMed] [Google Scholar]
- Sontag, E., V. Nunbhakdi-Craig, G.S. Bloom, and M.C. Mumby. 1995. A novel pool of protein phosphatase 2A is associated with microtubules and is regulated during the cell cycle. J. Cell Biol. 128:1131–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sontag, E., J.M. Sontag, and A. Garcia. 1997. Protein phosphatase 2A is a critical regulator of protein kinase C zeta signaling targeted by SV40 small t to promote cell growth and NF-kappaB activation. EMBO J. 16:5662–5671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sontag, E., V. Nunbhakdi-Craig, G. Lee, R. Brandt, C. Kamibayashi, J. Kuret, C.L. White, III, M.C. Mumby, and G.S. Bloom. 1999. Molecular interactions among protein phosphatase 2A, tau, and microtubules. Implications for the regulation of tau phosphorylation and the development of tauopathies. J. Biol. Chem. 274:25490–25498. [DOI] [PubMed] [Google Scholar]
- Standaert, M.L., G. Bandyopadhyay, L. Perez, D. Price, L. Galloway, A. Poklepovic, M.P. Sajan, V. Cenni, A. Sirri, J. Moscat, et al. 1999. a. Insulin activates protein kinases C-zeta and C-lambda by an autophosphorylation-dependent mechanism and stimulates their translocation to GLUT4 vesicles and other membrane fractions in rat adipocytes. J. Biol. Chem. 274:25308–25316. [DOI] [PubMed] [Google Scholar]
- Standaert, M.L., G. Bandyopadhyay, M.P. Sajan, L. Cong, M.J. Quon, and R.V. Farese. 1999. b. Okadaic acid activates atypical protein kinase C (zeta/lambda) in rat and 3T3/L1 adipocytes. An apparent requirement for activation of Glut4 translocation and glucose transport. J. Biol. Chem. 274:14074–14078. [DOI] [PubMed] [Google Scholar]
- Strnad, P., R. Windoffer, and R.E. Leube. 2001. In vivo detection of cytokeratin filament network breakdown in cells treated with the phosphatase inhibitor okadaic acid. Cell Tissue Res. 306:277–293. [DOI] [PubMed] [Google Scholar]
- Stuart, R.O., and S.K. Nigam. 1995. Regulated assembly of tight junctions by protein kinase C. Proc. Natl. Acad. Sci. USA. 92:6072–6076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki, A., T. Yamanaka, T. Hirose, N. Manabe, K. Mizuno, M. Shimizu, K. Akimoto, Y. Izumi, T. Ohnishi, and S. Ohno. 2001. Atypical protein kinase C is involved in the evolutionarily conserved par protein complex and plays a critical role in establishing epithelia-specific junctional structures. J. Cell Biol. 152:1183–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tehrani, M.A., M.C. Mumby, and C. Kamibayashi. 1996. Identification of a novel protein phosphatase 2A regulatory subunit highly expressed in muscle. J. Biol. Chem. 271:5164–5170. [DOI] [PubMed] [Google Scholar]
- Tokuyasu, K.T. 1986. Application of cryoultramicrotomy to immunocytochemistry. J. Microsc. 143(Pt 2):139–149. [DOI] [PubMed]
- Tripuraneni, J., A. Koutsouris, L. Pestic, P. De Lanerolle, and G. Hecht. 1997. The toxin of diarrheic shellfish poisoning, okadaic acid, increases intestinal epithelial paracellular permeability. Gastroenterology. 112:100–108. [DOI] [PubMed] [Google Scholar]
- Troxell, M.L., S. Gopalakrishnan, J. McCormack, B.A. Poteat, J. Pennington, S.M. Garringer, E.E. Schneeberger, W.J. Nelson, and J.A. Marrs. 2000. Inhibiting cadherin function by dominant mutant E-cadherin expression increases the extent of tight junction assembly. J. Cell Sci. 113:985–996. [DOI] [PubMed] [Google Scholar]
- Tsukita, S., and M. Furuse. 2000. Pores in the wall: claudins constitute tight junction strands containing aqueous pores. J. Cell Biol. 149:13–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong, V. 1997. Phosphorylation of occludin correlates with occludin localization and function at the tight junction. Am. J. Physiol. 273:C1859–C1867. [DOI] [PubMed] [Google Scholar]
- Wong, V., and B.M. Gumbiner. 1997. A synthetic peptide corresponding to the extracellular domain of occludin perturbs the tight junction permeability barrier. J. Cell Biol. 136:399–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, X.X., X. Du, C.S. Moreno, R.E. Green, E. Ogris, Q. Feng, L. Chou, M.J. McQuoid, and D.C. Pallas. 2001. Methylation of the protein phosphatase 2A catalytic subunit is essential for association of Balpha regulatory subunit but not SG2NA, striatin, or polyomavirus middle tumor antigen. Mol. Biol. Cell. 12:185–199. [DOI] [PMC free article] [PubMed] [Google Scholar]