

Abstract
Surfactant protein D (SP-D) is one of two collectins found in the pulmonary alveolus. On the basis of homology with other collectins, potential functions for SP-D include roles in innate immunity and surfactant metabolism. The SP-D gene was disrupted in embryonic stem cells by homologous recombination to generate mice deficient in SP-D. Mice heterozygous for the mutant SP-D allele had SP-D concentrations that were approximately 50% wild type but no other obvious phenotypic abnormality. Mice totally deficient in SP-D were healthy to 7 months but had a progressive accumulation of surfactant lipids, SP-A, and SP-B in the alveolar space. By 8 weeks the alveolar phospholipid pool was 8-fold higher than wild-type littermates. There was also a 10-fold accumulation of alveolar macrophages in the null mice, and many macrophages were both multinucleated and foamy in appearance. Type II cells in the null mice were hyperplastic and contained giant lamellar bodies. These alterations in surfactant homeostasis were not associated with detectable changes in surfactant surface activity, postnatal respiratory function, or survival. The findings in the SP-D-deficient mice suggest a role for SP-D in surfactant homeostasis.
Surfactant protein D (SP-D) is a 43-kDa glycoprotein (1) synthesized and secreted by pulmonary alveolar type II cells and nonciliated airway cells (2) and by cells of the gastric mucosa (3). SP-D, SP-A, and mannose binding protein (MBP) are collectins, members of the C-type lectin family (4). The collectins are composed of four domains: a short amino-terminal region with interchain disulfide bonds, a long collagen-like domain, a coiled-coil neck region and a calcium-dependent carbohydrate recognition domain. The basic structural unit of each collectin is a trimer based on the collagen-like triple helix, but the arrangement of multiple trimers into higher order oligomers varies (4). The close linkage of the mouse collectin genes on chromosome 14 suggests the collectins arose by ancestral gene duplication (5).
It is not known whether the genetic and structural relationships among the collectins lead to related functions. There are variable degrees of evidence for each of the collectins having a role in innate immunity (for review, see ref. 6). Humans with low levels of MBP secondary to gene mutations are predisposed to infections (7, 8), and mice deficient in SP-A secondary to gene targeting have delayed clearing of certain intratracheal bacterial challenges (9). Consistent with a possible role in innate immunity, SP-D binds to both microbes and phagocytic cells in vitro (6, 10), yet there is no direct evidence that SP-D has a role in host defense in vivo.
Although SP-D was initially called a SP because it was expressed in the alveolar type II cell and had striking biochemical similarities to SP-A (1), a role for SP-D in surfactant homeostasis has not been established. Some surfactant phospholipid is associated with SP-D purified from alveolar lavage (11), and SP-D will interact with surfactant phospholipids in vitro under certain circumstances (12–14). SP-D also binds to both type II cell apical membranes and alveolar macrophages (6), cells that participate in alveolar surfactant metabolism (15). To develop a model to test the role of SP-D, we have produced mice lacking in SP-D secondary to the disruption of the single copy mouse SP-D gene by homologous recombination. Initial characterization of the phenotype demonstrates a progressive alveolar surfactant accumulation and a striking increase in foamy alveolar macrophages and alteration in type II cell morphology. These findings differ markedly from the results of SP-A gene targeting (16, 17) and show that deletion of SP-D alters surfactant homeostasis.
MATERIALS AND METHODS
Generation of SP-D-Deficient Mice.
A murine 129 strain genomic library (Stratagene) was screened by using a 1.2-kb full-length SP-D cDNA to obtain a 15-kb genomic fragment containing all but the extreme 3′ end of the structural gene. A replacement-type targeting vector containing 1.2-kb and 4.3-kb homology regions was constructed by standard methods (Fig. 1a). Pgk-neo (1.8 kb) for positive selection replaced all of exon 2, including the translation start site for murine SP-D, and short segments of flanking intronic sequence (2.8 kb). Pgk-tk was inserted 5′ to the regions of homology for negative selection. The targeting vector was linearized by using a unique NotI site and electroporated into CB1–4 embryonic stem cells as described (18). After G418 (300 μg/ml)/1-(2-deoxy-2-fluoro-d-arabinofuranosyl-5-iodouracil (0.3 μM) selection for 7 days, surviving colonies were picked and screened for homologous recombination by PCR using primers specific for the targeted allele.
Figure 1.

Targeting strategy for SP-D gene disruption and confirmation of the expected null mutation by genotype analysis. (a) The thin line depicts the mouse SP-D gene, with the eight exons indicated by boxes. Pgk-neo replaced all of exon 2 that contains the translation start site for SP-D. The 600-bp probe used for Southern blot screening is located 5′ to the targeting vector. B, BstXI; r, EcoRI; H, HindIII; X, XbaI. (b) Southern blot of tail genomic DNA from wild type (+/+), heterozygous (+/−), and SP-D null (−/−) mice and embryonic stem clone DNA after BstXI digestion. The 6.2-kb BstXI band is the normal allele and the 3.1-kb BstXI band the mutant allele.
The presence of the targeted allele (a 1.3-kb PCR product) in five clones was confirmed by Southern blot hybridization of BstXI-digested genomic DNA to a 600-bp BstXI–XbaI probe corresponding to genomic sequence 5′ to the targeting construct. This probe yielded either the predicted 6.2-kb fragment characteristic of the wild-type allele or the 3.1-kb fragment from the targeted mutant allele. Two targeted embryonic stem cell clones containing a disrupted SP-D allele were injected separately into day 2.5 postcoital eight-cell to morula stage CD-1 zygotes, cultured overnight to blastocysts, and transferred to pseudopregnant B6D2 females. Chimeric offspring were bred with albino CD-1 female mice, and F1 agouti offspring were screened by PCR and Southern blot hybridization (Fig. 1b) for germ-line transmission of the mutant allele. Mice heterozygous for the SP-D mutation were intercrossed to produce mice of all three genotypes (+/+), (+/−), and (−/−).
Northern Blot Analysis.
Total RNA (10 μg) isolated from the lungs and livers of four 8-week-old mice of each genotype by the guanidine isothiocyanate method was hybridized with 32P-labeled cDNA probes for mouse SP-D, SP-A, SP-B, SP-C, MBP, and actin as described (2). Signals were quantified in a PhosphoImager (Molecular Dynamics) and normalized to actin.
Western Blot Analysis and Bronchoalveolar Lavage.
SDS/PAGE and Western blots were performed by using standard techniques and developed by using the ECL detection reagents (Amersham). All blots were performed with polyclonal antibodies to the surfactant apoproteins or MBP. To analyze tissue, the lungs of 8-week-old mice were homogenized in 10 mM Tris⋅HCl/100 mM NaCl, pH 7.4 (TBS), containing 0.25 M sucrose, 2 mM EDTA, 1 mM phenylmethylsulfonyl fluoride, 10 μM leupeptin, and 10 μM pepstatin A, and centrifuged at 200 × g at 4°C for 10 min to remove cellular debris. The supernatant was centrifuged at 120,000 × g at 4°C for 16 h and the pellet resuspended in TBS for SDS/PAGE. To analyze bronchoalveolar lavage (BAL), lungs were lavaged with four 1-ml aliquots of TBS/0.2 mM EGTA. The total BAL protein concentration was determined by using the Lowry method in the presence of 0.1% SDS to reduce turbidity (19). Western blots were performed without further fractionation of the BAL. Serial dilutions of whole BAL were analyzed for SP-A and SP-D content by using semiquantitative dot blots. Concentrations in the +/− and −/− BAL were normalized to the +/+ values. Serum obtained from seven 22-week-old mice of each genotype was analyzed by Western blot for MBP.
BAL Lipid Analysis.
The phospholipid content of the BAL, before and after removing cells by centrifugation at 150 × g for 5 min, was determined at 3, 8, and 22 weeks. The phospholipid content of lung homogenate before BAL was determined for mice at 1 week and after BAL at 3, 8, and 22 weeks of age. The lung homogenates and BAL were extracted into chloroform/methanol and the total phospholipid content was derived from the phosphorus concentration (20). To determine phospholipid composition, extracted BAL phospholipids were separated by TLC (21). Cholesterol concentration in BAL fluid was determined by the cholesterol oxidase method (Sigma).
Microscopy.
The lungs from two mice of each genotype before birth, at 21 days gestation (term), 3 weeks, 8 weeks, and 18 weeks postnatal age were fixed by intratracheal instillation of 2% glutaraldehyde/1% paraformaldehyde in 0.1 M sodium phosphate buffer (pH 7.4; 2 h at room temperature) and then postfixed overnight in 1.5% osmium tetroxide in Veronal acetate buffer at 4°C. They were stained en bloc in 1.5% uranyl acetate in maleate buffer and then quickly dehydrated in ice-cold acetone and propylene oxide. The tissue was finally infiltrated and embedded in LX 112 (Ladd Research Industries, Burlington, VT). Semithin sections were stained with toluidine blue and ultrathin sections were stained with 5% uranyl acetate and 0.8% lead citrate for electron microscopy. Immunochemistry on 2-μm cryosections was performed as described (2) with an antibody to mouse SP-D provided by J. R. Wright (Duke University, Durham, NC).
BAL Cell Analysis.
BAL fluid, in TBS containing 0.2 mM EGTA, from mice at 3, 8, and 22 weeks of age was centrifuged at 200 × g for 5 min at 4°C. The pellet was gently resuspended in 200 μl of lavage buffer for cell count. Cytospin slides were stained with Dif-Quik (Dade International, Miami, FL) for cell differential. Forty high-powered fields from two mice of each genotype were counted.
Surface Activity.
Pooled cell-free BAL fluid from mice at 22–24 weeks was incubated in 2.5 mM CaCl2 for 30 min and centrifuged at 60,000 × g for 60 min. The pellet was gently suspended in sample buffer (100 mM NaCl/1 mM EDTA/3.5 mM CaCl2/10 mM Tris⋅HCl, pH 6.9, at 37°C) at a concentration of 2.0 mg of phospholipid per ml. The sample was put into the chamber of a captive bubble surfactometer (22) and incubated at 37°C for 30 min. An air bubble of about 30 μl was formed and then expanded and compressed as described (23) to measure adsorption rate, surface tension-area isotherms, oscillatory behavior, and film stability. Height and diameter of the bubble were measured and used to calculate area and surface tension (24).
RESULTS
The SP-D gene was inactivated in embryonic stem cells by standard homologous recombination techniques (Fig. 1). Five of the 180 colonies that survived positive/negative selection contained the targeted mutant allele. The mutant allele deleted all of exon 2 and some flanking intronic sequence. Exon 2 contains the translation start site and encodes the leader sequence through to the first part of the collagen-like domain of SP-D. No differences in the analysis of SP-D deficient offspring from the 2 clones were detected, and data from both were pooled for this report. The genotype distribution of 680 F2 progeny of heterozygous F1 mice was 27% +/+, 49% +/−, and 24% −/−, consistent with the expected Mendelian ratio. The SP-D null homozygous and heterozygous mice maintained in a barrier facility were indistinguishable by appearance, weight, and activity from wild type littermates. No infection was detected with routine serological surveillance for viruses and regular autopsy for evidence of pneumonia. No viral inclusions were seen in the mice examined by electron microscopy. The oldest homozygous null mice are now 7 months of age.
SP-D mRNA levels in 8-week-old +/− mice were 40% of wild type (n = 4). Lungs from null mice had no full-length SP-D mRNA (1.35 kb), but low levels (<10% wild type) of a truncated message were detected (Fig. 2a). The truncated message, cloned by reverse transcription-coupled PCR, lacked exon 2 including the translation start site. There was no evidence of translation from an alternate start site, because no SP-D protein (full-length or truncated forms) was detected in whole lung homogenate (Fig. 2b) or in the BAL (Fig. 2c) of SP-D null mice by Western blot with three different polyclonal antibodies to SP-D. In addition, no SP-D was detected by immunohistochemistry in alveolar type II cells or Clara cells in the null mice (Fig. 3). SP-D levels in BAL were 39% of wild type in +/− mice (n = 8; P = 0.004). Thus, the mutated SP-D gene is a functionally null allele.
Figure 2.

Analysis of SP-D mRNA (a) and protein in the lungs (b) and BAL (c) of null mice. (a) Northern blot of lung RNA demonstrating the absence of a 1.35-kb full-length mRNA for SP-D in the lungs of −/− mice. The low level 1-kb mRNA lacking exon 2 is visible. (b) Western blot of lung tissue demonstrating the absence of SP-D in the −/− lung. (c) Western blot of BAL demonstrating the absence of SP-D in −/− BAL.
Figure 3.
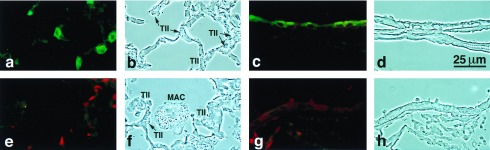
SP-D immunohistochemistry in null and wild-type mice. SP-D is detected in type II cells (a) and Clara cells (c) of wild-type mice but is not detected in SP-D null mice (e–h). Foamy alveolar macrophages are visible in the alveolus of the null (f).
The most striking phenotypic abnormality we have detected in the SP-D null mice is a progressive accumulation of surfactant phospholipid in the lung tissue and alveolar space. The phospholipid content of lungs from 1-week-old mice was similar in all three genotypes, but by 8 weeks the total phospholipid content of the lung tissue after lavage was increased 1.5-fold in −/− mice compared with +/+ mice, and by 18 weeks it was further increased to 3-fold over wild type (n = 4; P = 0.004). The phospholipid content of BAL (before removing cells by low-speed centrifugation) of 3-, 8-, and 18-week-old mice was increased 5-, 8-, and 8-fold, respectively, in null compared with wild type (Fig. 4A). Very little phospholipid was detected in the 200 × g pellet in the +/+ mice, but approximately 10% of the total BAL phospholipid was associated with the 250 × g pellet in null mice. Phospholipid levels in lung tissue and BAL were not significantly increased in heterozygous animals. The BAL phospholipid composition, analyzed by TLC in three mice from each genotype, was not significantly different. The cholesterol content of the cell-free BAL at 8 weeks was increased 6-fold in null compared with wild type mice (n = 5), approximately the same fold increase as the phospholipids.
Figure 4.

BAL analysis of SP-D +/+ (solid bars) and SP-D −/− (open bars). (A) Total BAL phospholipid per mouse at 3, 8, and 18 weeks (n = 8 for each genotype at each age; P < 0.001). (B) Total BAL protein per mouse at 3, 8, and 18 weeks (n = 8 for each genotype at each age; P < 0.01). (C) Number of alveolar macrophages per mouse at 3, 8, and 24 weeks (n = 4 to 7 for each genotype at each age; P < 0.05 at 3 and 24 weeks). Error bars indicate the SEM.
The total protein content of the BAL from −/− mice was increased 4-, 4-, and 2-fold compared with +/+ mice at 3, 8, and 18 weeks, respectively (Fig. 4B). Some of this increase was due to an accumulation of surfactant proteins other than SP-D. At 8 weeks of age, the SP-A protein levels in the BAL of SP-D −/− mice were increased to 2.4-fold over age-matched wild-type levels (n = 13; P = 0.045). SP-A levels were not significantly different from controls in +/− mice. SP-B levels in BAL were elevated approximately 6-fold in −/− mice compared with +/+ mice as estimated by semiquantitative Western blot analysis (n = 4). SP-C levels were not measured. Although SP-A and SP-B accumulated in the alveolar space, there was no evidence for altered gene expression, because SP-A, SP-B and SP-C lung mRNA levels were the same in all three genotypes (n = 4; data not shown). Similarly, there was no evidence for a contiguous gene effect or biologic feedback on MBP expression, because liver mRNA levels and serum MBP concentrations were similar in all three genotypes (data not shown).
Surfactant preparations from both null and wild-type mice formed surface films rapidly (n = 5 for each group). They reached nearly equilibrium tension at 0.3 sec after sudden expansion of the bubble. As shown in Fig. 5, preparations from both wild and null types initially formed highly compressible films, with minimum tensions averaging 13.2 and 17.4 mN/m, respectively. Upon cyclic reduction of bubble size at a frequency of 20 min−1, surface tension fell to very low values with all samples. By the second cycle, surface tension was less than 2.8 mN/m, and the dynamic minimum averaged 1.04 ± 0.99 mN/m for null and 1.1 ± 0.80 mN/m for wild types. Fig. 5 shows that after oscillation of the bubble, the compressibility of the films was dramatically reduced, resulting in a quasistatic minimum tension of 7 mN/m or less in all cases and averaging less than 2 mN/m for both wild and null types. No significant differences between groups in film stability before or after oscillation were detected.
Figure 5.

Effect of bubble oscillation on quasistatic surface tension–area isotherms of surfactants from null and wild-type mice. Open symbols (null) and solid symbols (wild type) represent averages of five preparations. Error bars indicate the standard errors of averages.
There were striking progressive histological changes in the lungs of the SP-D −/− mice (Fig. 6). A preliminary examination of term fetal lungs did not reveal any obvious differences (data not shown), but by 3 weeks of age there was a scattered accumulation of material in the alveolar lumen, most notably in the subpleural regions of the lung, and at high power, an increase in size of the type II cells was evident. By 8 weeks of age, these changes had progressed to involve more of the lung. Some alveoli appeared completely full of secretions, but many alveoli still appeared relatively unaffected. By 8 weeks, a marked increase in alveolar macrophage number and size was apparent, with many macrophages having a foamy appearance. The abnormality in type II cell morphology had also progressed with giant intracellular lamellar bodies evident even with the light microscope. A further progression of each of these abnormalities was evident by 18 weeks with the additional finding of a prominent cellular infiltrate around blood vessels associated with small airways. The infiltrate resembled bronchial-associated lymphoid tissue in location and appearance, but we have not yet positively identified the cells involved. Besides this peribronchial infiltrate, there was no histological evidence of lung inflammation or injury at any age, consistent with the apparent health of the mice. The histology of the lungs of the heterozygous mice did not differ significantly from the wild type at any age.
Figure 6.
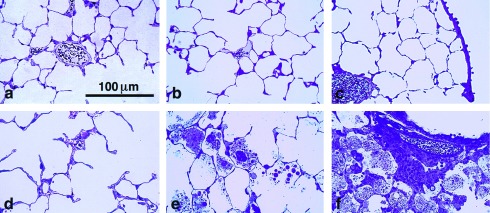
Lung histology demonstrating progressive accumulation of surfactant lipids in the alveolar space, foamy macrophages, and peribronchial infiltrate in SP-D null mice. (a–c) Sections from wild-type mice at 3, 8, and 18 weeks, respectively. (d–f) Sections from SP-D null mice at 3, 8, and 18 weeks, respectively.
The material accumulating in the alveolar space was better defined under the electron microscope (Fig. 7). Alveolar surfactant forms were rarely seen in situ in wild-type lungs but abundant secretions were always present in null mice. Many of the structures resembled surfactant forms seen in normal animals but the accumulation of structures resembling abnormally large lamellar body contents was most striking. Normal appearing tubular myelin and other vesicular structures were all present in increased amounts in the null mice. The type II cells were enlarged and appeared to be more numerous although formal morphometry has not yet been performed. Lamellar bodies appeared to be increased in number, and giant intracellular lamellar bodies were frequent. These changes in type II morphology were detected by 3 weeks and became more pronounced as the mice aged. Alveolar macrophages were grossly enlarged and stuffed with both membrane-bound phospholipid inclusions and cytoplasmic oil droplets, characteristic of foamy macrophages.
Figure 7.

Electron microscopy of an alveolar region demonstrating massive accumulation of intraalveolar surfactant and giant lamellar bodies (c) and stuffed macrophages (b) in the SP-D null mouse compared with the wild-type littermate (a). All micrographs are the same magnification. (Bar = 10 μm in c.)
Consistent with the qualitative impression given by the histology, greater than 95% of the cells in the BAL from each genotype were macrophages. The number of alveolar macrophages progressively increased in SP-D −/− mice compared with age- and litter-matched wild-type controls (Fig. 4C). At 3 weeks the number of alveolar macrophages recovered in the BAL was not significantly different (−/− 1.4-fold +/+), but at 8 and 24 weeks the number of alveolar macrophages in the SP-D-deficient mice was increased 4- and 10-fold compared with wild type (n = 4 to 7 at each age; P < 0.05 and < 0.001, respectively). Qualitatively, the size of the alveolar macrophages in the SP-D −/− mice increased with age (Fig. 8). Multinucleated alveolar macrophages were significantly increased in the BAL of −/− mice compared with +/+ mice at 3 and 22 weeks of age (2.3% vs. 0.4% at 3 weeks and 13.8% vs. 0.5% at 22 weeks; P < 0.01 at both ages).
Figure 8.

Cytospins of BAL cell pellets at 3 weeks (a and b) and 24 weeks (c and d) in wild-type (a and c) and SP-D null mice (b and d). The increased size of the macrophages in the null mice is visible. The arrows indicate multinucleated macrophages from null mice.
DISCUSSION
Disruption of the SP-D gene by deletion of exon 2 led to a functionally null allele and allowed us to breed mice deficient in SP-D. Although healthy and apparently free of infection to 7 months of age, SP-D-deficient mice have striking abnormalities in surfactant homeostasis and alveolar cell morphology. Most notably, there was a progressive accumulation of surfactant lipids and apoproteins in the alveolar space, hyperplasia of type II cells with massive enlargement of intracellular lamellar bodies, and an accumulation of foamy alveolar macrophages. These results were not predicted from preexisting in vitro work, which had failed to show any significant effect of SP-D on type II cell function (25) or surfactant activity (1). In fact, an extensive literature points to a role for the other pulmonary collectin SP-A in surfactant function and metabolism (for review, see ref. 26). In apparent contradiction to the in vitro work, the SP-A-deficient mouse has minimal disturbance in surfactant homeostasis (16, 17), and we now find major changes in surfactant metabolism associated with SP-D deficiency.
We can only speculate about the mechanism of the changes we observed because we have not yet established whether the alveolar surfactant pool is increased due to increased synthesis and secretion of surfactant or delayed clearance or both. The alveolar accumulation of SP-A and SP-B without a detectable change in mRNA levels suggests that a perturbation of alveolar clearance may be most likely, but further experiments are obviously required to test this idea. Whatever the abnormality, it should be noted that the accumulation of surfactant, although significant, is slow in developing, suggesting only a partial disturbance of surfactant homeostasis or the presence of effective counterregulatory mechanisms in the mouse. It is also possible the changes that we see are secondary and not reflective of a direct role of SP-D in surfactant metabolism. On the basis of the multiple immunomodulatory effects of the other collectins (for review, see ref. 6), it is at least reasonable to predict there might be changes in the cytokine or growth factor milieu of the alveoli as a consequence of SP-D deficiency. As recently described, changes in the alveolar cytokine milieu can have dramatic effects on surfactant homeostasis (27–29).
The similarities in pulmonary phenotype between the changes resulting from SP-D deficiency and a disruption in the granulocyte–macrophage colony-stimulating factor (GM-CSF)/GM-CSF receptor pathway are particularly intriguing (27, 29). With both SP-D deficiency and GM-CSF deficiency, there is a slow progressive accumulation of surfactant lipids and apoproteins in the alveolar space, an increase in number of foamy macrophages, and a peribronchial cellular infiltrate. The primary surfactant metabolic abnormality in the GM-CSF/GM-CSF receptor-deficient mice may be diminished surfactant clearance by alveolar macrophages (30, 31). Although SP-D has not been shown to increase surfactant clearance by the alveolar macrophage, it does bind to the surface of these cells (32). Several candidate receptors have been proposed to explain the binding of SP-D to alveolar macrophages including gp-340, a member of the scavenger receptor super family expressed on the surface of alveolar macrophages (33). This report is particularly interesting because scavenger receptors serve as a route for internalization of lipids in macrophages (34) and may mediate the macrophage proliferation induced by exposure to oxidized low density lipoproteins (35). The type II cell hyperplasia and lamellar body abnormalities seen in the SP-D-deficient mice were not reported in either the GM-CSF- or GM-CSF-receptor-deficient mice. Rather, chronic overexpression of GM-CSF in the alveolar space leads to type II cell hyperplasia (36). High levels of alveolar GM-CSF are also associated with alveolar macrophage proliferation and the appearance of multinucleated macrophages (36), both prominent features of the SP-D-deficient mice. Clearly, further work needs to be done to determine whether there is an association of SP-D and its receptor(s), the GM-CSF pathway, and surfactant clearance.
We found no evidence for a direct role for SP-D in the primary biophysical functions of surfactant. The surfactant isolated from null and wild-type mouse lavage liquid was similar in surface activity to that previously reported for rabbit (22, 23) and mouse (17). Despite the voluminous yield from null mice, the material performed like that from the wild type. In both cases it was highly functional in vitro, a result that is compatible with the apparently normal respiratory function of both groups. Although, these techniques do not allow us to exclude a role for SP-D in the formation and maintenance of the surfactant surface film, our results to date are more suggestive of a role in the cell signaling that normally maintains appropriate alveolar surfactant pool sizes.
Acknowledgments
We thank Drs. E. Meyers and J. Vanderbilt, University of California, San Francisco, for help with gene targeting, and Drs. J. R. Wright, Duke University, and F. McCormack, University of Cincinnati, for gifts of antibodies against SP-D and MBP. This work was supported by Grants HL 24075, HL 58047, and DK 47766 from the National Institutes of Health.
ABBREVIATIONS
- SP
surfactant protein
- MBP
mannose binding protein
- BAL
bronchoalveolar lavage
- GM-CSF
granulocyte–macrophage colony-stimulating factor
References
- 1. Persson A, Chang D, Rust K, Moxley M, Longmore W, Crouch E. Biochemistry. 1989;28:6361–6367. doi: 10.1021/bi00441a031. [DOI] [PubMed] [Google Scholar]
- 2.Wong C J, Akiyama J, Allen L, Hawgood S. Pediatr Res. 1996;39:930–937. doi: 10.1203/00006450-199606000-00002. [DOI] [PubMed] [Google Scholar]
- 3.Fisher J H, Mason R. Am J Respir Cell Mol Biol. 1995;12:13–18. doi: 10.1165/ajrcmb.12.1.7811466. [DOI] [PubMed] [Google Scholar]
- 4.Hoppe H J, Reid K B. Protein Sci. 1994;3:1143–1158. doi: 10.1002/pro.5560030801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Akiyama J, Poulain F, Vanderbilt J, Hawgood S. Am J Respir Crit Care Med. 1998;157:A561. (abstr.). [Google Scholar]
- 6.Wright J R. Physiol Rev. 1997;77:931–962. doi: 10.1152/physrev.1997.77.4.931. [DOI] [PubMed] [Google Scholar]
- 7.Summerfield J A. Biochem Soc Trans. 1993;21:473–477. doi: 10.1042/bst0210473. [DOI] [PubMed] [Google Scholar]
- 8.Summerfield J A, Ryder S, Sumiya M, Thursz M, Gorchein A, Monteil M A, Turner M W. Lancet. 1995;345:886–889. doi: 10.1016/s0140-6736(95)90009-8. [DOI] [PubMed] [Google Scholar]
- 9.LeVine A M, Bruno M D, Huelsman K M, Ross G F, Whitsett J A, Korfhagen T R. J Immunol. 1997;158:4336–4340. [PubMed] [Google Scholar]
- 10.Hartshorn K L, Crouch E C, White M R, Eggleton P, Tauber A I, Chang D, Sastry K. J Clin Invest. 1994;94:311–319. doi: 10.1172/JCI117323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kuroki Y, Shiratori M, Ogasawara Y, Tsuzuki A, Akino T. Biochim Biophys Acta. 1991;1086:185–190. doi: 10.1016/0005-2760(91)90006-4. [DOI] [PubMed] [Google Scholar]
- 12.Persson A V, Gibbons B J, Shoemaker J D, Moxley M A, Longmore W J. J Biol Chem. 1992;267:19846–19853. [Google Scholar]
- 13.Ogasawara Y, Kuroki Y, Akino T. J Biol Chem. 1992;270:21244–21249. [PubMed] [Google Scholar]
- 14.Taneva S, Voelker D R, Keough K M. Biochemistry. 1997;36:8173–8179. doi: 10.1021/bi963040h. [DOI] [PubMed] [Google Scholar]
- 15.Magoon M W, Wright J R, Baritussio A, Williams M C, Goerke J, Benson B J, Hamilton R L, Clements J A. Biochim Biophys Acta. 1983;750:18–31. doi: 10.1016/0005-2760(83)90200-x. [DOI] [PubMed] [Google Scholar]
- 16.Korfhagen T R, Bruno M D, Ross G F, Huelsman K M, Ikegami M, Jobe A H, Wert S E, Stripp B R, Morris R E, Glasser S W, et al. Proc Natl Acad Sci USA. 1996;93:9594–9599. doi: 10.1073/pnas.93.18.9594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ikegami M, Korfhagen T R, Bruno M D, Whitsett J A, Jobe A H. Am J Physiol. 1997;272:L479–L485. doi: 10.1152/ajplung.1997.272.3.L479. [DOI] [PubMed] [Google Scholar]
- 18.Li Y, Huang T T, Carlson E J, Melov S, Ursell P C, Olson J L, Noble L J, Yoshimura M P, Berger C, Chan P H, et al. Nat Genet. 1995;11:376–381. doi: 10.1038/ng1295-376. [DOI] [PubMed] [Google Scholar]
- 19.Lowry O H, Rosebrough N J, Farr A L, Randall R J. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 20.Bartlett G R. J Biol Chem. 1959;234:466–468. [PubMed] [Google Scholar]
- 21.Touchstone J C, Chen J C, Beaver K M. Lipids. 1979;15:61–62. [Google Scholar]
- 22.Putz G, Goerke J, Schürch S, Clements J A. J Appl Physiol. 1994;76:1417–1424. doi: 10.1152/jappl.1994.76.4.1417. [DOI] [PubMed] [Google Scholar]
- 23.Putz G, Goerke J, Clements J A. J Appl Physiol. 1994;77:597–605. doi: 10.1152/jappl.1994.77.2.597. [DOI] [PubMed] [Google Scholar]
- 24.Schoel W M, Schurch S, Goerke J. Biochim Biophys Acta. 1994;1200:281–290. doi: 10.1016/0304-4165(94)90169-4. [DOI] [PubMed] [Google Scholar]
- 25.Kuroki Y, Shiratori M, Murata Y, Akino T. Biochem J. 1991;279:115–119. doi: 10.1042/bj2790115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Johansson J, Curstedt T. Eur J Biochem. 1997;244:675–693. doi: 10.1111/j.1432-1033.1997.00675.x. [DOI] [PubMed] [Google Scholar]
- 27.Dranoff G, Crawford A D, Sadelain M, Ream B, Rashid A, Bronson R T, Dickersin G R, Bachurski C J, Mark E L, Whitsett J A, et al. Science. 1994;264:713–716. doi: 10.1126/science.8171324. [DOI] [PubMed] [Google Scholar]
- 28.Jain-Vora S, Wert S E, Temann U A, Rankin J A, Whitsett J A. Am J Respir Cell Mol Biol. 1997;17:541–551. doi: 10.1165/ajrcmb.17.5.2883. [DOI] [PubMed] [Google Scholar]
- 29.Nishinakamura R, Nakayama N, Hirabayashi Y, Inoue T, Aud D, McNeil T, Azuma S, Yoshida S, Toyoda Y, Arai K, et al. Immunity. 1995;2:211–22. doi: 10.1016/1074-7613(95)90046-2. [DOI] [PubMed] [Google Scholar]
- 30.Ikegami M, Ueda T, Hull W, Whitsett J A, Mulligan R C, Dranoff G, Jobe A H. Am J Physiol. 1996;270:L650–L658. doi: 10.1152/ajplung.1996.270.4.L650. [DOI] [PubMed] [Google Scholar]
- 31.Cooke K R, Nishinakamura R, Martin T R, Kobzik L, Brewer J, Whitsett J A, Bungard D, Murray R, Ferrara J L. Bone Marrow Transplant. 1997;20:657–662. doi: 10.1038/sj.bmt.1700958. [DOI] [PubMed] [Google Scholar]
- 32.Dong Q, Wright J R. Am J Physiol. 1998;274:L97–L105. doi: 10.1152/ajplung.1998.274.1.L97. [DOI] [PubMed] [Google Scholar]
- 33.Holmskov U, Lawson P, Teisner B, Tornoe I, Willis A C, Morgan C, Koch C, Reid K B. J Biol Chem. 1997;272:13743–13749. doi: 10.1074/jbc.272.21.13743. [DOI] [PubMed] [Google Scholar]
- 34.Nishikawa K, Arai H, Inoue K. J Biol Chem. 1990;265:5226–5231. [PubMed] [Google Scholar]
- 35.Sakai M, Miyazaki A, Hakamata H, Kodama T, Suzuki H, Kobori S, Shichiri M, Horiuchi S. J Biol Chem. 1996;271:27346–27352. doi: 10.1074/jbc.271.44.27346. [DOI] [PubMed] [Google Scholar]
- 36.Huffman Reed J A, Rice W R, Zsengeller Z K, Wert S E, Dranoff G, Whitsett J A. Am J Physiol. 1997;273:L715–L725. doi: 10.1152/ajplung.1997.273.4.L715. [DOI] [PubMed] [Google Scholar]