

Abstract
Adenovirus type 2 (Ad2) binds the coxsackie B virus Ad receptor and is endocytosed upon activation of the αv integrin coreceptors. Here, we demonstrate that expression of dominant negative clathrin hub, eps15, or K44A-dynamin (dyn) inhibited Ad2 uptake into epithelial cells, indicating clathrin-dependent viral endocytosis. Surprisingly, Ad strongly stimulated the endocytic uptake of fluid phase tracers, coincident with virus internalization but without affecting receptor-mediated transferrin uptake. A large amount of the stimulated endocytic activity was macropinocytosis. Macropinocytosis depended on αv integrins, PKC, F-actin, and the amiloride-sensitive Na+/H+ exchanger, which are all required for Ad escape from endosomes and infection. Macropinocytosis stimulation was not a consequence of viral escape, since it occurred in K44A-dyn–expressing cells. Surprisingly, 30–50% of the endosomal contents were released into the cytosol of control and also K44A-dyn–expressing cells, and the number of fluid phase–positive endosomes dropped below the levels of noninfected cells, indicating macropinosomal lysis. The release of macropinosomal contents was Ad dose dependent, but the presence of Ad particles on macropinosomal membranes was not sufficient for contents release. We conclude that Ad signaling from the cell surface controls the induction of macropinosome formation and leakage, and this correlates with viral exit to the cytosol and infection.
Keywords: adenovirus; clathrin; endocytosis; macropinocytosis; endosomal lysis
Introduction
Endocytosis is a major pathway of pathogen uptake into cells (Marsh and Helenius, 1989; Ireton and Cossart, 1998). Pathogen uptake follows the same routes as fluid, nutrient, membrane, and surface protein uptake and is driven by vesicle formation at the plasma membrane (Mellman, 1996; Gruenberg, 2001). Multiple types of morphologically and biochemically distinct endocytic mechanisms are known. Clathrin-dependent endocytosis contributes to the initiation, propagation, and downregulation of signaling, nutrient uptake, and the maintenance of membrane homeostasis (Wiley and Burke, 2001). It originates at specialized plasma membrane regions where the cytosolic adaptor protein complex 2 binds the endocytic receptors and phosphoinositides and mediates the formation of clathrin coats (Pearse et al., 2000). A variety of factors regulate vesicle formation and closure, including epsin (Chen et al., 1998), dynamin (dyn)* (McNiven et al., 2000), and eps15 (Benmerah et al., 1998), a tyrosine-phosphorylated substrate of the activated EGF receptor pathway (Fazioli et al., 1993). Less is known about other receptor-mediated uptake processes, such as caveolar endocytosis (Kurzchalia and Parton, 1999), constitutive nonclathrin uptake (Lamaze et al., 2001), and phagocytosis (Aderem and Underhill, 1999).
Macropinocytosis is the best studied type of receptor-independent endocytosis, closely related to the receptor-triggered phagocytosis. Macropinocytosis is a major endocytic pathway found in epithelial cells, fibroblasts, neutrophils, and macrophages (Swanson and Watts, 1995; Nichols and Lippincott-Schwartz, 2001). Typically, it is triggered by growth factor stimulation, such as EGF (Haigler et al., 1979), v-src (Veithen et al., 1996), phorbol esters (Araki et al., 1996), activated Rho family G proteins (West et al., 2000), or the p21-activated kinase (Dharmawardhane et al., 2000). Macropinosomes are dynamic structures formed by the closure of lamellipodia at ruffling membranes. They assure the endocytic removal of large membrane domains, alter the adhesive and communicative properties of the cell, and are involved in cell contraction and migration. Macropinocytosis has a key role in the entry of Salmonella and Shigella bacteria (Nhieu and Sansonetti, 1999), and it is crucial for antigen presentation of dendritic cells (DCs) and viral clearance (Lanzavecchia, 1996; Garrett et al., 2000; West et al., 2000).
Adenoviruses (Ads) are known for their versatility and efficiency of gene transfer, but cellular responses to the viral challenge are still largely unknown. Here, we demonstrate that binding of adenovirus type 2 (Ad2) to epithelial cells triggers macropinocytosis, coincident with the clathrin-mediated viral uptake. Ad-induced macropinocytosis did not depend on viral endocytosis, and macropinocytosis was not needed for viral uptake. Macropinocytosis required the activation of the Ad coreceptor αv integrin and downstream effectors, including PKC and F-actin, and was inhibited by Clostridium difficile toxin B (toxin B), which inactivates Rho family GTPases, and by amiloride, an inhibitor of the Na/H exchanger. Macropinosomal contents were efficiently released into the cytosol in normal infections and also in cells expressing dominant negative K44A-dyn2, which blocks viral uptake. EGF-triggered macropinosomes remained largely intact. We conclude that Ad triggers macropinosome formation and leakage and suggest that this is instrumental for viral escape from endosomes and for infection.
Results
Clathrin-mediated endocytosis of Ad2
Previous EM studies and K44A-dyn1 overexpression experiments suggested that Ad2 uptake into epithelial cells occurred by receptor-mediated endocytosis (for review see Greber, 2002). We first tested whether Ad2 endocytosis was clathrin dependent and expressed various dominant negative forms of key regulators of clathrin-mediated endocytosis. Entry of Texas red (TR)–labeled Ad2 was assessed by confocal laser scanning microscopy (CLSM) and EM. Expression of T7-tagged clathrin hubs, which inhibit the closure of the clathrin polyhedron (Liu et al., 1998), eGFP-tagged eps15-ΔEH2,3 lacking the EH domains 2 and 3 (Benmerah et al., 1998), or K44A-dyn2 mutants, all inhibited Ad2-TR uptake and transport to the nucleus compared with nontransfected cells at 75 min postinfection (p.i.) (Fig. 1, A and B). These cells typically restricted Ad2-TR entry at the plasma membrane region proximal to the nucleus, similar to K44A-dyn1–expressing cells (unpublished data; Fig. S1 available at http://www.jcb.org/cgi/ content/full/jcb.200112067/DC1). The eGFP-tagged eps15-DIIIΔ2 mutant lacking the adaptor protein complex 2 binding site had little effect on Ad2-TR targeting to the nucleus, establishing that clathrin-mediated endocytosis is the major entry pathway of Ad2. Accordingly, EMs readily depicted Ad2 particles within coated pits and coated vesicles of control cells at early time points of infection (Fig. 1 C).
Figure 1.
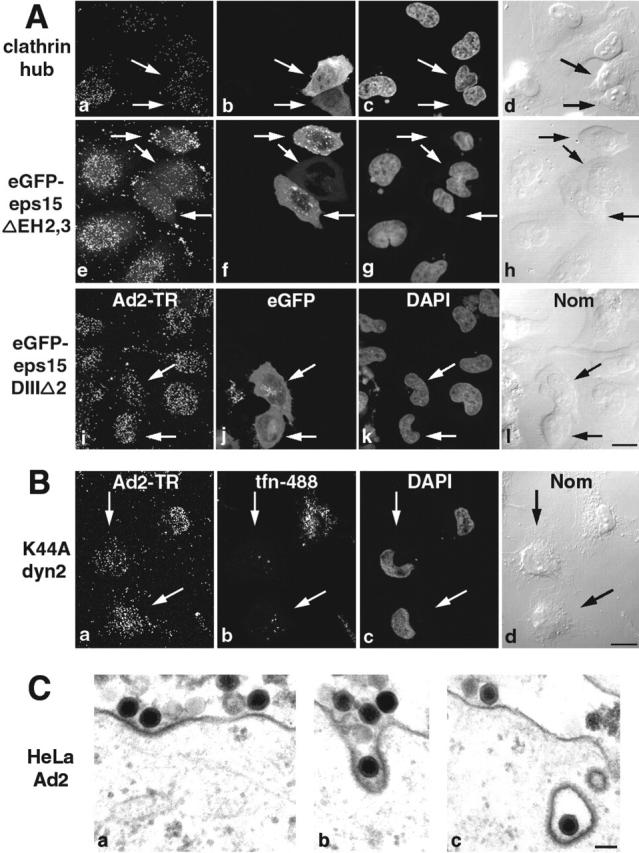
Clathrin-mediated uptake of Ad2 into HeLa cells. (A) Cells were transfected with T7-clathrin hub (a–d), eGFP–eps15ΔEH2,3 (e–h), or the control plasmid eGFP–eps15DIIIΔ2 (i–l), for 24 h and infected with Ad2-TR for 75 min. Transfected cells (arrows) were identified by mouse anti-T7 and FITC goat anti–mouse antibody (b) or eGFP expressions (f and j). The Ad2-TR optical stacks were projected in panels a, e, and i, and three merged sections across the middle of the cells show the transfected cells (b, f, and j) and DAPI stainings of nuclei (c, g, and k), respectively. DIC images (Nom) of cells are in panels d, h, and l. Bar, 20 μm. (B) Cells expressing K44A-dyn2 (arrows) were infected with Ad2-TR (a) and characterized by tfn-488 uptake (b). Arrows identifying cells lacking tfn uptake. Bar, 20 μm. (C) Transmission EM of incoming Ad2 at the plasma membrane (a), in a coated pit (b), and a coated vesicle (c). Bar, 100 nm. See also Fig. S1 available at http://www.jcb.org/cgi/content/full/jcb.200112067/DC1.
Ad2 transiently stimulates uptake of fluid phase markers
Viral infections have been known to permeabilize internal membranes and facilitate entry of nonphysiological agents such as antibiotics, HRP, luciferase (luc), or dextrans (Yoshimura, 1985; Defer et al., 1990; Carrasco, 1995). In particular, Ad2 or Ad5 infections delivered pseudomonas exotoxin (PE), PE coupled to EGF, or PE antitransferrin antibody conjugates into the cytosol of cultured cells and thus enhanced cell killing (Pastan et al., 1986). It has been speculated that cell killing was due to rupture of virus-containing endosomes.
We tested if Ad modulated specific endocytic processes or rather perforated the plasma membrane and first measured the extent of fluid phase endocytosis at different times p.i. Saturating amounts of Ad2 particles (30 μg/ml) were bound to HeLa cells in the cold followed by warming for different times in growth medium. Cells were pulsed with dex-FITC or HRP for 5 min, and extracellular dex-FITC was washed off at low pH in the cold (Fig. 2, A and B). Likewise, extracellular HRP was inactivated with the cell-impermeable methyl-ethyl-sulfonic acid (MESNA) at slightly alkaline pH without disrupting cell integrity (Fig. 2 B, inset). Flow cytometry indicated that a large majority of Ad-infected cells contained increased amounts of dex-FITC. This was most notable at 10 min p.i. when noninfected cells had a mean fluorescence value of 7.4 and Ad2-infected cells reached a mean value of 47.3 (Fig. 2 A). Cells not exposed to dex-FITC had a mean fluorescence of 0.3, indicating that the assay was specific for dex-FITC. Measured across the entire cell population, this corresponded to a 6.4-fold stimulation of fluid phase endocytosis. Similar results were obtained with HRP where the peak stimulation was at 10–15 min p.i. (Fig. 2 B). Notably, enhanced uptake of HRP and dex-FITC was detected until 50 min p.i., but no increase of HRP or dex uptake was measured with the noninfectious temperature-sensitive 1 Ad2 mutant (ts1), which was endocytosed with the same efficiency and kinetics as Ad2 (Fig. 2 A) (Greber et al., 1996). Likewise, Ad2 lacking penton base and fibers (and thus unable to attach to cells), did not increase fluid phase uptake, indicating that viral association with the target cells was required but not sufficient for fluid phase uptake stimulation (unpublished data). Ad2 infections did not affect transferrin (tfn) uptake as measured by flow cytometry up to 60 min p.i. (Fig. 2 C). The extent of fluid phase uptake stimulation depended on the viral dose and could be measured with as little as 20 viral particles bound per HeLa cell (Fig. 3 A). Ad-stimulated fluid phase uptake appeared to be a general phenomenon that was measured in many different host cells of Ad2, including human epithelial HeLa, A431, A549, and KB cells, African green monkey kidney TC7 cells, human melanoma cells, and murine fibrosarcoma L929 cells expressing coxsackie B virus Ad receptor (Fig. 3 B and Fig. 4 D; not depicted) and also observed with an E1-deleted replication-defective Ad5 (not depicted). Together, the results established that incoming Ad2 stimulated the uptake of fluid phase tracers without affecting clathrin-dependent endocytosis of tfn or virus.
Figure 2.
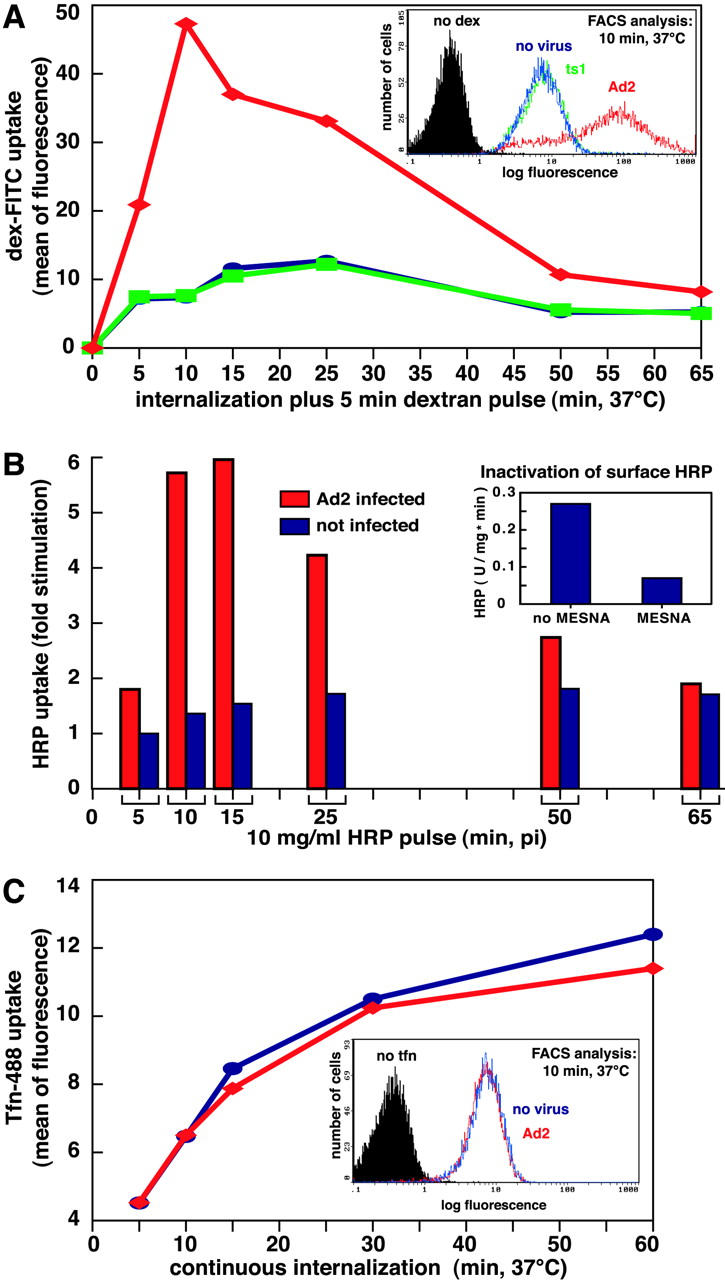
Ad transiently stimulates fluid phase but not tfn uptake. (A) FACS® analysis of dex-FITC uptake into Ad2-infected HeLa cells (♦, red), ts1-infected cells (▪, green), and noninfected cells (•, blue). Cells with cold bound Ad2 or ts1 (30 μg/ml) were warmed and pulsed with dex-FITC 5 min before washing and analysis by FACS®. A typical plot of cell number versus FITC fluorescence (log scale) is shown in the inset (10 min p.i.). (B) HRP uptake into Ad2-infected (red) and noninfected HeLa cells (blue). Cells were pulsed with HRP for 5 min (as in A), surface HRP was inactivated by MESNA treatment at pH 8.5 (inset), and intracellular HRP activity (units mg−1 min−1) was determined spectrophotometrically in cell lysates. Results are expressed as fold stimulation of HRP uptake with respect to noninfected cells at 5 min after warming. (C) Transferrin uptake. Infected and noninfected HeLa cells were starved in DME-BSA for 5 h, incubated with tfn-488 (10 μg/ml), acid washed in the cold at pH 5.5 to remove ∼90% of the surface-attached tfn (not depicted), and analyzed for green fluorescence by flow cytometry. Results are expressed as mean values of tfn-488. The inset depicts cell number as a function of tfn-488 fluorescence in log scale at 10 min p.i.
Figure 3.

Ad dose-dependent stimulation of pinocytosis. (A) HeLa cells were incubated with Ad2 in the cold for 60 min, washed, warmed for 5 min followed by a pulse of dex-FITC for 5 min, harvested, and analyzed by FACS®. Results are expressed as fold stimulation of dex uptake compared with noninfected cells. (B) HeLa, A431, TC7, A549, and KB cells were grown on plastic dishes or in suspension (susp), incubated with Ad2 (30 μg/ml) and dex-FITC, and analyzed by FACS® as described above.
Figure 4.
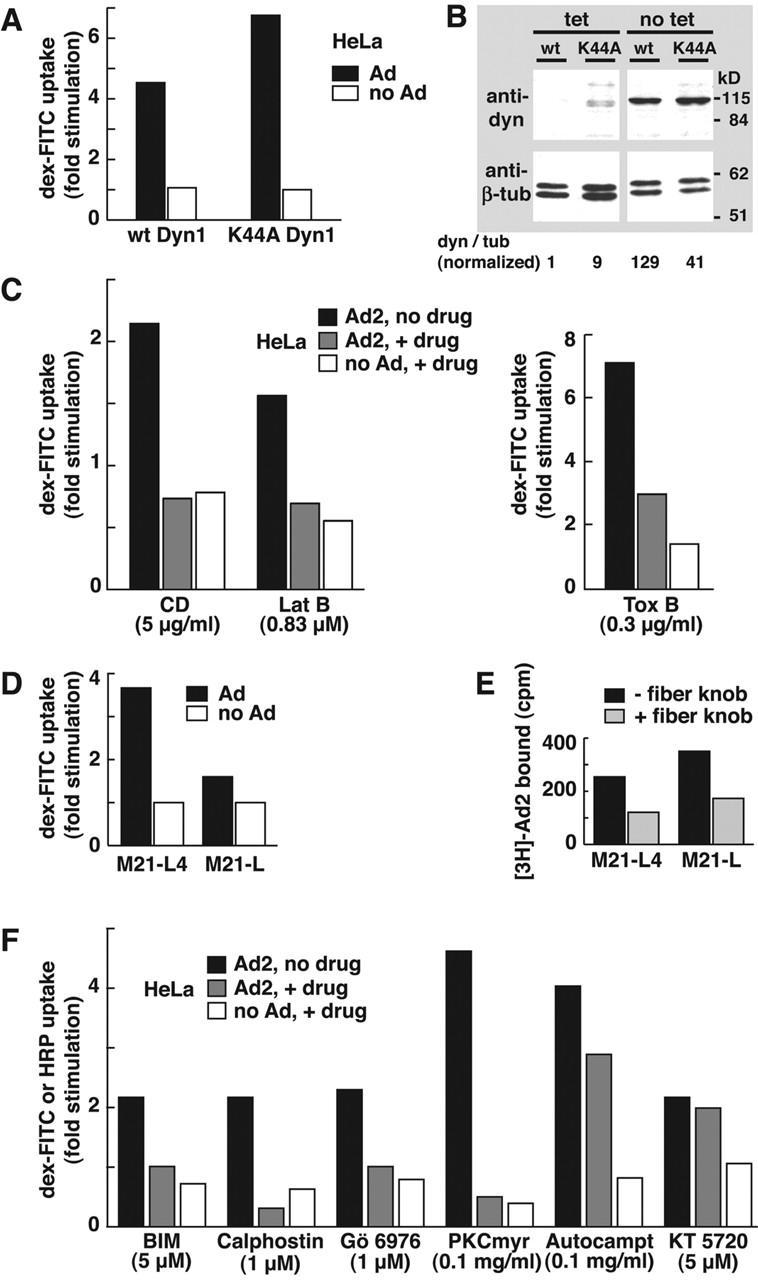
Actin- and integrin-dependent but dyn-independent stimulation of fluid phase uptake. (A) Dyn-independent stimulation of pinocytosis. Parallel dishes of HeLa cells were derepressed for dyn1 and K44A-dyn1 expressions and incubated with 50 μg/ml Ad2 in the cold, washed, pulsed with dex-FITC for 15 min, and chased for 15 min. Dex uptake was determined by FACS® analysis. (B) Derepression of dyn expression in HeLa cells. HeLa cells stably transformed with wild-type (wt) dyn1 or K44A-dyn1 cDNAs were incubated in the absence of tet for 90 h and analyzed by Western blotting using mouse anti-dyn and anti–β-tubulin antibodies (tub), respectively. (C) Actin dependence of pinocytosis stimulation. HeLa cells were pretreated with CD, latrunculin B (Lat B), or toxin B, or without drugs, incubated with Ad2 (30 μg/ml) in the cold, and analyzed for dex-FITC uptake 10 min p.i. Results are shown as fold stimulation normalized to noninfected, no drug–treated cells. (D) Stimulation of pinocytosis is integrin dependent. Ad2 (30 μg/ml) was bound to αv integrin–positive M21-L4 and αv integrin–negative M21 litter cells, and dex uptake was determined as above. (E) Ad2 binds specifically to both M21-L4 and M21 litter cells. 2.5 × 106 cells were incubated with recombinant Ad2 fiber knob (0.4 μg/ml) in DME-F12 containing 0.2% BSA at 37°C for 30 min followed by incubation with [3H]thymidine Ad2 in cold RPMI-BSA for 60 min. 3H was determined by liquid scintillation counting. (F) PKC is required for fluid phase uptake stimulation. HeLa cells were pretreated with BIM, calphostin, Gö 6976, PKC-myr, or the control inhibitors autocamptide (autocampt) and KT5720, followed by Ad2 binding in the cold. BIM- and calphostin-treated cells were warmed in the presence of HRP for 15 min and processed for intracellular HRP analysis. The other cells were pulsed with dex-FITC as described above.
Ad2-stimulated fluid phase uptake occurs without viral uptake
We next tested if fluid phase uptake stimulation required the large GTPase dyn. HeLa cells containing either the tetracycline (tet)-repressed dyn1 or the dominant negative K44A-dyn1 cDNA (Damke et al., 1994) were derepressed in tet-free medium for 90 h. Under these conditions, dyn1 was 129-fold and K44A-dyn1 41-fold induced with respect to β-tubulin (Fig. 4 B). Flow cytometry of dex-FITC indicated, however, that neither dyn1 nor K44A-dyn1 inhibited pinocytosis stimulation (Fig. 4 A). In fact, dex uptake was somewhat enhanced in K44A-dyn1 cells, and these cells had unchanged basal levels of fluid phase uptake in agreement with earlier reports (Damke et al., 1994). In addition, overexpression of the K44A mutant of the ubiquitous dyn2 had no apparent effect on Ad2-stimulated dex-FITC uptake in native HeLa cells. These results indicated that Ad-triggered fluid phase uptake occurred in the absence of viral internalization and suggested that it was induced from the cell surface.
Ad2-stimulated fluid uptake requires F-actin, integrins, and PKC
We next tested if fluid phase uptake was sensitive to inhibition of pinocytosis. Disruption of F-actin by latrunculin B (0.83 μM), which binds monomeric actin and inhibits actin polymerization (Coue et al., 1987), or by cytochalasin D (CD), which inhibits F-actin elongation (Sampath and Pollard, 1991), completely blocked Ad2-induced fluid phase uptake stimulation and inhibited infection (Fig. 4 C) (Li et al., 1998). Similar results were obtained by blocking the Rho GTPases, Rac, Rho, and Cdc42 with toxin B (Aktories, 1997), which inhibits Ad internalization and infection (Li et al., 1998), indicating that the increased fluid uptake was due to pinocytosis rather than perforation of the plasma membrane (Fig. 4 C). Pinocytosis stimulation required the Ad coreceptor αv integrin. The αv integrin lacking human M21 litter cells poorly stimulated pinocytosis, but the M21-L4 cells transfected with the αv cDNA robustly stimulated pinocytic uptake (Fig. 4 D). Both cells bound similar amounts of radiolabeled Ad2, and binding was largely inhibited by soluble fiber knobs (Fig. 4 E). Moreover, cyclic arginine-glycine-aspartate peptides inhibited the stimulation of dex uptake (unpublished data) and inhibited viral endocytosis (Wickham et al., 1993). We then tested if PKC, a downstream effector of αv integrins, was required for pinocytosis stimulation. PKC is implicated in triggering endocytosis and intracellular trafficking of integrins (Ng et al., 1999) and is involved in trafficking, endosomal escape, and infection of Ad2 but is not needed for Ad2 endocytosis itself (Nakano et al., 2000). HeLa cells were treated with a panel of PKC inhibitors, including a competitive inhibitor of ATP binding, bis-indolyl-maleimide (BIM), an effector binding site inhibitor, calphostin, and two rather narrow inhibitors of the classical PKC isoforms, Gö 6976 and a myristoylated pseudosubstrate of PKC α and β, PKC-myr. All of these agents completely blocked Ad2-stimulated pinocytosis and only slightly inhibited the basal uptake of dex-FITC (Fig. 4 F). A myristoylated control peptide directed against calmodulin kinase II (autocamptide) and the compound KT5720 binding the ATP-binding site of protein kinase A had no significant effects on Ad2-stimulated pinocytosis and did not inhibit endosomal escape (Nakano et al., 2000). We conclude that integrins, F-actin, Rho-GTPases, and PKC were required for Ad2-induced stimulation of pinocytosis.
Macropinocytosis accounts for Ad2-stimulated fluid phase uptake
So far, the results were consistent with Ad-stimulated macropinocytosis, i.e., the formation of distinct classes of nonclathrin-coated vesicles, typically at ruffling edges of spread cells (Swanson and Watts, 1995). To further test this hypothesis, we used scanning EM for surface analysis of human epidermoid carcinoma A431 or HeLa cells infected with Ad2. At 10 min p.i., infected cells had prominent ruffles, particularly near the borders, whereas the surface of noninfected cells or cells infected with ts1 was smooth (Fig. 5 A, a and b; not depicted). We then visualized macropinosomes of Ad2-infected HeLa cells using CLSM and two different markers, dex-FITC and the membrane dye 1,1′-dioctadecyl-6,6′-di(4-sulfophenyl)-3,3,3′,3′-tetramethylindocarbo-cyanine (SpDiIC-18). At 7 min p.i., >40% of infected cells contained numerous large dex-positive endosomes that were limited by the membrane dye (Fig. 5 B, a and b, arrows). These structures were also visible with differential interference contrast (DIC) optics (Fig. 5 B, c) but were not observed in noninfected cells (Fig. 5 B, d–f) or at 30 min p.i. (not depicted). Consistently, EM analyses demonstrated HRP labeling of both macropinosomes and smaller endocytic vesicles of Ad-infected cells (unpublished data), and live phase–contrast microscopy visualized the flow of macropinosomes in time-lapse mode (videos 1–3 available at http://www.jcb.org/cgi/content/full/jcb.200112067/DC1). Notably, both EGF stimulation and Ad2 infection gave rise to bright vesicular structures in A431 cells as early as 2 min p.i. (Fig. 6, A and B). A431 cells have high levels of cell surface EGF receptor (Wiley, 1988) and are known to display extensive surface ruffles and enhanced uptake of HRP upon EGF stimulation (Fig. 5 A, c) (Haigler et al., 1979). The Ad2-induced vesicles were motile and seemed to disappear (Fig. 6 A, arrows), but the EGF-triggered endosomes appeared to be more persistent (Fig. 6 B, arrows). Similar to the EGF-stimulated cells, the Ad-infected cells showed membrane ruffles that folded backward and lead to cell shape changes, albeit less extensive in nature than the EGF-stimulated cells (Fig. 5 A). In contrast, noninfected, non-EGF–treated cells contained little phase-bright vesicular structures but had extensive membrane extensions, typical of migratory cells (Fig. 6 C). Occasionally, bright dynamic structures were observed at the interface between adjacent cells, probably reflecting local cell shape changes (Fig. 6 C, arrow). Together, these results reinforce the conclusion that incoming Ad2 induces macropinocytosis.
Figure 5.

Ad2 induces membrane ruffles and macropinosomes. (A) Scanning EM of noninfected (a), Ad2-infected (b), and EGF-treated A431 cells (c). A431 cells were serum starved in DME-BSA for 14 h, incubated with Ad2 (30 μg/ml) or EGF (80 nM) in the cold for 1 h, and warmed in RPMI-BSA for 10 min (a and b) or 5 min (c), respectively. Bars, 10 μm. (B) Ad2-induced macropinosomes (arrows). Ad2 (20 μg/ml) was bound to HeLa cells in the cold and internalized for 5 min at 37°C, followed by a pulse with dex-FITC and SpDiIC-18 for 2 min. Cells were fixed in PFA and analyzed by CLSM. Noninfected cells were processed in parallel (d–f). Three merged consecutive sections are shown. Bar, 20 μm.
Figure 6.

Macropinosomes in Ad2-infected A431 cells. Serum-starved A431 cells infected with Ad2 (A), treated with 80 nM EGF (B) or untreated (C) were observed by phase–contrast microscopy from 2 to 15 min after warming. Note the appearance and disappearance of macropinosomes, visible as bright spots in Ad2- and EGF-treated cells (arrows). Time stamps indicate h:min:sec. Bar, 10 μm. The complete data can be found in videos 1–3 available at http://www.jcb.org/cgi/content/full/jcb.200112067/DC1.
Macropinocytosis correlates with Ad escape and infection
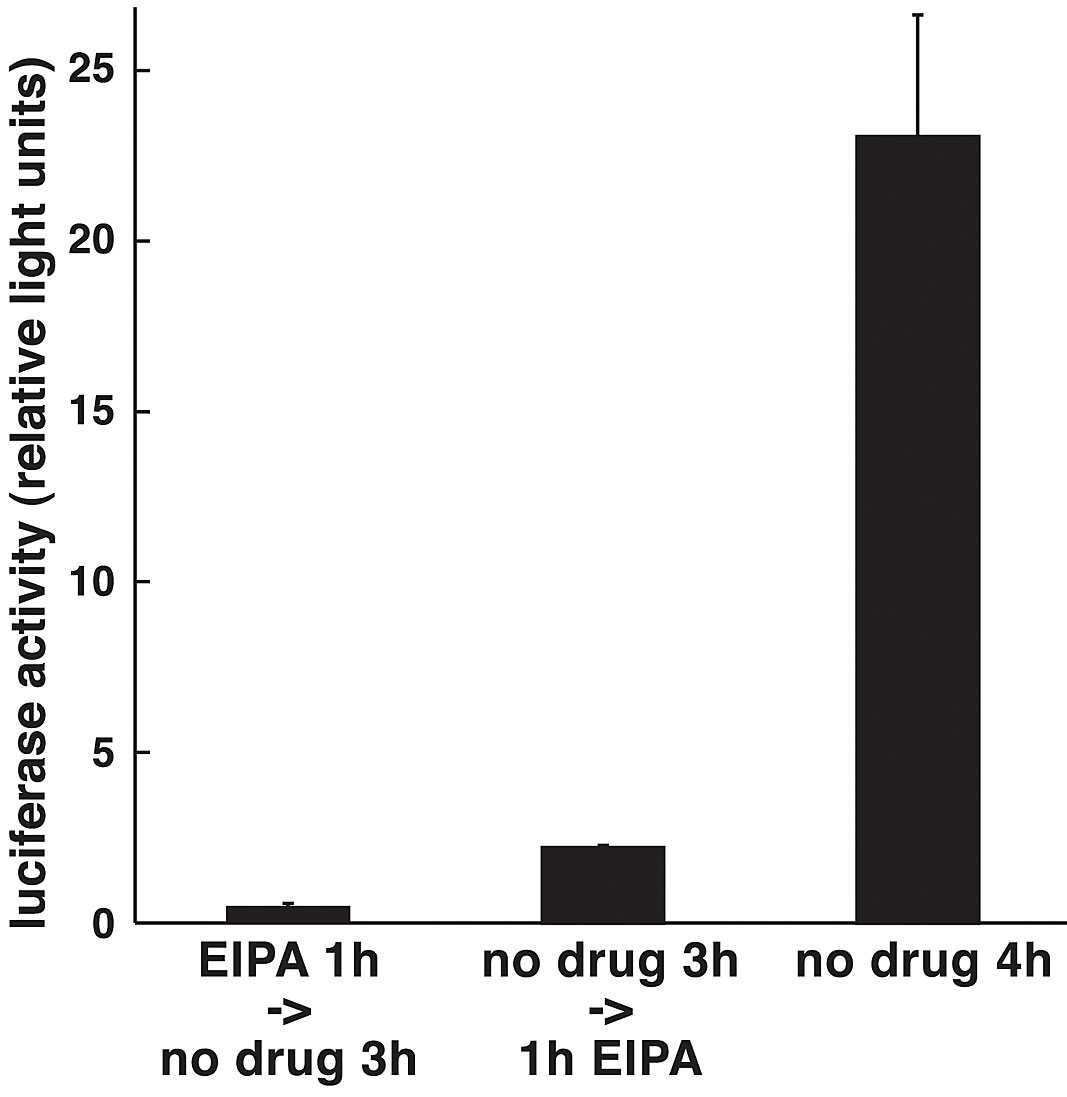
To corroborate the notion that macropinocytosis and Ad infection are linked, we tested if 5-(N-ethyl-N-isopropyl) amiloride (EIPA), a potent analogue of amiloride, affected virus entry and propagation. Amiloride is known to block the formation of macropinosomes in growth factor–stimulated cells without affecting coated pit-mediated endocytosis (West et al., 1989), probably by inhibiting aspects of endosomal trafficking (Gekle et al., 2001). Consistently, we found that EIPA inhibited Ad-induced macropinocytosis (Fig. 7 D), but it had no effect on viral uptake as measured by cell surface trypsinization of [35S]methionine-labeled Ad2 at low multiplicity of infection (m.o.i.) (i.e., ∼40 particles per cell) (Fig. 7 A). Quantitative EM analyses of very high m.o.i. infections (∼1,700 bound particles per cell) allowed us to assess if EIPA affected endosomal escape. EIPA severely inhibited viral escape from endosomes, since 57% of the particles were cytosolic in control cells compared with 26% in EIPA-treated cells (P < 0.01) (Fig. 7 B). The inhibition of cytosolic Ad2 localization occurred in the absence of apparent endosomal/lysosomal pH changes (unpublished data). Of more than 430 particles analyzed in EIPA-treated cells, none were seen at the nuclear membrane, consistent with the strong peripheral localization of Ad2-TR (100–200 particles per cell) (Fig. 7 C). The number of endosomal viruses was comparable, 22% in control and 25% in EIPA-treated cells. The fraction of plasma membrane-associated particles was significantly higher in EIPA-treated cells than in control cells (P < 0.01), and the overall number of particles at the plasma membrane of EIPA cells was lower than control cells (Fig. 7 B, c and d). This confirmed that EIPA inhibited virus escape from endosomes and suggested that viral recycling to the surface was possible in the presence of EIPA. Consistently, EIPA-treated cells had a larger fraction of particles in small endosomes and coated pits than control cells (Fig. 7 B, c and d), and EIPA-treated cells readily internalized fluorescent tfn (Fig. 7 C). This suggested that viral internalization via coated pits was unaffected. Importantly, however, EIPA strongly blocked the CMV-driven Ad expression of luc at 4 h p.i., although it also inhibited luc expression after viral entry (Fig. S2 available at http://www.jcb.org/cgi/content/full/jcb.200112067/DC1). We conclude that EIPA blocked Ad-induced macropinocytosis and inhibited viral escape from endosomes but did not affect viral uptake.
Figure 7.
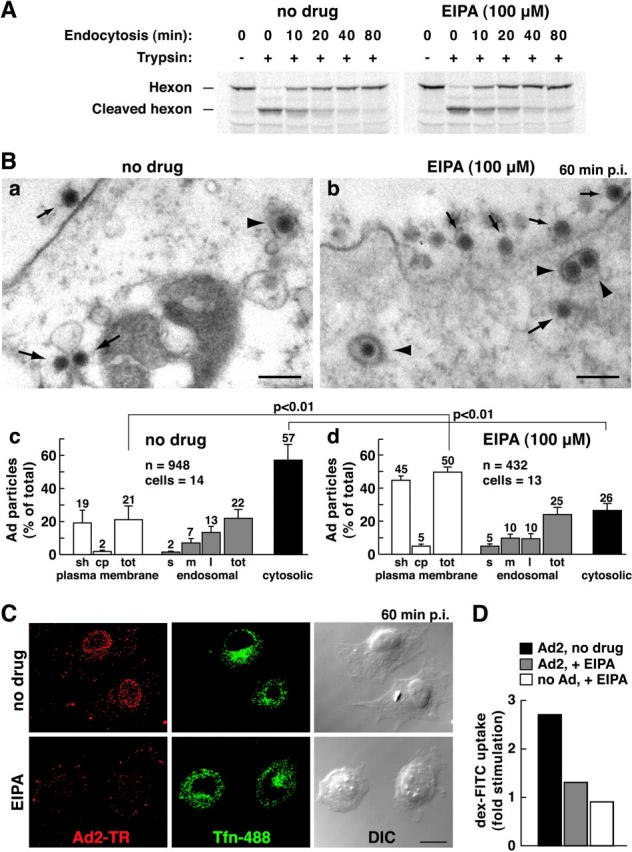
Inhibition of macropinocytosis by EIPA inhibits Ad2 escape from endosomes. (A) [35S]methionine-labeled Ad2 was internalized into EIPA-treated or untreated HeLa cells for different times followed by surface trypsinization (representative data from two independent experiments). (B) Thin section EM of incoming Ad2 in HeLa cells treated with EIPA (a) or without (b) 60 min p.i. Small arrows indicate extracellular virus, large arrows depict cytoplasmic Ad2, and arrowheads show Ad2 within coated vesicles. Bars, 200 nm. Quantification of Ad2 at the plasma membrane in intracellular vesicles, and in the cytosol of EIPA-treated (c) or untreated (d) HeLa cells. Viruses were scored in smooth (sh) and coated-pit (cp) regions of the plasma membrane (white bars), within small (s), medium (m), or large (l) endosomal vesicles (gray bars), and also in the cytosol (black bars), including the total (tot). Mean values are expressed as the percentage of total virus particles with corresponding SEM. n, Ad particles. (C) Nuclear targeting of Ad2-TR is blocked in EIPA-treated cells. HeLa cells were infected with Ad2-TR in the presence of tfn-488 for 50 min and chased for 10 min, fixed, and analyzed by CLSM. Projection of the entire optical stack for Ad2-TR and seven middle sections of tfn-488. Bar, 20 μm. (D) EIPA inhibits macropinocytosis stimulation. HeLa cells were pretreated with EIPA, bound with Ad2, pulsed with dex-FITC, and analyzed as in the legend to Fig. 4. See also Fig. S2 available at http://www.jcb.org/cgi/content/full/jcb.200112067/DC1.
Ad induces release of macropinosomal contents to the cytosol
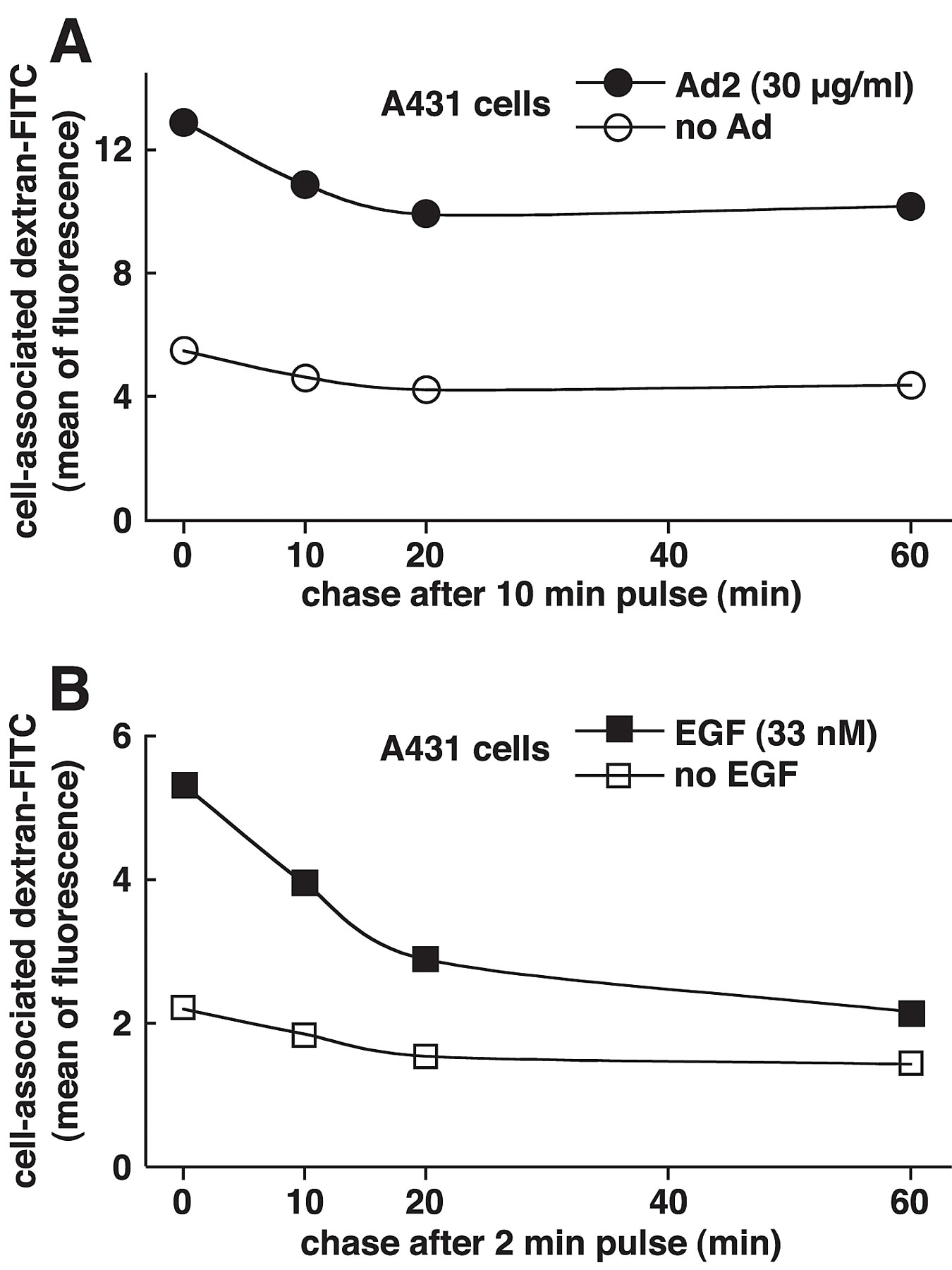
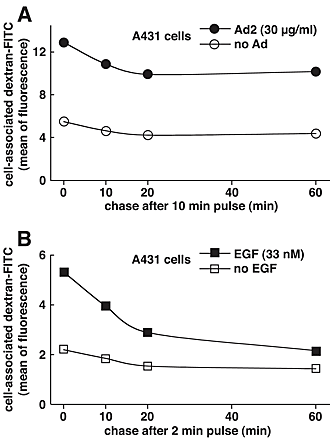
The release of fluid phase tracers from endosomes into the cytosol is a hallmark of Ad2 and Ad5 (species C) but not Ad3 and Ad7 (species B) infections (Defer et al., 1990) or ts1 entry (Suomalainen et al., 1999). Species B viruses and ts1 remain in the endocytic pathway until they arrive in late endosomes and lysosomes (Greber et al., 1996; Miyazawa et al., 2001), but Ad2 rapidly escapes from the endocytic pathway (Greber et al., 1993). Therefore, we developed an assay to directly measure the release of endosomal contents into the cytosol and coupled the SV40 large T antigen NLS peptide to FITC-labeled BSA (NLS-BSA-FITC). Microinjection into HeLa cells confirmed the quantitative nuclear targeting of NLS-BSA-FITC (unpublished data). NLS-BSA-FITC was allowed to endocytose together with Ad2, ts1, or no virus, and the FITC fluorescence was quantified by CLSM in the nucleus, and the cytoplasm 45 min p.i. Ad2-infected cells contained >50% of the total NLS-BSA-FITC in a homogeneous intranuclear pool (Fig. 8 A). However, ts1 or noninfected cells had no detectable signal in the nucleus but had occasional punctae over the nuclear area. These punctae accounted for ∼17% of the total signal and represented the background of the assay (Fig. 8 B). In all cases, the cytoplasmic signals complemented the nuclear signals, i.e., ∼75% of the NLS-BSA-FITC was cytoplasmic in ts1 and noninfected HeLa cells and <40% was cytoplasmic in Ad2-infected cells. This indicated that the assay reliably accounted for the bulk fraction of internalized NLS-BSA-FITC. Quantitation of the number of endosomes in single CLSM sections further indicated that the reduction of cytoplasmic NLS-BSA-FITC in Ad2-infected cells was largely due to a loss of endosomal structures (Fig. 8 C; videos 3–6 available at http://www.jcb.org/cgi/content/full/jcb.200112067/DC1). Ad2-infected cells contained 61 endosomes, whereas ts1 and noninfected cells contained 221 and 295 endosomes, respectively (P < 0.01). Possibly, the slight reduction of fluid phase marker-positive endosomes in ts1-infected cells was due to endosomal exclusion of NLS-BSA-FITC by the virus particle. We then tested if recycling of fluid phase contents to the extracellular medium accounted at least in part for the loss of endosomes in Ad2-infected A431 cells. FACS® analyses of dex-FITC–pulsed cells indicated that the Ad2-infected cells lost ∼15% of the initial dex-FITC amounts 60 min p.i. (Fig. S3 [panel A] available at http://www.jcb.org/cgi/content/full/jcb.200112067/DC1). This reduction reflected the recycling behavior of endosomes in noninfected cells. The situation was different in EGF-stimulated A431 cells, where 25% of the pulsed dex-FITC was lost after a 10-min chase and ∼60% was lost after 60 min (Fig. S3 [panel B]). In the absence of EGF, ∼30% of dex-FITC was lost at 60 min. These data confirmed that the EGF-generated macropinosomes recycled to the cell surface (Hewlett et al., 1994) and that this recycling was more efficient than Ad2-induced macropinosomes, which were triggered to release their contents into the cytosol.
Figure 8.

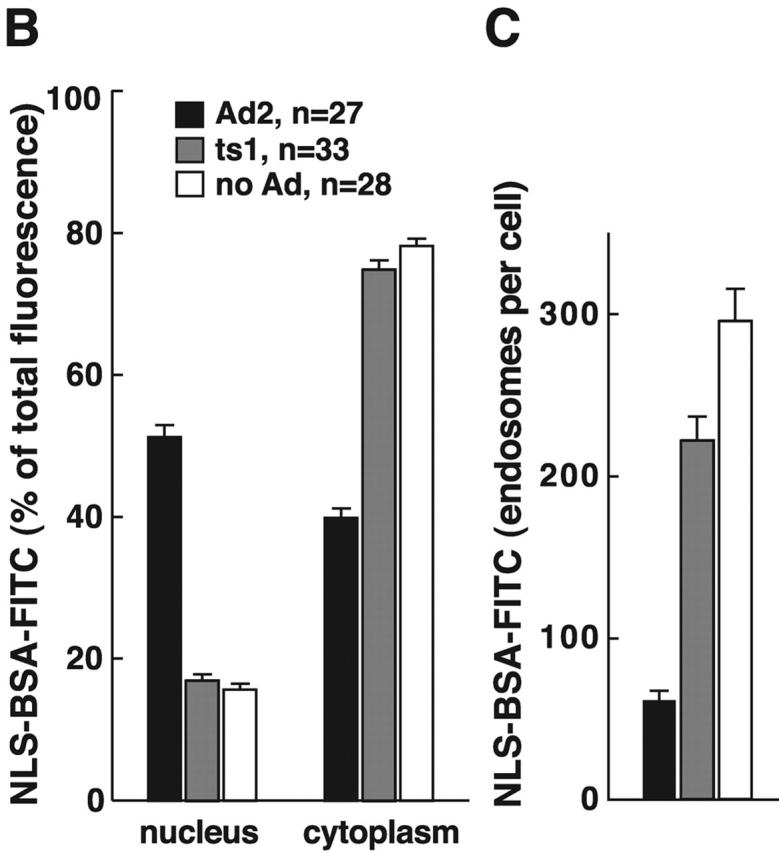
Ad2-triggered release of macropinosomal contents into the cytosol. (A) HeLa cells with prebound Ad2 (a–c), ts1 (d–f), or no virus (g–i) were pulsed with NLS-BSA-FITC for 15 min, chased for 30 min, fixed, and analyzed by CLSM. Mean projections (NIH image) of all of the optical planes, single sections across the middle of the cells, and corresponding DIC images are shown. Bar, 20 μm. (B) Quantitative subcellular analysis of endocytosed NLS-BSA-FITC in Ad2, ts1, and noninfected cells. Results are shown as mean fluorescence intensities in the percentage of total cell-associated FITC fluorescence. n, number of cells. (C) Quantification of NLS-BSA-FITC–positive endosomes. Endosomes of each cell were determined in all of the optical sections, and results are displayed as mean values per cell. The complete data can be found in videos 3–6 available at http://www.jcb.org/cgi/content/full/jcb.200112067/DC1.
We next analyzed if endosomal viruses were required to induce macropinosomal leakage and measured NLS-BSA-FITC nuclear transport in K44A-dyn2–expressing cells, where Ad is restricted to the surface. Similar amounts of NLS-BSA-FITC were found in the nucleus in both control and K44A-dyn2–expressing cells, i.e., 34 and 30%, respectively (Fig. 9 A). In contrast, no significant nuclear signal was detected in EGF-stimulated A431 cells (17%), unlike Ad2-infected A431 cells (31%) (Fig. 9 B), indicating that Ad2 uptake via clathrin-coated pits is not required to induce macropinosomal leakage. To address if Ad particles bound on macropinosomal membranes were involved in macropinosomal leakage, we quantitated the amounts of Ad2-TR in macropinosomes of control cells and K44A-dyn2–expressing cells using two different virus concentrations, 1 and 0.1 μg Ad2-TR per coverslip (Fig. 10 A). At 1 μg of virus, the K44A-dyn2–expressing cells contained ∼20 Ad2-TR fluorescence units per unit area of macropinosomes, whereas control cells had twice as much (Fig. 10 B). There were ∼46 fluorescence units on nonmacropinosomal membranes, ∼25% less than on control cells, consistent with earlier results obtained with K44A-dyn1–expressing cells (Nakano et al., 2000). Importantly, the K44A-dyn2 cells were as potent as control cells at inducing macropinosomal leakage (Fig. 10 C). However, a 10-fold lower input of Ad2 (0.1 μg, resulting in two- to threefold lower levels of cell-bound Ad) did not induce macropinosomal leakage in both K44A-dyn2 and control cells, as measured with the NLS-BSA-FITC assay, identical to noninfected cells (n = 20 and 14, respectively) (unpublished data). Importantly, the levels of macropinosomal virus in K44A-dyn2 and control cells were at 12 and 14 Ad2-TR fluorescence units, respectively, and these values were in the same range as those of K44A-dyn2–expressing cells inoculated with 1 μg Ad2-TR. We conclude that the presence of Ad2 on macropinosomal membranes is not sufficient for triggering macropinosomal contents release.
Figure 9.

Ad releases macropinosomal contents of K44A-dyn2–expressing cells, but EGF triggered macropinosomes remain largely intact. (A) Transiently transfected HeLa cells expressing K44A-dyn2 (identified by lack of tfn-488 uptake; not depicted) were pulsed with warm NLS-BSA-FITC with or without Ad2 and processed as described in the legend to Fig. 8. (B) A431 cells were pulsed with NLS-BSA-FITC with or without Ad2 (30 μg/ml) or EGF (33 nM) and processed as above. n, number of cells analyzed.
Figure 10.
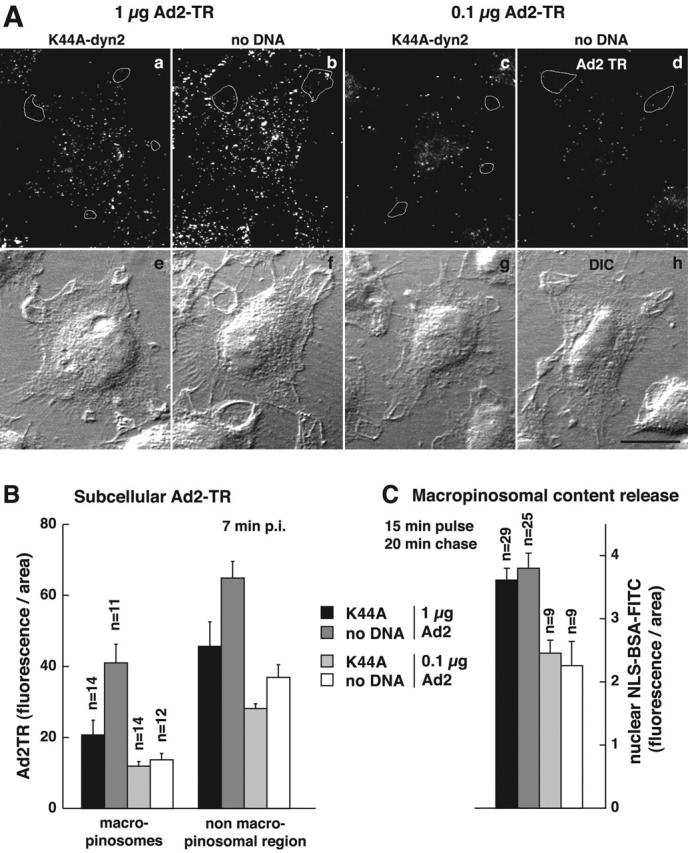
The presence of Ad particles in macropinosomes is not sufficient to elicit macropinosomal contents release. (A) K44A-dyn2–transfected and control HeLa cells were infected with 1 μg Ad per coverslip (a, b, e, and f) and 0.1 μg Ad per coverslip (c, d, g, and h) for 7 min, fixed, and analyzed by CLSM for subcellular Ad2-TR fluorescence (total projections in a–d and DIC images in e–h). Macropinosomes were identified by DIC optics, confirmed by filipin staining (not depicted), and marked by white tracings in one typical cell each (a–d). K44A-dyn2–expressing cells were identified by the absence of tfn–Alexa 495 staining (not depicted). Bar, 20 μm. (B) Quantification of Ad2-TR in macropinosomes. The amount of Ad2-TR fluorescence in macropinosomes and in nonmacropinosomal cell regions was determined using the MetaMorph software. The datasets were normalized per area. n, number of cells. Each cell contained several macropinosomes. (C) In a parallel experiment using Ad2, the amount of macropinosomal leakage was determined after a pulse of HeLa cells with NLS-BSA-FITC for 15 min followed by a 20-min chase in the absence of dye. K44A-dyn2–expressing cells were identified by the absence of tfn-rhodamine staining (not depicted). The datasets were normalized per area.
Discussion
Entry of a variety of viral agents, including Semliki Forest virus, poliovirus, vesicular stomatitis virus, and Ad has been known to enhance the delivery of antibiotics, protein toxins, HRP, luc, or dextrans to the cytosol (for a review see Carrasco, 1995). Here, we show that Ad2 infections trigger macropinocytosis, coincident with clathrin-mediated viral uptake and, most strikingly, that virus-induced macropinosomes release their soluble contents into the cytosol.
Ad-triggered macropinocytosis has several features of growth factor–stimulated macropinocytosis, including F-actin–dependent surface ruffling, formation of large endocytic vesicles, and a strong dependence on PKC. It is not sensitive to K44A-dyn1 or -dyn2, very similar to the macropinosomal uptake of Clamydia bacteria (Boleti et al., 1999). However, K44A-dyn1 or -dyn2 inhibited viral uptake in agreement with an earlier report (Wang et al., 1998). This underscores that macropinocytosis is not a pathway of viral uptake in epithelial cells expressing coxsackie B virus Ad receptor and αv integrins.
Macropinocytosis stimulation requires contact of virus with cell surface receptors but not viral uptake, implying that it is not merely a compensatory mechanism for lost endosomes. We have identified one critical surface receptor for Ad-triggered macropinocytosis, the αv integrin subunit, which has been implicated before in clathrin-dependent Ad endocytosis (Wickham et al., 1993). Integrins are crucial for bacterial invasion, and their activation is coupled to macropinocytosis and signaling to the cytoskeleton (Isberg et al., 2000). Downstream of Ad-activated αv integrin are the phosphatidylinositol 3-OH kinase (PI3K), the adaptor protein p130CAS docking PI3K, and the small GTPase Rac (for review see Greber, 2002). Intriguingly, p130CAS is part of a Rac-activating complex required for phagocytosis of apoptotic cells in both professional and nonprofessional phagocytes (Albert et al., 2000). Consistently, the small actin remodeling GTPases Rac and Cdc42 and also PKC are involved in Ad-induced macropinocytosis, downstream of αv integrins. These factors are required for growth factor–stimulated macropinocytosis (Haigler et al., 1979; Dharmawardhane et al., 2000) and constitutive macropinocytosis (West et al., 2000). Thus, PI3K is strongly implicated in Ad-stimulated macropinocytosis, consistent with the notion that Ad-induced macropinocytosis is sensitive to PI3K inhibitors (unpublished) and that PI3K is required for induced and constitutive macropinocytosis (Araki et al., 1996; Amyere et al., 2000).
A prominent feature of Ad-triggered macropinocytosis is that up to 50% of the macropinosomal contents are released. This is not observed with growth factor–stimulated macropinosomes, although low levels of macropinosomal leakage had been measured using highly sensitive assays, such as Hrp or antigen delivery to the cytosol for class I major histocompatibility (MHC) processing in DCs (Norbury et al., 1995; Rodriguez et al., 1999). Intriguingly, Ad2 particles in endosomes, including macropinosomes, are not sufficient for macropinosomal lysis. This was demonstrated by the observation that K44A-dyn2–expressing cells released macropinosomal contents, but they contained macropinosomal virus in similar amounts as control cells infected with a lower m.o.i., and these latter cells did not release measurable amounts of macropinosomal contents. This indicates that viral signals from the surface are involved in controlling endosomal leakiness. The idea that a large fraction of the released endosomal contents is indeed derived from macropinosomes is supported by the notion that K44A-dyn blocks clathrin-dependent endocytosis of many receptors, caveolar uptake, and also phagocytosis (for review see McNiven et al., 2000).
Interestingly, macropinocytosis and Ad infection were sensitive to the macropinocytosis inhibitor EIPA, which had no effect on Ad uptake but inhibited exit to the cytosol. Likewise, PKC inhibition or ts1 infection blocked macropinocytosis and viral escape to the cytosol but not viral uptake via clathrin-coated membranes (see above; Greber et al., 1996; Nakano et al., 2000). Furthermore, Ad5 retargeted to the high affinity Fc receptor (CD64) effectively transduced hematopoietic cells and elicited a robust fluid phase uptake stimulation (unpublished data; Ebbinghaus et al., 2001). These data suggest that macropinosomes are either part of the productive Ad entry pathway, i.e., they could be involved in viral exit to the cytosol, or there is a more general mechanism of endosomal leakage.
Precisely how Ad controls the machinery of macropinosomal leakage is presently unknown. Possibly, the mechanisms could be similar to bacterial lysis of cellular membranes; they could, e.g., involve pore forming cytolysins and bacterially encoded phospholipases. Since the Ad genome neither contains cytolysins nor phospholipases, it is possible that Ad utilizes cellular phospholipases to break open endosomal membranes. However, additional mechanisms are conceivable. They include a volume control mechanism via the activation of water channels documented, e.g., in DCs (de Baey and Lanzavecchia, 2000), or a combination of low endosomal pH and penton base-mediated integrin activation (Seth et al., 1984; Wickham et al., 1994). Another possibility is that Ad-containing endosomes fuse with macropinosomes and Ad particles are then released by virtue of a destabilized macropinosomal membrane.
One implication of our study is that binding of a viral vector to and endocytosis into a target cell are not sufficient for successful gene delivery. For example, an Ad5 with a genetically engineered fiber targeting to the tfn receptor was shown to mediate only a small increase of gene expression in tfn receptor-positive cells and was difficult to propagate, suggesting poor gene delivery (Xia et al., 2000). Productive infections with species C Ad apparently require additional vector properties, including the ability to elicit membrane ruffling, macropinosome formation, and lysis and the induction of a migratory phenotype. We anticipate that macropinocytosis has both pro- and antiviral effects. The destruction of macropinosomal membranes may help viral escape. Additionally, it may transiently impair MHC class II immunopresentation and thus provide an advantage to pathogens as suggested by recent work on unrelated viral agents, including Varizella zoster virus and lymphocytic choriomeningitis virus infecting DCs (Abendroth et al., 2001; Kunz et al., 2001). On the other hand, professional antigen-presenting cells such as DCs use macropinocytosis to accumulate extracellular antigen (Lanzavecchia, 1996). These cells also allow the leakage of internalized antigen from endosomes to the cytosol, resulting in the loading of antigen on to MHC class I molecules (Mellman and Steinman, 2001). Thus, enhanced macropinosomal leakage may enhance immune defense.
Materials and methods
Cells, viruses, and proteins
Cells were grown in DME (GIBCO-BRL) containing 10% FBS (Hyclone) at low passage number as described (Suomalainen et al., 1999). HeLa cells expressing dyn1 or K44A-dyn1 in tet-off mode were supplied by Dr. C. Lamaze (Pasteur Institute, Paris, France) (Damke et al., 1994). Dyn overexpression was confirmed by Western blotting of ∼3 × 104 cell equivalents lysed in hot SDS followed by ECL-enhanced detection and reprobing the blotted membrane with a mouse anti–β-tubulin antibody. Human epidermoid carcinoma A431 containing amplified and rearranged copies of the EGF receptor gene were obtained from Dr. J. Pavlovic (University of Zürich), and human melanoma M21 litter (negative for surface-expressed αv integrins) and M21-L4 cells (transfected with an αv cDNA and positive for surface-expressed αv integrins) were from Dr. D. Cheresh (The Scripps Research Institute, La Jolla, CA) (Felding-Habermann et al., 1992).
Ad2 and ts1 were grown and isolated as described (Greber et al., 1996). Labeling of Ad2 with TR was as published (Nakano and Greber, 2000). [35S]methionine-labeled Ad2 and cold synchronized infections were performed as described (Greber et al., 1993). Using [3H]thymidine-labeled Ad2 (1,200 cpm/μl and 1.2 × 109 particles/μl), we estimated that ∼2% of the input virus specifically attached to human epithelial cells under our cold binding conditions. Ad2 binding to cells was effectively competed by preincubating cells with soluble fiber knob at 37°C for 30 min as described (Nakano and Greber, 2000). Cell lysates were prepared in hot SDS (2%), sheared in a 20G clinical syringe, and radioactivity was determined by fluid scintillation counting (Ready Safe; Beckman Coulter) with a Beckman Coulter Scintillation System LS 3801. Ad5-luc (lacking E1 and E3) expressed luc from a cytomegalovirus promotor inserted into the E1 region as constructed from a pPoly plasmid backbone (Chartier et al., 1996) and the pTG-H5 DL324 plasmid (supplied by Dr. Sandro Rusconi, University of Fribourg, Fribourg, Switzerland).
cDNAs, proteins, and chemicals
cDNAs encoding hub fragments of clathrin heavy chain tagged with a phage T7 epitope were obtained from Dr. F. Brodsky (University of California, San Francisco, CA) (Liu et al., 1998), eGFP-eps15ΔEH2,3 and eGFP-eps15DIIIΔ2 cDNAs were from Dr. A. Benmerah (Institut National de la Santé et de la Recherche Médicale E9925, Paris, France) (Benmerah et al., 1999), the K44A-dyn2 expression plasmid was from Dr. C. Lamaze (Pasteur Institute), and a cytomegalovirus major promotor–controlled eGFP expression plasmid was obtained from Dr. S. Zimmermann (University of Zürich). Transient transfections of HeLa cells were performed with TransFast (Promega). The mouse monoclonal LM142 antibody recognizing αv integrins (Wickham et al., 1993) was used to verify surface levels of αv integrins on melanoma cells. Mouse monoclonal anti-dyn1 antibody (Hudy-1) was from Upstate Biotechnology (Lucerna AG), mouse anti–β-tubulin (N357) was from Amersham Biosciences, mouse monoclonal anti-T7 tag was from Novagen (Axon Lab), human tfn–Alexa 488 and tfn-rhodamine was from Molecular Probes, and toxin B (0.5 mg/ml) was from Drs. F. Hofmann and K. Aktories (University of Freiburg, Freiburg, Germany). Cells were pretreated with inhibitors in growth medium at 37°C for 30 min followed by cold synchronized infections in the presence of drugs as described earlier (Nakano et al., 2000). The PKC inhibitor Gö 6976 (1 μM) was purchased from Calbiochem (Juro Supply), and all the other inhibitors were used as described (Nakano et al., 2000). The membrane dye SpDiIC-18 (0.05 mg/ml; Molecular Probes) was dissolved in DMSO (1 mg/ml) and stored at −20°C until use, the Na+/H+exchanger inhibitor EIPA (100 μM; Alexis Corporation) was used in RPMI medium throughout, and NLS-BSA-FITC was produced as described (Trotman et al., 2001). Filipin (100 μg/ml for 2 h in RPMI medium; Sigma-Aldrich) was used on cells fixed in PFA for 1 h.
HRP and dextran uptake
Cells were incubated with Ad2 in the cold, washed with cold RPMI-BSA, and warmed in RPMI-BSA containing HRP (10 mg/ml; Sigma-Aldrich) (P8250, 175 U/mg) at 37°C for 15 min, followed by three washes in cold RPMI, 0.2% BSA, and 0.02 M Hepes, pH 7.4 (RPMI-BSA). Surface-attached HRP was inactivated by incubation with cell-impermeable methyl-ethyl-sulfonic acid (MESNA; 20 mM, freshly prepared in 0.05 M NaCl, 0.05 M Tris-HCl, pH 8.6 [Sigma-Aldrich]) at 4°C for 10 min. After three washes in PBS, MESNA was inactivated by N-ethyl-maleimide (NEM; 20 mM in PBS [Sigma-Aldrich]) at 4°C for 5 min, and cells were washed three times and lysed in MNT buffer (30 mM 2-N-morpholino-ethanesulfonic acid, 20 mM Tris, 100 mM NaCl, pH 7.5) containing 0.5% Empigen BB (Calbiochem) and 0.2% BSA (cell culture grade; Sigma-Aldrich). The lysate was passed three times through a 20G syringe, and HRP activity was determined by incubating 0.1 ml cell lysate with 0.9 ml HRP reaction mix containing 3 mg/ml o-dianisidine (Sigma-Aldrich), 0.06% H2O2 (Sigma-Aldrich) in 0.05 M sodium phosphate–citric acid buffer, pH 5.0, and spectrophotometric recording at 405 nm at 25°C (Ultrospec 2000) (Amersham Biosciences). The reaction kinetics were determined using Amersham Biosciences application software.
Uptake of lysine-fixable dex-FITC (1 mg/ml; Molecular Probes) into attached cells containing cold bound Ad2 occurred in warm RPMI-BSA on a horizontal shaker in a water bath at 37°C for the indicated times. KB suspension cells (2.5 × 106) were incubated with Ad2 in the cold, washed, resuspended in 0.25 ml RPMI-BSA, warmed for 5 min, and supplemented with 0.25 ml of 2 mg/ml dex-FITC. Dex uptake was stopped by adding 3 vol of ice-cold RPMI and three washes with cold PBS. Surface-bound dex was washed off in cold 0.1 M sodium acetate, 0.05 M NaCl, pH 5.5, for 10 min. For FACS® analyses, cells were detached with 2 mg/ml trypsin in PBS containing 50 mM EDTA (GIBCO-BRL) on ice for 25 min, transferred into 6 ml polypropylene tubes (no 2063; Falcon, Becton Dickinson) containing 2 ml 7% FCS/PBS, pelleted at 290 g and resuspended in 2% FCS/PBS. At least 10,000 viable cells were counted per sample in a flow fluorocytometer (Epics XL; Beckman Coulter) using emission filters of 520–550 nm (green fluorescence), 580–620 nm (rhodamine fluorescence), and 620–680 nm (TR fluorescence) as described (Ebbinghaus et al., 2001). Most experiments were performed three times, but at least twice, with similar results.
Release of endosomal contents
Cells on 12-mm glass coverslips were serum-starved for 4 h in DME-BSA, incubated with Ad2 in the cold, washed in BSA-free RPMI, placed onto 30 μl of RPMI containing 40 mg/ml NLS-BSA-FITC, and incubated in a humid chamber on a water bath at 37°C for 15 min, and then incubated in RPMI, 0.2% BSA for 30 min, fixed in PFA, and analyzed by CLSM. NLS-BSA-FITC quantifications in the nucleus and the cytoplasm were performed on projections of the entire optical sections using the MetaMorph software (Universal Imaging Corp.) and a semiautomated routine to identify cell borders and nuclear regions (Nakano and Greber, 2000). Endosomes were counted in individual CLSM sections of single cells using an automated procedure (Trotman et al., 2001) and displayed using the public domain NIH image analysis software (developed at the U.S. National Institutes of Health and available at http://rsb.info.nih.gov/nih-image/) and contrast enhancement (Adobe Photoshop 5.5®).
Microscopy
For scanning EM, cells grown on round coverslips were fixed in 2% glutaraldehyde in 0.1 M cacodylate buffer (pH 7.4) for 30 min, washed in buffer, postfixed in 2% OsO4/cacodylate (at 4°C for 30 min), washed, dehydrated in acetone, critical point dried, coated with palladium-gold in a sputter coater, and observed with a Hitachi S4000 field emission scanning EM.
Phase–contrast microscopy of live cells was performed on a homemade observation chamber (Suomalainen et al., 2001) using a 63× oil immersion objective (NA 1.32; Leica). Movies were assembled on a Macintosh computer using the NIH image program. CLSM, transmission EM, and statistical analyses were performed as described (Nakano et al., 2000).
Online supplemental material
Videos and supplemental figures are available at http://www.jcb.org/cgi/content/full/jcb.200112067/DC1. Fig. S1 shows K44A-dyn1 inhibiting Ad2 entry (see also Fig. 1). Fig. S2 displays luc expression of transgenic Ad5 in the presence and absence of EIPA (see also Fig. 7). Fig. S3 shows EGF-induced but not Ad2-induced macropinosomal contents recycled to the cell surface. Videos 1–3 show phase–contrast images of serum-starved A431 cells infected with Ad2 (video 1), treated with EGF (video 2), or nontreated (video 3) (see also Fig. 6). Videos 3–6 show CLSM sections of NLS-BSA-FITC and thresholded endosomes of Ad2-infected HeLa cells (video 4), ts1-infected cells (video 5), and noninfected cells (video 6) (see also Fig. 8).
Supplemental Material
Acknowledgments
We thank Ari Helenius and Ernst Hafen for discussions and Michel Nakano for help with image acquisition and processing.
Financial support was obtained from the Swiss National Science Foundation and the Kanton Zürich (UFG).
The online version of this article contains supplemental material.
Footnotes
Abbreviations used in this paper: Ad, adenovirus; BIM, bis-indolyl-maleimide; CLSM, confocal laser scanning microscopy; CD, cytochalasin D; DC, dendritic cell; DIC, differential interference contrast; dyn, dynamin; EIPA, 5-(N-ethyl-N-isopropyl) amiloride; luc, luciferase; MHC, major histocompatibility; m.o.i., multiplicity of infection; PE, pseudomonas exotoxin; p.i., post infection; PI3K, phosphatidylinositol 3-OH kinase; SpDiIC-18, 1,1′-dioctadecyl-6,6′-di(4-sulfophenyl)-3,3,3′,3′-tetramethylindocarbo-cyanine; TR, Texas red; ts1, temperature-sensitive Ad2 mutant 1; tet, tetracycline; tfn, transferrin.
References
- Abendroth, A., G. Morrow, A.L. Cunningham, and B. Slobedman. 2001. Varicella-zoster virus infection of human dendritic cells and transmission to T cells: implications for virus dissemination in the host. J. Virol. 75:6183–6192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aderem, A., and D.M. Underhill. 1999. Mechanisms of phagocytosis in macrophages. Annu. Rev. Immunol. 17:593–623. [DOI] [PubMed] [Google Scholar]
- Aktories, K. 1997. Rho proteins: targets for bacterial toxins. Trends Microbiol. 5:282–288. [DOI] [PubMed] [Google Scholar]
- Albert, M.L., J.I. Kim, and R.B. Birge. 2000. alphavbeta5 integrin recruits the CrkII-Dock180-rac1 complex for phagocytosis of apoptotic cells. Nat. Cell Biol. 2:899–905. [DOI] [PubMed] [Google Scholar]
- Amyere, M., B. Payrastre, U. Krause, P.V. Smissen, A. Veithen, and P.J. Courtoy. 2000. Constitutive macropinocytosis in oncogene-transformed fibroblasts depends on sequential permanent activation of phosphoinositide 3-kinase and phospholipase C. Mol. Biol. Cell. 11:3453–3467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki, N., M.T. Johnson, and J.A. Swanson. 1996. A role for phosphoinositide 3-kinase in the completion of macropinocytosis and phagocytosis by macrophages. J. Cell Biol. 135:1249–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benmerah, A., C. Lamaze, B. Begue, S.L. Schmid, A. Dautry-Varsat, and N. Cerf-Bensussan. 1998. AP-2/Eps15 interaction is required for receptor-mediated endocytosis. J. Cell Biol. 140:1055–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benmerah, A., M. Bayrou, N. Cerf-Bensussan, and A. Dautry-Varsat. 1999. Inhibition of clathrin-coated pit assembly by an Eps15 mutant. J. Cell Sci. 112:1303–1311. [DOI] [PubMed] [Google Scholar]
- Boleti, H., A. Benmerah, D.M. Ojcius, N. Cerf-Bensussan, and A. Dautry-Varsat. 1999. Chlamydia infection of epithelial cells expressing dynamin and Eps15 mutants: clathrin-independent entry into cells and dynamin-dependent productive growth. J. Cell Sci. 112:1487–1496. [DOI] [PubMed] [Google Scholar]
- Carrasco, L. 1995. Modification of membrane permeability by animal viruses. Adv. Virus Res. 45:61–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chartier, C., E. Degryse, M. Gantzer, A. Dieterle, A. Pavirani, and M. Mehtali. 1996. Efficient generation of recombinant adenovirus vectors by homologous recombination in Escherichia coli. J. Virol. 70:4805–4810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, H., S. Fre, V.I. Slepnev, M.R. Capua, K. Takei, M.H. Butler, P.P. Di Fiore, and P. De Camilli. 1998. Epsin is an EH-domain-binding protein implicated in clathrin-mediated endocytosis. Nature. 394:793–797. [DOI] [PubMed] [Google Scholar]
- Coue, M., S.L. Brenner, I. Spector, and E.D. Korn. 1987. Inhibition of actin polymerization by latrunculin A. FEBS Lett. 213:316–318. [DOI] [PubMed] [Google Scholar]
- Damke, H., T. Baba, D.E. Warnock, and S.L. Schmid. 1994. Induction of mutant dynamin specifically blocks endocytic-coated vesicle formation. J. Cell Biol. 127:915–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Baey, A., and A. Lanzavecchia. 2000. The role of aquaporins in dendritic cell macropinocytosis. J. Exp. Med. 191:743–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Defer, C., M.-T. Belin, M.-L. Caillet-Boudin, and P. Boulanger. 1990. Human adenovirus-host interactions: comparative study with members of subgroups B and C. J. Virol. 64:3661–3673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dharmawardhane, S., A. Schurmann, M.A. Sells, J. Chernoff, S.L. Schmid, and G.M. Bokoch. 2000. Regulation of macropinocytosis by p21-activated kinase-1. Mol. Biol. Cell. 11:3341–3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebbinghaus, C., A. Al-Jaibaji, E. Operschall, A. Schoeffel, I. Peter, U.F. Greber, and S. Hemmi. 2001. Functional and selective targeting of adenovirus to high affinity Fcg receptor I positive cells using a bispecific hybrid adaptor. J. Virol. 75:480–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fazioli, F., L. Minichiello, B. Matoskova, W.T. Wong, and P.P. Di Fiore. 1993. eps15, a novel tyrosine kinase substrate, exhibits transforming activity. Mol. Cell. Biol. 13:5814–5828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felding-Habermann, B., B.M. Mueller, C.A. Romerdahl, and D.A. Cheresh. 1992. Involvement of integrin alpha V gene expression in human melanoma tumorigenicity. J. Clin. Invest. 89:2018–2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett, W.S., L.M. Chen, R. Kroschewski, M. Ebersold, S. Turley, S. Trombetta, J.E. Galan, and I. Mellman. 2000. Developmental control of endocytosis in dendritic cells by Cdc42. Cell. 102:325–334. [DOI] [PubMed] [Google Scholar]
- Gekle, M., R. Freudinger, and S. Mildenberger. 2001. Inhibition of Na+-H+ exchanger-3 interferes with apical receptor-mediated endocytosis via vesicle fusion. J. Physiol. 531:619–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greber, U.F. 2002. Signalling in viral entry. Cell. Mol. Life Sci. 59:608–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greber, U.F., M. Willetts, P. Webster, and A. Helenius. 1993. Stepwise dismantling of adenovirus 2 during entry into cells. Cell. 75:477–486. [DOI] [PubMed] [Google Scholar]
- Greber, U.F., P. Webster, J. Weber, and A. Helenius. 1996. The role of the adenovirus protease in virus entry into cells. EMBO J. 15:1766–1777. [PMC free article] [PubMed] [Google Scholar]
- Gruenberg, J. 2001. The endocytic pathway: a mosaic of domains. Nat. Rev. Mol. Cell Biol. 2:721–730. [DOI] [PubMed] [Google Scholar]
- Haigler, H.T., J.A. McKanna, and S. Cohen. 1979. Rapid stimulation of pinocytosis in human carcinoma cells A-431 by epidermal growth factor. J. Cell Biol. 83:82–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewlett, L.J., A.R. Prescott, and C. Watts. 1994. The coated pit and macropinocytic pathways serve distinct endosome populations. J. Cell Biol. 124:689–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ireton, K., and P. Cossart. 1998. Interaction of invasive bacteria with host signaling pathways. Curr. Opin. Cell Biol. 10:276–283. [DOI] [PubMed] [Google Scholar]
- Isberg, R.R., Z. Hamburger, and P. Dersch. 2000. Signaling and invasin-promoted uptake via integrin receptors. Microbes Infect. 2:793–801. [DOI] [PubMed] [Google Scholar]
- Kunz, S., N. Sevilla, D.B. McGavern, K.P. Campbell, and M.B. Oldstone. 2001. Molecular analysis of the interaction of LCMV with its cellular receptor α-dystroglycan. J. Cell Biol. 155:301–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurzchalia, T.V., and R.G. Parton. 1999. Membrane microdomains and caveolae. Curr. Opin. Cell Biol. 11:424–431. [DOI] [PubMed] [Google Scholar]
- Lamaze, C., A. Dujeancourt, T. Baba, C.G. Lo, A. Benmerah, and A. Dautry-Varsat. 2001. Interleukin 2 receptors and detergent-resistant membrane domains define a clathrin-independent endocytic pathway. Mol. Cell. 7:661–671. [DOI] [PubMed] [Google Scholar]
- Lanzavecchia, A. 1996. Mechanisms of antigen uptake for presentation. Curr. Opin. Immunol. 8:348–354. [DOI] [PubMed] [Google Scholar]
- Li, E., D. Stupack, G.M. Bokoch, and G.R. Nemerow. 1998. Adenovirus endocytosis requires actin cytoskeleton reorganization mediated by Rho family GTPases. J. Virol. 72:8806–8812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, S.H., M.S. Marks, and F.M. Brodsky. 1998. A dominant-negative clathrin mutant differentially affects trafficking of molecules with distinct sorting motifs in the class II major histocompatibility complex (MHC) pathway. J. Cell Biol. 140:1023–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh, M., and A. Helenius. 1989. Virus entry into animal cells. Adv. Virus Res. 36:107–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNiven, M.A., H. Cao, K.R. Pitts, and Y. Yoon. 2000. The dynamin family of mechanoenzymes: pinching in new places. Trends Biochem. Sci. 25:115–120. [DOI] [PubMed] [Google Scholar]
- Mellman, I. 1996. Endocytosis and molecular sorting. Annu. Rev. Cell Dev. Biol. 12:575–625. [DOI] [PubMed] [Google Scholar]
- Mellman, I., and R.M. Steinman. 2001. Dendritic cells: specialized and regulated antigen processing machines. Cell. 106:255–258. [DOI] [PubMed] [Google Scholar]
- Miyazawa, N., R.G. Crystal, and P.L. Leopold. 2001. Adenovirus serotype 7 retention in a late endosomal compartment prior to cytosol escape is modulated by fiber protein. J. Virol. 75:1387–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano, M.Y., and U.F. Greber. 2000. Quantitative microscopy of fluorescent adenovirus entry. J. Struct. Biol. 129:57–68. [DOI] [PubMed] [Google Scholar]
- Nakano, M.Y., K. Boucke, M. Suomalainen, R.P. Stidwill, and U.F. Greber. 2000. The first step of adenovirus type 2 disassembly occurs at the cell surface, independently of endocytosis and escape to the cytosol. J. Virol. 74:7085–7095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng, T., D. Shima, A. Squire, P.I. Bastiaens, S. Gschmeissner, M.J. Humphries, and P.J. Parker. 1999. PKCalpha regulates beta1 integrin-dependent cell motility through association and control of integrin traffic. EMBO J. 18:3909–3923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nhieu, G.T., and P.J. Sansonetti. 1999. Mechanism of Shigella entry into epithelial cells. Curr. Opin. Microbiol. 2:51–55. [DOI] [PubMed] [Google Scholar]
- Nichols, B.J., and J. Lippincott-Schwartz. 2001. Endocytosis without clathrin coats. Trends Cell Biol. 11:406–412. [DOI] [PubMed] [Google Scholar]
- Norbury, C.C., L.J. Hewlett, A.R. Prescott, N. Shastri, and C. Watts. 1995. Class I MHC presentation of exogenous soluble antigen via macropinocytosis in bone marrow macrophages. Immunity. 3:783–791. [DOI] [PubMed] [Google Scholar]
- Pastan, I., P. Seth, D. FitzGerald, and M. Willingham. 1986. Adenovirus entry into cells: some new observations on an old problem. Concepts in Viral Pathogenesis II. A.L. Notkins and M.B.A. Oldstone, editors. Springer-Verlag New York, Inc., New York. 141–146.
- Pearse, B.M., C.J. Smith, and D.J. Owen. 2000. Clathrin coat construction in endocytosis. Curr. Opin. Struct. Biol. 10:220–228. [DOI] [PubMed] [Google Scholar]
- Rodriguez, A., A. Regnault, M. Kleijmeer, P. Ricciardi-Castagnoli, and S. Amigorena. 1999. Selective transport of internalized antigens to the cytosol for MHC class I presentation in dendritic cells. Nat. Cell Biol. 1:362–368. [DOI] [PubMed] [Google Scholar]
- Sampath, P., and T.D. Pollard. 1991. Effects of cytochalasin, phalloidin, and pH on the elongation of actin filaments. Biochemistry. 30:1973–1980. [DOI] [PubMed] [Google Scholar]
- Seth, P., M.C. Willingham, and I. Pastan. 1984. Adenovirus-dependent release of 51Cr from KB cells at an acidic pH. J. Biol. Chem. 259:14350–14353. [PubMed] [Google Scholar]
- Suomalainen, M., M.Y. Nakano, K. Boucke, S. Keller, R.P. Stidwill, and U.F. Greber. 1999. Microtubule-dependent minus and plus end–directed motilities are competing processes for nuclear targeting of adenovirus. J. Cell Biol. 144:657–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suomalainen, M., M.Y. Nakano, K. Boucke, S. Keller, and U.F. Greber. 2001. Adenovirus-activated PKA and p38/MAPK pathways boost microtubule-mediated nuclear targeting of virus. EMBO J. 20:1310–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson, J.A., and C. Watts. 1995. Macropinocytosis. Trends Cell Biol. 5:424–428. [DOI] [PubMed] [Google Scholar]
- Trotman, L.C., N. Mosberger, M. Fornerod, R.P. Stidwill, and U.F. Greber. 2001. Import of adenovirus DNA involves the nuclear pore complex receptor CAN/Nup214 and histone H1. Nat. Cell Biol. 3:1092–1100. [DOI] [PubMed] [Google Scholar]
- Veithen, A., P. Cupers, P. Baudhuin, and P.J. Courtoy. 1996. v-Src induces constitutive macropinocytosis in rat fibroblasts. J. Cell Sci. 109:2005–2012. [DOI] [PubMed] [Google Scholar]
- Wang, K.N., S. Huang, A. Kapoormunshi, and G. Nemerow. 1998. Adenovirus internalization and infection require dynamin. J. Virol. 72:3455–3458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West, M.A., M.S. Bretscher, and C. Watts. 1989. Distinct endocytotic pathways in epidermal growth factor-stimulated human carcinoma A431 cells. J. Cell Biol. 109:2731–2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West, M.A., A.R. Prescott, E.L. Eskelinen, A.J. Ridley, and C. Watts. 2000. Rac is required for constitutive macropinocytosis by dendritic cells but does not control its downregulation. Curr. Biol. 10:839–848. [DOI] [PubMed] [Google Scholar]
- Wickham, T.J., P. Mathias, D.A. Cheresh, and G.R. Nemerow. 1993. Integrin alpha v beta 3 and integrin alpha v beta 5 promote adenovirus internalization but not virus attachment. Cell. 73:309–319. [DOI] [PubMed] [Google Scholar]
- Wickham, T.J., E.J. Filardo, D.A. Cheresh, and G.R. Nemerow. 1994. Integrin αvβ5 selectively promotes adenovirus-mediated cell membrane permeabilization. J. Cell Biol. 127:257–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiley, H.S. 1988. Anomalous binding of epidermal growth factor to A431 cells is due to the effect of high receptor densities and a saturable endocytic system. J. Cell Biol. 107:801–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiley, H.S., and P.M. Burke. 2001. Regulation of receptor tyrosine kinase signaling by endocytic trafficking. Traffic. 2:12–18. [DOI] [PubMed] [Google Scholar]
- Xia, H., B. Anderson, Q. Mao, and B.L. Davidson. 2000. Recombinant human adenovirus: targeting to the human transferrin receptor improves gene transfer to brain microcapillary endothelium. J. Virol. 74:11359–11366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura, A. 1985. Adenovirus-induced leakage of co-endocytosed macromolecules into the cytosol. Cell Struct. Funct. 10:391–404. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}