

Abstract
Agrin is a nerve-derived factor that directs neuromuscular synapse formation, however its role in regulating interneuronal synaptogenesis is less clear. Here, we examine agrin's role in synapse formation between cholinergic preganglionic axons and sympathetic neurons in the superior cervical ganglion (SCG) using agrin-deficient mice. In dissociated cultures of SCG neurons, we found a significant decrease in the number of synapses with aggregates of presynaptic synaptophysin and postsynaptic neuronal acetylcholine receptor among agrin-deficient neurons as compared to wild-type neurons. Moreover, the levels of pre- and postsynaptic markers at the residual synapses in agrin-deficient SCG cultures were also reduced, and these defects were rescued by adding recombinant neural agrin to the cultures. Similarly, we observed a decreased matching of pre- and postsynaptic markers in SCG of agrin-deficient embryos, reflecting a decrease in the number of differentiated synapses in vivo. Finally, in electrophysiological experiments, we found that paired-pulse depression was more pronounced and posttetanic potentiation was significantly greater in agrin-deficient ganglia, indicating that synaptic transmission is also defective. Together, these findings indicate that neural agrin plays an organizing role in the formation and/or differentiation of interneuronal, cholinergic synapses.
Keywords: superior cervical ganglia; synaptogenesis; neuronal acetylcholine receptors; agrin knockout; compound action potential
Introduction
Synaptogenesis is critical for the formation, regeneration, and plasticity of the nervous system. Despite its importance, the signals that induce synapse formation and that regulate the differentiation of synaptic specializations are largely unknown. This process is best understood for the cholinergic neuromuscular junction, where a motoneuron-derived factor called agrin directs multiple aspects of synaptic differentiation (Burden, 1998; Sanes and Lichtman, 1999). Motoneurons express a neural-specific isoform of agrin that contains inserts at the z splice site (z+) and that is highly active in synaptogenesis, whereas muscle expresses an isoform that lacks these inserts (z−) and that is significantly less active (for review see Burden, 1998). Agrin induces synaptogenesis by signaling through the muscle specific receptor tyrosine kinase (MuSK),* and mice with a targeted disruption of either neural (z+) agrin or MuSK fail to form synaptic specializations at neuromuscular contacts, and consequently die at birth (DeChiara et al., 1996; Gautam et al., 1996; Glass et al., 1996). Thus, agrin/MuSK signaling is essential for both pre- and postsynaptic differentiation at the developing neuromuscular junction.
Analogous signaling factors, which direct the formation of interneuronal synapses in the central and peripheral nervous systems, are yet to be identified, although several candidates have been proposed (Garner et al., 2002). Interestingly, agrin is highly expressed in the developing central nervous system (CNS) and peripheral nervous system (PNS) during the period of synapse formation (Ruegg et al., 1992; Hoch et al., 1993; Ma et al., 1995), and agrin protein is also localized in some synaptic sites in the retina (Kroger et al., 1996; Mann and Kroger, 1996). However, glutamatergic synapses were still observed in the brains of neural agrin-deficient mice, and hippocampal and cortical neurons derived from these animals still formed glutamatergic and GABAergic synapses in culture (Li et al., 1999; Serpinskaya et al., 1999). On the other hand, antisense knockdown of agrin expression inhibited synaptogenesis in hippocampal neuron cultures (Ferreira, 1999; Bose et al., 2000), and it has been argued that compensatory mechanisms in the agrin-null animals might mask a requirement for agrin. Thus, the role of agrin in synaptogenesis in the CNS is currently controversial.
Agrin could function at cholinergic, interneuronal synapses in the PNS, which share a number of similarities with the neuromuscular junction. Consistent with this, agrin is concentrated at interneuronal synapses in superior cervical ganglion (SCG) neuronal cultures, and the sympathetic neurons expressed agrin isoforms that are neural specific (z+) and that occur as a type II transmembrane protein (Gingras and Ferns, 2001). We have now tested agrin's role in sympathetic synapse formation, and find that synaptogenesis is impaired in neural agrin-deficient SCG cultures, with significantly fewer synaptophysin-labeled nerve terminals and synaptic aggregates of the neuronal acetylcholine receptor (AChR) being formed. The synaptic defect in agrin−/− cultures was not due to impaired neuronal development, and was rescued by the addition of recombinant, neural (z+) agrin protein. In addition, we observed a decreased matching of pre- and postsynaptic differentiation in the SCG of agrin-deficient embryos, and defects in synaptic transmission. Together, these findings indicate that agrin plays an important role in regulating the formation of interneuronal, sympathetic synapses.
Results
Synaptogenesis is impaired in agrin-deficient SCG neuronal cultures
To investigate whether agrin plays a role in cholinergic, sympathetic synapse formation, we made use of agrin-deficient mice generated by Gautam et al. (1996), which lack the neural (z+) isoform of agrin, but still express low levels of z− agrin. We began by comparing synapse formation in wild-type (wt) and agrin−/− neuronal cultures derived from the SCG. In this well-characterized culture system (Higgins et al., 1991), it has been found that SCG neurons treated with heart cell–conditioned media or ciliary neurotrophic factor form numerous cholinergic synapses (Furshpan et al., 1976; Sah et al., 1987; Francis and Landis, 1999). Moreover, agrin is specifically concentrated at these synapses in vitro (Gingras and Ferns, 2001). As agrin−/− mice die at birth, we generated neuronal cultures from the SCG of wt and agrin−/− embryos at embryonic day (E)18.5. Synaptogenesis was then analyzed after 7 d in vitro (DIV 7) by immunostaining for pre- and postsynaptic markers; synaptophysin in the presynaptic nerve terminal and the neuronal AChR in the postsynaptic site. In wt cultures, we found that there were numerous synaptophysin-positive nerve terminals abutting the neuronal cell bodies and proximal dendrites, and these typically colocalized with aggregates of neuronal AChR containing the α5-subunit (Fig. 1 A). The terminals also costained for vesicular acetylcholine transporter (unpublished data), confirming that these are cholinergic synapses (Gingras and Ferns, 2001). However, in agrin−/− cultures, we observed significantly fewer synaptophysin puncta and α5-AChR aggregates on the neuronal cell bodies and proximal dendrites (Fig. 1 A). Identical results were obtained using another antibody to the neuronal AChR, which recognizes both the β2 and β4 subunits (Fig. 1 B; Forsayeth and Kobrin, 1997).
Figure 1.

Synapse number is reduced in agrin−/− SCG cultures. (A) SCG neurons from E18.5 embryos were cultured for 7 d, and immunostained for synaptophysin and α5-AChR. In wt SCG cultures, we observed numerous synaptophysin-labeled nerve terminals and colocalized aggregates of α5-AChR on the neuronal cell bodies (arrows) and proximal dendrites. In contrast, significantly fewer synaptic contacts were detected in the agrin−/− cultures. (B) A similar reduction was evident in the number of β2/β4-AChR puncta in agrin−/− cultures. (C) The number of synaptophysin and α5-AChR puncta/neuron were quantified according to their size, and expressed as a percentage of wt values. We found a significant decrease in the numbers of medium, large, and total synaptophysin puncta/neuron in agrin−/− cultures as compared with wt (P < 0.0003, Mann-Whitney test; n =13). The number of α5-AChR puncta was also significantly reduced in agrin−/− cultures (P < 0.0003, Mann-Whitney test; n = 8). Bars, 20 μm.
To quantify the relative levels of synaptogenesis in wt and agrin−/− cultures, the number of synaptophysin and AChR puncta on neuronal cell bodies and proximal dendrites were counted in a blinded fashion. We found that the total numbers of synaptophysin-labeled terminals and AChR clusters were decreased by 47 ± 2% and 49 ± 3%, respectively, in agrin−/− cultures as compared with +/+ or +/− cultures (Fig. 1 C) (mean decrease ± SEM; P < 0.0003, Mann-Whitney test; n = 13 and 8, respectively). We also quantified puncta according to size (medium 0.5–1.5 μm in diameter, and large >1.5 μm), and found that the decrease in agrin−/− cultures was most pronounced for the larger class of puncta. No significant difference was noted between synaptogenesis in agrin+/+ and +/− cultures, and therefore both were considered as wt. These findings indicate that there is a significant impairment of synaptogenesis in neural (z+) agrin−/− cultures.
We also found that the residual synaptophysin and α5-AChR immunostained puncta on agrin−/− neurons were generally less intense than those on wt neurons (Figs. 1 A and 2 A). Using Metamorph image analysis software, we generated intensity profiles and this analysis showed that the peak intensity of both synaptophysin and α5-AChR puncta was typically lower on agrin−/− neurons as compared with wt (Fig. 2 B). Indeed, we found that the total intensity of synaptophysin and α5-AChR puncta located on the neuronal cell body of agrin−/− neurons was significantly reduced as compared with wt (5 ± 2% and 5 ± 2% of wt levels, respectively; mean ± SEM, Mann-Whitney test, P < 0.014; n = 4; Fig. 2 C); this reflects a decrease in both the size and intensity of the synaptic puncta. A decrease was also seen for puncta located on the neurite network of agrin−/− cultures, which includes both dendrites and neurites/axons (42 ± 15% and 40 ± 9% of wt levels for synaptophysin and α5-AChR, respectively; mean ± SEM, Mann-Whitney test, P < 0.014, n = 4; Fig. 2 C). Thus, the decreased levels of synaptophysin and α5-AChR at synapses in agrin−/− cultures is more striking for contacts made on the neuronal cell bodies than for those on the neurites, which in some cases probably reflect axonal/axonal contacts or axonal/substrate contacts. Together, these findings indicate that both pre- and postsynaptic differentiation is impaired in neural agrin−/− SCG cultures.
Figure 2.

Synaptic differentiation is impaired in agrin−/− SCG cultures. A) Representative examples of wt and agrin−/− neurons show that the intensity of the α5-AChR puncta is typically reduced in agrin−/− cultures (arrows). (B) Intensity profiles corresponding to the lines drawn in A are shown for synaptophysin and α5-AChR. The peak intensity of the synaptophysin and α5-AChR puncta is lower on agrin−/− neurons than on wt (arrows). (C) The total intensity of synaptophysin and α5-AChR puncta was measured and compared for wt and agrin−/− cultures. We found a 95 ± 2% decrease in the total intensity of synaptophysin and α5-AChR immunostained puncta located on the cell body of agrin−/− neurons as compared with wt, reflecting a decrease in both the average size and intensity of the puncta (mean decrease ± SEM; P < 0.014; Mann-Whitney test, n = 4). Decreases of 58 ± 15 and 60 ± 9% were also observed for synaptophysin and α5-AChR immunostained puncta on the dendrites and neurites (mean decrease ± SEM; P < 0.014; Mann-Whitney test, n = 4). Bar, 20 μm.
Impaired synaptogenesis in agrin-deficient cultures is not due to defects in neuronal differentiation or neurite outgrowth
The decreased level of synaptogenesis in agrin−/− SCG cultures could reflect a role for agrin in synapse formation, or alternatively, in some other aspect of neuronal development. Therefore, we compared the general development of wt and agrin−/− SCG cultures, and observed no differences in the efficiency of plating or survival of agrin−/− neurons as compared with wt controls (unpublished data). To assay neuronal differentiation, we immunostained the cultures with a MAP-2 antibody to specifically label the soma and dendrites, and observed no difference in dendritic morphology between wt and agrin−/− neurons (Fig. 3 A) or in the average dendrite length (Fig. 3 B). Similarly, we assayed neurite outgrowth by immunostaining for neurofilament (NF-200), and observed no obvious differences in the pattern or density of neurites in the wt and agrin−/− cultures (Fig. 3 A) or in the total intensity of neurofilament immunostaining (101 ± 16% of wt values). Counts of neurite numbers were also comparable, indicating that neurite outgrowth was not defective in agrin−/− neurons (Fig. 3 C). Finally, we found that addition of exogenous agrin protein (z+ or z−) in the culture media of agrin−/− cultures did not affect neurite density.
Figure 3.

Neuronal development is comparable in wt and agrin−/− SCG cultures. (A) wt and agrin−/− SCG neurons (DIV 10) were immunostained for MAP-2 to identify the soma and the dendrites (arrows), and for neurofilament (NF-200) to label neurites. We observed no obvious differences in dendritic morphology or in the pattern of neurite outgrowth between wt and agrin−/− cultures. (B) Quantification of the length of the dendrites after MAP-2 immunostaining showed no significant difference between wt and agrin−/− neurons. (C) Quantification of neurite numbers, by counting the number of neurites intersecting a randomly chosen line, revealed no significant difference between wt and agrin−/− cultures. In addition, treatment of agrin−/− cultures with exogenous, z+ or z− agrin protein did not affect neurite numbers. (D) Quantification of the numbers of synaptophysin and α5-AChR puncta/neuron at DIV 7, 10, and 15 showed that there is a similar impairment of synaptogenesis at all three time points in agrin−/− cultures. Thus, there is no recovery of the synaptic defect with development. Bars: (MAP-2) 20 μm; (NF-200) 40 μm.
We also examined whether the defect in synaptogenesis was maintained with further development in vitro. To do this, we quantified the number of synaptophysin and α5-AChR puncta on wt and agrin−/− neurons in cultures ranging in age from 7–15 d in vitro. At all stages of development in vitro, we found that the number of synaptic puncta in the agrin−/− cultures was ∼50% of wt levels (Fig. 3 D). Thus, we observe no recovery of the synaptic defect in agrin−/− cultures, at least over this time period.
Agrin antibodies also inhibit synaptogenesis
In a complementary approach, we inhibited agrin function in SCG neuron cultures using acute application of a polyclonal antibody generated against the COOH terminus of rat agrin (Sugiyama et al., 1994). Rat SCG cultures were treated for 48 h with the agrin function-blocking antibody (16 μg/ml) or with preimmune serum, and synaptogenesis was then assayed by immunostaining for synaptophysin and α5-AChR (Fig. 4). We found that treatment with the anti-agrin antibody resulted in a significant decrease in the total numbers of both synaptophysin and α5-AChR puncta. Interestingly, the decrease was most evident for the larger class of puncta, suggesting that the agrin block may primarily affect the growth and maturation of synapses (60 ± 6% and 50 ± 7% decreases, respectively; mean decrease ± SEM, Mann-Whitney test, P < 0.005; n =11 and 5, respectively). In control experiments, we found that dendritic length and neurite numbers in agrin antibody-treated cultures were comparable to that in control cultures (101 ± 2% and 100 ± 4% of wt values, respectively). Thus, a transient block of agrin function also impairs interneuronal synaptogenesis in the SCG cultures.
Figure 4.
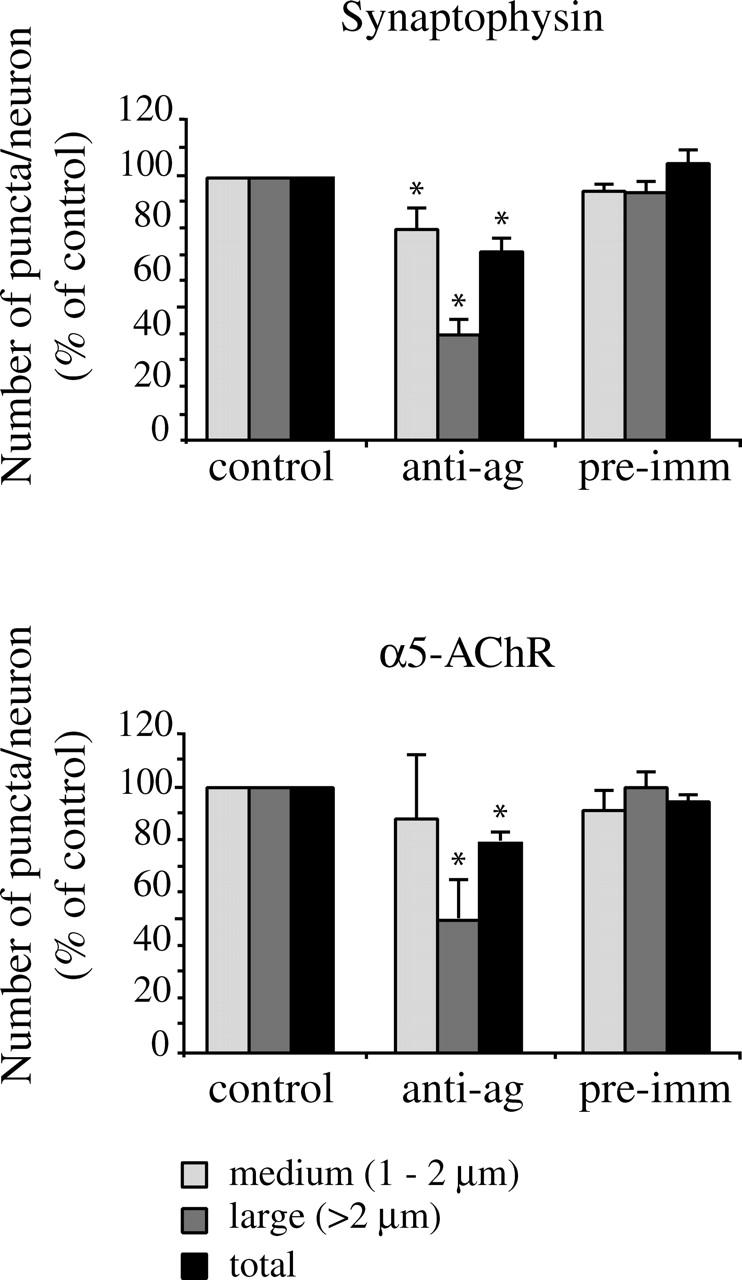
Agrin function-blocking antibody inhibits synaptogenesis. SCG cultures were treated with agrin antibody for 48 h, and then immunostained for synaptophysin and α5-AChR. Treatment with agrin antibody resulted in a significant decrease in the total number of synaptophysin-labeled puncta (28 ± 6%), with an even greater effect on the large class of puncta (60 ± 6%; mean decrease ± SEM; P < 0.0001; Mann-Whitney test; n = 11). A similar pattern was observed for α5-AChR immunostained puncta, with decreases of 50 ± 7% for large puncta, and 21 ± 2% for total puncta (mean decrease ± SEM; P < 0.005; Mann-Whitney test; n = 5). Treatment with the preimmune serum had no effect on numbers of synaptophysin or α5-AChR puncta.
Neural agrin rescues the synaptic defect in agrin-deficient SCG cultures
To further confirm the direct role of agrin in interneuronal synaptogenesis, and to compare the activity of the different isoforms, we tested whether recombinant agrin protein could rescue the synaptic defect in agrin−/− cultures. The neural agrin−/− cultures were treated with 200 pM soluble, COOH-terminal fragments of either z+ or z− agrin (C-Ag4, 8 and C-Ag0, 0; Ferns et al., 1993). We found that treatment with z+ agrin for 15 or 48 h restored the number of synaptophysin-positive nerve terminals and synaptic aggregates of α5-AChR on agrin−/− neurons to wt levels (Figs. 5 and 6 A). In contrast, we observed no rescue after treatment with z− agrin, even after 48 h. Thus, we observe a neural isoform-specific rescue of both pre- and postsynaptic markers. Potentially, this could reflect either the formation of new synapses, or the coordinated pre- and postsynaptic differentiation of existing, but immature synaptic contacts. Interestingly, neither z+ nor z− agrin isoforms had any effect on synaptogenesis in wt cultures (unpublished data).
Figure 5.

Neural agrin rescues the synaptic defect in agrin−/− SCG cultures. Agrin−/− SCG cultures were treated with 200 pM of z+ or z− agrin from DIV 5–7, and then immunostained for synaptophysin and α5-AChR. Treatment with soluble z+ agrin increased the number of synaptophysin and α5-AChR puncta on the cell body (arrow) and proximal dendrites (arrowhead) of agrin−/− neurons, as compared with untreated agrin−/− controls. In contrast, treatment with z− agrin produced no obvious change in the numbers of synaptophysin or α5-AChR puncta. Bar, 20 μm.
Figure 6.
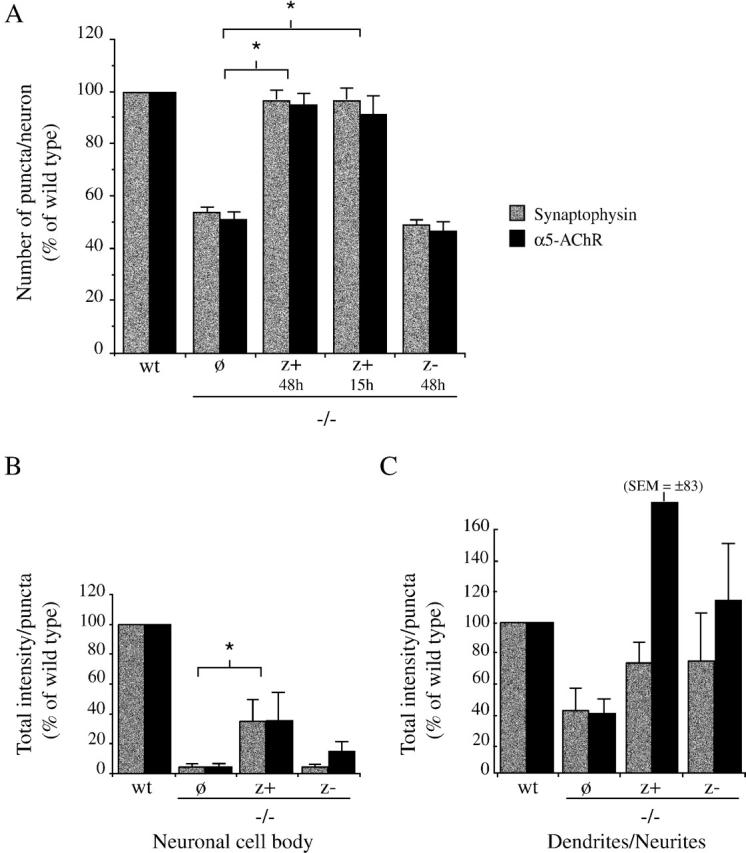
Neural agrin rescues the number and level of differentiation of synapses in agrin−/− cultures. (A) Agrin−/− SCG neurons were treated with soluble z+ or z− agrin, and the number of synaptophysin and α5-AChR puncta were quantified. Treatment with z+ agrin for 15 or 48 h significantly increased the number of synaptophysin and α5-AChR puncta on agrin−/− neurons as compared with untreated agrin−/− controls (P < 0.0001, Mann-Whitney test; n = 10 (synaptophysin [SP]) and 5 (AChR). In contrast, treatment with z− agrin for 48 h had no effect on the number of puncta on agrin−/− neurons. (B and C) The total intensity of the synaptophysin and α5-AChR puncta were determined for puncta on the cell body (B), and on the dendrites and neurites (C). (B) Treatment with z+ agrin significantly increased the total intensity of the synaptophysin and α5-AChR puncta on the neuronal cell body of agrin−/− neurons as compared with untreated agrin−/− neurons (to 35 ± 14% and 36 ± 17% of wt levels, respectively; mean ± SEM; P < 0.014; Mann-Whitney test; n = 4). Treatment with z− agrin had no significant effect. (C) Analysis of synaptophysin and α5-AChR puncta localized on the dendrites and neurites revealed some increase in the total intensity of the puncta in both z+ and z− agrin-treated cultures, but these effects were not statistically significant.
To assay whether exogenous agrin affects synaptic differentiation in agrin−/− cultures, we measured the total intensity of synaptophysin and α5-AChR puncta in each of the treatment conditions (Fig. 6, B and C). We found that the add-back of soluble z+ agrin significantly increased the total intensity of synaptophysin and α5-AChR puncta on agrin−/− neuronal cell bodies, although the levels only reached 35 ± 14 and 36 ± 17% of those on wt neurons, respectively (mean ± SEM; Mann-Whitney test; P < 0.014; n = 4). In contrast, treatment with z− agrin did not affect puncta intensity. Interestingly, both z+ and z− agrin treatments were associated with slight increases in the total intensity of puncta located on the dendrites and neurites, but the effects were not statistically significant (Fig. 6 C). Thus, agrin may have a differential effect on synaptic contacts on the neuronal cell bodies as compared to neurite/neurite or neurite/substrate contacts. Together, these findings demonstrate that exogenous, z+ agrin partially rescues the defect in synaptic differentiation in agrin−/− cultures, providing further support for a synaptic function for neural agrin.
Coordination of pre- and postsynaptic differentiation is impaired in neural agrin−/− SCG
To assay agrin's role in sympathetic synapse formation in vivo, we compared synaptogenesis in the SCG of wt and agrin−/− animals. As agrin-null animals die at birth, this analysis was limited to E18.5 embryos, and only a small number of immature synapses have formed in the ganglia at this stage (Rubin, 1985a, 1985b). Cryosections of the SCG were first immunostained for neurofilament, to localize the incoming axons and to confirm that the levels of innervation of the wt and agrin−/− ganglia were comparable. In both wt and agrin−/− ganglia, the innervation was patchy and immature, but we observed some punctate, synaptophysin labeling of nerve terminals in regions with neurofilament labeling (Fig. 7 A). Synaptogenesis was then assayed by immunostaining for synaptophysin in the nerve terminal and the neuronal AChR in the postsynaptic site. We found that a proportion of the synaptophysin labeled terminals colocalized with postsynaptic aggregates of β2/β4-AChR, indicating that some differentiated synapses form by E18.5 in both the wt and agrin−/− SCG (Fig. 7 B). We also observed some punctate synaptophysin and β2/β4-AChR immunostaining that did not colocalize with the other marker, which presumably reflects synaptophysin staining in axons as well as terminals, uninnervated β2/β4-AChR clusters, or asynchronous differentiation of immature synaptic contacts. A similar, partial overlap of pre- and postsynaptic markers is also seen during the initial stages of normal neuromuscular synapse formation (Lupa and Hall, 1989; Lin et al., 2001; Yang et al., 2001).
Figure 7.
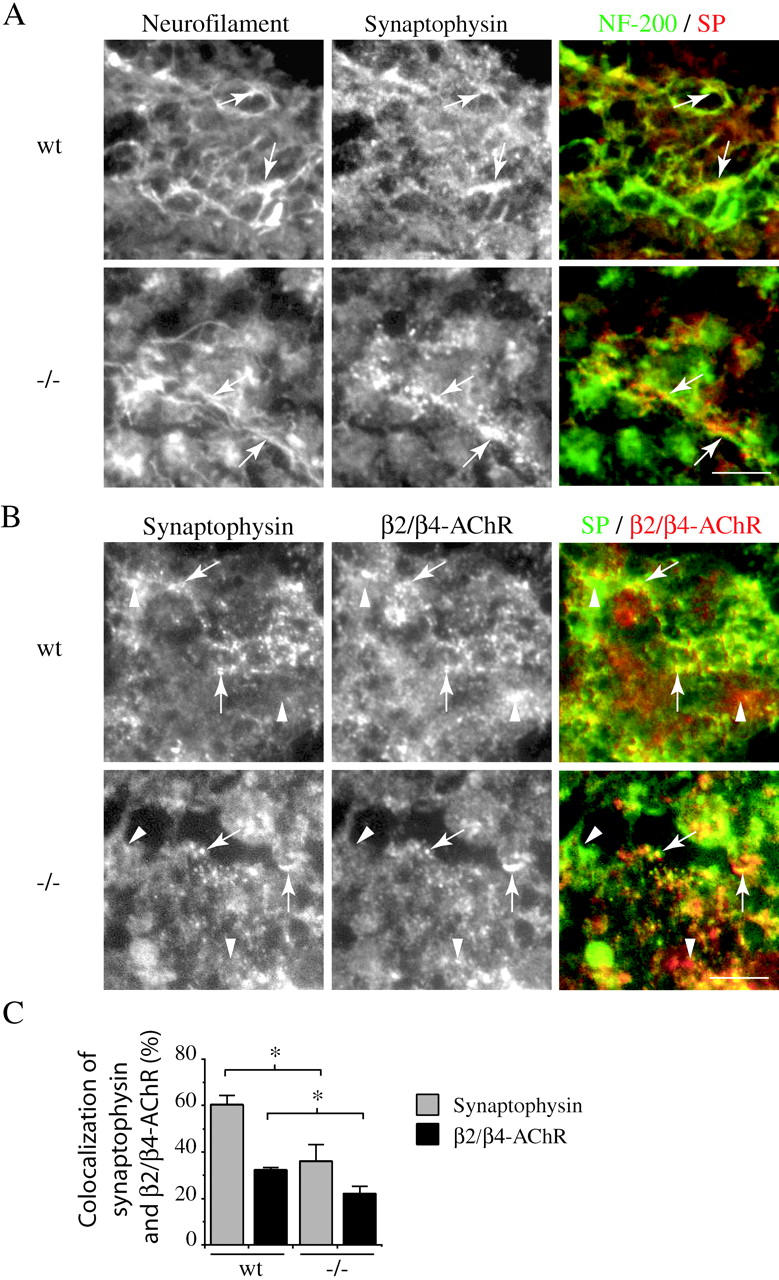
Innervation and synaptogenesis in wt and agrin−/− SCG. Frozen sections of E18.5 wt and agrin−/− SCG were immunostained for neurofilament, synaptophysin, and the β2/β4 subunits of the AChR. (A) In both wt and agrin−/− ganglia, we observed some punctate, synaptophysin labeling of nerve terminals in regions with neurofilament labeling (arrows). (B) A proportion of the SP-labeled terminals also colocalized with postsynaptic aggregates of β2/β4-AChR (arrows), indicating that some differentiated synapses are present at E18.5 in both the wt and agrin−/− SCG. We also observed some regions immunolabeled by synaptophysin that were devoid of β2/β4-AChR immunolabeling and vice-versa (arrowheads). (C) To assess the fraction of the total staining that reflects differentiated synapses with aligned pre- and postsynaptic markers, we quantified the overlap between synaptophysin and β2-/β4-AChR staining. In wt ganglia, we found that 60 ± 4% of synaptophysin labeling overlapped with β2-/β4-AChR puncta, and only 32 ± 1% of β2-/β4-AChR staining overlapped with synaptophysin puncta. In agrin−/− ganglia, the degree of overlap was significantly reduced to 36 ± 7 and 22 ± 3%, respectively (mean ± SEM, Mann-Whitney test; P < 0.029; n = 4). Thus, we find that there is a 30–40% decrease in the degree of overlap of synaptophysin and β2-/β4-AChR puncta in agrin−/− ganglia, suggesting that coordinated, pre- and postsynaptic differentiation is impaired. Bars, 50 μm.
To compare the levels of pre- and postsynaptic markers in wt and agrin−/− SCG, we measured the number, area, and total intensity of synaptophysin and β2/β4-AChR puncta. We found that there was no significant difference in any of these measures of total synaptophysin and β2/β4-AChR immunostaining between wt and agrin−/− ganglia. We then assessed the degree of overlap between the pre- and postsynaptic markers, to determine what fraction of the total staining actually reflects differentiated synapses. In wt ganglia, we found that 60 ± 4% of synaptophysin labeling overlapped with β2/β4-AChR puncta, and only 32 ± 1% of β2/β4-AChR staining overlapped with synaptophysin puncta (n = 4; Fig. 7 C). Interestingly, in agrin−/− ganglia, the degree of overlap was significantly reduced to 36 ± 7 and 22 ± 3%, respectively (mean ± SEM, Mann-Whitney test; P < 0.029; n = 4). Therefore, we find that the total levels of synaptophysin and β2/β4-AChR immunostaining in agrin−/− SCG are comparable to that in wt, but that there is a 30–40% decrease in their degree of overlap. This finding suggests that coordinated, pre- and postsynaptic differentiation is significantly impaired in the agrin−/− SCG, and consequently that there is a decreased number of differentiated synapses as compared with wt.
Synaptic transmission is defective in agrin−/− SCG
To determine whether synaptic transmission in sympathetic ganglia is defective in agrin−/− animals, we stimulated the preganglionic nerve and recorded from the postganglionic trunk. Fig. 8 A shows postganglionic compound action potentials (a reflection of the number of sympathetic neurons firing action potentials) from SCG of an E18.5 agrin−/− animal and from a wt littermate in response to suprathreshold stimulation of the preganglionic nerve. In agreement with the immunostaining data, these results demonstrate that preganglionic axons make functional, synaptic connections with sympathetic neurons in agrin-deficient animals. These compound action potentials were reversibly blocked by 100 μM hexamethonium as expected for cholinergic nicotinic synapses.
Figure 8.

Abnormalities in synaptic transmission in agrin−/− SCG. (A) Recordings of compound action potentials from the postganglionic trunk of SCG upon stimulation of the preganglionic nerve, from agrin−/− animal and wt littermate. The recordings show the stimulus artifact (Stim), the preganglionic action potential (Pre) and the postganglionic compound action potential (Post). Each panel shows two records superimposed. The panels on the left shows examples from PPD; 1 and 2 are the first and second responses of the pair. The panels on the right shows examples from PTP; C refers to control before the 10 Hz tetanus and P refers to the potentiated response 10 s after a 10 s train. (B) Quantification of the compound action potentials amplitudes relative to control (1st pulse). PPD was more pronounced in agrin−/− ganglia than in wt (44 and 14% decreases, respectively; P < 0.002), whereas PTP was enhanced as compared with wt (85 and 17% increases, respectively; P < 0.0003; n = 8 for wt animals and n = 6 for agrin−/− animals). These findings demonstrate that synaptic transmission in agrin−/− ganglia is defective, and are consistent with a decreased safety factor at agrin-deficient synapses.
We used paired-pulse depression (PPD) and posttetanic potentiation (PTP) to investigate the functional efficacy of these synapses. For PPD, we gave a pair of suprathreshold stimuli to the preganglionic nerve separated by 100 ms. We found that the compound action potential evoked by the second stimulus was depressed by ∼34% in SCG from agrin−/− animals; in ganglia from wt littermates, the depression was <15% (Fig. 8, A and B). For PTP, we applied a 10 Hz train to the preganglionic nerve for 10 s. 10 s after the train, the compound action potential was potentiated 185% in ganglia from agrin−/− animals, but only by 117% in ganglia from wt animals (Fig. 8, A and B). These results indicate clear differences in synaptic transmission in SCG from agrin−/− animals compared to that in wt littermates, and suggest that the safety factor for synaptic transmission (size of the excitatory postsynaptic potential relative to the threshold to evoke an action potential) is lower in agrin−/− than in wt ganglia. Importantly, this functional impairment of synapses in agrin−/− SCG is consistent with the reduced synaptic differentiation that we observed by immunostaining of agrin−/− SCG and SCG cultures.
Discussion
Agrin has a well-characterized role in organizing the formation of the neuromuscular junction, but its potential role in interneuronal synapse formation has been controversial (Li et al., 1999; Serpinskaya et al., 1999; Bose et al., 2000). Here, we show that agrin regulates the formation of cholinergic synapses in the sympathetic system, demonstrating that agrin also functions as a synaptogenic factor at some neuronal synapses.
Synaptically localized agrin regulates sympathetic synapse formation
Our findings provide strong evidence that agrin plays a central role in sympathetic synapse formation. In vitro, we find that synaptogenesis is significantly impaired in neural agrin-deficient SCG cultures, and after acute treatment of wt SCG cultures with an agrin function-blocking antibody. Similarly, in vivo, we find decreased coordination of pre- and postsynaptic differentiation in neural agrin−/− SCG, and a corresponding defect in synaptic function that results in impaired electrical transmission through the ganglia.
Our findings further suggest that agrin acts directly at the sympathetic synapse to regulate synaptogenesis. First, the impairment of synapse formation in agrin−/− cultures occurred in the absence of any detectable changes in general neuronal development, in agreement with earlier studies that reported normal differentiation of agrin−/− neurons (Li et al., 1999; Serpinskaya et al., 1999). Second, we found that the remaining synapses in agrin−/− cultures had reduced levels of clustered pre- and postsynaptic markers, suggesting that synaptic differentiation is also inhibited in the absence of neural agrin. Third, we found that the defect in synaptogenesis in agrin−/− cultures could be rescued by the addition of a soluble, agrin COOH-terminal fragment (Ferns et al., 1993), and that this was specific to the neural (z+) isoform, making it unlikely that agrin has a more general function as an extracellular matrix protein. Fourth, agrin is specifically localized at sympathetic synapses in SCG cultures (Gingras and Ferns, 2001). Together, these findings suggest that agrin acts at the synapse to orchestrate interneuronal, sympathetic synapse formation.
Neural agrin function in sympathetic synaptogenesis
Our findings show that sympathetic synapse formation is impaired, but not abolished, in neural agrin-deficient SCG and SCG cultures. These findings are somewhat reminiscent of the defects in neuromuscular synapse formation in agrin−/− mice, where there is a severe deficit in coordinated pre- and postsynaptic differentiation at nerve-muscle contacts, but a few rudimentary synaptic contacts are still observed (Gautam et al., 1996). In addition, substantial postsynaptic differentiation is evident in agrin−/− muscle at early stages of innervation, but this is progressively lost (Lin et al., 2001; Yang et al., 2001). Thus, it is currently unclear whether neural agrin functions to initiate neuromuscular synapse formation or to stabilize and promote the differentiation of synapses. Agrin could also act in either of these ways during sympathetic synapse formation, although our findings favor the idea that agrin is more critical for synaptic differentiation and maturation than for initiation of synaptogenesis. For example, in agrin−/− SCG cultures, we observed an ∼50% decrease in synapse number, but an even greater decrease in the levels of synaptic markers at the remaining synapses, and in agrin−/− SCG in vivo, we observed decreased matching of pre- and postsynaptic differentiation. The largest synaptic contacts were also the most affected in agrin−/− cultures and in the agrin antibody block experiments, consistent with an inhibition of synaptic maturation. Moreover, the rescue of the synaptic defect in agrin−/− cultures with soluble agrin likely reflects the differentiation of rudimentary synaptic contacts rather than the initiation of new synapse formation.
The physiological experiments also indicate that preganglionic axons form functional synapses with sympathetic neurons in SCG of agrin-deficient animals; however, clear differences exist between the properties of these synapses and those in wt littermates. We found that PPD was much more pronounced and PTP was significantly greater in agrin-deficient ganglia compared with those from control littermates. The simplest explanation for these results is that the safety factor for synaptic transmission is reduced at synapses in agrin-deficient ganglia and many preganglionic-evoked excitatory postsynaptic potentials are subthreshold. Our immunocytochemical results suggest that synapses in agrin-deficient ganglia are less differentiated than those in control ganglia and likely have a reduction in postsynaptic nAChRs density; both features could account for a reduction in the safety factor. If we assume that the amplitude of the potentiated compound action potential corresponds to the maximum response when all postganglionic sympathetic neurons are firing action potentials, then we estimate that >85% of the sympathetic neurons fire action potentials in control animals in response to low frequency stimulation of the preganglionic nerve; in contrast, ∼50% fire action potentials in agrin−/− ganglia. Together with the in vitro results, these findings indicate that neural agrin is required for the normal differentiation of sympathetic synapses, and correspondingly, for efficient synaptic transmission.
Agrin's mechanism of action at sympathetic synapses
The molecular mechanism by which agrin regulates sympathetic synapse formation is unclear, but could parallel that at the neuromuscular junction. In particular, the neural (z+) isoform of agrin appears to be the most active at both synapses, as we observed a significant impairment of synaptogenesis in neural agrin−/− SCG cultures, and the rescue of this defect was z+ agrin-specific. The MuSK receptor tyrosine kinase is therefore a candidate receptor for agrin at sympathetic synapses, and interestingly, a recent study has reported its expression in neural tissue as well as muscle (Ip et al., 2000). However, it is currently unclear whether MuSK is expressed in the preganglionic or postganglionic neurons, or if z+ agrin signals via pre- and/or postsynaptic receptors.
Agrin could also have a structural function at the sympathetic synapse. Agrin occurs as a type II transmembrane protein in sympathetic preganglionic neurons in vivo (Burgess et al., 2000) and in sympathetic neurons in vitro (Gingras and Ferns, 2001), and could act as an adhesion protein that interacts with a receptor on the apposing synaptic membrane. This would be somewhat analogous to the cadherins or neurexins/neuroligins that are proposed to align the pre- and postsynaptic membranes and facilitate the development of CNS synapses (Shapiro and Colman, 1999; Davis, 2000). Many adhesion proteins also have signaling functions (Aplin et al., 1998; Davis, 2000), and transmembrane agrin could signal via an adhesion type receptor, or even act as a receptor itself. Potential partners that are known to bind agrin include dystroglycan (Ruegg, 2001), which is concentrated at sympathetic synapses (Zaccaria et al., 2000; Gingras and Ferns, 2001), integrins (Martin and Sanes, 1997; Burkin et al., 2000), and IgCAMs (Cole and Halfter, 1996; Bixby et al., 2002). Finally, we cannot discount the possibility that agrin acts in less direct ways to induce or maintain sympathetic synapses, such as by binding other growth or differentiation factors (Daggett et al., 1996; Cotman et al., 1999), or inducing gene expression (Ji et al., 1998; Hilgenberg et al., 1999).
In conclusion, we have found that synaptic differentiation and function are significantly impaired in neural agrin-deficient SCG and SCG cultures, demonstrating that neural agrin plays an important organizing role in sympathetic synapse formation. Thus, our findings establish agrin as a synaptogenic signal at interneuronal synapses.
Materials and methods
Neural agrin-deficient (−/−) mice
For these studies, we used neural agrin-deficient mice generated by Gautam et al. (1996) that lack the z+ isoform of agrin, but still express low levels of the z− agrin isoform. The neural agrin-deficient (−/−) animals develop normally in utero, but die perinatally due to an inability to move and breathe. The colony was maintained by breeding heterozygous animals, and all breeding animals and experimental embryos were individually genotyped by PCR, as previously described (Serpinskaya et al., 1999).
Neuronal cultures
SCG were dissected from E18.5 agrin−/− and wt littermates, and dissociated enzymatically and mechanically as previously described (Gingras and Ferns, 2001). Briefly, ganglia were incubated for 45 min in Hank's Balanced Salt Solution/trypsin 3× (1 mg/ml; Worthington Biochemical Corporation) and gently triturated with a fire-polished Pasteur pipette. Dissociated cells were washed in plating media, isolated by centrifugation (800 rpm, 4 min), and resuspended in growth media. Neurons were then plated at a density of ∼35 cells/mm2 on laminin-coated aclar coverslips in a modified petri dish. Plating media consisted of Leibovitz's L-15 medium supplemented with cofactors, fresh vitamins, penicillin-streptomycin, and 10% normal horse serum, and growth medium (1 ml/dish) consisted of DME supplemented with cofactors, fresh vitamins, penicillin-streptomycin, 2.5S NGF (25 ng/ml), 5% normal rat serum, and ciliary neurotrophic factor (0.02 μg/ml), a gift from Drs. P. Richardson and R. Dunn (McGill University, Montreal, Quebec, Canada). Cultures were maintained in 5% CO2 at 37°, and treated with 1-β-D-arabinofuranoside (Ara-C, 5 mM; Sigma-Aldrich) for the first 2–3 d to eliminate nonneuronal cells.
For agrin rescue experiments, SCG cultures generated from wt and agrin−/− littermates were treated for 15 or 48 h with soluble recombinant z+ (C-Ag 4,8) or z- (C-Ag 0,0) agrin (Ferns et al., 1993) at 200 pM concentration. Untreated wt and agrin−/− cultures were used as controls, and the phenotype was then analyzed immunohistochemically as described below.
For the agrin antibody block experiments, neuronal cultures were prepared from the SCG of newborn rats as described above (Sprague Dawley). At DIV 5, the cultures were treated with a polyclonal anti-agrin antibody (16 μg/ml; Sugiyama et al., 1994) or preimmune serum for 48 h, and then immunostained as below. In control experiments, we found that this antibody significantly inhibited agrin-induced clustering of the AChR in C2 muscle cell cultures (unpublished data).
Immunohistochemistry
Antibodies.
To localize presynaptic nerve terminals, we immunostained cultures with a monoclonal antibody to the synaptic vesicle protein, synaptophysin (SVP-38; Sigma-Aldrich). To localize the AChR, we used a polyclonal antibody to the β2/β4 subunits (Forsayeth and Kobrin, 1997), a gift from J. Forsayeth (Elan Pharmaceuticals, San Francisco, CA), and rat mAb210 (Bio/Can Scientific), which recognizes the rodent AChR α5 subunit. The β2/β4 antiserum worked best on unfixed tissue and was used for immunostaining of cryosections, whereas mAb210 worked best on fixed tissue and was used for immunostaining and quantification of synapses in SCG cultures. Monoclonal anti–MAP-2 (Sigma-Aldrich) was used to label the soma and the dendrites, and a polyclonal anti-neurofilament 200 kD (Sigma-Aldrich) to label axons and neurites. To immunostain for agrin, we used a polyclonal antibody that recognizes all agrin isoforms (Sugiyama et al., 1994), a gift from J. Sugiyama and Z. Hall (University of California, San Francisco, CA). Rhodamine- and fluorescein-conjugated secondary antibodies were all species specific (Jackson ImmunoResearch Laboratories, Inc.; Bio/Can Scientific).
For analysis of the SCG, ganglia were dissected from E18.5 wt and agrin−/− littermates, embedded fresh in OCT compound, and frozen using 2-methyl-butane cooled in liquid nitrogen. Frozen sections (∼12 μm) of the SCG were mounted on poly-l-lysine–coated slides (0.1 mg/ml; Sigma-Aldrich), and blocked with 10% normal donkey serum/phosphate buffer saline for 30 min. Sections were then incubated with primary antibodies (see above) for 1 h at room temperature, rinsed several times in PBS, and reincubated with rhodamine- or fluorescein-conjugated secondary antibodies. After rinsing in PBS, the slides were coverslipped using Immuno Floure mounting media (ICN). As controls, some cryosections were processed without the primary antibody or with preimmune serum. Crossreactivity tests were also conducted with all different combinations of the secondary antibodies. The sections were observed with a Nikon Eclipse E600 fluorescence microscope, and digital images were taken with a SensSys air-cooled camera system (CCD) and analysed using the Metamorph 4.6 image analysis system.
For analysis of SCG cultures, for most of the antibodies the cultures were fixed with 4% paraformaldehyde/4% sucrose/ 0.1 M phosphate buffer at room temperature. For labeling with the β2/β4 antibody, the cells were fixed more lightly in 1% paraformaldehyde/4% sucrose/0.1 M phosphate buffer. After fixation, the cells were permeabilized in a solution of 0.1% Triton X-100 for 10 min, and then immunostained as described above. To assess for surface expression of agrin and α5-AChR, we incubated the live cultures with the anti-agrin and mAb210 antibodies for 30 min at 37°C. After rinsing in growth media, the cultures were fixed and stained as above.
Quantification
To quantify the number of synaptophysin and α5-AChR puncta in SCG cultures, in each experiment 10 neurons/condition were selected randomly using phase optics. The number of puncta on the cell bodies and proximal dendrites (<50 μm from soma) of each neuron were then counted under fluorescence optics. These puncta were classified in arbitrary categories of medium or large (mouse: medium = 0.5 to 1.5 μm, large = >1.5 μm), and total puncta numbers were then calculated. To normalize the counts between experiments, we expressed the number of puncta on the agrin−/− neurons as a percentage of that in the wt.
To quantify the intensity of the puncta in SCG cultures, we used the Metamorph imaging system. First, we randomly selected isolated neurons or pairs of neurons under phase optics, therefore avoiding any complex groups of neuronal cell bodies. For each synaptic protein, digital images of the immunostained neurons were captured using identical exposure times for wt and agrin−/− neurons. We then quantified the intensity of puncta on the neuronal cell body and most proximal dendrites, by selecting a box specifically around the cell body. A threshold was selected to eliminate background staining, and the total intensity of puncta above this background was then measured. The measurement of total puncta intensity reflects both the size and intensity of the puncta. Similarly, to quantify puncta on the dendrites and neurites, we selected a larger box containing the cell body and proximal 50 μm of the dendrites, and quantified as above. This selected area also contained neurites and axons, and the cell body was excluded from this second analysis due to its higher level of background staining. To compare neurite numbers between agrin−/− and wt cultures, we quantified neurofilament immunostaining in a similar fashion (eight coverslips for each, in two independent experiments).
To quantify the intensity of the synaptic puncta in the SCG, we always processed and immunostained wt and agrin−/− ganglia in parallel. We analyzed eight wt and eight agrin−/− ganglia (n = 4 embryos) by taking cryosections through the central 2/3 of each ganglia (total of 63 wt and 60 agrin−/− sections), and immunostaining as above. We then captured two to three digital images that covered the entire cryosection, using identical exposure times for wt and agrin−/−. Using wt sections, a threshold was selected to eliminate diffuse background staining, and the intensity of selected puncta was measured within a 130 × 100-μm box centered on each digital image (for both wt and agrin−/−). We then measured the degree of overlap between the synaptophysin and α5-AChR puncta.
Electrophysiological experiments
All recordings were done on SCG isolated from E18 agrin−/− embryos or their wt littermates and perfused continuously with oxygenated Ringer solution at 22°C. We used suction electrodes to stimulate the preganglionic nerve (cervical sympathetic trunk) and record compound action potentials from the postganglionic trunk as it exits the ganglion. The preganglionic nerve was stimulated with an S88 stimulator (Grass Instruments) connected to a SIU5 stimulus isolation unit (Grass Instruments). Signals from the postganglionic electrodes were amplified with an AC differential amplifier (DP-301, Warner Instruments), filtered (low band pass = 100 Hz and high band pass = 1,000 Hz), digitized at 44 KHz by a pulse code modulation unit (PCM701; Sony) and stored on a videocassette recorder (Sony). For offline analysis, first we acquired the stored signals at 2K Hz using Patchkit (Alembic Software), an A/D card (Omega) and a Pentium 3-based PC computer, then analyzed the records with Igor (Wavemetrics).
When recording with suction electrodes, the shunt between the electrode and the nerve trunk varies from preparation to preparation; this makes it difficult to compare absolute amplitudes of compound action potentials among animals. Therefore, we evaluated synaptic transmission by measuring PPD and PTP. To measure PPD, we delivered two suprathreshold stimuli to the preganglionic nerve separated by 100 ms and expressed the amplitude of the second compound action potential as a percentage of the first. To measure PTP, first we delivered a 10 Hz train of suprathreshold stimuli to the preganglionic nerve for 10 s, waited 10 s, and then we gave 1 suprathreshold stimuli and expressed the amplitude of the compound action potential as a percentage of the control measured before the train. No differences were noted between recordings from the SCG of agrin+/+ and +/− embryos, and both were considered as the controls for the quantitative analysis.
Acknowledgments
We would like to thank Drs. Fabio Rupp and Josh Sanes (Washington University, St. Louis, MO) for supplying the neural agrin-deficient mice, and Drs. J. Forsayeth (Elan Pharmaceuticals), J. Sugiyama, and Z. Hall (University of California San Francisco, San Francisco, CA) for their gifts of antibodies. We also thank Francois-Joseph Lapointe for his valuable help with statistics, and the Ferns lab for their helpful comments on the manuscript.
This work was supported by a Canadian Institutes of Health Research (CIHR)/Neuromuscular Research Partnership grant (13237) and a CIHR scholarship to M. Ferns, a CIHR grant to E. Cooper, and a doctoral fellowship to J. Gingras.
Footnotes
Abbreviations used in this paper: AChR, acetylcholine receptor; CNS, central nervous system; DIV, days in vitro; E, embryonic day; MuSK, muscle specific receptor tyrosine kinase; PNS, peripheral nervous system; PPD, paired-pulse depression; PTP, posttetanic potentiation; SCG, superior cervical ganglia; SP, synaptophysin; wt, wild-type.
References
- Aplin, A.E., A. Howe, S.K. Alahari, and R.L. Juliano. 1998. Signal transduction and signal modulation by cell adhesion receptors: the role of integrins, cadherins, immunoglobulin-cell adhesion molecules, and selectins. Pharmacol. Rev. 50:197–263. [PubMed] [Google Scholar]
- Bixby, J.L., K. Baerwald-De La Torre, C. Wang, F.G. Rathjen, and M.A. Ruegg. 2002. A neuronal inhibitory domain in the N-terminal half of agrin. J. Neurobiol. 50:164–179. [DOI] [PubMed] [Google Scholar]
- Bose, C.M., D. Qiu, A. Bergamaschi, B. Gravante, M. Bossi, A. Villa, F. Rupp, and A. Malgaroli. 2000. Agrin controls synaptic differentiation in hippocampal neurons. J. Neurosci. 20:9086–9095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burden, S.J. 1998. The formation of neuromuscular synapses. Genes Dev. 12:133–148. [DOI] [PubMed] [Google Scholar]
- Burgess, R.W., W.C. Skarnes, and J.R. Sanes. 2000. Agrin isoforms with distinct amino termini. Differential expression, localization, and function. J. Cell Biol. 151:41–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkin, D.J., J.E. Kim, M. Gu, and S.J. Kaufman. 2000. Laminin and alpha7beta1 integrin regulate agrin-induced clustering of acetylcholine receptors. J. Cell Sci. 113:2877–2886. [DOI] [PubMed] [Google Scholar]
- Cole, G.J., and W. Halfter. 1996. Agrin: an extracellular matrix heparan sulfate proteoglycan involved in cell interactions and synaptogenesis. Perspect. Dev. Neurobiol. 3:359–371. [PubMed] [Google Scholar]
- Cotman, S.L., W. Halfter, and G.J. Cole. 1999. Identification of extracellular matrix ligands for the heparan sulfate proteoglycan agrin. Exp. Cell Res. 249:54–64. [DOI] [PubMed] [Google Scholar]
- Daggett, D.F., M.W. Cohen, D. Stone, K. Nikolics, H. Rauvala, and H.B. Peng. 1996. The role of an agrin-growth factor interaction in ACh receptor clustering. Mol. Cell. Neurosci. 8:272–285. [DOI] [PubMed] [Google Scholar]
- Davis, G.W. 2000. The making of a synapse: target-derived signals and presynaptic differentiation. Neuron. 26:551–554. [DOI] [PubMed] [Google Scholar]
- DeChiara, T.M., D.C. Bowen, D.M. Valenzuela, M.V. Simmons, W.T. Poueymirou, S. Thomas, E. Kinetz, D.L. Compton, E. Rojas, J.S. Park, et al. 1996. The receptor tyrosine kinase MuSK is required for neuromuscular junction formation in vivo. Cell. 85:501–512. [DOI] [PubMed] [Google Scholar]
- Ferns, M.J., J.T. Campanelli, W. Hoch, R.H. Scheller, and Z. Hall. 1993. The ability of agrin to cluster AChRs depends on alternative splicing and on cell surface proteoglycans. Neuron. 11:491–502. [DOI] [PubMed] [Google Scholar]
- Ferreira, A. 1999. Abnormal synapse formation in agrin-depleted hippocampal neurons. J. Cell Sci. 112:4729–4738. [DOI] [PubMed] [Google Scholar]
- Forsayeth, J.R., and E. Kobrin. 1997. Formation of oligomers containing the beta3 and beta4 subunits of the rat nicotinic receptor. J. Neurosci. 17:1531–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis, N.J., and S.C. Landis. 1999. Cellular and molecular determinants of sympathetic neuron development. Annu. Rev. Neurosci. 22:541–566. [DOI] [PubMed] [Google Scholar]
- Furshpan, E.J., P.R. MacLeish, P.H. O'Lague, and D.D. Potter. 1976. Chemical transmission between rat sympathetic neurons and cardiac myocytes developing in microcultures: evidence for cholinergic, adrenergic, and dual-function neurons. Proc. Natl. Acad. Sci. USA. 73:4225–4229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garner, C.C., R.G. Zhai, E.D. Gundelfinger, and N.E. Ziv. 2002. Molecular mechanisms of CNS synaptogenesis. Trends Neurosci. 25:243–251. [DOI] [PubMed] [Google Scholar]
- Gautam, M., P.G. Noakes, L. Moscoso, F. Rupp, R.H. Scheller, J.P. Merlie, and J.R. Sanes. 1996. Defective neuromuscular synaptogenesis in agrin-deficient mutant mice. Cell. 85:525–535. [DOI] [PubMed] [Google Scholar]
- Gingras, J., and M. Ferns. 2001. Expression and localization of agrin during sympathetic synapse formation in vitro. J. Neurobiol. 48:228–242. [DOI] [PubMed] [Google Scholar]
- Glass, D.J., D.C. Bowen, T.N. Stitt, C. Radziejewski, J. Bruno, T.E. Ryan, D.R. Gies, S. Shah, K. Mattsson, S.J. Burden, et al. 1996. Agrin acts via a MuSK receptor complex. Cell. 85:513–523. [DOI] [PubMed] [Google Scholar]
- Higgins, D., P.J. Lein, D.J. Osterhout, and M.I. Johnson. 1991. Tissue culture of mammalian autonomic neurons. In Culturing Nerve Cells. G. Banker and K. Goslin, editors. The MIT Press, Cambridge, MA. 453 pp.
- Hilgenberg, L.G., C.L. Hoover, and M.A. Smith. 1999. Evidence of an agrin receptor in cortical neurons. J. Neurosci. 19:7384–7393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoch, W., M. Ferns, J.T. Campanelli, Z.W. Hall, and R.H. Scheller. 1993. Developmental regulation of highly active alternatively spliced forms of agrin. Neuron. 11:479–490. [DOI] [PubMed] [Google Scholar]
- Ip, F.C., D.G. Glass, D.R. Gies, J. Cheung, K.O. Lai, A.K. Fu, G.D. Yancopoulos, and N.Y. Ip. 2000. Cloning and characterization of muscle-specific kinase in chicken. Mol. Cell. Neurosci. 16:661–673. [DOI] [PubMed] [Google Scholar]
- Ji, R.R., C.M. Bose, C. Lesuisse, D. Qiu, J.C. Huang, Q. Zhang, and F. Rupp. 1998. Specific agrin isoforms induce cAMP response element binding protein phosphorylation in hippocampal neurons. J. Neurosci. 18:9695–9702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroger, S., S.E. Horton, and L.S. Honig. 1996. The developing avian retina expresses agrin isoforms during synaptogenesis. J. Neurobiol. 29:165–182. [DOI] [PubMed] [Google Scholar]
- Li, Z., L.G. Hilgenberg, D.K. O'Dowd, and M.A. Smith. 1999. Formation of functional synaptic connections between cultured cortical neurons from agrin-deficient mice. J. Neurobiol. 39:547–557. [DOI] [PubMed] [Google Scholar]
- Lin, W., R.W. Burgess, B. Dominguez, S.L. Pfaff, J.R. Sanes, and K.F. Lee. 2001. Distinct roles of nerve and muscle in postsynaptic differentiation of the neuromuscular synapse. Nature. 410:1057–1064. [DOI] [PubMed] [Google Scholar]
- Lupa, M.T., and Z.W. Hall. 1989. Progressive restriction of synaptic vesicle protein to the nerve terminal during development of the neuromuscular junction. J. Neurosci. 9:3937–3945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, E., R. Morgan, and E.W. Godfrey. 1995. Agrin mRNA variants are differentially regulated in developing chick embryo spinal cord and sensory ganglia. J. Neurobiol. 26:585–597. [DOI] [PubMed] [Google Scholar]
- Mann, S., and S. Kroger. 1996. Agrin is synthesized by retinal cells and colocalizes with gephyrin. Mol. Cell. Neurosci. 8:1–13. [DOI] [PubMed] [Google Scholar]
- Martin, P.T., and J.R. Sanes. 1997. Integrins mediate adhesion to agrin and modulate agrin signaling. Development. 124:3909–3917. [DOI] [PubMed] [Google Scholar]
- Rubin, E. 1985. a. Development of the rat superior cervical ganglion: ingrowth of preganglionic axons. J. Neurosci. 5:685–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin, E. 1985. b. Development of the rat superior cervical ganglion: initial stages of synapse formation. J. Neurosci. 5:697–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruegg, M.A. 2001. Molecules involved in the formation of synaptic connections in muscle and brain. Matrix Biol. 20:3–12. [DOI] [PubMed] [Google Scholar]
- Ruegg, M.A., K.W. Tsim, S.E. Horton, S. Kroger, G. Escher, E.M. Gensch, and U.J. McMahan. 1992. The agrin gene codes for a family of basal lamina proteins that differ in function and distribution. Neuron. 8:691–699. [DOI] [PubMed] [Google Scholar]
- Sah, D.W., R.H. Loring, and R.E. Zigmond. 1987. Long-term blockade by toxin F of nicotinic synaptic potentials in cultured sympathetic neurons. Neuroscience. 20:867–874. [DOI] [PubMed] [Google Scholar]
- Sanes, J.R., and J.W. Lichtman. 1999. Development of the vertebrate neuromuscular junction. Annu. Rev. Neurosci. 22:389–442. [DOI] [PubMed] [Google Scholar]
- Serpinskaya, A.S., G. Feng, J.R. Sanes, and A.M. Craig. 1999. Synapse formation by hippocampal neurons from agrin-deficient mice. Dev. Biol. 205:65–78. [DOI] [PubMed] [Google Scholar]
- Shapiro, L., and D.R. Colman. 1999. The diversity of cadherins and implications for a synaptic adhesive code in the CNS. Neuron. 23:427–430. [DOI] [PubMed] [Google Scholar]
- Sugiyama, J., D.C. Bowen, and Z.W. Hall. 1994. Dystroglycan binds nerve and muscle agrin. Neuron. 13:103–115. [DOI] [PubMed] [Google Scholar]
- Yang, X., S. Arber, C. William, L. Li, Y. Tanabe, T.M. Jessell, C. Birchmeier, and S.J. Burden. 2001. Patterning of muscle acetylcholine receptor gene expression in the absence of motor innervation. Neuron. 30:399–410. [DOI] [PubMed] [Google Scholar]
- Zaccaria, M.L., M.E. De Stefano, C. Gotti, T.C. Petrucci, and P. Paggi. 2000. Selective reduction in the nicotinic acetylcholine receptor and dystroglycan at the postsynaptic apparatus of mdx mouse superior cervical ganglion. J. Neuropathol. Exp. Neurol. 59:103–112. [DOI] [PubMed] [Google Scholar]