

Abstract
Duchenne muscular dystrophy results from the lack of dystrophin, a cytoskeletal protein associated with the inner surface membrane, in skeletal muscle. The absence of dystrophin induces an abnormal increase of sarcolemmal calcium influx through cationic channels in adult skeletal muscle fibers from dystrophic (mdx) mice. We observed that the activity of these channels was increased after depletion of the stores of calcium with thapsigargin or caffeine. By analogy with the situation observed in nonexcitable cells, we therefore hypothesized that these store-operated channels could belong to the transient receptor potential channel (TRPC) family. We measured the expression of TRPC isoforms in normal and mdx adult skeletal muscles fibers, and among the seven known isoforms, five were detected (TRPC1, 2, 3, 4, and 6) by RT-PCR. Western blot analysis and immunocytochemistry of normal and mdx muscle fibers demonstrated the localization of TRPC1, 4, and 6 proteins at the plasma membrane. Therefore, an antisense strategy was used to repress these TRPC isoforms. In parallel with the repression of the TRPCs, we observed that the occurrence of calcium leak channels was decreased to one tenth of its control value (patch-clamp technique), showing the involvement of TRPC in the abnormal calcium influx observed in dystrophic fibers.
Keywords: calcium channels; dystrophy; sarcoplasmic reticulum; capacitative entry; TRP channel
Introduction
Duchenne muscular dystrophy (DMD)* is a progressive neuromuscular disease affecting 1/3,500 male births. It is due to a defect in the p21 band of the X chromosome (Monaco et al., 1986) affecting dystrophin, a 427-kD protein located at the cytoplasmic face of the sarcolemma (Hoffman et al., 1987). In normal skeletal muscle cells, dystrophin is associated with actin and with a complex of membrane glycoproteins, linking the cytoskeleton and the ECM, and therefore protecting the membrane against mechanical damage during muscle contraction. The absence of dystrophin in DMD patients leads to muscle degeneration and progressive weakness. It has been proposed that an alteration of the homeostasis of intracellular Ca might be responsible for this muscle degeneration. This was based on two facts: (1) total Ca (free and bound calcium) content is doubled in muscle biopsies from DMD patients and in mdx muscles (a murine model of DMD) and (2) the influx of Ca through the cellular membrane is increased (Bertorini et al., 1982; Tutdibi et al., 1999; for review see Gillis, 1999). The functional repercussions on the resting cytosolic concentration of free Ca ([Ca2+]i) in muscle fibers are still disputed (Gailly et al., 1993; Pressmar et al., 1994; Hopf et al., 1996a; Leijendekker et al., 1996; Collet et al., 1999), but [Ca2+] seems increased at least in the submembrane compartment (Mallouk et al., 2000). It has been shown that spontaneous proteolysis was higher in mdx than in control muscle (MacLennan et al., 1991), that it could be reduced to normal rate if the external Ca concentration was reduced, and exaggerated by increasing Ca (Turner et al., 1988). This led to the hypothesis that dystrophin-lacking fibers suffer from a chronic Ca overload, resulting in activation of Ca-dependent proteases (MacLennan et al., 1991). The present study was designed to identify the membrane ion channels responsible for the abnormal Ca influx observed in muscle fibers of mdx mice. These channels were previously characterized by Franco-Obregon and Lansman (1994). We found that they were activated by depletion of the internal stores of Ca. By analogy to the situation known in nonexcitable cells (see Discussion), we studied whether transient receptor potential channel (TRPC) isoforms were involved in the influx. Using an antisense strategy, we show that TRPC1 and/or TRPC4 proteins are constitutive of the voltage-independent Ca channels through which an increased amount of Ca penetrates into the muscle cells in mdx mice. This finding gives a new possible trail in the search of drug therapies of DMD.
Results
Voltage-independent Ca channels in adult skeletal muscle fibers
By using the patch-clamp technique (cell-attached configuration at −60-mV holding potential), we detected spontaneous Ca channel activity in collagenase-isolated flexor digitorum brevis (FDB) fibers maintained for 48 h in culture. In the conditions used (110 mM CaCl2 in the pipette), these channels had a conductance of 8 pS. The current-voltage relationship showed no sign of voltage activation or inactivation and was identical in mdx and normal fibers (Fig. 1, A and B). Reversal potential was similar in C57 (19 ± 4 mV, n = 5) and in mdx fibers (20 ± 3 mV, n = 6). The channel had a poor selectivity with respect to Ba2+, Mn2+, and Na+ (unpublished data). To evaluate how much Ca entered through the cation channel, we calculated the relative selectivity of divalent over monovalent cations using the Goldman-Hodgkin-Katz equation modified for divalent cations (Lee and Tsien, 1984). With 110 mM CaCl2 in the electrode and assuming K+ to be the predominant intracellular cation, the channel was slightly more permeable to Ca than to K+ with P Ca/P K = 1.3. This value is in the range observed previously (Franco and Lansman, 1990b). All these characteristics are similar to those of the mechanosensitive channels already described (Franco-Obregon and Lansman, 1994) and are similar in C57 and mdx fibers. However, the occurrence of the channel (number of patches in which a Ca current is recorded/number of patches sampled) was significantly increased by a factor of 1.6 in mdx compared with C57 (87 vs. 54%) and the open probability (Po) was approximately doubled in mdx fibers (0.07 ± 0.06, n = 14 vs. 0.13 ± 0.1, n = 14; Fig. 2). The distribution of open and closed times was best fitted with a single exponential function (Fig. 1 C). The open time constant (τ0) and closed time constant (τc) were similar in both strains (τ0 = 16.1 ± 3.3 and 14.6 ± 3.1, τc = 15.5 ± 3.6 and 12.15 ± 2.2 in C57 and mdx, respectively; n = 14). Together, these data confirm that the total Ca conductance is larger in mdx fibers compared with C57 ones and suggest that this could lead to an abnormal overload of Ca in mdx fibers.
Figure 1.

Characterization of voltage-independent Ca channels in C57 and mdx fibers. (A) Examples of current traces (patch clamp; attached cell configuration). The closed state of the channel is marked by c. (B) Current- voltage relationships. (C) Distribution of open and closed times.
Figure 2.

Thapsigargin and caffeine-activated single-channel currents in normal and mdx adult skeletal muscle fibers. (A) Original traces of single-channel inward currents recorded cell-attached membrane patch (at –60 mV) before (control) and after stimulation by 1 μM thapsigargin of wild-type and mdx fibers. The closed and open states of the channel are marked by c and o1 and o2, respectively. (B) Histogram showing the fraction of patches on normal (open bars) and mdx (filled bars) fibers with channel activity in control condition and after application of 1 μM thapsigargin or 10 mM caffeine. (C) Mean channel Po on normal C57 and mdx fibers. (D) Quantity of Ca charge in C57 and mdx fibers obtained by integrating recordings of cell-attached patches. The asterisk denotes significant difference (P < 0.05) between control and treated C57 fibers and between control and treated mdx fibers. The number of patches for each type of fibers in each condition is indicated in the histograms.
Dependence on stores depletion
The following experiments were designed to examine whether the activity of the voltage-independent Ca channels was dependent on Ca stores depletion. Thapsigargin and caffeine were used to deplete the stores by inhibiting the Ca-ATPase (sarco/ER-Ca ATPase) and stimulating the ryanodine receptors, respectively. If the effect of caffeine on the release of Ca in skeletal muscle is well documented, the effect of thapsigargin has been less studied in this tissue where it is well-known that Ca pools are difficult to deplete (see Discussion). Therefore, this point is first studied here.
The results given in the previous section were obtained on depolarized cells (fibers maintained 3–5 min in potassium aspartate solution, as described previously [Franco-Obregon and Lansman, 1994]). This was necessary to correctly impose a holding potential (cell-attached configuration). Therefore, to mimic as much as possible the patch-clamp experiments, the effect of thapsigargin on Ca stores was studied in fibers depolarized in potassium aspartate solution (also containing 10 mM EGTA; see Materials and methods). Depolarization in this solution induced a [Ca2+]i transient that was taken as an index of the amount of releasable Ca (Fig. 3). After 5 min in this solution in the absence or presence of 1 μM thapsigargin, fibers were repolarized for 5 min in a Krebs solution (to prepare for the next depolarization) containing 50 μM EGTA to avoid any refilling of the stores during this period of time. On return in potassium aspartate solution and in the absence of thapsigargin, this produced a peak of [Ca2+]i ∼200 nM (from the resting level just before stimulation to the peak of [Ca2+]i, n =10). No difference could be observed between C57 and mdx fibers (n = 5 for each). Fibers treated with 1 μM thapsigargin showed an increase of resting [Ca2+]i from their basal level (60 ± 9 nM) to 111 ± 24 nM (n = 10). Upon depolarization, the peak of [Ca2+]i was significantly reduced by a factor of 6 (n = 10). This effect was observed in both C57 and mdx fibers. In conclusion, thapsigargin-treated fibers had lost 85% of their depolarization-releasable content of Ca. This result contrasts with the milder effect of thapsigargin observed by Kurebayashi and Ogawa (2001), and is probably due to the fact that thapsigargin-induced depletion is obtained in depolarized fibers.
Figure 3.
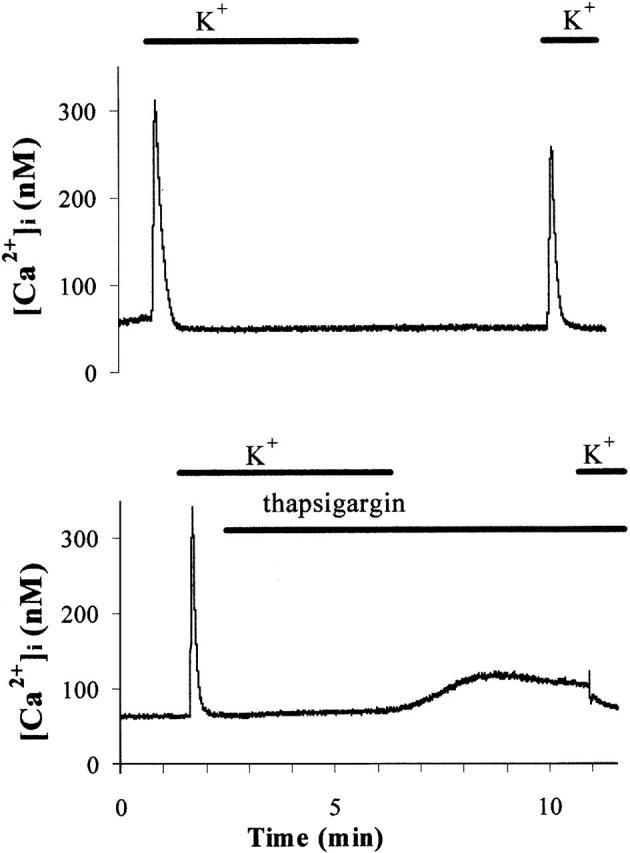
Representative examples of [Ca2 + ]i measurements in C57 muscle fibers. Fibers were incubated for 5 min in potassium aspartate (10 mM EGTA) solution in the presence or absence of 1 μM thapsigargin. Next, they were repolarized for 5 min in a Krebs solution (50 μM EGTA). Finally, depolarization in potassium aspartate solution induced a [Ca2+]i transient that was taken as an index of the amount of releasable Ca.
Store depletion by thapsigargin or caffeine increased the Po of the Ca channels by a factor of ∼3. For example, in C57 fibers, 1 μM thapsigargin significantly increased the Po from 0.07 ± 0.06 (n = 14) to 0.24 ± 0.1 (n = 14; Fig. 2 C). The same effect was obtained with the application of caffeine (Po = 0.15 ± 0.04, n = 10). Both drugs significantly increased the quantity of charge passing through the channels (integration of the current over a period of 120 s of observation; Fig. 2 D) but did not affect the occurrence of channels activity (Fig. 2 B). All these effects were present and similar in C57 and mdx fibers; however, the differences observed in control (not depleted) fibers (Fig. 2, control conditions, wild-type vs. mdx) persisted in thapsigargin- and caffeine-treated cells. Finally, the application of thapsigargin significantly increased the τ0 from 14.6 ± 3.1 ms (n = 14) to 25.2 ± 2.3 ms (n = 14, P < 0.05) in mdx fibers but not in C57 fibers (from 16.1 ± 3.3 ms to 17.8 ± 1.9 ms, n = 14 for each). Thapsigargin did not affect the τc, which stayed close to 12 ms in both fiber types whatever the experimental conditions used. These results show that the voltage-independent Ca channels observed in C57 and mdx fibers are activated by stores depletion. Channel activity was inhibited by 60% by La3+ (in mdx, Po diminished from 0.11 ± 0.015 [n = 24] to 0.04 ± 0.01 [n = 30]) but not by 100 μM 2-aminoethoxydiphenyl borate (unpublished data).
Expression and localization of mammalian TRPC isoforms in C57 and mdx fibers
The mRNA expression of the seven known mammalian TRPC isoforms was examined in adult skeletal muscle fibers using RT-PCR technique. Fig. 4 A shows that TRPC1, 2, 3, 4, and 6 mRNAs are detected in wild-type and mdx adult skeletal muscle fibers, whereas TRPC5 and TRPC7 isoforms were not. As a control, these two isoforms were found in another tissue (mouse brain).
Figure 4.


Expression and localization of TRPC isoforms. (A) Detection of the presence of TRPC mRNA by RT-PCR in C57 and mdx fibers. Brain (+) has been used as a positive control to prove the absence of TRPC5 and TRPC7. (B) Localization of endogenously expressed TRPC1, 2, 3, 4, and 6 in C57 and mdx fibers. Triton-soluble (a) or Triton-insoluble (b) proteins were separated on 8% SDS-PAGE, transferred onto PVDF membrane, and processed for Western blot analysis with specific pAbs. Sol 8 cells (++) have been used as a positive control for TRPC2. (C) Localization of Trp1, 2, 3, 4, and 6 proteins in mdx muscle fibers immunostained with specific antibodies (f, staining with the secondary antibody as a negative control). The corresponding light micrographs are presented for each case. Bar, 10 μm.
The expression of TRPC proteins and their localization in the cells were studied by Western blot analysis after partitioning proteins in “Triton-soluble” (supernatant) or “Triton-insoluble” (pellet) fractions. TRPC1, 2, 3, 4, and 6 proteins were endogenously expressed in both mdx and C57 skeletal muscle fibers (Fig. 4 B), thus paralleling the results obtained in PCR analyses. TRPC1, 4, and 6 proteins were localized in the supernatant fraction, suggesting a possible plasma membrane localization. TRPC2 was barely detectable and both TRPC2 and TRPC3 isoforms were detected only in the pellet fraction. Immunocytochemistry experiments confirmed the localization of TRPC1, 4, and 6 in the plasma membrane (Fig. 4 C). The function of TRPC2 and 3 isoforms, absent from the plasma membrane, was not investigated further. Western blots realized in the presence of respective control antigen peptides resulted in the disappearance of specific bands (not depicted).
Repression of TRPC in transfected mdx fibers
To examine the role of endogenous TRPC in the Ca currents observed, we tried to specifically repress the expression of these proteins and to study the functional repercussions. As the occurrence of Ca channels was higher in mdx than in C57 fibers, we chose to repress the expression in this type of fibers. We transiently transfected mdx fibers with antisense and sense oligonucleotides whose sequence was chosen in the most conserved region of the mRNA sequences of the different TRPCs. 48 h after transfection, the efficiency, assessed by the percentage of fluorescent fibers, was ∼60%.
Mdx fibers treated with sense or antisense oligonucleotides were submitted to fractionation, SDS-PAGE, and Western blot analysis. TRPC1 and 4 isoforms were significantly repressed by the treatment with antisense oligonucleotides (residual expression of 40 and 50%, respectively), but TRPC6 expression was not modified (Fig. 5, A and B). Note that only 60% of the studied fibers were successfully transfected.
Figure 5.
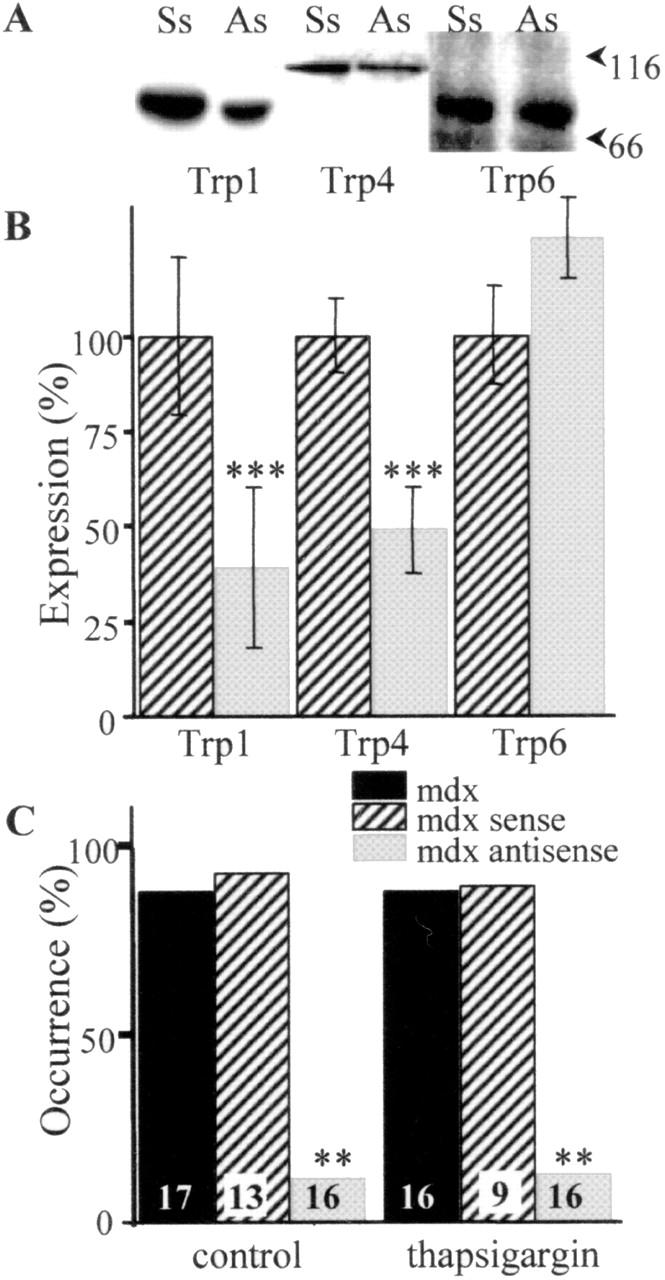
Repression of Trp in mdx fibers transfected with sense (Ss) or antisense (As) oligonucleotides. (A) Total transfected mdx fiber proteins were separated on 8% SDS-PAGE and blotted onto PVDF membrane. Western blots were performed with anti-Trp1, 4, and 6 pAbs. The standard molecular mass markers (kD) are indicated. (B) The intensities of the reactions were measured by integrated densitometry and were normalized for the protein load assayed by Coomassie blue staining. n = 6. ***, P < 0.0001. (C) Single-channel Ca activity in mdx fibers transfected with antisense or sense. Occurrence of channel activity in absence or presence of 1 μM thapsigargin. **, P < 0.001. The number of patches for each type of fibers in each condition is indicated in the histograms.
We examined the role of TRPC proteins by recording single-channel activity in mdx fibers transfected with antisense or sense oligonucleotides. In this case, only successfully transfected (and therefore fluorescent) fibers were studied. Fig. 5 C shows that the occurrence of Ca channel activity, in the absence or in the presence of thapsigargin, was strongly reduced in mdx fibers transfected with antisense oligonucleotides, but not in mdx fibers transfected with sense oligonucleotides or in nontransfected fibers. All the other characteristics of the channels (Po and quantity of charge, τ0, and τc) stayed identical in all mdx fibers transfected or not transfected.
As the repression of the TRPC protein expression led to an important decrease of the occurrence of Ca activity, we investigated whether the repression was specific and did not affect the expression of other proteins. To do so, we studied sodium currents in the same population of transfected mdx fibers. Sodium currents were elicited by 50-ms depolarizing pulses from a holding potential of −70 to −30 mV. Single openings generally occurred at the beginning of the test pulse. Average unitary currents showed the characteristics of sodium macroscopic currents with rapid onset and inactivation (Fig. 6). In both sense- and antisense-transfected mdx fibers, peak current amplitudes of average currents, recorded at –30 mV, were identical. Single-channel conductance was calculated as the slope of linear regression of single-channel current-voltage curves between−60 and 0 mV (Fig. 6 B). It was close to 18 pS in the mdx fibers transfected with sense and antisense oligonucleotides. This value of conductance is similar to the one obtained previously by others in FDB fibers (Desaphy et al., 2001).
Figure 6.
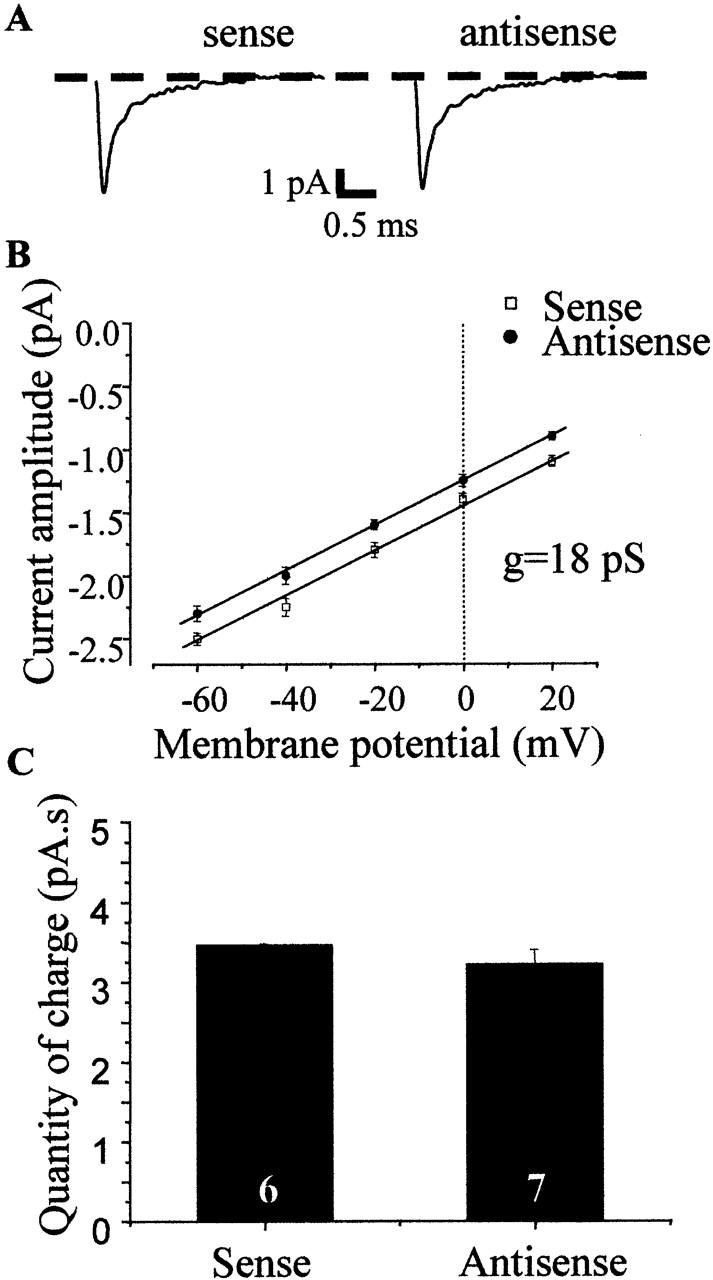
Sodium currents in mdx fibers transfected with sense or antisense. (A) Average currents constructed from 100 traces elicited in the same patches. (B) Single-channel current amplitudes plotted as a function of test potential for transfected mdx fibers. n ≥ 3. g, unitary conductance value. (C) Quantity of charge in mdx fibers (obtained by integrating recordings of cell-attached patches over a period of 120 s).
Discussion
Store-dependent entry of Ca has been largely studied in nonexcitable cells (Putney, 1986; Berridge, 1995; Parekh and Penner, 1997). However, in the last few years it became clear that excitable cells such as smooth muscle cells (Gibson et al., 1998) and skeletal myotubes (Hopf et al., 1996a) also do present capacitative entry of Ca. In both cases, IP3 receptors are expressed and Ca stores can be easily depleted by agonist stimulation. Recently, Kurebayashi and Ogawa (2001) demonstrated, for the first time, the presence of such influx in adult skeletal muscle fibers. This was a surprise because adult muscle fibers do not seem to exchange much Ca with the extracellular medium. Indeed, skeletal muscle fibers have huge amounts of sarcoplasmic reticulum that is extremely rich in Ca pumps (sarco/ER-Ca ATPase) and contains a high buffering capacity (calsequestrin). On the other hand, Ca extrusion through the plasma membrane is very slow, so almost all of the Ca released during contraction is immediately restored after stimulation. Accordingly, twitch contractions can thus be produced repeatedly for hours in the absence of extracellular Ca. Nevertheless, the authors show that the store-dependent influx is activated even by a partial depletion of the stores and must therefore be functional in physiological conditions.
The importance of an abnormal influx of Ca has been noticed in skeletal myotubes from DMD patients (Imbert et al., 1996) and in myotubes and adult fibers from mdx mice (Turner et al., 1988; Haws and Lansman, 1991; Tutdibi et al., 1999; Mallouk et al., 2000). This influx seems to implicate various spontaneously active and/or mechanosensitive cationic channels (Imbert et al., 1996; Vandebrouck et al., 2001), whereas the voltage-dependent Ca channels do not seem to be involved (Imbert et al., 2001). In myotubes, Hopf et al. (1996a) described a Ca leak channel whose activity was increased in mdx and which was activated by store depletion. The identity of this channel was unknown; it might be similar to the voltage-independent Ca channels found in adult fibers (Franco and Lansman, 1990a, and this paper) although its Po was ∼30 times lower than in adult fibers. In this paper, we show that the voltage-independent Ca channels that are abnormally activated in adult mdx skeletal muscle fibers are sensitive to Ca stores depletion by thapsigargin or caffeine. Indeed, store depletion increases the Po of these channels by keeping them opened for a longer time (increased τ0, same τc). The Ca channel studied here is similarly permeable to Ba2+, Ca2+, and Mn2+, as is the store-dependent channel from endothelial cells (Luckhoff and Clapham, 1994).
The identification of the channel involved in store-dependent Ca entry has been largely investigated in nonexcitable cells. Candidate molecules for this pathway probably belong to the TRPC protein family (for review see Clapham et al., 2001). This has been shown by heterologous expression of TRPC proteins. Using this methodology, TRPC1–5 have been shown to be possibly activated by store depletion (Groschner et al., 1998; Philipp et al., 1998; Vannier et al., 1999; Warnat et al., 1999; Liu et al., 2000). However, other authors showed that heterologous expression of TRPC3–6 also gave channels whose activation was independent of store depletion (Zitt et al., 1997; Hofmann et al., 1999; Schaefer et al., 2000; Wu et al., 2000). Therefore, different groups tried to evaluate the functional role of TRPC proteins by repressing their expression by transfection with antisense nucleotide sequences. This strategy has been used to clearly demonstrate that TRPC4 is part of the native Ca-release activated Ca channel in adrenal cells (Philipp et al., 2000), that TRPC1 and/or TRPC3 are involved in the thapsigargin-induced entry of Ca in HEK-293 cells (Wu et al., 2000), and that TRPC2 protein is involved in store-operated influx of Ca in fibroblasts (Gailly and Colson-Van Schoor, 2001). As the voltage-independent Ca channels found in C57 and mdx adult fibers were store dependent, we hypothesized that they might be constituted of TRPC proteins. By using the most conserved coding sequence in the different TRPC genes, we repressed their expression by transfection with antisense oligonucleotides. Among the seven known TRPC isoforms, five were endogenously expressed at the mRNA level, and three were present at the plasma membrane (TRPC1, 4, and 6). After 48-h repression with antisense oligonucleotides, we observed a 50–60% decrease in the amount of TRPC expressed. As transfection efficiency was also ∼60%, it seems reasonable to think that these proteins were almost absent from the transfected fibers. In contrast, TRPC6 expression was not diminished in transfected cells. This might be due to the ternary configuration of the mRNA or to a slow turnover of the protein. In parallel with these observations, we found that the channel occurrence was reduced by a factor of 10 in transfected fibers (selected by their fluorescein tag). The behavior of the channel was unchanged (Po, τ0, τc, activation by stores depletion) but its presence became very rare. Therefore, we conclude that the abnormal influx of Ca observed in adult mdx fibers occurs through store-dependent Ca channels constituted, at least partially, of TRPC proteins. The remaining occurrence might be due to an incomplete repression of the TRPC1 and 4 isoforms, or to the presence of TRPC6 protein that was not repressed. Further study will be necessary to precisely identify which TRPC isoform(s) is (are) involved in this process, TRPC1 and/or TRPC4. However, it is interesting to note that the TRPC1 channel is known to have a dystrophin homology domain (Lockwich et al., 2000). Therefore, the lack of dystrophin and the consequent disorganization of the cytoskeleton observed in dystrophic fibers might induce a disregulation of this protein, leading to an increased influx of Ca.
The mechanisms coupling depletion of the stores to subsequent entry of Ca remain unknown. Almost all cells presenting this kind of Ca entry release Ca from the ER through IP3 receptors. Furthermore, recent studies strongly suggest the involvement of the IP3 receptor in capacitative Ca entry (Kiselyov et al., 1998; Boulay et al., 1999; Putney, 1999; Ma et al., 2000). Skeletal muscle sarcoplasmic reticulum releases Ca essentially through ryanodine receptors, which might therefore be involved in the coupling process (Kiselyov et al., 2001). However, skeletal muscle also contains low amounts of IP3 receptors (Valdivia et al., 1992), and Liberona et al. (1998) observed that dystrophic muscle cell lines (human and mouse) have a two- to threefold increase in basal levels of IP3 and an increased number of IP3 receptors (Jaimovich et al., 2000). This might also be involved in the abnormal regulation of these Ca channels.
In this paper, we have thus identified the molecular nature of the channels involved in the abnormal influx of Ca observed in adult dystrophic muscle fibers. The originality of our paper stands in the fact that TRPCs are involved in capacitative Ca entry in excitable cells and, as shown for the first time, that a disregulation of their activity is involved in a neuromuscular disorder. Hopefully, this might give a new target in the search of drug therapies of DMD.
Materials and methods
Isolation of fibers
Adult (2- to 3-mo-old) wild-type (C57) and dystrophin-deficient (mdx) mice were killed by cervical dislocation. This protocol has been approved by the Animal Ethics Committee of the University of Louvain. FDB muscles were removed and incubated for 36 min at 37°C in Krebs solution containing (mM): 124 NaCl, 1.2 MgCl2, 5.9 KCl, 11.5 glucose, 11.5 Hepes-Na, 1.5 CaCl2, and 0.2% collagenase type IV (Sigma-Aldrich). The muscles were then removed from the collagenase-containing solution, washed twice in Krebs buffer, and suspended in Ham's F12/DME (Sigma-Aldrich) supplemented with 2% FBS (Sigma-Aldrich) and 1% antibiotics (penicillin-streptomycin; Sigma-Aldrich).
Single fibers were mechanically dissociated by passing the muscle repeatedly through fire-polished Pasteur pipettes. Dissociated fibers were plated onto tissue culture dishes coated with ECM basement membrane (Harbor Bio-Products) and allowed to adhere to the bottom of the dish for 2 h.
Measurements of cytosolic [Ca2+]
Muscle fibers were loaded for 1 h at RT with the membrane-permeant Ca indicator Fura-PE3/AM (Calbiochem) 1μM and Pluronic F-127 0.004%. Fura-PE3/AM was preferred over Fura-2/AM because it is stable during long lasting experiments, with little or no compartmentation (Hopf et al., 1996b). Fibers were illuminated through an inverted microscope (40× objective; Nikon) alternatively at 340 and 380 nm (20 Hz), and the fluorescent light emitted at 510 nm was measured. The ratio R340/380 of the fluorescence intensity emitted at the two excitation wavelengths was calculated, and cytosolic concentration of calcium ([Ca2+]i) was determined by using a cuvette calibration (Hopf et al., 1996b).
PCR amplification
Total RNA was extracted from C57 and mdx tibialis anterior muscles using the Tripure RNA extraction reagent (Roche). First-strand cDNA was synthesized from 2 μg of total RNA in a final volume of 20 μl using oligo dTPrimer (Thermoscript RT-PCR System; Life Technologies). PCR reaction was conducted using 2-μl aliquots of the RT reaction and specific primers for TRPC1–7 mouse sequences. These primers were chosen in a region known to cross a sizeable intron (Zhu et al., 1996), thus eliminating contribution from genomic DNA amplification. Primers used for amplification of TRPC1, 3, 4, 5, and 6 fragments were those designed by Garcia and Schilling (1997). For TRPC7 amplification, we used primers described by Okada et al. (1999), and for TRPC2, the primers we designed in a previous study (Gailly and Colson-Van Schoor, 2001). PCR was performed with Expand High Fidelity Taq polymerase (Roche) for 35 cycles (Idaho rapid light cycler), consisting of denaturation at 94°C for 30 s, annealing at 53 to 62°C (optimized for each TRPC fragment amplified) for 1 min, extension at 72°C for 1 min, and a final extension at 72°C for 6 min. The expected sizes of the different PCR products were 387, 345, 317, 415, 340, 327, and 701 bp for TRPC1–7, respectively.
Transient transfection of adult skeletal muscle fibers with TRPC antisense oligonucleotides
C57 and mdx fibers were plated onto dishes 2–12 h before transfection. They were transiently transfected with degenerated 5′ fluorescein-labeled TRPC antisense or sense oligonucleotides (Genset). The antisense sequence was chosen in a region conserved in all TRPC isoforms, as follows: Fluo 5′-aagtarswvadsyanaryttbghhckwgcaaayttccaytc-3′. The sense sequence was the reverse of the antisense sequence and was used as a control. Transient transfection was performed with 50-nM antisense or sense oligonucleotides and oligofectAMINE™ reagent (Life Technologies) in Ham's F12/DME without serum. Fibers were incubated for 4 h at 30°C in the presence of oligonucleotides before adding Ham's F12/DME with 6% FBS, and the culture was maintained at 30°C for 48 h. Transfection efficiency was determined by the fluorescence of cells. Electrophysiological recordings of transfected (fluorescent) cells and Western blot analyses were performed 48 h after transfection.
Western blotting
Dissociated fibers were harvested by centrifugation and resuspended in lysis buffer (20 mM Na2HPO4, pH 7.2, 150 mM NaCl, 10% glycerol, 1 mM EDTA, 0.5% Triton X-100) containing protease inhibitor cocktail (Roche). Fibers were incubated for 1 h on ice and centrifuged at 10,000 g for 15 min at 4°C, generating Triton-soluble (supernatant) and Triton-insoluble (pellet) proteins. In some experiments, dissociated mdx fibers were directly lysed and boiled for 5 min in 2× SDS-loading sample buffer containing 1% (vol/vol) 2-mercaptoethanol (Laemmli, 1970).
Proteins were separated by 8% SDS-PAGE and transferred onto PVDF membranes (Amersham). After incubation of these membranes in Tris-buffered saline containing 0.05% Tween 20 and 5% skimmed milk (wt/vol), rabbit anti-TRPC1, 3, 4, and 6 (1/600) pAbs (Alomone Labs) or anti-TRPC2 (1/100) pAb (a gift of Dr. H. Florman [University of Massachusetts Medical School, Worcester, MA]; Jungnickel et al., 2001) were added for 2 h at RT, and bound antibodies were detected by peroxidase-conjugated anti–rabbit IgG serum (1/10,000 for 1 h at RT). The immunoreactive bands were detected using a chemiluminescence ECL+ Western blotting detection kit (Amersham Biosciences) and quantified by densitometry after correction for gel loading assayed by Coomassie blue staining and/or actin content (detected using an anti-β actin antibody; Sigma-Aldrich).
Immunocytochemistry
The following protocol was used: fixation in 4% PFA for 20 min; permeabilization in 0.2% Triton/PBS for 15 min; preincubation in 10% normal goat serum/PBS for 1 h; incubation in 100-fold diluted TRPC1, 2, 3, 4, or 6 antiserum for 1 h; and incubation in 200-fold diluted FITC-labeled anti–rabbit goat antiserum for 1 h. Immunostained cells were observed under an inverted microscope (Axiovert T-100; Carl Zeiss MicroImaging, Inc.; excitation, 488 nm; emission, 505 nm). Images were acquired with a CCD camera (model 1300-v; Princeton Instruments) and analyzed with MetaFluor® software.
Electrophysiological methods
Single-channel Ca activity was recorded from cell-attached patches using the technique described by Hamill and co-workers (1981). Patch electrodes were pulled on a DMZ-Universal puller (Zeitz-Instruments) in three stages from borosilicate glass capillaries (1.5 mm diameter; Harvard Apparatus, Inc.) to a tip diameter of 1–2 μm. Patch electrodes had a resistance of 2–5 MΩ. Cells were viewed under phase-contrast with an inverted microscope (Axiovert S-100; Carl Zeiss MicroImaging, Inc.).
Single channel activity was recorded at a constant holding potential of −60 mV and at RT using a patch-clamp amplifier (EPC-9; HEKA). Current records were filtered with a Bessel filter at 3 kHz and digitized at 10 kHz. Data were analyzed using Pulse-Fit, Pulse-Tools, and Origin 6.1 software. The intrapipette solution contained the following (mM): 110 CaCl2, 10 Hepes, and 0.01 4.4'-diisothyocyanate stilbene-2.2' disulfonic acid (DIDS). The bathing solution was an isotonic potassium aspartate solution containing the following (mM): 150 potassium aspartate, 5 MgCl2, 10 EGTA, and 10 Hepes, pH 7.4. The osmolarity of each solution was adjusted to 320–330 mosmol.l−1 by adding glucose. The potassium aspartate bathing solution was used to render null the resting membrane potential so that the patch potential would be equal to the applied voltage command. DIDS was used to block possible chloride conductances.
Thapsigargin (Sigma-Aldrich) was dissolved in DMSO and diluted 1:2,000 into the bath to a final concentration of 1 μM. The solvent alone had no effect on channel activity. Caffeine (Merck Eurolab) was used at a final concentration of 10 mM.
Sodium currents were recorded at RT in cell-attached configuration. The intrapipette solution contained the following (mM): 150 NaCl, 1 MgCl2, 1 CaCl2, and 10 Hepes, pH 7.3. The bath solution contained Cs+ ions as the main cations in order to inhibit potassium currents and to depolarize the fiber: 145 CsCl, 5 EGTA, 1 MgCl2, 10 Hepes, and 5 glucose, pH 7.3. After 5–10 min of incubation in this solution, the intrinsic resting membrane potential of the FDB muscle fibers was consistently close to 0 mV. Capacitance currents were almost totally cancelled by the compensation circuit of the amplifier. Sodium currents were elicited by 50-ms depolarizing pulses from a holding potential of −70 to −30 mV applied every 0.5 s.
Statistics
Data are presented as mean ± SD. x2 test, ANOVA, or t tests were used to determine statistical significance.
Acknowledgments
We thank Dr. H. Florman for his generous gift of anti-TRPC2 antibody, and Profs. J.M. Gillis and J. Lebacq (University of Louvain, Belgium) for their critical comments on the manuscript.
This work was supported by the Association française contre les myopathies, the Association belge contre les maladies neuro-musculaires, and by a grant from the General Direction of Scientific Research of the French Community of Belgium (ARC 00/05-260).
C. Vandebrouck and D. Martin contributed equally to this work.
Footnotes
Abbreviations used in this paper: DMD, Duchenne muscular dystrophy; FDB, flexor digitorum brevis; mdx, murine X-linked dystrophy; Po, open probability; τc, closed time constant; τ0, open time constant; TRPC, transient receptor potential channel.
References
- Berridge, M.J. 1995. Capacitative calcium entry. Biochem. J. 312:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertorini, T.E., S.K. Bhattacharya, G.M. Palmieri, C.M. Chesney, D. Pifer, and B. Baker. 1982. Muscle calcium and magnesium content in Duchenne muscular dystrophy. Neurology. 32:1088–1092. [DOI] [PubMed] [Google Scholar]
- Boulay, G., D.M. Brown, N. Qin, M. Jiang, A. Dietrich, M.X. Zhu, Z. Chen, M. Birnbaumer, K. Mikoshiba, and L. Birnbaumer. 1999. Modulation of Ca2+ entry by polypeptides of the inositol 1,4, 5-trisphosphate receptor (IP3R) that bind transient receptor potential (TRP): evidence for roles of TRP and IP3R in store depletion-activated Ca2+ entry. Proc. Natl. Acad. Sci. USA. 96:14955–14960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapham, D.E., L.W. Runnels, and C. Strubing. 2001. The TRP ion channel family. Nat. Rev. Neurosci. 2:387–396. [DOI] [PubMed] [Google Scholar]
- Collet, C., B. Allard, Y. Tourneur, and V. Jacquemond. 1999. Intracellular calcium signals measured with indo-1 in isolated skeletal muscle fibres from control and mdx mice. J. Physiol. 520:417–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desaphy, J.F., S. Pierno, C. Leoty, A.L. George, Jr., A. De Luca, and D.C. Camerino. 2001. Skeletal muscle disuse induces fibre type-dependent enhancement of Na+ channel expression. Brain. 124:1100–1113. [DOI] [PubMed] [Google Scholar]
- Franco, A., Jr., and J.B. Lansman. 1990. a. Calcium entry through stretch-inactivated ion channels in mdx myotubes. Nature. 344:670–673. [DOI] [PubMed] [Google Scholar]
- Franco, A., Jr., and J.B. Lansman. 1990. b. Stretch-sensitive channels in developing muscle cells from a mouse cell line. J. Physiol. 427:361–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco-Obregon, A., Jr., and J.B. Lansman. 1994. Mechanosensitive ion channels in skeletal muscle from normal and dystrophic mice. J. Physiol. 481:299–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gailly, P., and M. Colson-Van Schoor. 2001. Involvement of trp-2 protein in store-operated influx of calcium in fibroblasts. Cell Calcium. 30:157–165. [DOI] [PubMed] [Google Scholar]
- Gailly, P., B. Boland, B. Himpens, R. Casteels, and J.M. Gillis. 1993. Critical evaluation of cytosolic calcium determination in resting muscle fibres from normal and dystrophic (mdx) mice. Cell Calcium. 14:473–483. [DOI] [PubMed] [Google Scholar]
- Garcia, R.L., and W.P. Schilling. 1997. Differential expression of mammalian TRP homologues across tissues and cell lines. Biochem. Biophys. Res. Commun. 239:279–283. [DOI] [PubMed] [Google Scholar]
- Gibson, A., I. McFadzean, P. Wallace, and C.P. Wayman. 1998. Capacitative Ca2+ entry and the regulation of smooth muscle tone. Trends Pharmacol. Sci. 19:266–269. [DOI] [PubMed] [Google Scholar]
- Gillis, J.M. 1999. Understanding dystrophinopathies: an inventory of the structural and functional consequences of the absence of dystrophin in muscles of the mdx mouse. J. Muscle Res. Cell Motil. 20:605–625. [DOI] [PubMed] [Google Scholar]
- Groschner, K., S. Hingel, B. Lintschinger, M. Balzer, C. Romanin, X. Zhu, and W. Schreibmayer. 1998. Trp proteins form store-operated cation channels in human vascular endothelial cells. FEBS Lett. 437:101–106. [DOI] [PubMed] [Google Scholar]
- Hamill, O.P., A. Marty, E. Neher, B. Sakmann, and F.J. Sigworth. 1981. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Arch. 391:85–100. [DOI] [PubMed] [Google Scholar]
- Haws, C.M., and J.B. Lansman. 1991. Developmental regulation of mechanosensitive calcium channels in skeletal muscle from normal and mdx mice. Proc. R. Soc. Lond. B Biol. Sci. 245:173–177. [DOI] [PubMed] [Google Scholar]
- Hoffman, E.P., R.H. Brown, Jr., and L.M. Kunkel. 1987. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 51:919–928. [DOI] [PubMed] [Google Scholar]
- Hofmann, T., A.G. Obukhov, M. Schaefer, C. Harteneck, T. Gudermann, and G. Schultz. 1999. Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature. 397:259–263. [DOI] [PubMed] [Google Scholar]
- Hopf, F.W., P. Reddy, J. Hong, and R.A. Steinhardt. 1996. a. A capacitative calcium current in cultured skeletal muscle cells is mediated by the calcium-specific leak channel and inhibited by dihydropyridine compounds. J. Biol. Chem. 271:22358–22367. [DOI] [PubMed] [Google Scholar]
- Hopf, F.W., P.R. Turner, W.F. Denetclaw, Jr., P. Reddy, and R.A. Steinhardt. 1996. b. A critical evaluation of resting intracellular free calcium regulation in dystrophic mdx muscle. Am. J. Physiol. 271:C1325–C1339. [DOI] [PubMed] [Google Scholar]
- Imbert, N., C. Vandebrouck, B. Constantin, G. Duport, C. Guillou, C. Cognard, and G. Raymond. 1996. Hypoosmotic shocks induce elevation of resting calcium level in Duchenne muscular dystrophy myotubes contracting in vitro. Neuromuscul. Disord. 6:351–360. [DOI] [PubMed] [Google Scholar]
- Imbert, N., C. Vandebrouck, G. Duport, G. Raymond, A. Hassoni, B. Constantin, M. Cullen, and C. Cognard. 2001. Calcium currents and transients in co-cultured contracting normal and Duchenne muscular dystrophy human myotubes. J. Physiol. 534:343–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaimovich, E., R. Reyes, J.L. Liberona, and J.A. Powell. 2000. IP3 receptors, IP3 transients, and nucleus-associated Ca2+ signals in cultured skeletal muscle. Am. J. Physiol. Cell Physiol. 278:C998–C1010. [DOI] [PubMed] [Google Scholar]
- Jungnickel, M.K., H. Marrero, L. Birnbaumer, J.R. Lemos, and H.M. Florman. 2001. Trp2 regulates entry of Ca2+ into mouse sperm triggered by egg ZP3. Nat. Cell Biol. 3:499–502. [DOI] [PubMed] [Google Scholar]
- Kiselyov, K., X. Xu, G. Mozhayeva, T. Kuo, I. Pessah, G. Mignery, X. Zhu, L. Birnbaumer, and S. Muallem. 1998. Functional interaction between InsP3 receptors and store-operated Htrp3 channels. Nature. 396:478–482. [DOI] [PubMed] [Google Scholar]
- Kiselyov, K., D.M. Shin, N. Shcheynikov, T. Kurosaki, and S. Muallem. 2001. Regulation of Ca2+-release-activated Ca2+ current (Icrac) by ryanodine receptors in inositol 1,4,5-trisphosphate-receptor-deficient DT40 cells. Biochem. J. 360:17–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurebayashi, N., and Y. Ogawa. 2001. Depletion of Ca2+ in the sarcoplasmic reticulum stimulates Ca2+ entry into mouse skeletal muscle fibres. J. Physiol. 533:185–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laemmli, U.K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 227:680–685. [DOI] [PubMed] [Google Scholar]
- Lee, K.S., and R.W. Tsien. 1984. High selectivity of calcium channels in single dialysed heart cells of the guinea-pig. J. Physiol. 354:253–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leijendekker, W.J., A.C. Passaquin, L. Metzinger, and U.T. Ruegg. 1996. Regulation of cytosolic calcium in skeletal muscle cells of the mdx mouse under conditions of stress. Br. J. Pharmacol. 118:611–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberona, J.L., J.A. Powell, S. Shenoi, L. Petherbridge, R. Caviedes, and E. Jaimovich. 1998. Differences in both inositol 1,4,5-trisphosphate mass and inositol 1,4,5-trisphosphate receptors between normal and dystrophic skeletal muscle cell lines. Muscle Nerve. 21:902–909. [DOI] [PubMed] [Google Scholar]
- Liu, X., W. Wang, B.B. Singh, T. Lockwich, J. Jadlowiec, B. O'Connell, R. Wellner, M.X. Zhu, and I.S. Ambudkar. 2000. Trp1, a candidate protein for the store-operated Ca2+ influx mechanism in salivary gland cells. J. Biol. Chem. 275:3403–3411. [DOI] [PubMed] [Google Scholar]
- Lockwich, T.P., X. Liu, B.B. Singh, J. Jadlowiec, S. Weiland, and I.S. Ambudkar. 2000. Assembly of Trp1 in a signaling complex associated with caveolin-scaffolding lipid raft domains. J. Biol. Chem. 275:11934–11942. [DOI] [PubMed] [Google Scholar]
- Luckhoff, A., and D.E. Clapham. 1994. Calcium channels activated by depletion of internal calcium stores in A431 cells. Biophys. J. 67:177–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, H.T., R.L. Patterson, D.B. van Rossum, L. Birnbaumer, K. Mikoshiba, and D.L. Gill. 2000. Requirement of the inositol trisphosphate receptor for activation of store-operated Ca2+ channels. Science. 287:1647–1651. [DOI] [PubMed] [Google Scholar]
- MacLennan, P.A., A. McArdle, and R.H. Edwards. 1991. Effects of calcium on protein turnover of incubated muscles from mdx mice. Am. J. Physiol. 260:E594–E598. [DOI] [PubMed] [Google Scholar]
- Mallouk, N., V. Jacquemond, and B. Allard. 2000. Elevated subsarcolemmal Ca2+ in mdx mouse skeletal muscle fibers detected with Ca2+-activated K+ channels. Proc. Natl. Acad. Sci. USA. 97:4950–4955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monaco, A.P., R.L. Neve, C. Colletti-Feener, C.J. Bertelson, D.M. Kurnit, and L.M. Kunkel. 1986. Isolation of candidate cDNAs for portions of the Duchenne muscular dystrophy gene. Nature. 323:646–650. [DOI] [PubMed] [Google Scholar]
- Okada, T., R. Inoue, K. Yamazaki, A. Maeda, T. Kurosaki, T. Yamakuni, I. Tanaka, S. Shimizu, K. Ikenaka, K. Imoto, and Y. Mori. 1999. Molecular and functional characterization of a novel mouse transient receptor potential protein homologue TRP7. Ca2+-permeable cation channel that is constitutively activated and enhanced by stimulation of G protein-coupled receptor. J. Biol. Chem. 274:27359–27370. [DOI] [PubMed] [Google Scholar]
- Parekh, A.B., and R. Penner. 1997. Store depletion and calcium influx. Physiol. Rev. 77:901–930. [DOI] [PubMed] [Google Scholar]
- Philipp, S., J. Hambrecht, L. Braslavski, G. Schroth, M. Freichel, M. Murakami, A. Cavalie, and V. Flockerzi. 1998. A novel capacitative calcium entry channel expressed in excitable cells. EMBO J. 17:4274–4282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philipp, S., C. Trost, J. Warnat, J. Rautmann, N. Himmerkus, G. Schroth, O. Kretz, W. Nastainczyk, A. Cavalie, M. Hoth, and V. Flockerzi. 2000. TRP4 (CCE1) protein is part of native calcium release-activated Ca2+-like channels in adrenal cells. J. Biol. Chem. 275:23965–23972. [DOI] [PubMed] [Google Scholar]
- Pressmar, J., H. Brinkmeier, M.J. Seewald, T. Naumann, and R. Rudel. 1994. Intracellular Ca2+ concentrations are not elevated in resting cultured muscle from Duchenne (DMD) patients and in MDX mouse muscle fibres. Pflügers Arch. 426:499–505. [DOI] [PubMed] [Google Scholar]
- Putney, J.W., Jr. 1986. Identification of cellular activation mechanisms associated with salivary secretion. Annu. Rev. Physiol. 48:75–88. [DOI] [PubMed] [Google Scholar]
- Putney, J.W., Jr. 1999. TRP, inositol 1,4,5-trisphosphate receptors, and capacitative calcium entry. Proc. Natl. Acad. Sci. USA. 96:14669–14671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer, M., T.D. Plant, A.G. Obukhov, T. Hofmann, T. Gudermann, and G. Schultz. 2000. Receptor-mediated regulation of the nonselective cation channels TRPC4 and TRPC5. J. Biol. Chem. 275:17517–17526. [DOI] [PubMed] [Google Scholar]
- Turner, P.R., T. Westwood, C.M. Regen, and R.A. Steinhardt. 1988. Increased protein degradation results from elevated free calcium levels found in muscle from mdx mice. Nature. 335:735–738. [DOI] [PubMed] [Google Scholar]
- Tutdibi, O., H. Brinkmeier, R. Rudel, and K.J. Fohr. 1999. Increased calcium entry into dystrophin-deficient muscle fibres of MDX and ADR-MDX mice is reduced by ion channel blockers. J. Physiol. 515:859–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdivia, C., D. Vaughan, B.V. Potter, and R. Coronado. 1992. Fast release of 45Ca2+ induced by inositol 1,4,5-trisphosphate and Ca2+ in the sarcoplasmic reticulum of rabbit skeletal muscle: evidence for two types of Ca2+ release channels. Biophys. J. 61:1184–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandebrouck, C., G. Duport, C. Cognard, and G. Raymond. 2001. Cationic channels in normal and dystrophic human myotubes. Neuromuscul. Disord. 11:72–79. [DOI] [PubMed] [Google Scholar]
- Vannier, B., M. Peyton, G. Boulay, D. Brown, N. Qin, M. Jiang, X. Zhu, and L. Birnbaumer. 1999. Mouse trp2, the homologue of the human trpc2 pseudogene, encodes mTrp2, a store depletion-activated capacitative Ca2+ entry channel. Proc. Natl. Acad. Sci. USA. 96:2060–2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnat, J., S. Philipp, S. Zimmer, V. Flockerzi, and A. Cavalie. 1999. Phenotype of a recombinant store-operated channel: highly selective permeation of Ca2+. J. Physiol. 518:631–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, X., G. Babnigg, and M.L. Villereal. 2000. Functional significance of human trp1 and trp3 in store-operated Ca2+ entry in HEK-293 cells. Am. J. Physiol. Cell Physiol. 278:C526–C536. [DOI] [PubMed] [Google Scholar]
- Zhu, X., M. Jiang, M. Peyton, G. Boulay, R. Hurst, E. Stefani, and L. Birnbaumer. 1996. trp, a novel mammalian gene family essential for agonist-activated capacitative Ca2+ entry. Cell. 85:661–671. [DOI] [PubMed] [Google Scholar]
- Zitt, C., A.G. Obukhov, C. Strubing, A. Zobel, F. Kalkbrenner, A. Luckhoff, and G. Schultz. 1997. Expression of TRPC3 in Chinese hamster ovary cells results in calcium-activated cation currents not related to store depletion. J. Cell Biol. 138:1333–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]