

Abstract
The neurotropic virus, herpes simplex type 1 (HSV-1), inhibits the excitability of peripheral mammalian neurons, but the molecular mechanism of this effect has not been identified. Here, we use voltage-clamp measurement of ionic currents and an antibody against sodium channels to show that loss of excitability results from the selective, precipitous, and complete internalization of voltage-activated sodium channel proteins from the plasma membrane of neurons dissociated from rat dorsal root ganglion. The internalization process requires viral protein synthesis but not viral encapsulation, and does not alter the density of voltage-activated calcium or potassium channels. However, internalization is blocked completely when viruses lack the neurovirulence factor, infected cell protein 34.5, or when endocytosis is inhibited with bafilomycin A1 or chloroquine. Although it has been recognized for many years that viruses cause cell pathology by interfering with signal transduction pathways, this is the first example of viral pathology resulting from selective internalization of an integral membrane protein. In studying the HSV-induced redistribution of sodium channels, we have uncovered a previously unknown pathway for the rapid and dynamic control of excitability in sensory neurons by internalization of sodium channels.
Keywords: voltage-gated sodium channels; sensory neurons; sodium channel regulation; calcium channels; potassium channels
Introduction
Herpes simplex virus type 1 (HSV-1)* is a neurotropic virus that, in vivo, forms a latent infection in dorsal root ganglion (DRG) and other sensory ganglia and is the causative agent for the common cold sore. Recurrent infections throughout the host's life occur when the virus “creeps” back down the neuron to the original site of infection. HSV infection is associated with abnormal sensations around the site of initial infection, including tingling parasthesia, loss of touch and pain sensations, and formication (Andoh et al., 1995), which have been attributed to alterations in neuronal excitability. In vitro, an HSV infection of DRG neurons eliminates excitability in most neurons, and in neurons that remain excitable, the action potentials are smaller in amplitude and longer in duration than those in uninfected neurons (Fukuda and Kurata, 1981; Mayer, 1986). Viral protein synthesis is necessary for the loss of excitability (Oakes et al., 1981). The molecular mechanism underlying the loss of excitability and the viral proteins or processes involved in the pathology are unknown.
We used dissociated rat DRG neurons for investigating virally induced loss of excitability and the ionic currents underlying these changes. Dissociated DRG neurons in vitro are a good model for studying ionic changes, as they maintain their excitability in culture and their ionic currents have been well characterized (for review see Pearce and Duchen, 1994). Here, we report a profound, rapid, and selective loss of voltage-dependent sodium currents 20–24 h after infection. The cellular regulation of sodium channel expression and turnover is not well understood. Using a sodium channel antibody and confocal microscopy, we visualized a change in the distribution of sodium channels; specifically a loss of sodium channels from the plasma membrane that exactly correlated with the loss of excitability. Although sodium channel redistribution during development has been reported (Dargent et al., 1994; Paillart et al., 1996; Vabnick et al., 1996), there is little description of the molecular mechanism underlying these changes in channel distribution. Here, we examined the role of membrane internalization in the process of sodium channel removal from the plasma membrane. We show that blocking internalization with endosome-neutralizing agents such as bafilomycin A1 (Bowman et al., 1988) prevented the HSV-induced loss of sodium currents.
The viral proteins that interfere with normal sodium channel trafficking are unknown. HSV expresses a neurovirulence factor encoded by the infected cell protein (ICP) 34.5 gene that has been shown to be important for enhancing viral pathogenicity in neurons, and increased the ability of HSV-1 to cause death in infected animals (Poon and Roizman, 1997). Because of these features, HSV genomes lacking the neurovirulence factor ICP 34.5 are often used as the backbone for viral vectors used in gene therapy. We tested the role of the neurovirulence factor on sodium channel density during an HSV infection. We found that infection with mutant viruses lacking both copies of the ICP34.5 gene prevented the characteristic loss of sodium currents observed during wild-type infections.
Results
Current-clamp experiments confirmed the observation that neurons infected with the nonsyncytial strain of HSV-1, wild-type virus HSV-1 Glasgow strain 17 syn+ (wt HSV 17+), for 24 h had either no action potential or small, broad action potentials. The shape of the residual action potentials resembled that of the calcium action potentials seen in many DRG neurons after the addition of the sodium-channel blocker TTX, or in all DRG neurons in sodium-free solutions. We investigated the molecular changes responsible for the changes in excitability and action potential shape by isolating the major classes of voltage-activated currents.
Sodium currents
For this study, we used dissociated rat DRG neurons without neurites. We recorded from neurons either immediately after dissociation (4–12 h) or from replated neurons for comparison with HSV-infected neurons that do not produce neurites. Fig. 1 a shows a sodium current recorded from an uninfected DRG neuron and the response of a neuron infected with HSV-1 for 24 h using a voltage step from −80 to +10 mV. At this time after infection, only 24% (51/210) of neurons showed a sodium current, and the normalized amplitude of the residual current in the 51 responding neurons was smaller than in uninfected neurons (HSV, −0.048 ± 0.008 nA/pF [n = 51]; control, −0.14 ± 0.006 nA/pF [n = 171]). The time course of the loss of sodium current is illustrated in Fig. 1 b. No change in the sodium current amplitude was noted over the first 2–20 h after infection. The sodium current reduction was maximal after 24 h of infection and maintained until at least 48 h. The cell capacitance and input resistance of uninfected, control neurons did not significantly differ from infected neurons at any of the measured time points after infection (input resistance, HSV 24 h, 84 ±10 mΩ [n = 35]; control, 121 ± 20 mΩ [n = 37]; cell capacitance, HSV 24 h, 30 ± 2 pF [n = 35]; and control 33 ± 3 pF [n = 37]).
Figure 1.

Sodium currents from HSV-1–infected and uninfected DRG neurons. (a) Sodium currents evoked by a voltage step from −80 to 10 mV in control, uninfected DRG neurons and 24 h after infection with HSV-1. (b) Temporal effects of HSV-1 infection on normalized sodium currents in DRG neurons. Adult rat DRG neurons were infected with 5 plaque-forming units HSV-1/DRG neuron in vitro. At intervals during HSV-1 infection, sodium currents were recorded and normalized for cell capacitance. The mean-normalized sodium current ± SEM is plotted against time after infection. DRG neurons infected with HSV-1 for 24–30 or 48 h had significantly smaller normalized currents than neurons infected for less time (Kruskal-Wallis statistical test followed by the Kolmogorov-Smirnov pairwise comparison; P < 0.0001).
We investigated the characteristics of the remaining sodium current in HSV-1–infected neurons and compared them with the currents in control, uninfected DRG neurons. The loss or reduction of the sodium currents was not associated with any shift in the activation characteristics of either the total sodium current or the isolated TTX-sensitive (TTX-S) and TTX-resistant (TTX-R) currents (Fig. 2, c and d). No difference in the voltage dependence of activation was noted for the total sodium current, which is dominated by the rapidly activated TTX-S component. Single Boltzmann curves fitted to the voltage-conductance plots yielded mean V50 values of −23.6 ± 1.4 mV and −20.1 ± 1.3 mV, and slope factors of 7.4 ± 0.3 mV and 9.1 ± 0.3 mV for control (n = 16) and HSV-1 infected (n = 13) neurons. Furthermore, a prepulse potential to −120 mV with a subsequent step to a test potential of +10 mV did not reveal currents in infected neurons that showed no current with a step from −80 to +10 mV.
Figure 2.

TTX-S and TTX-R sodium currents of uninfected and HSV-1–infected DRG neurons. (a) The total sodium current (INa) was evoked after a prepulse to −120 mV with a voltage step to −20 mV. The TTX-R sodium current was evoked after a prepulse to −50 mV also with a voltage step to −20 mV. The TTX-S trace was obtained by subtraction. (b) The normalized mean sodium current ± SEM from neurons with a measurable current (nA/pF). The remaining TTX-S and TTX-R sodium currents of infected neurons were significantly smaller than the TTX-S and TTX-R sodium currents of control cells (P < 0.05 t test). (c) Averaged mean normalized sodium current ± SEM plotted against command potential for control (•; n = 16) and HSV-1–infected neurons (▪; n = 13). (d) Representative normalized TTX-S (▪) and TTX-R (•) sodium currents plotted against command potential from a neuron infected for 24 h (open) and an uninfected neuron (filled).
Electrophysiologically separated TTX-R and TTX-S current sodium currents are illustrated in Fig. 2 a. When the analysis was restricted to DRG neurons that showed sodium currents, the normalized amplitudes of both the TTX-R and TTX-S currents were significantly less (P < 0.05) in infected neurons than in uninfected neurons (Fig. 2 b).
Calcium currents
Low voltage–activated (LVA) and high voltage–activated (HVA) calcium currents in uninfected neurons and in neurons infected with HSV-1 for 24 h were compared. The two components of the calcium current were separated on the basis of their activation and inactivation properties as described in Materials and methods (Fig. 3 a). HVA calcium currents were recorded in all infected and uninfected neurons studied, and the normalized mean amplitude in infected neurons (−0.043 ± 0.009 nA/pF) did not significantly differ from that in uninfected neurons (−0.056 ± 0.001 nA/pF; Fig. 3 b). Similarly, the percentage of neurons with LVA calcium currents (uninfected, 25% [9/36]; infected, 17%, [4/24]) and the mean-normalized current amplitudes (uninfected, −0.032 ± 0.007 nA/pF; infected, −0.034 ± 0.015 nA/pF) were not significantly altered by HSV-1 infection (Fig. 3 b).
Figure 3.

Effect of HSV-1 infection on calcium currents in DRG neurons. (a) Leak-subtracted raw traces illustrating the separation of LVA and HVA calcium channel currents. (b) The mean-normalized peak HVA and LVA calcium current ± SEM of uninfected or DRG neurons infected with HSV-1 for 24 h. The HVA- and LVA-type calcium currents did not differ significantly between the uninfected and the infected neurons (t test, P > 0.3). (c) Two families of calcium channel currents evoked by a range of command potentials between −80 and 40 mV from an uninfected and infected neuron. The current traces were leak current–subtracted. (d) The graph shows the mean-normalized conductance ± SEM from control (•; n = 36) and infected (▪; n = 27) neurons and the normalized calcium currents ± SEM plotted against prepulse potentials from control (○; n = 9) and infected (□; n = 8) neurons. The continuous lines were obtained by fitting Boltzmann functions to the mean-normalized data.
No obvious kinetic differences were noted between calcium currents in uninfected and HSV-1–infected neurons evoked by voltage steps from −80 mV to test potentials of −70 mV to +20 mV (Fig. 3 c). The activation and availability curves constructed for the total calcium currents (Fig. 3 d) also showed no significant change in voltage sensitivities after HSV-1 infection (activation V50 – uninfected, −16.9 ± 1.1 mV [n = 36]; infected, −14.7 ± 1.5 mV [n = 27]; availability V50 – uninfected, −48.2 ± 0.7 mV [n = 9]; infected, −45.6 ± 0.9 mV [n = 8]).
Outwardly rectifying potassium currents
The broad action potentials recorded in current-clamp mode could reflect a reduction in delayed rectifier potassium currents. Potassium currents were accordingly investigated under voltage-clamp conditions. Although several distinct types of potassium currents have been described for DRG neurons, we separated the outward potassium current into two components: (1) a low threshold current that inactivated during a 40-ms depolarizing voltage step from −80 to −50 mV, and (2) a relatively sustained current that showed little or no inactivation over this period. The two currents were isolated by subtraction of the current evoked by a voltage step from −50 to +10 mV from the current evoked by a step from −80 mV to the same test potential (Fig. 4 a). After a 24-h infection with HSV-1, all neurons studied showed both inactivating and “sustained” outward currents, and no significant differences (P > 0.2) were noted in the amplitudes of the normalized current amplitudes (Fig. 4 b). Current-voltage relationships for the total potassium currents constructed from the tail currents after a 15-ms depolarizing pulse to a range of test potentials (from −90 to +40 mV; traces, Fig. 4 c) are shown in Fig. 4 d. No significant difference in voltage sensitivity of activation was noted between uninfected and HSV-1–infected neurons (V50 uninfected, −26.8 ± 1.9 mV [n = 25]; HSV, −23.7 ± 1.7 mV [n = 11]). Similarly, the availability curves (Fig. 4 d) constructed from the current responses evoked by a voltage step to +45 mV after a 1-s prepulse to a range of potentials (from −120 to 0 mV) showed no difference between uninfected and HSV-1–infected neurons (V50 uninfected, −64.7 ± 1.2 mV [n = 25]; HSV-1, −63.8 ± 1.5 mV [n = 11]).
Figure 4.

Effect of HSV-1 infection on potassium currents of DRG neurons. (a) Inactivating and noninactivating delayed rectifier type potassium currents from an uninfected DRG neuron (leak subtracted). The total potassium currents were evoked from −80 mV with a step potential to +10 mV, and the noninactivating potassium currents were evoked after a prepulse to −50 mV from a test potential of +10 mV. The inactivating potassium current could be separated by subtraction (dashed line). (b) The normalized mean noninactivating and inactivating type potassium current amplitude ± SEM is shown for control and infected neurons. There was no significant difference between the normalized potassium current amplitudes of uninfected or HSV-1–infected neurons (t test, P > 0.3). (c) A family of outward potassium currents in neurons. Neurons were held at −80 mV. The activation protocol consisted of a series of 15-ms depolarizing voltage steps between −90 and +40 mV. Note the outward tail currents that occur when the membrane is stepped back to −40 mV. (d) The activation and steady-state inactivation curves of uninfected and HSV-1–infected neurons. The graph shows the normalized mean conductance ± SEM plotted against the command potential. The activation and steady-state inactivation curves of control (•; n = 25) and infected (▪; n = 11) neurons are shown. The curves are obtained by fitting the data points with Boltzmann functions. (e) A family of inwardly rectifying potassium currents were evoked from an HSV-1–infected neuron by a range of 900-ms potentials between −50 and −140 mV, and from a holding potential of −70 mV. (f) The graph shows the mean-normalized sustained current amplitude ± SEM of uninfected (•) or infected (▪) DRG neurons. There was no significant difference between the normalized current amplitudes (t test, P > 0.1).
Inwardly rectifying potassium currents
The possibility that the virally induced alterations in neuron excitability were the result of an alteration in the inwardly rectifying potassium currents was investigated. Inwardly rectifying potassium currents were recorded from uninfected neurons and neurons infected with HSV-1 for 24 h. A family of inwardly rectifying potassium currents was evoked with steps from −50 to −140 mV from the holding potential of −70 mV. There was no significant difference in the normalized amplitude of the slowly activating, inwardly rectifying potassium currents in uninfected (0.016 ± 0.0017 nA/pF [n = 22]) and HSV-1–infected (0.022 ± 0.0057 nA/pF [n = 7]) neurons (Fig. 4 e). The tail current amplitudes were measured upon repolarization to −70 mV and the normalized currents plotted against the command potential (Fig. 4 f). There was no significant difference in the voltage sensitivity of activation between uninfected and HSV-1–infected neurons (V50 uninfected, −94.1 ± 1.4 mV [n = 9]; HSV-1, −91.2 ± 1.6 [n = 7]; P > 0.08). In addition, a proportion of the neurons tested showed the IIR fast inwardly rectifying currents that were rapidly activated by hyperpolarization. There was no significant difference (P > 0.3) in the normalized rapidly activated, inwardly rectifying potassium current amplitudes evoked at −120mV of HSV-infected (8/15, −0.021 ± 0.004 nA/pF) and uninfected neurons (8/12, −0.025 ± 0.003 nA/pF).
Localization of sodium channel α subunits in DRG neurons
The loss of sodium conductance after 24 h of HSV infection could be a result of a redistribution of the sodium channels of DRG neurons. Sodium channel distribution was studied in HSV-infected and uninfected DRG neurons by immunofluorescence. Total-cell (left) and cross sectional (right) confocal images of the sodium channel α subunit staining pattern are shown for two examples of uninfected DRG neurons (Fig. 5 a), and for two examples of neurons infected with HSV-1 for 24 h (Fig. 5 b). The uninfected DRG neurons showed a diffuse, bright staining pattern with clear plasma membrane staining. The HSV-1–infected neurons showed duller staining without obvious plasma membrane localization. The mean density of fluorescence was quantified for HSV-1–infected and uninfected neurons from the total-cell images as shown in Fig. 5 c. The background mean fluorescence density was measured in the area between cells. Sodium channel staining was significantly less throughout HSV-1–infected neurons than in control, uninfected neurons. Control staining was performed by omitting the primary antibody; no staining was observed when the primary antibody was omitted.
Figure 5.
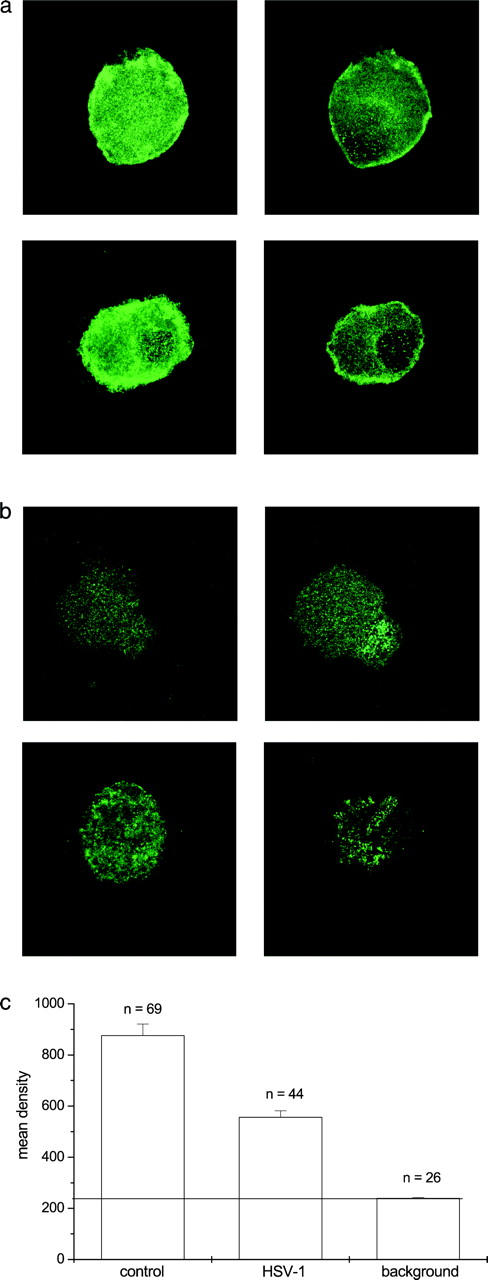
The distribution of sodium channels in HSV-1–infected and uninfected DRG neurons. (a) The left panel is the collapsed image of several serial scans showing the total neuron distribution of sodium channel α subunits of uninfected neurons, and the right panel is a single z-axis image showing the cross section of an uninfected neuron demonstrating plasma membrane staining. (b) The left panel is the collapsed image of several serial scans showing the total neuron distribution of sodium channel α subunits of an HSV-1–infected neuron, and the right panel is a single z-axis image showing the cross section of the infected neurons. (c) The mean fluorescence intensity of stained HSV-1–infected and uninfected neurons. The mean intensity of fluorescence was significantly reduced in infected neurons (t test, P < 0.00001).
Internalization of sodium channels
The membrane internalization inhibitors, bafilomycin A1 (1 μM) and chloroquine (100 μM), were used to further investigate the possibility that sodium channels were internalized after HSV-1 infection. HSV does not depend on endocytosis or acid pH for penetration (Morgan et al., 1968). The neurons were treated with the internalization inhibitors 1 h after viral infection to allow viral adsorption, and were also present for the subsequent 24-h incubation. Representative sodium current traces from HSV-1–infected and uninfected neurons treated with bafilomycin A1 are shown in Fig. 6 a. Fig. 6 b illustrates the normalized sodium current amplitudes after treatment with either 1 μM bafilomycin A1 or 100 μM chloroquine. All HSV-1–infected DRG neurons treated with the internalization inhibitors had sodium currents. Furthermore, the amplitudes of the currents in bafilomycin A1– and chloroquine-treated, HSV-1–infected neurons were significantly larger than in untreated, HSV-1–infected cells (P < 0.001), but were not significantly different to the amplitudes seen in control, uninfected DRG neurons. In addition, the normalized sodium current amplitudes of uninfected, untreated DRG neurons did not differ significantly from either uninfected, internalization inhibitor–treated neurons, or HSV-infected, internalization inhibitor–treated neurons. Only HSV-infected DRG neurons that were not treated with internalization inhibitors showed a dramatic and significant reduction in normalized sodium current amplitudes.
Figure 6.
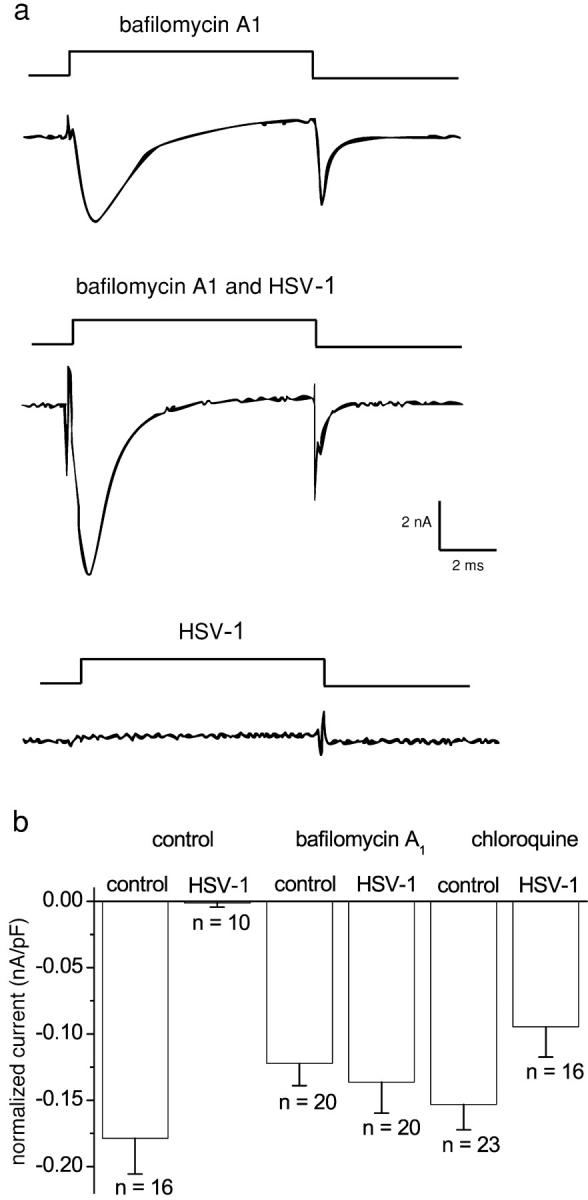
The effect of internalization inhibitors on sodium currents in uninfected and HSV-1–infected DRG neurons. (a) Sodium current traces from HSV-1–infected and uninfected neurons treated with bafilomycin A1 contrasts with the lack of current in an HSV-1– infected neuron. (b) The mean-normalized current amplitude ± SEM of uninfected and infected neurons treated with the inhibitors of internalization, 1 μM bafilomycin A1 or 100 μM chloroquine. The current amplitudes of infected or uninfected neurons treated with internalization inhibitors did not differ significantly from each other (P > 0.5). However, all were significantly larger than the current amplitudes of infected neurons (P > 0.0001). Statistical differences were examined by the Kruskal-Wallis statistical test followed by the Kolmogorov-Smirnov pairwise comparison.
In a further set of experiments, DRG neurons were treated with 25 mM ammonium chloride, which is known to neutralize the acidic endosomal/lysosomal compartments and to block membrane internalization and turnover. All HSV-1 neurons treated in this way had sodium currents 24 h after infection, and once again the normalized amplitudes were similar in control and HSV-1–infected cells (unpublished data).
Role of HSV-1 infection
A late process in viral maturation is the envelopment and egress of the virus. We investigated the effect of brefeldin A, a compound that inhibits viral envelopment and egress by collapsing the trans-Golgi network, but has no effect on the internalization processes of the host cell (Koyama and Uchida, 1994). DRG neurons were infected with HSV-1 for 1 h to allow for virus adsorption and penetration. Infected and control cells were incubated in brefeldin A for 24 h. Uninfected neurons treated with 1 μM brefeldin A possessed sodium currents of normal amplitude. Furthermore, exposure to brefeldin A did not affect the loss of sodium currents after HSV-1 infection (Fig. 7).
Figure 7.
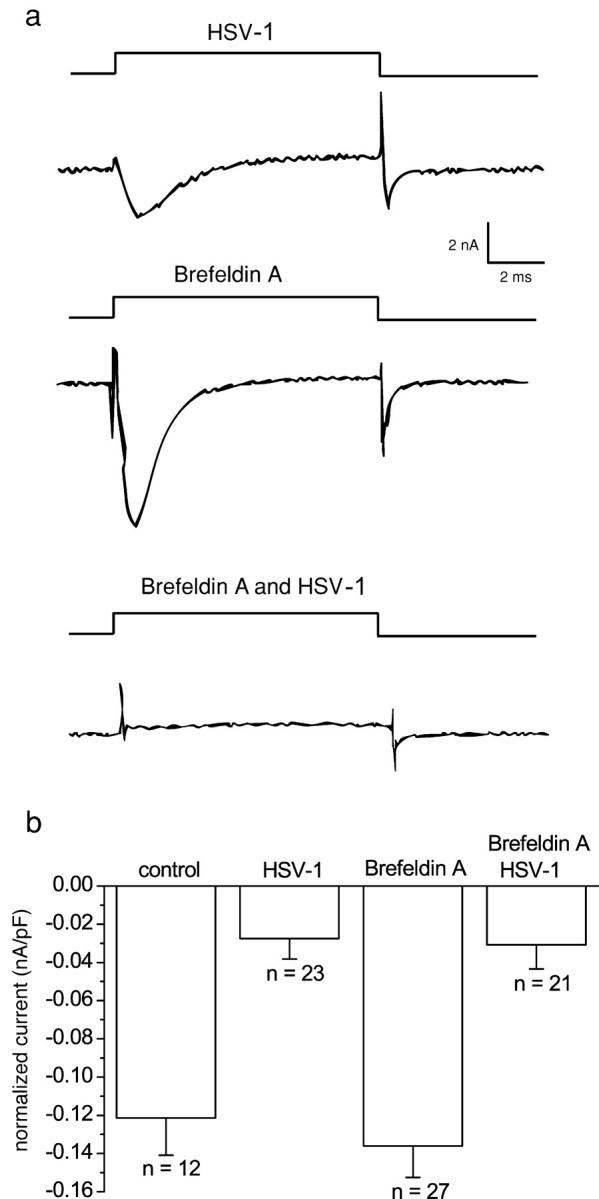
The effect of trans-Golgi network collapse on normalized sodium current amplitude of control and HSV-1–infected DRG neurons. (a) Sodium currents of DRG neurons either treated with brefeldin A and/or infected with wt HSV-1. (b) Mean-normalized sodium current amplitude ± SEM for HSV-1–infected and uninfected DRG neurons with or without brefeldin A. Treatment with brefeldin A did not significantly alter the amplitude of sodium currents in infected or uninfected neurons. The normalized amplitudes of brefeldin A–treated HSV-1 infected and HSV-1–infected DRG neurons were both significantly smaller than the amplitudes of brefeldin A–treated or untreated control DRG neurons (Kruskal-Wallis test followed by the Kolmogorov-Smirnov pairwise comparison; P < 0.001). 70% of wt HSV-1–infected DRG neurons had no sodium current, whereas 71% of cells infected with wt HSV-1 and treated with brefeldin A had no sodium current.
One trivial explanation for the loss of sodium currents is that the virally induced cessation of host protein synthesis changes the balance in sodium channel turnover. To mimic this process, DRG neurons were exposed to 50 μg/ml cycloheximide for 24 h. Such treatment inhibits protein synthesis in DRG neuron cultures by >97% (unpublished data). DRG neurons treated in this way showed no loss of sodium currents as measured by either the percentage of cells with currents (100%) or the normalized amplitudes (0.15 ± 0.01 nA/pF [n = 82] compared with control neurons 0.13 ± 0.01 nA/pF [n = 59]).
HSV-1 gene expression is a tightly regulated temporal cascade beginning with viral immediate early genes that encode mainly transcription factors, followed by viral early genes that encode proteins predominantly with a role in DNA replication, and finally, viral late genes that encode late proteins involved in viral maturation (Honess and Roizman, 1974). Inhibition of viral DNA synthesis with the viral-specific inhibitor acyclovir results in accumulation of early proteins in the infected cells and no expression of late proteins (Holland et al., 1980). We treated infected neurons with acyclovir to determine whether late proteins play a role in the virally induced loss of sodium currents. All infected neurons treated with acyclovir had a sodium current. Representative currents elicited by a step depolarization from −80 to +10 mV are shown in Fig. 8 a. TTX-S– and TTX-R–normalized current amplitudes of uninfected acyclovir-treated and HSV-1–infected acyclovir-treated neurons did not differ significantly (P > 0.1; Fig. 8, b and c), and were significantly larger than the corresponding normalized currents of infected neurons (P < 0.0001 in all cases).
Figure 8.
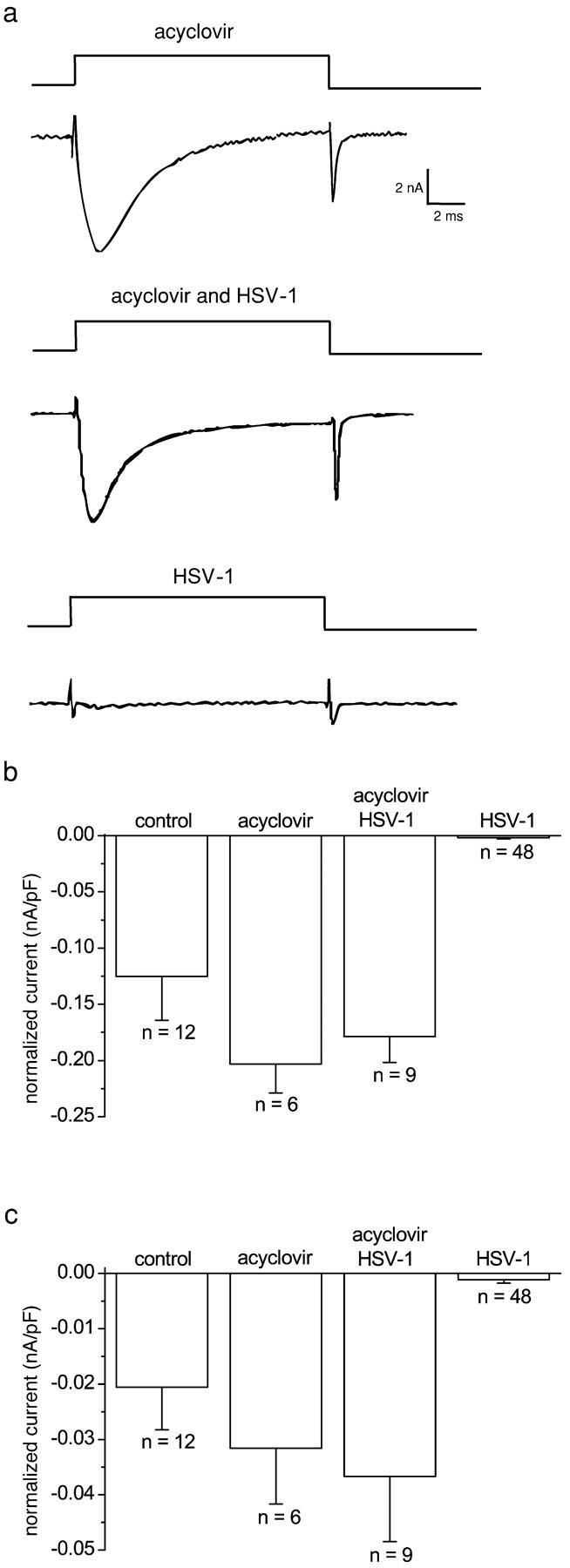
The effect of acyclovir on sodium currents of HSV-1–infected and uninfected DRG neurons. (a) Sodium currents in neurons treated with acyclovir and/or HSV-1 infected. The effect of acyclovir on TTX-S– and TTX-R–normalized (b and c, respectively) sodium current amplitudes. The normalized sodium currents in HSV-1–infected neurons were significantly smaller than those in infected neurons treated with acyclovir (Kruskal-Wallis statistical test followed by the Kolmogorov-Smirnov pairwise comparison; P < 0.0005).
The late protein ICP34.5 that encodes the neurovirulence factor could confer the loss of sodium conductance phenotype to infected neurons. We investigated the role of this late protein in the loss of sodium currents by infecting DRG neurons with an HSV-1 mutant lacking both copies of the ICP34.5 gene. The ICP34.5 mutant virus–infected neurons did not extend neurites, like the wild type infected neurons. These neurons showed no loss of either TTX-S or TTX-R sodium currents (Fig. 9).
Figure 9.

TTX-S and TTX-R sodium currents in DRG neurons infected with HSV lacking ICP 34.5. Graphs a and c show the mean-normalized TTX-S and TTX-R sodium current amplitudes ± SEM of uninfected neurons or neurons infected with HSV lacking ICP 34.5. There was no significant difference in the normalized current amplitude (analysis of variance followed by Tukey's post hoc analysis; P > 0.4). Graphs b and d show the mean-normalized conductance ± SEM from uninfected neurons (○; n = 8) and neurons infected with HSV lacking ICP 34.5 (□; n = 5). The continuous lines were obtained by fitting Boltzmann functions to the mean-normalized data.
Discussion
The cold sore–causing virus HSV-1 is a common human pathogen. About 70% the population has been exposed to HSV by their mid-twenties, and by old age almost the entire population has been infected. This ubiquitous neurotropic virus forms a latent infection in sensory neurons, where it remains throughout the host's life. We were interested in the molecular mechanisms underlying the neurological phenomena reported by patients with an initial or recurrent HSV infection. We report a dramatic, rapid, and selective decrease of sodium conductance that explains the loss of excitability of HSV-1–infected DRG neurons (Fukuda and Kurata, 1981; Oakes et al., 1981; Mayer et al., 1986). Most (∼70%) infected neurons had no measurable sodium current, but when present, its amplitude was smaller in infected neurons than in uninfected neurons. Both TTX-S and TTX-R sodium channel subtypes were similarly affected, as separation of any residual sodium currents revealed that both were of reduced amplitude in infected neurons. A hyperpolarizing prepulse to −120 mV did not reveal sodium currents in HSV-1–infected neurons lacking sodium currents. These findings showed that the loss of sodium currents reflected a loss of functional sodium channels rather than a shift in the voltage dependences of activation or availability (steady-state inactivation). A loss of sodium conductance explains the loss of excitability in vitro and may account for the loss of touch and tingling “anesthesia-like” neurological sensations that patients with recurrent herpes infections describe. In addition to the observations of hypoalgesia during infections, rats inoculated with HSV in the hind paw showed an increased nociceptive threshold indicative of hypoalgesia (Andoh et al., 1995). They described a 24-h lag period before the detection of viral DNA in the DRG somata; this lag time is presumably required for viral axonal transport (2.2 μm/s; Tomishima et al., 2001). The onset of hypoalgesia or increased nociceptive threshold described in rats occurs 24 h after the detection of HSV DNA in the soma. This correlates well with our observation that sodium conductance is lost 24 h after in vitro infection of dissociated rat DRG neurons.
In contrast to sodium currents, the calcium and potassium currents were unaffected after HSV-1 infection. The results described here differ from the findings of Mayer (1986) who showed that the inwardly rectifying potassium currents were lost when DRG neurons were infected with nonsyncytial strains of HSV-1. In the present study, DRG neurons infected with the nonsyncytial strain, wt HSV 17+, showed inwardly rectifying potassium currents of similar amplitude to those of uninfected neurons. It is possible that the disparity between our results is due to sampling differences, as it has recently been shown that the inwardly rectifying potassium currents are found predominantly in large-diameter DRG neurons (Scroggs et al., 1994), and care was taken in our study to include larger neurons in the sample.
We investigated possible mechanisms for the loss of sodium conductance in virally infected cells. Direct observation of the localization of sodium channels in DRG neurons infected with HSV-1 for 24 h showed a decrease in sodium channel immunoreactivity. The redistribution of sodium channels was demonstrated by a notable reduction in plasma membrane staining. The regulation of sodium channel expression and turnover is not well understood. Here, we show that infected neurons retained their sodium conductance when internalization was inhibited with bafilomycin A1 or chloroquine that neutralize endosomes. Internalization may occur in an ubiquitin-dependent manner, as voltage-gated sodium channels contain a conserved ubiquitination site (Abriel et al., 2000), and many cellular proteins of HSV-infected cells are targeted for destruction by modification of the ubiquitin–proteosome pathway (Everett, 2000). Further experiments will be required to determine the role of ubiquitination in the down-regulation of sodium channels in sensory neurons. We suggest the density of voltage-gated sodium channels in the plasma membrane is dynamic, and could control excitability in adult sensory neurons in response to appropriate stimuli. This redistribution of sodium channels may occur by a similar mechanism to those seen in developmental reorganization of sodium channels at the nodes of Ranvier or in activity-induced internalization and degradation in cultured fetal neurons (Dargent et al., 1994; Paillart et al., 1996; Vabnick et al., 1996). Interestingly, this effect of HSV-1 seemed to be dependent on the presence of sodium in the culture media, although the underlying mechanism is unclear (Bevan and Storey, 2002). It is worth noting that in addition to the loss of sodium channel staining in the plasma membrane after 24 h of HSV infection, the cytoplasmic sodium channel density was also decreased. We did not observe any neurons “in the act” of internalization, and this is possibly a reflection of the fact that the sodium channels were degraded rapidly after internalization. To clarify the fate of internalized sodium channels, further analysis of labeled sodium channel proteins would be required.
One possible explanation for the loss of surface sodium channels is that HSV-1 infection inhibits synthesis and/or export of channels to the plasma membrane by virus-induced inhibition of host cell protein synthesis, while allowing normal internalization of the surface channels. Although there are no data available for DRG neurons, it has been estimated that surface sodium channels turn over with a half-life of 50 h (Schmidt and Catterall, 1986), which is slower than the sodium current loss observed in our experiments. Furthermore, the observation that inhibiting DRG neuronal protein synthesis with cycloheximide for 24 h had no effect on sodium current density makes this explanation unlikely and suggests that HSV-1 must accelerate the normal process of internalization.
We investigated the viral proteins and processes that occur during an infection that could interfere with the dynamic regulation of sodium channels. Taking advantage of the fact that late gene expression is strictly dependent on viral DNA replication (Holland et al., 1980), we were able to infect neurons with HSV in the presence of a viral DNA replication inhibitor (acyclovir) that allows the accumulation of HSV early proteins without the expression of late genes. We found that loss of sodium conductance was dependent on a viral late protein or process. However, the loss of sodium conductance was not dependent on the late process of viral envelopment or egress because brefeldin A, which collapses the trans Golgi apparatus and prevents viral maturation, did not prevent the loss of sodium conductance. Furthermore, we have shown that the HSV neurovirulence factor, late protein ICP34.5, was necessary for the virally induced loss of sodium conductance. This is consistent with reports that viral late proteins are abundant in the neurons 24 h after infection. Late genes are expressed at 12 h after infection of nonneuronal cells in vitro (Honess and Roizman, 1974), and in neurons in vivo, late proteins are expressed 24 h after detection of viral DNA at the DRG somata (Margolis et al., 1992). The molecular mechanisms underlying the effects of ICP34.5 on the internalization of sodium channels is unclear. It has recently been shown that ICP34.5 binds to protein phosphatase 1, which has an essential role in intracellular trafficking (Peters et al., 1999; Mao and Rosenthal, 2002). The internalization of another ion channel (ROMK1) is increased when protein phosphatase activity is inhibited (Sterling et al., 2002). The role of phosphatase 1 on the regulation of sodium channel density is an interesting topic for further study. Although it is possible that ICP34.5 is directly responsible for the loss of sodium conductance, there are many putative effects of infection with ICP34.5 deletion mutants, and we cannot rule out the possibility that the failure to affect sodium conductance could be because of reduced replication efficiency (MacLean et al., 1991), changes in the apoptotic status of the neuron (Poon and Roizman, 1997), or a decrease in the level of latency-associated transcripts (Spivack et al., 1995). The properties of the ICP34.5 mutant are particularly interesting, as a viral vector generated from an ICP34.5-null backbone has been used in human gene therapy (Rampling et al., 2000), and it is of particular importance that such a vector does not alter the excitability of the recipient neurons.
In summary, infection of sensory neurons with HSV-1 led to the internalization of sodium channels from the plasma membrane that resulted in a loss of excitability. In contrast, the voltage-dependent properties of potassium and calcium currents were unaffected by viral infection. We suggest that a late protein, possibly ICP34.5, has a role in targeting sodium channels for degradation, potentially by ubiquitination or by increasing the rate of internalization by interacting with protein phosphatase 1. By observing the electrophysiological changes during a viral infection, we have revealed a novel level of regulation of excitability in adult DRG neurons by the rapid internalization of sodium channels.
Materials and methods
Preparation of DRG neurons
DRG neurons were prepared from adult (∼200 g) male or female Sprague-Dawley rats (Charles River Laboratories) as described by Bevan and Winter (1995). In brief, adult rats were asphyxiated with CO2 as approved by the Home Office, and spinal ganglia were removed aseptically from all levels of the spinal cord. Ganglia were incubated in 0.125% collagenase type IV (Worthington Biochemical Corporation) for 3 h in Ham's F-14 medium (Imperial) with 2 mM l-glutamine and 10 μg/ml penicillin at 37°C in a humidified incubator gassed with 3% CO2 in air. Neurons were dissociated mechanically by trituration with a flame-polished Pasteur pipette. The DRG neurons were centrifuged through 2 ml of 15% bovine albumin in Ham's F-14 media and the pellet was resuspended in Ham's F-14 with 4% Ultroser G (GIBCO BRL), 2 mM l-glutamine, 10 μg/ml penicillin and streptomycin, and 50 ng/ml NGF (Promega).
For culture, the neurons were plated onto sterile 13-mm glass coverslips previously coated with 10 μg/ml poly-d-ornithine and 5 μg/ml laminin (GIBCO BRL), and were maintained at 37°C in a humidified incubator gassed with 3% CO2 in air overnight. In experiments where neurons were replated, the cells were first plated onto 35-mm petri dishes (Nunc) previously coated with poly-d-ornithine and laminin at a density of 2 × 104 neurons/plate. In order that all the neurons studied had the same morphology (i.e., no neurite outgrowth) neurons were studied either 4–12 h after dissociation or 2–12 h after replating. The replating step provided a population of neurons that were spherical and without obvious neurites. When the neurons were needed for study, they were aspirated from the petri dish and plated onto 13-mm coverslips coated with poly-d-ornithine and laminin.
Viral DNA replication was inhibited by addition of 50 μM acyclovir (Calbiochem) to the cultures for 24 h. Sodium channel internalization was investigated by incubating the neurons in medium containing internalization inhibitors (either 1 μM bafilomycin A1 or 100 μM chloroquine; Bowman et al., 1988). Protein synthesis was inhibited with cycloheximide (50 μg ml−1). Viral envelopment and egress was inhibited with 1 μM brefeldin A (Koyama and Uchida, 1994). The inhibitors were added to the culture medium 1 h after infection with HSV, to allow time for viral attachment and adsorption, and were present for the subsequent 23 h of infection.
Electrophysiology
DRG neurons were voltage-clamped in the whole-cell configuration with a patch-clamp amplifier (Axopatch 200A; Axon Instruments, Inc.). An IBM-compatible personal computer (model 486; Dell) was used for data acquisition and pulse generation using pCLAMP software (Axon Instruments, Inc.). Borosilicate patch pipettes were pulled from 1.5-mm external diameter glass fiber-filled capillaries (model GC150-F10; Clark Electromedical) in a three-stage pull using a micropipette puller (P-80 Flaming/Brown; Sutter Instrument Co.). The pipettes were fire polished and had a final resistance of 2–5 MΩ when filled. Pipette and membrane capacitance were compensated through the use of the circuitry on the amplifier, and series resistance compensation (∼80%) was used. The DRG neurons were 20–50 μm in diameter. DRG neurons vary significantly in sodium channel expression depending on neuron size, so we studied a wide distribution of neuron sizes for both infected and uninfected populations. Sodium currents were recorded using a pipette solution composed of the following (mM): 130 CsCl, 10 NaCl, 10 EGTA, 1 CaCl2, 1 MgCl2, and 10 Hepes, pH 7.2, and an extracellular solution composed of 30 mM NaCl, 100 mM choline chloride, 20 mM tetraethylammonium chloride, 5 mM MgCl2, 5 mM Hepes, 40 mM sucrose, and 10 μM CaCl2, pH 7.4.
Except where stated, the total sodium current was evoked by a 13-ms voltage step from the holding potential (−80 mV) to a command potential of +10 mV. The TTX-S and TTX-R sodium currents were separated electrophysiologically on the basis of their different inactivation properties (Elliott and Elliott, 1993). In this protocol, the combined TTX-R and TTX-S sodium current was evoked by a 10-ms depolarizing command step to +10 mV from a prepulse potential of −120 mV, and then the TTX-R sodium current was evoked from a prepulse potential of −50 mV. The TTX-S sodium current was obtained by subtracting the TTX-R current from the total combined sodium current. Activation curves were obtained from families of sodium currents generated after a prepulse to either −120 mV (TTX-S) or −50 mV (TTX-R) followed by 10-ms duration, 5-mV step depolarizations from −50 to +25 mV.
Calcium currents were recorded using an intracellular, pipette-filling solution composed of the following (mM): 130 CsCl, 25 Hepes, 1 Na-GTP, 3 Mg-ATP, and 10 EGTA, pH 7.2, and an external bath solution containing (mM): 120 tetraethylammonium Cl, 10 Hepes, 10 glucose, and 5 CaCl2, pH 7.2. The LVA and HVA calcium currents were separated electrophysiologically on the basis of their inactivation properties. The HVA calcium current was measured by first applying a depolarizing step to −40 mV that inactivated the LVA current, followed by a second depolarizing step to 0 mV that evoked only the HVA calcium current. Activation curves were obtained from calcium currents evoked by a series of 360-ms duration pulses to command potentials ranging from −80 to +40 mV. The steady-state availability characteristics of the calcium channel currents were investigated with a protocol that elicited currents with a 320-ms duration command pulse to −10 mV, after 320-ms duration prepulses that ranged from −100 to +15 mV.
Delayed rectifier-type potassium currents were recorded with a pipette solution containing (mM) 130 KCl, 10 KF, 5 EGTA, 2 MgCl2, and 10 Hepes, pH 7.4, and a bath solution containing (mM) 130 choline chloride, 10 Hepes, 10 glucose, 1.2 NaHCO3, 3 KCl, 0.6 MgCl2, and 2.5 CoCl2, pH 7.2. A variety of voltage protocols was used to investigate the outward potassium currents. The total potassium current was evoked with a 40-ms depolarizing step to 10 mV from a prepulse potential of −80 mV. Noninactivating-type potassium currents were evoked after a 60-ms prepulse to −50 mV by a test pulse of 40-ms duration to +10 mV. Activation curves were obtained from potassium currents evoked by 15-ms duration pulses to potentials ranging from −90 to + 40 mV from a prepulse potential of −120 mV, after which the voltage was stepped back to −40 mV. Currents evoked by a step to +45 mV after a series of prepulses from −120 to 0 mV were used to study the steady-state availability of the inactivating component of the potassium currents.
The inwardly rectifying potassium currents were recorded using a pipette solution containing the following (mM): 130 KCl, 11 EGTA, 1 CaCl2, and 7.5 Hepes, and a bath solution of (mM) 140 NaCl, 4 KCl, 2 MgCl2, 10 Hepes, and 10 glucose. A hyperpolarizing step from −70 to −120 mV for 900-ms was used to generate a single inwardly rectifying potassium current, and families of currents were generated by a series of 1-s duration hyperpolarizing steps from −50 to −140 mV.
All experiments were performed at RT, typically 22°C. All current traces were leak-subtracted except for the recordings of inward rectifier currents. The currents recorded were normalized for cell size (capacitance is proportional to membrane area; nA/pF). All chemicals were supplied by Sigma-Aldrich unless otherwise stated. Results are presented as mean ± SEM. Results were statistically tested with Tukey's post hoc analysis after analysis of variance, t test (parametric tests), or the Kruskal-Wallis statistical test followed by the Kolmogorov-Smirnov pairwise comparison (nonparametric test). In each experiment, treated neurons were compared with control neurons from the same preparations and platings to avoid possible differences between batches of cells. Possible differences in proportions of neurons expressing currents were analyzed by the χ2 test. The results were considered significantly different when P < 0.05.
Preparation and use of viral stocks
Wt HSV 17+ (Brown et al., 1973) was used in this study. Wt HSV 17+ was routinely propagated in cells of the baby hamster kidney C 13 line (MacPhearson and Stoker, 1962), which were grown in DME (GIBCO BRL) with 10% FCS (GIBCO BRL) and 1% penicillin and streptomycin (GIBCO BRL). Wt HSV 17+ was collected from the culture after a series of rapid-freeze thaw cycles and was titrated on Vero cells (an African green monkey kidney cell line) grown in RPMI 1640 with 10% FCS, 1% penicillin, 1% streptomycin and 2% sodium bicarbonate (GIBCO BRL).
Infection of neurons with wt HSV 17+ was titrated either with an mAb against immediate early protein ICP-4 or by observation of morphological characteristics. DRG neurons infected with wt HSV 17+ showed no neurite outgrowth after 24 h of infection. An infection of 5 plaque-forming units (pfu)/DRG was sufficient to infect 96% of the neurons in the culture. This was the lowest amount of virus that infected essentially all of the neurons in culture: 1 pfu/neuron resulted in 55% infection efficiency, and 50 pfu/neuron provided no further improvement (97%) than 5 pfu/neuron. We were convinced that all DRG neurons were infected by 5 pfu/neuron, as infected neurons did not grow neurites, and it was clear that essentially all of the population was infected. Also, indirect immunofluorescence of viral proteins or infection with a virus that contains the LacZ gene were used to show that 5 pfu/neuron was sufficient to infect the majority of neurons in the population. In addition, we restricted all experiments to wt HSV 17+-treated neurons without neurites to maximize the probability that only infected neurons were studied.
Immunohistochemistry
DRG neurons were fixed in 4% PFA in PBS at 4°C for 20 min. The primary antibodies, ICP4 antibody (Autogen Bioclear UK Ltd.) and a pan-sodium α subunit antibody directed against a highly conserved region of the intracellular III-IV loop of the channel (Alamone Labs), were diluted in PBS with 10% FCS, 0.1% Triton X-100, and 0.02% sodium azide, and incubated with the DRG samples overnight at 4°C. After incubation with biotinylated antibody (Jackson ImmunoResearch Laboratories) followed by a wash and incubation with streptavidin-fluorescein for 1 h at RT, the samples were mounted in Citifluor (CITIFLUOR Ltd.) and visualized under a fluorescence objective (excitation 470–490 nM) of a brightfield microscope (Eclipse 800; Nikon). The mean density of fluorescence was measured with Image Pro software from Data Cell. Images were obtained by a confocal microscope (TCS Spuv; Leica) with a UV krypton laser.
Acknowledgments
N. Storey was supported by a collaborative Medical Research Council CASE studentship awarded to D. Latchman and S. Bevan.
Footnotes
Abbreviations used in this paper: DRG, dorsal root ganglion; HSV-1, herpes simplex virus type 1; HVA, high voltage–activated; ICP, infected cell protein; LVA, low voltage–activated; pfu, plaque-forming units; TTX-R, TTX resistant; TTX-S, TTX sensitive; wt HSV 17+, wild-type virus HSV-1 Glasgow strain 17 syn+.
References
- Andoh, T., K. Shiraki, M. Kurokawa, and Y. Kuraishi. 1995. Paresthesia induced by cutaneous infection with herpes simplex virus in rats. Neurosci Lett. 190:101–104. [DOI] [PubMed] [Google Scholar]
- Abriel, H., E., Kamynina, J.D. Horisberger, and O. Staub. 2000. Regulation of the cardiac voltage-gated Na+ channel (H1) by the ubiquitin-protein ligase Nedd4. FEBS Lett. 446:377–380. [DOI] [PubMed] [Google Scholar]
- Bevan, S., and N. Storey. 2002. Modulation of sodium channels in primary afferent neurons. Novartis Found. Symp. 241:144–153. [PubMed] [Google Scholar]
- Bevan, S., and J. Winter. 1995. Nerve growth factor (NGF) differentially regulates the chemosensitivity of adult rat cultured sensory neurons. J. Neurosci. 15:4918–4926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman, E.J., A. Siebers, and K. Altendorf. 1988. Bafilomycins: a class of inhibitors of membrane ATPases from microorganisms, animal cells, and plant cells. Proc. Natl. Acad. Sci. USA. 85:7972–7976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, S.M., D.A. Ritchie, and J.H. Subak-Sharp. 1973. Genetic studies with herpes simplex virus type 1. The isolation of temperature-sensitive mutants, their arrangement into complementary groups and recombination analysis leading to a linkage map. J. Gen. Virol. 18:329–346. [DOI] [PubMed] [Google Scholar]
- Dargent, B., C. Paillart, E. Carlier, G. Alcaraz, M.F. Martin-Eauclaire, and F. Couraud. 1994. Sodium channel internalization in developing neurons. Neuron. 13:683–690. [DOI] [PubMed] [Google Scholar]
- Elliott, A.A., and J.R. Elliott. 1993. Characterization of TTX-sensitive and TTX-resistant sodium currents in small cells from adult rat dorsal root ganglia. J. Physiol. 463:39–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett, R.D. 2000. ICP0 induces the accumulation of colocalizing conjugated ubiquitin. J. Virol. 74:9994–10005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda, J., and T. Kurata. 1981. Loss of membrane excitability after herpes simplex virus infection in tissue-cultured nerve cells from adult mammals. Brain Res. 211:235–241. [DOI] [PubMed] [Google Scholar]
- Honess, R.W., and B. Roizman. 1974. Regulation of herpes macromolecular synthesis. I. Cascade regulation of the synthesis of three groups of viral proteins. J. Virol. 14:8–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland, L.E., K.P. Anderson, C.J.R. Shipman, and E.K. Wagner. 1980. Viral DNA synthesis is required for the efficient expression of specific herpes simplex virus type 1 mRNA species. Virology. 101:10–24. [DOI] [PubMed] [Google Scholar]
- Koyama, A., and T. Uchida. 1994. Inhibition by brefeldin A of the envelopment of nucleocaspids in herpes simplex type 1-infected Vero cells. Arch. Virol. 135:305–317. [DOI] [PubMed] [Google Scholar]
- MacLean, A.R., M. ul-Fareed, L. Robertson, J. Harland, and S.M. Brown. 1991. Herpes simplex virus type 1 deletion variants 1714 and 1716 pinpoint neurovirulence-related sequences in Glasgow strain 17+ between immediate early gene 1 and the ‘a’ sequence. J. Gen. Virol. 71:631–639. [DOI] [PubMed] [Google Scholar]
- MacPhearson, I., and M. Stoker. 1962. Polyoma transformation of hamster cell clones: an investigation of genetic factors affecting cell competence. Virology. 16:147–151. [DOI] [PubMed] [Google Scholar]
- Margolis, T.P., F. Sedarati, A.T. Dobson, L.T. Feldman, and J.G. Stevens. 1992. Pathways of viral gene expression during acute neuronal infection with HSV-1. Virology. 189:150–160. [DOI] [PubMed] [Google Scholar]
- Mao, H., and K.S. Rosenthal. 2002. An N-terminal arginine rich cluster and a proline-alanine-threonine repeat region determines the cellular localization of the herpes simplex virus type-1 ICP34.5 protein and its ligand, protein phosphatase 1. J. Biol. Chem. 277:11423–11431. [DOI] [PubMed] [Google Scholar]
- Mayer, M.L. 1986. Selective block of inward but not outward rectification in rat sensory neurones infected with herpes simplex virus. J. Physiol. 375:327–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer, M.L., M.H. James, R.J. Russell, J.S. Kelly, and C.A. Pasternak. 1986. Changes in excitability induced by herpes simplex viruses in rat dorsal root ganglion neurons. J. Neurosci. 6:391–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan, C., H.M. Rose, and. B. Mednis. 1968. Electron microscopy of herpes simplex virus. I. Entry. J Virol. 2:507–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakes, S.G., R.W. Petry, R.J. Ziegler, and R.S. Pozos. 1981. Electrophysiological changes of HSV-1–infected dorsal root ganglia neurons in culture. J. Neuropathol. Exp. Neurol. 40:380–389. [DOI] [PubMed] [Google Scholar]
- Pearce, R.J., and M.R. Duchen. 1994. Differential expression of membrane currents in dissociated mouse primary sensory neurons. Neuroscience. 63:1041–1056. [DOI] [PubMed] [Google Scholar]
- Peters, C., P.D. Andrews, M.J. Stark, S. Cesaro-Tadic, A. Glatz, A. Podtelejnikov, M. Mann, and A. Mayer. 1999. Control of the terminal step of intracellular membrane fusion by protein phosphatase 1. Science. 285:1084–1087. [DOI] [PubMed] [Google Scholar]
- Paillart, C., J.L. Boudier, J.A. Boudier, H. Rochat, F. Couraud, and B. Dargent. 1996. Activity-induced internalization and rapid degradation of sodium channels in cultured fetal neurons. J. Cell Biol. 134:499–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon, A.P., and B. Roizman. 1997. Differentiation of the shutoff of protein synthesis by virion host shutoff and mutant γ (1)34.5 genes of herpes simplex virus 1. J. Virol. 229:98–105. [DOI] [PubMed] [Google Scholar]
- Rampling, R., G. Cruickshank, V. Papanastassiou, J. Nicoll, D. Hadley, D. Brennan, R. Petty, A. MacLean, J. Harland, E. McKie, R. Mabbs, and M. Brown. 2000. Toxicity evaluation of replication-competent herpes simplex virus (ICP 34.5 null mutant 1716) in patients with recurrent malignant glioma. Gene Ther. 7:859–866. [DOI] [PubMed] [Google Scholar]
- Schmidt, J.W., and W.A. Catterall. 1986. Biosynthesis and processing of the α subunit of the voltage-sensitive sodium channel in rat brain neurons. Cell. 46:437–444. [DOI] [PubMed] [Google Scholar]
- Scroggs, R.S., S.M. Todorovic, E.G. Anderson, and A.P. Fox. 1994. Variation in IH, IIR, and ILEAK between acutely isolated adult rat dorsal root ganglion neurons of different size. J. Neurophysiol. 71:271–279. [DOI] [PubMed] [Google Scholar]
- Spivack, J.G., M.U. Fareed, T. Valyi-Nagy, T.C. Nash, J.S. O'Keefe, R.M. Gesser, E.A. McKie, A.R. MacLean, N.W. Fraser, and S.M. Brown. 1995. Replication, establishment of latent infection, expression of the latency-associated transcripts and explant reactivation of herpes simplex virus type 1 γ 34.5 mutants in a mouse eye model. J.Gen.Virol. 76:321–332. [DOI] [PubMed] [Google Scholar]
- Sterling, H., D.H. Lin, R.M. Gu, K. Dong, S.C. Hebert, and W.H. Wang. 2002. Inhibition of protein-tyrosine phosphatase stimulates the dynamin-dependent endocytosis of ROMK1. J. Biol. Chem. 277:4317–4323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomishima, M.J., G.A. Smith, and L.W. Enquist. 2001. Sorting and transport of α herpesviruses in axons. Traffic. 2:429–436. [DOI] [PubMed] [Google Scholar]
- Vabnick, I., S.D. Novakovic, S.R. Levinson, M. Schachner, and P. Shrager. 1996. The clustering of axonal sodium channels during development of the peripheral nervous system. J. Neurosci. 16:4914–4922. [DOI] [PMC free article] [PubMed] [Google Scholar]