

Abstract
The COOH-terminal A168–170 region of the giant sarcomeric protein titin interacts with muscle-specific RING finger-1 (MURF-1). To investigate the functional significance of this interaction, we expressed green fluorescent protein fusion constructs encoding defined fragments of titin's M-line region and MURF-1 in cardiac myocytes. Upon expression of MURF-1 or its central region (containing its titin-binding site), the integrity of titin's M-line region was dramatically disrupted. Disruption of titin's M-line region also resulted in a perturbation of thick filament components, but, surprisingly, not of the NH2-terminal or I-band regions of titin, the Z-lines, or the thin filaments. This specific phenotype also was caused by the expression of titin A168–170. These data suggest that the interaction of titin with MURF-1 is important for the stability of the sarcomeric M-line region.
MURF-1 also binds to ubiquitin-conjugating enzyme-9 and isopeptidase T-3, enzymes involved in small ubiquitin-related modifier–mediated nuclear import, and with glucocorticoid modulatory element binding protein-1 (GMEB-1), a transcriptional regulator. Consistent with our in vitro binding data implicating MURF-1 with nuclear functions, endogenous MURF-1 also was detected in the nuclei of some myocytes. The dual interactions of MURF-1 with titin and GMEB-1 may link myofibril signaling pathways (perhaps including titin's kinase domain) with muscle gene expression.
Keywords: MURF-1; titin; GMEB-1; cardiac myocyte; SUMO-3
Introduction
Numerous structural and regulatory proteins are assembled and maintained in an exquisitely precise order within the myofibrils of striated muscle, yet the molecular mechanisms responsible for this phenomenon are not well understood. The principal components of muscle sarcomeres, the basic contractile units of myofibrils, include parallel arrays of actin-containing thin filaments that overlap with myosin-containing thick filaments. A third filament system is formed by single molecules of titin (connectin), the largest vertebrate protein identified to date (mol wt ∼3–3.7 MD) (Maruyama et al., 1977; Wang et al., 1979; Labeit and Kolmerer, 1995; Bang et al., 2001). Titin molecules from adjacent sarcomeres overlap in the Z-line, and those from opposite half sarcomeres overlap in the M-line, thus forming a continuous filament system within myofibrils (Obermann et al., 1996; Gregorio et al., 1999). The majority (∼90%) of the titin molecule is comprised of repeating modular domains from the fibronectin (FN)* type III and the immunoglobulin (Ig) superfamilies. Additionally, there are 17 unique insertions distributed along the length of titin, including a serine/threonine kinase domain in its M-line region (Labeit et al., 1992), the function of which remains elusive.
To dissect the functional properties of titin's domains, recent investigations have focused on searching for novel titin-binding proteins. One of these proteins is muscle-specific RING finger-1 (MURF-1) (Centner et al., 2001), also recently identified as striated muscle RING zinc finger (SMRZ) (Dai and Liew, 2001) and RING finger 28 (RNF 28) (see http://www.gene.ucl.ac.uk/cgi-bin/nomenclature/searchgenes.pl). This protein binds to the titin domains A168–169–170 (A168–170) located directly NH2-terminal to the titin kinase domain (Centner et al., 2001). Two proteins with a high degree of homology to MURF-1 also have been identified: MURF-2 (RNF 29) and MURF-3 (RNF 30) (Spencer et al., 2000; Centner et al., 2001). The three MURFs are members of the RING finger-B-box-coiled-coil (RBCC) family, a class of proteins that have critical roles in cellular processes including signal transduction, gene transcription, ubiquitination, and differentiation (for reviews see Freemont, 2000; Borden, 2000). Structurally, the MURFs contain a Zn-binding RING finger domain at their extreme NH2-terminal end, a MURF family–specific conserved region, a B-box domain, coiled-coil motifs, and an acidic tail (Spencer et al., 2000; Centner et al., 2001; Dai and Liew, 2001).
To date, little insight into the cellular roles of the MURFs is available. In vitro binding studies revealed that MURF family members homo- and hetero-oligomerize (Centner et al., 2001). MURF-3 appears to associate with microtubules and have a role in myogenic differentiation and microtubule stabilization (Spencer et al., 2000). MURF-1 (SMRZ) recently has been shown to interact with small ubiquitin-related modifier-3 (SUMO-3/SMT3b; Dai and Liew, 2001), a member of a ubiquitin-related class of proteins implicated in subcellular targeting and nuclear import (for review on SUMO proteins see Melchior, 2000). Consistent with this finding, MURF-1 is detected in nuclei (Dai and Liew, 2001; this study). In a different study, it was determined that MURF-1 appears to be the only MURF family member that interacts directly with titin, within the M-line region of the sarcomere (Centner et al., 2001). Despite these recent studies, the exact physiological role(s) of the MURF family members, particularly MURF-1 and -2, have remained elusive.
To determine which regions of MURF-1 target to myofibrils and/or nuclear sites, and as an initial approach to decipher the cellular properties of MURF-1, we expressed green fluorescent protein (GFP) fusion constructs encoding defined regions of MURF-1 and its titin-binding site, A168–170, in live cardiac myocytes. Our data suggest that the interaction of titin with the central region of MURF-1 is important for maintaining the integrity of titin's M-line structure. In turn, titin's COOH-terminal (M-line) region appears to be necessary for thick filament integrity, but, surprisingly, not for the integrity of titin's NH2-terminal or I-band region, the thin filaments, or the Z-lines.
Intriguingly, endogenous MURF-1 also was detected in the nuclei of some myocytes. Consistent with this observation, in vitro interaction studies revealed that MURF-1 binds to ubiquitin-conjugating enzyme 9 (Ubc9) and isopeptidase T-3 (ISOT-3), enzymes involved in SUMO modification of target proteins, a posttranslational modification that occurs in the nucleus. Our in vitro interaction studies also demonstrated that MURF-1 is capable of binding to glucocorticoid modulatory element binding protein-1 (GMEB-1), a nuclear protein implicated in transcriptional regulation (Theriault et al., 1999; Jimenez-Lara et al., 2000; Zeng et al., 2000b). Therefore, our data suggest MURF-1 has an important role in titin filament M-line structure (and perhaps in titin kinase-based signaling processes) as well as a nuclear function (potentially in the control of muscle gene expression). Future studies will likely provide insights into how the dual functions of MURF-1 are associated, as well as how myofibrillogenesis and the regulation of muscle gene expression are linked.
Results
MURF-1 has multiple subcellular localizations in cardiac myocytes
Previous immunolocalization studies revealed that in adult striated muscle, MURF-1 is distributed diffusely throughout the cytoplasm, is assembled at the Z-line region, and is at the M-line region where its binding partner, titin A168–170, is located (Centner et al., 2001). We used primary cultures of fetal rat and embryonic chick cardiac myocytes in studies designed to investigate the cellular role of the interaction of MURF-1 with titin. When these cells were stained with anti–MURF-1 antibodies, MURF-1 was detected diffusely throughout the cytoplasm, and was assembled at the M-line region (Fig. 1; data from rat myocytes shown). Costaining for α-actinin, which stains all myofibrils, revealed that in some myocytes, MURF-1 assembled in only a few myofibrils, whereas in other myocytes, it was assembled in all myofibrils (Fig. 1 a, arrows). Consistent with this observation, immunolocalization studies on isolated rat cardiac and skeletal muscle myofibrils also demonstrated that MURF-1 was detected at the M-line region in only a portion of myofibrils (30–50%; unpublished data). Interestingly, although MURF-1 was detected at the M- and the Z-line in adult cardiac tissue sections (Centner et al., 2001), MURF-1 was not detected at the Z-line in rat fetal or chick embryonic cardiac myocytes. The significance of this is not known, but may be due to developmental differences. MURF-1 staining was also observed diffuse in the cytoplasm (Fig. 1, c and e, arrowheads) and at varying intensities in the nuclei of some cardiac myocytes, as shown by colocalization with DAPI staining (Fig. 1, e and f); this was confirmed by scanning at different focal planes by deconvolution microscopy (unpublished data). MURF-1's nuclear localization is consistent with a recent study that detected MURF-1 (SMRZ) fusion proteins in the nuclei of C2C12 skeletal muscle cells (Dai and Liew, 2001). Staining with the secondary antibody alone yielded negligible background in all experiments (unpublished data).
Figure 1.
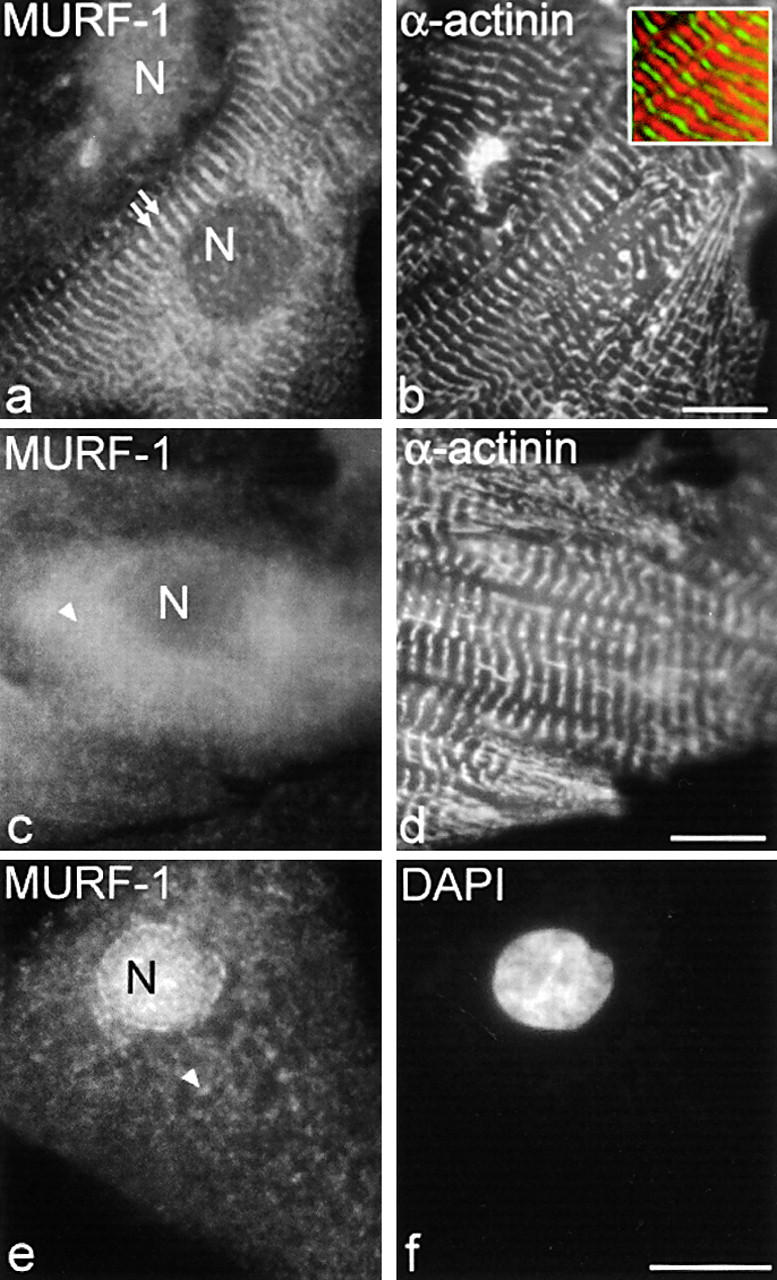
MURF-1 is detected in the M-line region of the sarcomere, diffuse in the cytoplasm, and in nuclei in fetal rat cardiac myocytes. Rat cardiac myocytes were labeled with polyclonal anti–MURF-1 antibodies followed by Texas red–conjugated secondary antibodies (a, c, and e), and with monoclonal α-actinin antibodies followed by Cy2-conjugated secondary antibodies (b and d). DAPI stain was added to identify nuclei (f). Note, inset in b is a merged image of MURF-1 (red) and α-actinin (green) staining. MURF-1 staining in the M-line region (a, double arrows), diffuse (c and e, arrowheads), and/or in nuclei with varying staining intensities (a and e). N, nucleus. Bars, 10 μm.
Expression of MURF-1 or titin A168–170 disrupts the integrity of titin's M-line region and the organization of thick filament components.
We generated a GFP–MURF-1 fusion construct for expression studies in chick cardiac myocytes (Fig. 2). Confirming the immunolocalization studies, GFP–MURF-1 assembled in a portion of myofibrils at the M-line region and colocalized with anti-titin A168–170 staining (unpublished data). GFP–MURF-1 was also diffusely distributed and in distinct aggregates (of varying size and intensity) in the cytoplasm of transfected myocytes (Fig. 3 c, arrowheads), as well as in contaminating fibroblasts that normally do not express MURF-1 (unpublished data). These aggregates are likely a result of homo-oligomerization, which has been reported previously with MURF proteins (Spencer et al., 2000; Centner et al., 2001). Titin staining also colocalized with some GFP–MURF-1 aggregates in the cytoplasm of myocytes (Fig. 3, c and d, arrowheads) perhaps because the MURF-1 aggregates provided additional binding sites for titin during myofibril assembly or turnover. GFP–MURF-1 also was observed in the nuclei of some myocytes (unpublished data). Strikingly, the majority of the GFP–MURF-1–transfected cells exhibited severe disruption of the COOH-terminal region of titin compared with cells expressing GFP alone (Fig. 3, b and d, arrows).
Figure 2.

The COOH-terminal region of titin, revealing its Ig, FN III, and Ser/Thr kinase domains (modified from Centner et al., 2001). Most of the full-length titin molecule is composed of repeating Ig (dark gray) and FN III (red) modules, but also contains 17 unique domains, including a Ser/Thr kinase domain at the M-line region (light gray). The MURF-1 binding site resides within the Ig and FN domains A168–170, just NH2-terminal to the kinase domain. Bars above the titin molecule denote constructs used. Schematic structure of MURF-1 shown under titin, including its RING domain (orange), MURF family conserved region (purple), B-box domain (green), coiled-coil domains (yellow), and tail region (blue). The titin A168–170 binding site is located within the central region of MURF-1. The GFP–MURF-1 deletion constructs are shown under full-length MURF-1.
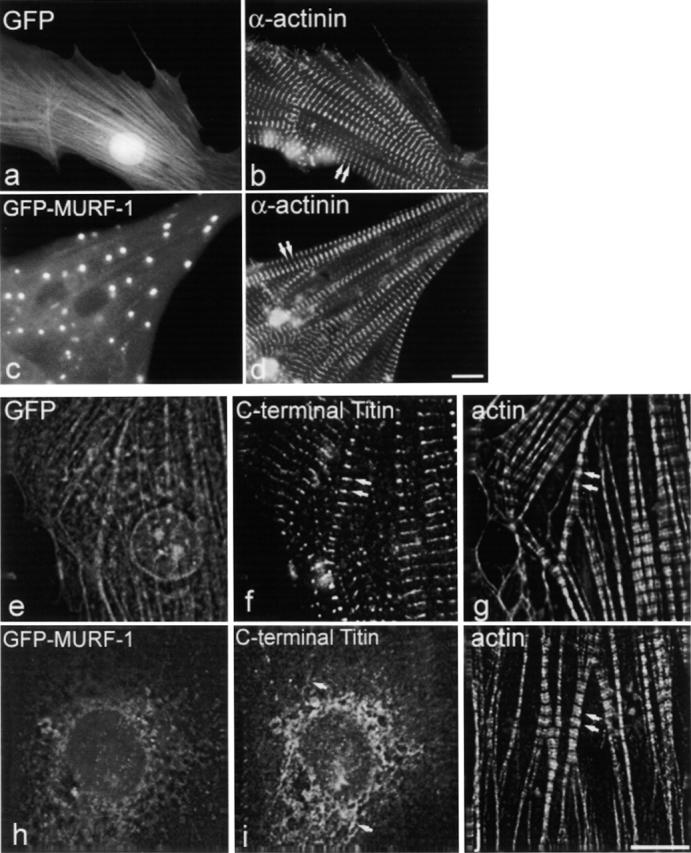
Figure 3.

Expression of GFP–MURF-1 results in severe disruption of titin's M-line structure. Cells expressing GFP–MURF-1 (c) or GFP alone (a) were stained with anti-titin A168–170 antibodies followed by Texas red– conjugated secondary antibodies (b and d). In most myocytes, GFP–MURF-1 expression resulted in a severe disruption of the M-line region of titin (d) compared with myocytes expressing GFP alone (b). Note, titin staining colocalized with many of the GFP–MURF-1 aggregates (c and d, arrowheads). The disruption of titin appears to be localized to its COOH-terminal region. Triple-labeling studies in GFP- (e) and GFP–MURF-1–expressing (h) cells revealed that epitopes from the NH2-terminal region of titin (j) appeared regular and striated in the same myofibrils exhibiting disrupted COOH-terminal titin staining (i). Double arrows mark regular, striated titin and single arrows mark disrupted titin. Bars, 10 μm.
Surprisingly, triple-labeling studies revealed that although titin's COOH-terminal region was disrupted in GFP–MURF-1–expressing myocytes, titin's NH2-terminal region appeared in a regular, striated pattern in the identical myofibrils (Fig. 3, i and j). However, staining transfected cells for myosin, myosin-binding protein C (MyBP-C), and myomesin demonstrated that GFP–MURF-1 expression also perturbed thick filament integrity in a large percentage of transfected myocytes (Fig. 4). Specifically, ∼80–90% of cells transfected with MURF-1 had disrupted COOH-terminal titin structure, whereas ∼60–70% of the cells had perturbed thick filament structure (Fig. 6). This difference in percentages of cells affected suggests that the thick filament perturbation may be a secondary effect of the disruption of titin's M-line region. Unexpectedly, the structure of the actin (thin) filaments, the I-band region of titin, and the Z-lines (as assessed by the distribution of phalloidin, titin N2A, and α-actinin staining, respectively) appeared in typical striated patterns in the majority of GFP–MURF-1–transfected cells (Figs. 5 and 6), even though the COOH-terminal region of titin was disrupted (Fig. 5, i and j).
Figure 4.

Expression of GFP–MURF-1 also perturbs the organization of thick filament components. Transfected myocytes were stained with antibodies to myosin (b and d), MyBP-C (f and h), and myomesin (j and l). In many GFP–MURF-1–expressing myocytes, staining for thick filament components was perturbed (d, h, and l), compared with myocytes transfected with GFP alone (b, f, and j). Single arrows mark perturbed thick filament component staining, and double arrows mark regular, striated thick filament staining. Note, size and intensity of the GFP–MURF-1 cytoplasmic aggregates vary from cell to cell (c, g, and k, arrowheads). Bar, 10 μm.
Figure 6.

Quantification of GFP–MURF-1– and GFP-expressing myocytes exhibiting disrupted staining patterns for sarcomeric components. Myocytes expressing GFP–MURF-1 (gray bars) or GFP alone (white bars) were stained for various sarcomeric components, and the number of cells exhibiting perturbed staining patterns was counted. The results indicate that the COOH-terminal region of titin (A168–170 and AB5) was severely disrupted in most GFP–MURF-1–expressing cells, whereas its I-band (N2A) and NH2-terminal region (T11) were not affected. The thick filament components myosin, myomesin, and C-protein were also disrupted in a large majority of GFP–MURF-1–expressing cells, but thin filament and Z-line components appear relatively unaffected. Data are presented as the mean percentage of total myocytes with disrupted staining ± SD. Means were obtained by counting >50 myocytes from more than two experiments and the results are representative of >10 experiments.
Figure 5.
Expression of GFP–MURF-1 does not appear to affect the integrity of thin filament or Z-line components. Myocytes expressing GFP–MURF-1 (c) or GFP alone (a) were stained for Z-lines with saromeric α-actinin antibodies (b and d) and show regular, striated staining. Triple-labeling studies in GFP–MURF-1–transfected cells (h), using Texas red–conjugated phalloidin (j) and antibodies to titin A168–170 (i), determined that thin filament integrity is not affected upon disruption of COOH-terminal titin in identical myofibrils. GFP-transfected cells (e) exhibited normal actin filament (g) and COOH-terminal titin (f) staining. Double arrows mark regular, striated staining. Single arrows mark disrupted titin staining. Bars, 10 μm.
Because MURF-1 was shown to interact with the COOH-terminal titin domains A168–A170 (Centner et al., 2001), we also performed expression studies with titin GFP fusion constructs encoding the A168–170 region or its flanking regions for comparison (Fig. 2). When expressing titin A168-170–GFP in myocytes, both the M-line region of titin (Fig. 7 b) and the thick filaments (Fig. 7 h; data shown for myomesin, similar results were obtained for myosin and MyBP-C) were perturbed. Consistent with the results from the GFP–MURF-1 expression studies, Z-line and thin filament structure appeared in their characteristic patterns in the titin A168-170–GFP–transfected cells (Fig. 7, n and t). In contrast, myocytes expressing other titin M-line regions (the Ig domains located COOH-terminal to the A168–170 region, M1-M2-M3–GFP or M8-M9-M10–GFP) had regular, striated staining patterns for titin, thick filament, Z-line, and thin filament components (Fig. 7, d, j, p, and v; data shown for M8-M9-M10-GFP). Furthermore, myocytes expressing GFP fusion constructs encoding titin's unique Ser/Thr kinase domain, as well as those expressing a constitutively activated mutant form of this domain, exhibited typical titin, thick filament, thin filament, and Z-line staining (Fig. 7, f, l, r, and x; data shown for titin kinase–GFP). In conclusion, the disruption of sarcomeric M-line integrity was specific to the expression of titin A168–170 and its ligand, MURF-1. These data suggest that the interaction of titin with MURF-1 has an important role in maintaining the structure of sarcomeric M-line components in cardiac myocytes. Our data also surprisingly indicate that disruption of the integrity of the COOH-terminal region of titin perturbs thick filament structure but does not appear to affect the structure of titin's I-band or NH2-terminal regions, the thin filaments, or the Z-lines.
Figure 7.
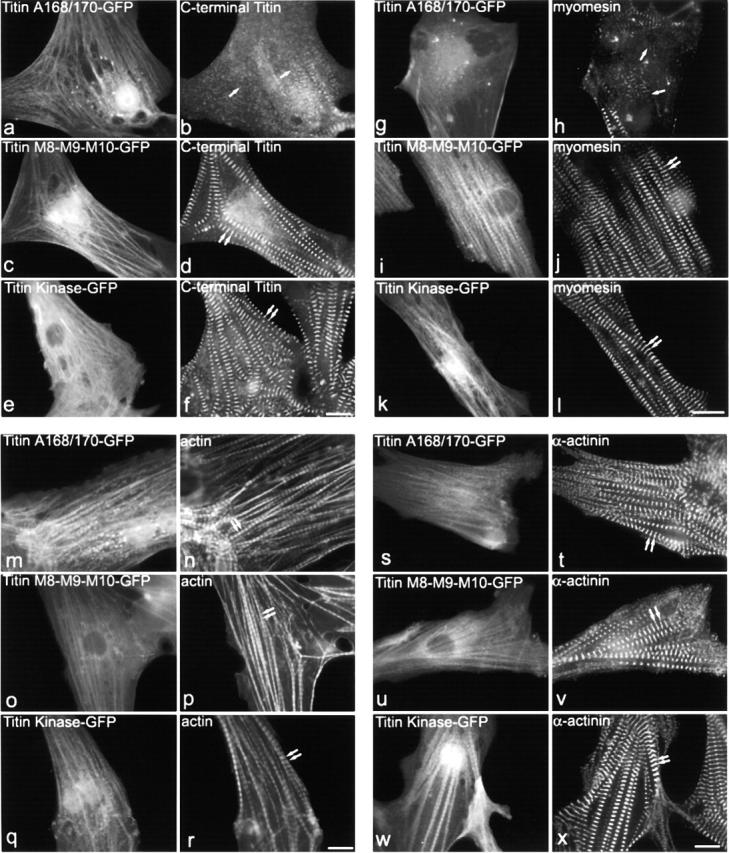
Expression of titin domains A168–170 also disrupts titin M-line region and thick filament structure, but not thin filaments or Z-lines. Myocytes expressing titin A168–170 or other Ig domains from the M-line region of titin were stained for various sarcomeric components. Costaining of A168-170–GFP expressing myocytes with anti– C-terminal titin region antibodies revealed a severe disruption of titin (a and b), compared with myocytes transfected with M8-M9-M10-GFP (c and d) or titin kinase–GFP (e and f). Costaining of titin A168-170–transfected myocytes with antibodies against thick filament components, including myomesin (g and h), reveals that the integrity of the thick filaments is perturbed compared with myocytes transfected with titin M8-M9-M10-GFP (i and j) or titin kinase–GFP (k and l). Myocytes transfected with titin A168-170–GFP (m, n, s, and t), titin M8-M9-M10–GFP (o, p, u, and v), or titin kinase–GFP (q, r, w, and x) were stained with Texas red–conjugated phalloidin (n, p, and r) or antibodies to sarcomeric α-actinin (t, v, and x). Double arrows mark regular, striated staining. Single arrows mark disrupted staining. Bars, 10 μm.
MURF-1's central region targets to the M-line and maintains M-line structure, whereas the RING domain targets to nuclei
To investigate the regions of MURF-1 involved in its subcellular targeting and maintaining the structure of titin's COOH-terminal region, we generated five GFP–MURF-1 deletion constructs (Fig. 2). One construct encoded the MURF-1 NH2-terminal RING domain alone (RING), which contains the SUMO-3/SMT3b binding site (Dai and Liew, 2001). Another construct encoded only the COOH-terminal acidic tail region (Tail). Three constructs contained the central region of MURF-1, which binds to titin A168–170 (Centner et al., 2001): one encoded the NH2-terminal RING domain plus the central region (Tailless); one encoded the central region plus the COOH-terminal tail (RINGless); and one encoded the MURF-1 central region alone (Central). Neither RING nor Tail were observed to assemble at the sarcomeric M-line region or form cytoplasmic aggregates. Strikingly, however, RING–GFP was detected in the nuclei of all transfected myocytes observed (Table I). Because aberrant nuclear localization of GFP fusion proteins is known to occur, we quantified the nuclear localization of each GFP–MURF-1 fusion protein in transfected myocytes (Table I). RING–GFP localized to nuclei in 100% of observed cells, compared with GFP alone, which was observed in nuclei in 40–65% of cells. Consistent with this, RINGless–GFP never exhibited nuclear localization, but Tailless-GFP (containing the RING domain) was detected in nuclei in 40–65% of cells (Table I). Finally, Tail–GFP was detected in ≤10% of myocyte nuclei, suggesting that it does not contain a fully functional nuclear targeting site. These data, together with the observations that MURF-1's RING domain binds to the nuclear protein SUMO-3 (Dai and Liew, 2001) and other RING proteins have been reported in nuclei (for review see Borden, 2000), are consistent with the RING domain targeting MURF-1 to the nuclei of cardiac myocytes.
Table I. Subcellular localization of GFP–MURF-1 deletion constructs in cardiac myocytesa .
| GFP–MURF-1 deletion construct | Cytoplasmic aggregates | Striated at M-line region | Nuclei |
|---|---|---|---|
| % | % | % | |
| GFP alone | 0 | 0 | 40–65 |
| RING | 0 | 0 | 100 |
| Tail | 0 | 0 | ≤10 |
| Tailless | 90–95 | 30–40 | 40–65 |
| RINGless | 90–95 | 30–40 | 0 |
| Central | 90–95 | 10–20 | 0 |
Represents the total percentage of transfected myocytes observed to contain GFP fusion proteins in the specified localization.
In contrast, all three constructs encoding MURF-1's central region (Tailless, RINGless, and Central) assembled at the M-line region in a portion of myofibrils of some myocytes (Table I). Additionally, the expressed proteins all formed cytoplasmic aggregates in the majority of transfected cells, most likely due to their coiled-coil domains. These data indicate that MURF-1's central region, containing the titin-binding site, targets the protein to the sarcomeric M-line region, whereas the RING domain appears to target it to nuclei.
Next, we costained transfected myocytes with anti–COOH-terminal titin antibodies to identify the region(s) of MURF-1 involved in M-line structure. Expression of each of the three MURF-1 fragments containing its central region, shown above to contain an M-line targeting site, resulted in a phenotype identical to that observed when full-length MURF-1 was expressed. Specifically, a marked disruption of titin A168–170 staining was observed in the vast majority of cells expressing Central, Tailless, or RINGless (Fig. 8, f, h, and j), compared with regular, striated titin staining in myocytes expressing RING or Tail (Fig. 8, b and d). These studies reveal that MURF-1 contains two distinct targeting and functional domains. Its central region, containing the titin-binding site, targets it to the M-line and participates in maintaining the integrity of titin's COOH-terminal region. The RING domain, containing the binding site for SUMO-3, appears to be involved in MURF-1 nuclear targeting.
Figure 8.

The central portion of MURF-1 is involved in maintaining the structure of COOH-terminal titin. Myocytes expressing defined regions of MURF-1 were stained with anti-titin A168–170 antibodies. Expression of the RING–GFP (a and b) or Tail–GFP (c and d) proteins did not appear to affect the integrity of COOH-terminal titin. Expression of GFP fusion proteins of the central MURF region (Central; i and j), the RING plus central regions (Tailless; g and h), and the central plus tail regions (RINGless; e and f) severely disrupted titin's COOH-terminal region (single arrow marks disrupted titin A168–170 staining; double arrows mark regular, striated titin A168–170 staining). Note that titan staining colocalized with many of the GFP–MURF-1 aggregates (g–j, arrowheads). Bar, 10 μm.
MURF family members interact with Ubc9 and ISOT-3, but only MURF-1 interacts with the transcriptional modulator GMEB-1
Previous binding studies revealed that MURF-1 interacts with titin A168–A170 and with itself (by oligomerization), and also hetero-oligomerizes with MURF-2 and -3 (Centner et al., 2001). Other in vitro interaction studies determined that MURF-1 also binds to SUMO-3/SMT3b (Dai and Liew, 2001). We searched for additional MURF binding proteins in human heart and fetal mouse cDNA libraries by yeast two-hybrid (Y2H) screening. These novel screens demonstrated that both MURF-1 and -2 baits interact with Ubc9 and ISOT-3 prey clones (Fig. 9 A). Ubc9 is an enzyme involved in SUMO modification; it specifically catalyzes the formation of an isopeptide bond between SUMO and a target protein (Desterro et al., 1997; Johnson et al., 1997; Tatham et al., 2001). ISOT-3 is a member of an enzyme family responsible for cleaving isopeptide bonds. It has been proposed that Ubc9 and isopeptidases regulate the dynamics of SUMO modification (for review see Melchior, 2000; Muller et al., 2001). Further Y2H studies revealed that MURF-3 also interacts with Ubc9 (Fig. 9 A).
Figure 9.
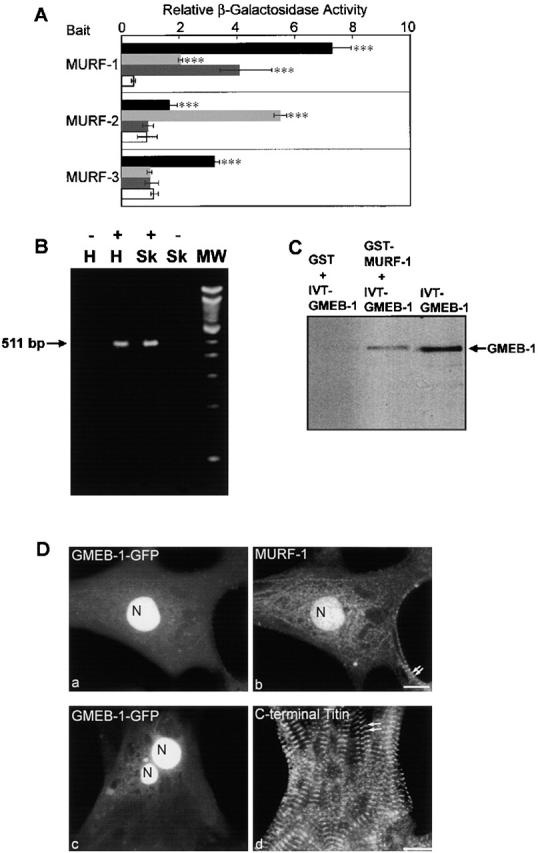
MURF family members interact with SUMO modifying enzymes ISOT-3 and Ubc9, but only MURF-1 interacts with the transcriptional regulator GMEB-1. (A) Y2H screens using full-length cDNAs of individual MURF family members as baits identified ISOT-3 (light gray) and Ubc9 (black) as MURF-binding proteins. However, GMEB-1 (dark gray) was found to interact only with MURF-1. β-Galactosidase assays were performed to confirm positive clones, and the levels were compared with colonies transformed with each prey construct and the empty bait vector (white). Data are presented as mean levels of β-galactosidase from triplicate experiments ± SD. ***, P > 0.001. (B) RT-PCR analysis of human heart total RNA revealed that GMEB-1 mRNA transcripts are detectable in heart (H) and skeletal (Sk) tissues. Lane 1, no reverse transcriptase control in human heart RNA (−); lane 2, 511-bp GMEB-1 PCR product amplified from human heart RNA (+); lane 3, 511-bp GMEB-1 PCR product amplified from human skeletal RNA (+); lane 4, no reverse transcriptase control in human skeletal RNA (−). (C) GMEB-1 specifically binds to MURF-1 in GST pull-down assays. GMEB-1 was translated in vitro (lane 3). When incubated with bacterially expressed GST–MURF-1 fusion peptides, GMEB-1 and MURF-1 binding to glutathione–sepharose 4B beads was detectable (lane 2). Lane 1 contains no detectable binding of GMEB-1 to the beads alone. IVT, in vitro translated. (D) GMEB-1–GFP targets to the nuclei of cardiac myocytes (a and c). MURF-1 staining also was present in some of the nuclei that contained GMEB-1–GFP (b). Note, MURF-1 is also detected at the M-line region in the same myocytes (b, double arrows). Expression of GMEB-1–GFP in cardiac myocytes does not appear to affect the integrity of the COOH-terminal region of titin (d, staining with anti-titin A168–170 antibodies). Double arrows mark regular, striated titin staining. N, nuclei. Bars, 10 μm.
Interestingly, another MURF-1 binding partner that we identified through Y2H screens is GMEB-1 (Fig. 9 A). GMEB-1 is a nuclear protein that regulates transcription in response to changes in cellular glucocorticoid levels (Zeng et al., 1998, 2000b; Jimenez-Lara et al., 2000). Remarkably, GMEB-1 binding to MURF-2 or -3 was not detected in the Y2H system (Fig. 9 A), although these proteins are highly homologous to MURF-1. The interaction of MURF-1 with GMEB-1 was confirmed by glutathione-S-transferase (GST) pull-down assays under stringent conditions (Fig. 9 C). Previously, GMEB-1 mRNA was found in skeletal muscle but not in cardiac tissue by RT-PCR studies (Zeng et al., 2000a). However, it was detected in heart by Northern and Western blot analysis (Theriault et al., 1999; Jimenez-Lara et al., 2000). We performed RT-PCR analysis on heart and skeletal cDNA and detected GMEB-1 mRNA transcripts in both tissues (Fig. 9 B). (The discrepancy between our study and the Zeng et al., 2000a study might be due to differences in PCR conditions.) GMEB-1–GFP fusion proteins localized to nuclei in transfected myocytes (Fig. 9 D, c), consistent with previous studies in COS and HeLa cells (Theriault et al., 1999; Jimenez-Lara et al., 2000). In a few myocytes, GMEB-1 also was detected diffusely throughout the cytoplasm (Fig. 9 D, c), but assembly at the M-line region of the sarcomere was never observed. Costaining myocytes expressing GMEB-1–GFP with anti–MURF-1 antibodies demonstrated that GMEB-1–GFP and MURF-1 were both present in the same nuclei of some myocytes (Fig. 9 D, a and b), which also supports their potential interaction. Finally, cells expressing GMEB-1–GFP exhibited regular striated staining patterns for titin, thin filament, thick filament, and Z-line components, indicating that the overall sarcomeric integrity in the GMEB-1–transfected cells was not affected (Fig. 9 D, d; data shown for COOH-terminal titin staining). These data suggest that MURF-1 specifically interacts with GMEB-1. However, unlike exogenous expression of MURF-1, exogenous expression of GMEB-1 does not affect the integrity of the M-line region of cardiac sarcomeres.
Discussion
In recent years, there has been significant progress in deciphering the structure and functions of the giant sarcomeric protein titin. It appears that titin, the only known protein to span the entire half sarcomere, has multiple cellular roles. Titin contains elastic elements in its I-band region responsible for passive tension generated upon stretch; thus it functions as a molecular spring to maintain the structural integrity of contracting myofibrils. Additionally, titin may act as a “molecular blueprint” to orchestrate the assembly and organization of the thick filaments as well as other structural and regulatory components of sarcomeres. The presence of a unique Ser/Thr kinase domain at the COOH-terminal end of titin also suggests that it may participate in signal transduction pathways (for reviews with original citations see Labeit et al., 1997; Gregorio et al., 1999; Trinick and Tskhovrebova, 1999; Gregorio and Antin, 2000).
To decipher the roles of titin in sarcomeric structure, recent studies have focused on dissecting the properties of individual titin regions and their potential ligands (for reviews see McElhinny et al., 2000; Sanger and Sanger, 2001). Here, we aimed to investigate the functional significance of the interaction of titin's COOH-terminal Ig domains A168–170 (located directly NH2-terminal to the titin Ser/Thr kinase domain) with MURF-1, a RING finger protein. Expression of MURF-1 or titin A168–170 in primary cultures of embryonic chick cardiac myocytes severely disrupted the integrity of titin's M-line region. The region of MURF-1 that is responsible for this phenotype was mapped to its central region, which previously has been shown to contain its titin-binding site (Centner et al., 2001). The most plausible explanation for our observations is that a dominant-negative phenotype occurred. That is, upon expression of the central region of MURF-1 or titin A168–170, the fusion proteins likely interfered with the interaction of endogenous MURF-1 with titin. Surprisingly, expression of the full-length MURF-1 molecule also resulted in this striking phenotype, suggesting that endogenous MURF-1 levels are tightly regulated. Our data suggest that MURF-1 and its interaction with titin A168–170 are critical for maintaining the stability of titin's COOH-terminal region.
In turn, it appears that the interaction of titin with MURF-1 plays a critical role in maintaining the stability of the thick filaments. Although all of the sarcomeric components comprising the M-line region have not been elucidated, the thick filaments appear to be laterally associated with titin via their interactions with MyBP-C (along the A-band), and myomesin (at the M-line) (Houmeida et al., 1995; Obermann et al., 1997). In fact, it has been proposed that titin specifies the number and location of thick filament components (Whiting et al., 1989; Trinick, 1994; Houmeida et al., 1995). It is striking that expression of titin A168–170 in cardiac myocytes perturbed M-line titin and thick filament structure, yet expression of other titin COOH-terminal domains (M1-M2-M3, M8-M9-M10, titin kinase domain, or a mutant, constitutively active kinase domain) had no effect on M-line or thick filament integrity. These results specifically implicate titin A168–170 and MURF-1 in the regulation of sarcomeric M-line and thick filament organization. We hypothesize that MURF-1 forms a complex with titin A168–170 that functions in the assembly of M-line and thick filament components during myofibrillogenesis, as well as in myofibril turnover.
Intriguingly, although titin's COOH-terminal region and the thick filaments were disrupted in MURF-1 or titin A168-170–transfected myocytes, the structural integrity of the thin filaments remained intact. These data are consistent with the observation that thin filament assembly occurs independently of the formation of the thick filaments (Antin et al., 1981; Schultheiss et al., 1990; Epstein and Fischman, 1991; Holtzer et al., 1997; Ehler et al., 1999; Gregorio and Antin, 2000; Rudy et al., 2001). Because titin's NH2-terminal and I-band regions were also intact, these data indicate that certain regions of the titin filament can be “selectively” perturbed. Previous studies have shown that titin interacts with various Z-line components (α-actinin and T-cap/telethonin) and thin filament components (actin) (Funatsu et al., 1993; Jin, 1995; Granzier et al., 1997; Linke et al., 1997; Sorimachi et al., 1997; Gregorio et al., 1998; Mues et al., 1998; Young et al., 1998). From our studies, it appears that these associations along the titin molecule stabilize its NH2-terminal and I-band regions even when the structure of its COOH-terminal end is perturbed. Consistent with this idea, the NH2-terminal end of titin becomes organized during myofibril assembly before the COOH-terminal end of titin and other M-line components (Fürst et al., 1989; Schultheiss et al., 1990; Komiyama et al., 1993; van der Loop et al., 1996; Ehler et al., 1999; Rudy et al., 2001). A possible explanation for this observation is that the less organized M-line region of titin filaments may not be concentrated enough for a signal to be detected by immunofluorescence microscopy (Ehler et al., 1999). Therefore, in our study, it is likely that titin's COOH-terminal region lost its stable interactions and was “less organized,” whereas the more NH2-terminal regions remained “bolted” to other Z- and I-band components. Interestingly, another study has demonstrated that the thick filaments remain intact when the thin filaments are perturbed upon expression of titin's I-band, N2B region (Linke et al., 1999), in the same cell type used in our studies. Thus, titin also may function to keep the thick filaments aligned in the absence of thin filaments.
In addition to sarcomeric M-line localization, endogenous MURF-1 was detected in the nuclei of cardiac myocytes. The observation that MURF-1 assembles in only some myofibrils also indicates that its cellular levels are tightly regulated. An excellent candidate for regulating MURF-1 levels, its localization pattern, and nuclear import is SUMO-3. SUMO-3 binds to MURF-1's RING domain (Dai and Liew, 2001), the region of MURF-1 that appears to be involved in its nuclear localization (this study). We found that two enzymes potentially involved in regulating the conjugation of SUMO with its target proteins, Ubc9 and ISOT-3, interact with MURF-1 and other MURF family members. Although the biochemical pathways and classes of enzymes involved in “SUMO modification” are parallel to those in ubiquitination, the two processes may be functionally distinct. In fact, SUMO modification has been implicated in regulating the levels and localization patterns of target proteins, including nuclear localization (for review see Melchior, 2000). All three MURF proteins exhibit multiple cellular localization patterns (Spencer et al., 2000; Centner et al., 2001; unpublished data), consistent with them being potential SUMO targets. Interestingly, SUMO-3 has recently been shown to be a key component of a new class of acute and reversible cellular stress response pathways (Saitoh and Hinchey, 2000). Further studies are needed to determine whether MURF-1 levels and/or localization patterns change in response to cellular stress, and whether titin plays a role in sensing stress response pathways in cardiac myocytes.
Strongly implicating MURF-1 with nuclear functions are the data demonstrating its specific interaction with GMEB-1 and their colocalization in the nuclei of some myocytes. GMEB-1 was first characterized as a component of a complex that binds to the GME in liver cells, thereby modulating transcription in response to glucocorticoid levels (Zeng et al., 1998; Theriault et al., 1999). Recent studies indicate that GMEB-1 is expressed in a wide range of cell types and may have roles in development and differentiation (Zeng et al., 2000a). We confirmed that GMEB-1 mRNA transcripts are present in striated muscle tissue and found that GMEB-1–GFP fusion proteins targeted to the nuclei of transfected cardiac myocytes. Given that MURF-1 is involved both in sarcomeric M-line structure (via its interaction with titin) and potentially in gene expression (via its interaction with GMEB-1), we speculate that MURF-1 acts as a link between gene expression and myofibril signaling pathways. Currently, it remains unclear what factors might modulate the MURF-1–based myofibril-to-nuclear signaling pathways. One idea is that the titin kinase domain could play a role in this process, because the MURF-1 binding site is located adjacent to this domain (Centner et al., 2001).
In conclusion, we demonstrated that the central region of MURF-1 targets to the M-line region and participates in its structural integrity in cardiac myocytes, most likely through its interaction with titin's COOH-terminal A168–170 region. Interestingly, MURF-1 is an RBCC protein, a family whose members have been referred to as “builders of master scaffolds” because many are involved in the formation of multiprotein complexes (for review see Borden, 2000). Thus, perhaps additional factors, such as other MURF family members, are involved in regulating M-line and thick filament structure through their interactions with MURF-1. It is also striking that RING finger and RBCC proteins have been implicated in ubiquitination pathways (for review see Freemont, 2000). Consistent with this, recombinant MURF-1 protein was recently reported to have ubiquitin ligase activity (Bodine et al., 2001). Moreover, MURF-1−/− mice were resistant to skeletal muscle atrophy, suggesting that MURF-1 regulates the degradation of critical muscle proteins (Bodine et al., 2001). These results and the results from our study support our hypothesis that MURF-1 is involved in a novel pathway responsible for titin structure and/or turnover. Intriguingly, because MURF-1 also is a nuclear component that binds to the transcriptional modulator GMEB-1, MURF-1 may participate in both the regulation of myofibril assembly and structure as well as muscle gene expression. In support of our hypothesis, rat skeletal muscle MURF-1 mRNA levels increased 10-fold in response to glucocorticoid exposure (Bodine et al., 2001), which regulates GMEB-1 transcriptional activity (Theriault et al., 1999; Zeng et al., 2000b). Future studies are required to elucidate whether the dual localization of MURF-1 to myofibrils and nuclei is a result of dynamic SUMO modification.
Materials and methods
Y2H interaction studies
For a survey of potential MURF-1 interactions, a full-length MURF-1 cDNA fragment was amplified from human skeletal muscle cDNA by PCR (Saiki et al., 1985) and inserted into pAS2-1 (Matchmaker system II; CLONTECH Laboratories, Inc.) to obtain a GAL4–BD fusion. For screening, bait constructs were transformed into Saccharomyces cerevisiae strain CG1945, PJ69-4A (pAS2-1 bait), or L40c (BTM117c bait). For some screens, the pAS2C-1 vector also was used (provided by T. Maeda, University of Tokyo). Subsequently, the cells were transformed with a human skeletal muscle cDNA library into the pGAD10 prey vector (HL4010AB; CLONTECH Laboratories, Inc.), essentially as described by the manufacturer. Cells were plated onto SD/Leu−/Trp−/His− plates and incubated at 30°C until colonies appeared (after ∼5 d). In screens where the pAS2-1 bait vector was used, the plates were supplemented with 1.5 mM 3-amino-1,2,4-triazole (Sigma-Aldrich). Transformants were picked, restreaked onto SD/Leu−/Trp−/His− plates, and screened for β-galactosidase activity. β-Galactosidase activity of the cells was measured either by colony lift filter assays using X-gal or in liquid culture using chlorophenol red β-d-galactopyranoside, as described by the manufacturer (CLONTECH Laboratories, Inc.). β-Galactosidase–positive colonies were processed to lose the bait plasmid, and prey clones were recovered by electroporation of yeast DNA in Escherichia coli and sequenced. To confirm binding, transformants were retransformed into yeast with either the bait or the vector. In addition, the inserts of the bait and the prey vector were swapped and cotransformed into yeast; identical results were obtained (unpublished data).
RT-PCR analysis of GMEB-1 mRNA transcripts
To determine the tissue distribution of GMEB-1 mRNA transcripts, commercially available mRNAs (Stratagene) were reverse transcribed using random hexamer primers and SuperScript reverse transcriptase according to the manufacturer (Stratagene). To ensure that PCR products were not amplified from contaminating genomic DNA, RNA also was incubated without SuperScript reverse transcriptase. PCR amplifications were performed essentially as described by Centner et al. (2000), using GMEB-1–specific primers designed to amplify a 511-bp product (forward primer: CGGAGGAGGGTGTAAAGAAAGACTC; reverse primer: GGGTGAGATGACTGTGAACTGAGG). PCR products were verified by sequencing.
In vitro translation and GST pull-down experiments
In vitro transcription and translation were performed in the presence of [35S]methionine (Amersham Pharmacia Biotech) using a TNT T7–coupled reticulocyte lysate system according to the manufacturer's instructions (Promega). His6–GST double-tagged fusion proteins were obtained by cloning into a modified pET9D vector. The constructs were transformed into BL21-DE3 (CLONTECH Laboratories, Inc.) cells. Whole-cell lysates in coating buffer (2× PBS, 1% Triton X-100) were prepared as previously described (Studier et al., 1990). The GST fusion protein was immobilized on glutathione–Sepharose 4B beads (Amersham Pharmacia Biotech) by incubating ∼200 μl of the lysate with 50 μl of beads (50% slurry) for 1 h at 4°C. The beads were washed three times with coating buffer and resuspended in binding buffer (20 mM Tris, pH 7.4, 100 mM KCl, 1 mM EDTA, 1% Triton X-100, plus protease inhibitors). 5 μl of in vitro–translated 35S-labeled proteins were added to 50 μl of beads coated with 20 μg of bound GST fusion proteins in 300 μl binding buffer. The mixture was incubated for 1.5 h at 4°C, washed three times with binding buffer, and resuspended in SDS sample buffer. The protein complexes were fractionated by SDS-PAGE using 15% gels. The gels were fixed (20% methanol, 10% acetic acid), stained with Coomassie blue, treated with Amplify (Amersham Pharmacia Biotech), dried, and exposed using BioMax MR-1 film (Eastman Kodak Co.). The results of the Coomassie blue staining confirmed that equal amounts of each GST fusion protein were bound to the beads in the different samples (unpublished data).
Cell culture and transfection procedures
For myocyte expression studies, cDNAs containing the entire open reading frame of MURF-1 (residues 1–565) and subfragments of MURF-1, corresponding to its 82 NH2-terminal residues, central 243 residues, and 85 COOH-terminal residues (Fig. 2), were amplified by PCR, using total human skeletal cDNA as a template. The MURF-1 cDNA sequence has been deposited in the GenBank/EMBL/DDBJ data library under accession no. AJ291713 (Centner et al., 2001). The full-length MURF-1 was cloned into pEGFP-N1 (CLONTECH Laboratories, Inc.). The primers used to generate the MURF-1 fragments were: 475S/720R for RING, 1525R/685S for RINGless, 475S/1413R for Tailless, 685S/1413R for Central, and 1370S/1525R for Tail, and the fragments were cloned into pEGFP-C1 (CLONTECH Laboratories, Inc.). Similarly, titin fragments corresponding to the MURF-1 binding site, A168–A170, the COOH-terminally located kinase domain, and M-line Ig domains were amplified from total human cDNA and cloned into pEGFP-C1. The full-length human cardiac titin accession no. in the GenBank/EMBL/DDBJ data library is X90568, and primers (listed as base pairs) used to generate the titin fragments were: 73419S/74402R for A168–170, 74254S/75345R for titin kinase, 74397S/75242R for mutated activated titin kinase, 75343S/76280R for M1-M2-M3, and 79177S/80918R for M8-M9-M10. Recombinant pEGFP-C1 constructs were purified using Qiagen columns (QIAGEN) before transfection into myocytes. Plasmids were verified by sequencing. To rule out any potential artifacts resulting from the GFP tag, pCMVmyc-MURF1 constructs, as well as constructs encoding MURF-1 with the GFP tag at its COOH-terminal end, were generated (Gregorio et al., 1998) and transfected into cardiac myocytes. Identical results were obtained (unpublished data).
Cardiac myocytes were prepared from 6-d embryonic chick hearts and cultured as described previously (Gregorio and Fowler, 1995). Isolated cells were plated in 35-mm tissue culture dishes containing 12-mm round coverslips (106 cells/dish). 15–20% of the cells in our primary cultures are fibroblasts. 24 h after plating, cultured myocytes were washed two times in OptiMEM, placed in 800 μl fresh OptiMEM, and returned to the incubator while DNA liposome complexes were prepared using LipofectAmine Plus reagents. In brief, 1 μg plasmid was mixed with 6 μl PLUS Reagent in 100 μl serum-free OptiMEM and incubated at room temperature for 15 min. Next, 4 μl of LipofectAmine reagent were added to 100 μl of OptiMEM and mixed with the DNA–PLUS reagent solution. After 15 min, the DNA–lipid complexes were added dropwise to the culture dish. 3 h later, 1 ml of minimal essential medium (10% FBS; Hyclone Laboratories, Inc.) was added to the dish. 3–6 d later, cells were gently washed with PBS and fixed with 2% formaldehyde in PBS for 10 min. Coverslips were washed and stored in PBS at 4°C until staining. Over 200 transfected cells per construct were analyzed. Our transfection efficiencies ranged from 10–40%. All tissue culture reagents (except where noted) were purchased from Life Technologies.
Indirect immunofluorescence microscopy
Primary cultures of rat cardiac myocytes were isolated and maintained as previously described (Gustafson et al., 1987). Transfected cardiac myocytes were essentially stained as described by Gregorio et al. (1998). Cells were fixed in 2% formaldehyde–PBS for 10 min, washed in PBS, and permeabilized in 0.2% Triton X-100–PBS for 15 min. The coverslips were preincubated in 2% BSA, 1% normal donkey serum–PBS for 1 h to minimize nonspecific binding of antibodies. For double-labeling protocols, cells were incubated with affinity-purified rabbit polyclonal antibodies specific to MURF-1 (5–10 μg/ml) (Centner et al., 2001), followed by Cy2-conjugated goat anti–rabbit IgG (1:600), and incubated with monoclonal sarcomeric anti–α-actinin antibodies (1:1,500) (EA-53; Sigma-Aldrich) followed by goat anti–mouse Texas red–conjugated IgG (1:600). For staining GFP-transfected cells, well-characterized antibodies were used against various sarcomeric components, including monoclonal anti-titin T11 antibodies (1:1,000; Sigma-Aldrich), monoclonal anti-myosin antibodies F59 (1:10; provided by F. Stockdale, Stanford University, Stanford, CA), rabbit anti-titin M-line–specific antibodies (1:100; Centner et al., 2001), anti-titin N2A antibodies (10 μg/ml; Centner et al., 2000), rabbit anti–MyBP-C antibodies (1:50; Linke et al., 1999), monoclonal anti-titin AB5 (1:3 of cultured supernatant; provided by John Trinick, University of Leeds, Leeds, UK) (Whiting et al., 1989), monoclonal anti-myomesin B4 antibodies (1:50 of cultured supernatant; provided by J.C. Perriard, Swiss Federal Institute of Technology, Zurich, Switzerland) (Grove et al., 1984), and Texas red–conjugated phalloidin (1:200; Molecular Probes Inc.). The staining was followed by incubation with Texas red–conjugated goat anti–mouse IgG plus IgM (1:600) or Texas red–conjugated donkey anti–rabbit antibodies (1:700) for 45 min. To identify nuclei in some experiments, fixed cells were also incubated in a DAPI stain (10 μg/ml; Sigma-Aldrich) for 5 min at room temperature before the final washing steps. Note, because of the intense diffuseness of soluble titin–GFP fusion proteins in transfected cells, we extracted soluble proteins with cytoskeletal (myofibril) stabilization buffer before fixation in some experiments (Gregorio and Fowler, 1995). For triple-labeling studies, a cascade blue–conjugated secondary antibody was used (1:200). All coverslips were mounted on slides using Aqua Poly/Mount (Polysciences, Inc.) and subsequently analyzed on a Zeiss Axiovert microscope using a 100× (NA 1.3) objective, and micrographs were recorded as digital images on a SenSys cooled HCCD (Photometrics). For triple-labeling studies, transfected cells were analyzed on a DeltaVision Deconvolution Model D-OL Olympus microscope with a 60× objective (1.4 NA) using a Photometrics Series 300 CCD camera (Applied Precision). Images were processed for presentation using Adobe Photoshop® 6.0. All secondary antibodies were purchased from Jackson ImmunoResearch Laboratories, except the Cy2-conjugated antibodies, which were purchased from Pierce Chemical Co.
Acknowledgments
The authors would like to thank Thomas Centner (European Molecular Biology Laboratory, Heidelberg, Germany) for performing the MURF-1/GMEB-1 pull-down assays, Dietmar Labeit and Christian Witt (Universitätsklinikum Mannheim) for providing antibodies, Ryan Mudry (University of Arizona) for deconvolution microscopy and assembling the figures, Adam Geach (University of Arizona) for excellent technical assistance and for preparing Fig. 2, Joseph Bahl and Yewen Wu (University of Arizona) for providing us with rat cardiac myocytes, Mary Martin and Kathleen Kunke (University of Arizona) for initiating this project, and all the members of the Gregorio laboratory for thoughtful discussions. We also acknowledge Frank White and Steven Kazmierski (University of Arizona) for assistance with the RT-PCR analysis, and Ronald Heinmark (University of Arizona) for control primers.
This work was supported by the National Institutes of Health (grants HL57461 and HL03985 to C.C. Gregorio; HL07249 to A.S. McElhinny), the American Heart Association (0120586Z to A.S. McElhinny), the Deutsche Forschungsgemeinschaft (La668/5-2 to S. Labeit), and a Core Research for Evolutional Science and Technology grant 11B-1 from the Ministry of Health and Welfare (H. Sorimachi).
Footnotes
Abbreviations used in this paper: FN, fibronectin; GFP, green fluorescent protein; GMEB-1, glucocorticoid modulatory element binding protein-1; GST, glutathione-S-transferase; Ig, immunoglobulin; ISOT-3, isopeptidase T-3; MURF-1, muscle-specific RING finger-1; MyBP-C, myosin-binding protein C; RBCC, RING finger-B-box-coiled-coil; RNF, RING finger; SMRZ, striated muscle RING zinc finger; SUMO, small ubiquitin-related modifier; Ubc9, ubiquitin-conjugating enzyme 9; Y2H, yeast two-hybrid.
References
- Antin, P.B., S. Forry-Schaudies, T.M. Friedman, S.J. Tapscott, and H. Holtzer. 1981. Taxol induces postmitotic myoblasts to assemble interdigitating microtubule-myosin arrays that exclude actin filaments. J. Cell Biol. 90:300–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bang, M.L., T. Centner, F. Fornoff, A.J. Geach, M. Gotthardt, M. McNabb, C.C. Witt, D. Labeit, C.C. Gregorio, H. Granzier, and S. Labeit. 2001. The complete gene sequence of titin, expression of an unusual approximately 700-kDa titin isoform, and its interaction with obscurin identify a novel Z-line to I-band linking system. Circ. Res. 89:1065–1072. [DOI] [PubMed] [Google Scholar]
- Bodine, S.C., E. Latres, S. Baumhuerer, V.K. Lai, L. Nunez, B.A. Clarke, W.T. Poueymirou, F.J. Panaro, E. Na, K. Dharmarajan, Z.Q. Pan, D.M. Valenzuela, T.M. DeChiara, T.N. Stirr, G.D. Yancopoulos, and D.J. Glass. 2001. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science. 294:1704–1708. [DOI] [PubMed] [Google Scholar]
- Borden, K.L. 2000. RING domains: master builders of molecular scaffolds? J. Mol. Biol. 295:1103–1112. [DOI] [PubMed] [Google Scholar]
- Centner, T., F. Fougerousse, A. Freiburg, C. Witt, J.S. Beckmann, H. Granzier, K. Trombitas, C.C. Gregorio, and S. Labeit. 2000. Molecular tools for the study of titin's differential expression. Adv. Exp. Med. Biol. 481:35–49. [DOI] [PubMed] [Google Scholar]
- Centner, T., J. Yano, E. Kimura, A.S. McElhinny, K. Pelin, C.C. Witt, M.L. Bang, K. Trombitas, H. Granzier, C.C. Gregorio, et al. 2001. Identification of muscle specific ring finger proteins as potential regulators of the titin kinase domain. J. Mol. Biol. 306:717–726. [DOI] [PubMed] [Google Scholar]
- Dai, K.S., and C.C. Liew. 2001. A novel human striated muscle RING zinc finger protein, SMRZ, interacts with SMT3b via its RING domain. J. Biol. Chem. 276:23992–23999. [DOI] [PubMed] [Google Scholar]
- Desterro, J.M., J. Thomson, and R.T. Hay. 1997. Ubch9 conjugates SUMO but not ubiquitin. FEBS Lett. 417:297–300. [DOI] [PubMed] [Google Scholar]
- Ehler, E., B.M. Rothen, S.P. Hammerle, M. Komiyama, and J.C. Perriard. 1999. Myofibrillogenesis in the developing chicken heart: assembly of Z-disk, M-line and the thick filaments. J. Cell Sci. 112:1529–1539. [DOI] [PubMed] [Google Scholar]
- Epstein, H.F., and D.A. Fischman. 1991. Molecular analysis of protein assembly in muscle development. Science. 251:1039–1044. [DOI] [PubMed] [Google Scholar]
- Freemont, P.S. 2000. RING for destruction? Curr. Biol. 10:84–87. [DOI] [PubMed] [Google Scholar]
- Funatsu, T., E. Kono, H. Higuchi, S. Kimura, S. Ishiwata, T. Yoshioka, K. Maruyama, and S. Tsukita. 1993. Elastic filaments in situ in cardiac muscle: deep-etch replica analysis in combination with selective removal of actin and myosin filaments. J. Cell Biol. 120:711–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fürst, D.O., M. Osborn, and K. Weber. 1989. Myogenesis in the mouse embryo: differential onset of expression of myogenic proteins and the involvement of titin in myofibril assembly. J. Cell Biol. 109:517–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granzier, H., M. Kellermayer, M. Helmes, and K. Trombitas. 1997. Titin elasticity and mechanism of passive force development in rat cardiac myocytes probed by thin-filament extraction. Biophys. J. 73:2043–2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregorio, C.C., and P.B. Antin. 2000. To the heart of myofibril assembly. Trends Cell Biol. 10:355–362. [DOI] [PubMed] [Google Scholar]
- Gregorio, C.C., and V.M. Fowler. 1995. Mechanisms of thin filament assembly in embryonic chick cardiac myocytes: tropomodulin requires tropomyosin for assembly. J. Cell Biol. 129:683–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregorio, C.C., K. Trombitas, T. Centner, B. Kolmerer, G. Stier, K. Kunke, K. Suzuki, F. Obermayr, B. Herrmann, H. Granzier, et al. 1998. The NH2 terminus of titin spans the Z-disc: its interaction with a novel 19-kD ligand (T-cap) is required for sarcomeric integrity. J. Cell Biol. 143:1013–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregorio, C.C., H. Granzier, H. Sorimachi, and S. Labeit. 1999. Muscle assembly: a titanic achievement? Curr. Opin. Cell Biol. 11:18–25. [DOI] [PubMed] [Google Scholar]
- Grove, B.K., V. Kurer, C. Lehner, T.C. Doetschman, J.C. Perriard, and H.M. Eppenberger. 1984. A new 185,000-dalton skeletal muscle protein detected by monoclonal antibodies. J. Cell Biol. 98:518–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafson, T.A., J.J. Bahl, B.E. Markham, W.R. Roeske, and E. Morkin. 1987. Hormonal regulation of myosin heavy chain and alpha-actin gene expression in cultured fetal rat heart myocytes. J. Biol. Chem. 262:13316–13322. [PubMed] [Google Scholar]
- Holtzer, H., T. Hijikata, Z.X. Lin, Z.Q. Zhang, S. Holtzer, F. Protasi, C. Franzini-Armstrong, and H.L. Sweeney. 1997. Independent assembly of 1.6m long bipolar MHC filaments and I-Z-I bodies. Cell Struc. Func. 22:83–93. [DOI] [PubMed] [Google Scholar]
- Houmeida, A., J. Holt, L. Tskhovrebova, and J. Trinick. 1995. Studies of the interaction between titin and myosin. J. Cell Biol. 131:1471–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez-Lara, A.M., M.J. Heine, and H. Gronemeyer. 2000. Cloning of a mouse glucocorticoid modulatory element binding protein, a new member of the KDWK family. FEBS Lett. 468:203–210. [DOI] [PubMed] [Google Scholar]
- Jin, J.-P. 1995. Cloned rat cardiac titin class I and class II motifs. J. Biol. Chem. 270:6908–6916. [PubMed] [Google Scholar]
- Johnson, E.S., I. Schwienhorst, R.J. Dohmen, and G. Blobel. 1997. The ubiquitin-like protein Smt3p is activated for conjugation to other proteins by an Aos1p/Uba2p heterodimer. EMBO J. 16:5509–5519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komiyama, M., K. Kouchi, K. Maruyama, and Y. Shimada. 1993. Dynamics of actin and assembly of connectin (titin) during myofibrillogenesis in embryonic chick cardiac muscle cells in vitro. Dev. Dyn. 196:291–299. [DOI] [PubMed] [Google Scholar]
- Labeit, S., and B. Kolmerer. 1995. Titins, giant proteins in charge of muscle ultrastructure and elasticity. Science. 270:293–296. [DOI] [PubMed] [Google Scholar]
- Labeit, S., M. Gautel, A. Lakey, and J. Trinick. 1992. Towards a molecular understanding of titin. EMBO J. 11:1711–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labeit, S., B. Kolmerer, and W.A. Linke. 1997. The giant protein titin: Emerging roles in physiology and pathophysiology. Circ. Res. 80:290–294. [DOI] [PubMed] [Google Scholar]
- Linke, W.A., M. Ivemeyer, S. Labeit, H. Hinssen, J.C. Ruegg, and M. Gautel. 1997. Actin-titin interaction in cardiac myofibrils: probing a physiological role. Biophys. J. 73:905–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linke, W.A., D.E. Rudy, T. Centner, M. Gautel, C. Witt, S. Labeit, and C.C. Gregorio. 1999. I-band titin in cardiac muscle is a three-element molecular spring and is critical for maintaining thin filament structure. J. Cell Biol. 146:631–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruyama, K., R. Matsubara, Y. Natori, S. Nonomura, S. Kimura, K. Ohashi, F. Murakami, S. Handa, and G. Eguchi. 1977. Connectin, an elastic protein of muscle. J. Biochem. (Tokyo). 82:317–337. [PubMed] [Google Scholar]
- McElhinny, A.S., S. Labeit, and C.C. Gregorio. 2000. Probing the functional roles of titin ligands in cardiac myofibril assembly and maintenance. Adv. Exp. Med. Biol. 481:67–86. [DOI] [PubMed] [Google Scholar]
- Melchior, F. 2000. SUMO—nonclassical ubiquitin. Annu. Rev. Cell Dev. Biol. 16:591–626. [DOI] [PubMed] [Google Scholar]
- Muller, S., C. Hoege, G. Pyrowolakis, and S. Jentsch. 2001. SUMO, ubiquitin's mysterious cousin. Nat. Rev. Mol. Cell Biol. 2202–2210. [DOI] [PubMed]
- Mues, A., P.F. van der Ven, P. Young, D.O. Furst, and M. Gautel. 1998. Two immunoglobulin-like domains of the Z-disc portion of titin interact in a conformation-dependent way with telethonin. FEBS Lett. 428:111–114. [DOI] [PubMed] [Google Scholar]
- Obermann, W.M., M. Gautel, F. Steiner, P.F. van der Ven, K. Weber, and D.O. Fürst. 1996. The structure of the sarcomeric M band: localization of defined domains of myomesin, M protein, and the 250-kD carboxy terminal region of titin by immunoelectron microscopy. J. Cell Biol. 134:1441–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obermann, W.M., M. Gautel, K. Weber, and D.O. Fürst. 1997. Molecular structure of the sarcomeric M band: mapping of titin and myosin binding domains in myomesin and the identification of a potential regulatory phosphorylation site in myomesin. EMBO J. 16:211–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudy, D.E., T.A. Yatskievych, P.B. Antin, and C.C. Gregorio. 2001. Assembly of thick, thin, and titin filaments in chick precardiac explants. Dev. Dyn. 221:61–71. [DOI] [PubMed] [Google Scholar]
- Saiki, R.K., S.J. Scharf, F. Faloona, G.T. Mullis, and H.A. Erlich. 1985. Enzymatic amplification of beta-globin genomic sequences and restriction site analysis for diagnosis of sickle cell anemia. Science. 230:1350–1354. [DOI] [PubMed] [Google Scholar]
- Saitoh, H., and J. Hinchey. 2000. Functional heterogeneity of small ubiquitin-related protein modifiers SUMO-1 versus SUMO-2/3. J. Biol. Chem. 275:6252–6258. [DOI] [PubMed] [Google Scholar]
- Sanger, J.W., and J.M. Sanger. 2001. Fishing out proteins that bind to titin. J. Cell Biol. 154:21–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultheiss, T., Z. Lin, M.-H. Lu, J. Murray, D.A. Fischman, K. Weber, M. Masaki, M. Imamura, and H. Holtzer. 1990. Differential distribution of subsets of myofibrillar proteins in cardiac nonstriated and striated myofibrils. J. Cell Biol. 110:1159–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorimachi, H., A. Freiburg, B. Kolmerer, S. Ishiura, G. Stier, C.C. Gregorio, D. Labeit, W.A. Linke, K. Suzuki, and S. Labeit. 1997. Tissue-specific expression and alpha-actinin binding properties of the Z-disc titin: implications for the nature of vertebrate Z-discs. J. Mol. Biol. 270:688–695. [DOI] [PubMed] [Google Scholar]
- Spencer, J.A., S. Eliazer, R.L. Ilaria, Jr., J.A. Richardson, and E.N. Olson. 2000. Regulation of microtubule dynamics and myogenic differentiation by MURF, a striated muscle RING-finger protein. J. Cell Biol. 150:771–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studier, F.W., A.H. Rosenberg, J.J. Dunn, and J.W. Dubendorff. 1990. Use of T7 RNA polymerase to direct expression of cloned genes. Methods Enzymol. 185:60–89. [DOI] [PubMed] [Google Scholar]
- Tatham, M.H., E. Jaffray, O.A. Vaughan, J.M. Desterro, C. Botting, J.H. Naismith, and R.T. Hay. 2001. Polymeric chains of SUMO-2 and SUMO-3 are conjugated to protein substrates by SAE1/SAE2 and Ubc9. J. Biol. Chem. 276:35368–35374. [DOI] [PubMed] [Google Scholar]
- Theriault, J.R., S.J. Charette, H. Lambert, and J. Landry. 1999. Cloning and characterization of hGMEB1, a novel glucocorticoid modulatory element binding protein. FEBS Lett. 452:170–176. [DOI] [PubMed] [Google Scholar]
- Trinick, J. 1994. Titin and nebulin protein rulers in muscle? Trends Biochem. Sci. 19:405–408. [DOI] [PubMed] [Google Scholar]
- Trinick, J., and L. Tskhovrebova. 1999. Titin: a molecular control freak. Trends Cell Biol. 9:377–380. [DOI] [PubMed] [Google Scholar]
- van der Loop, F.T., P.F. van der Ven, D.O. Furst, M. Gautel, G.J. Van Eys, and F.C. Ramaekers. 1996. Integration of titin into the sarcomeres of cultured differentiating human skeletal muscle cells. Eur. J. Cell Biol. 69:301–307. [PubMed] [Google Scholar]
- Wang, K., J. McClure, and A. Tu. 1979. Titin: major myofibrillar components of striated muscle. Proc. Natl. Acad. Sci. USA. 76:3698–3702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiting, A., J. Wardale, and J. Trinick. 1989. Does titin regulate the length of muscle thick filaments? J. Mol. Biol. 205:263–268. [DOI] [PubMed] [Google Scholar]
- Young, P., C. Ferguson, S. Banuelos, and M. Gautel. 1998. Molecular structure of the sarcomeric Z-disk: two types of titin interactions lead to an asymmetrical sorting of alpha-actinin. EMBO J. 17:1614–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng, H., D.A. Jackson, H. Oshima, and S.S. Simons, Jr. 1998. Cloning and characterization of a novel binding factor (GMEB-2) of the glucocorticoid modulatory element. J. Biol. Chem. 273:17756–17762. [DOI] [PubMed] [Google Scholar]
- Zeng, H., S. Kaul, and S.S. Simons, Jr. 2000. a. Genomic organization of human GMEB-1 and rat GMEB-2: structural conservation of two multifunctional proteins. Nucleic Acids Res. 28:1819–1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng, H., S.Y. Plisov, and S.S. Simons, Jr. 2000. b. Ability of the glucocorticoid modulatory element to modify glucocorticoid receptor transactivation indicates parallel pathways for the expression of glucocorticoid modulatory element and glucocorticoid response element activities. Mol. Cell. Endocrinol. 162:221–234. [DOI] [PubMed] [Google Scholar]