

Abstract
Întegrins, matrix metalloproteases (MMPs), and the cytokine TGF-β have each been implicated in homeostatic cell behaviors such as cell growth and matrix remodeling. TGF-β exists mainly in a latent state, and a major point of homeostatic control is the activation of TGF-β. Because the latent domain of TGF-β1 possesses an integrin binding motif (RGD), integrins have the potential to sequester latent TGF-β (SLC) to the cell surface where TGF-β activation could be locally controlled. Here, we show that SLC binds to αvβ8, an integrin expressed by normal epithelial and neuronal cells in vivo. This binding results in the membrane type 1 (MT1)-MMP–dependent release of active TGF-β, which leads to autocrine and paracrine effects on cell growth and matrix production. These data elucidate a novel mechanism of cellular homeostasis achieved through the coordination of the activities of members of three major gene families involved in cell–matrix interactions.
Keywords: integrins; transforming growth factor β; metalloprotease; cell cycle; homeostasis
Introduction
Cellular homeostasis is maintained in the organism through the correct responses to extra-, intra-, and intercellular signals (Potter, 1974). Imbalances in these signals can result in disruption of cellular homeostasis, leading to changes in cell mass and/or tissue organization. The cellular homeostatic machinery consists of secreted, cell surface, and intracellular molecules that together maintain cellular differentiation and the balance between quiescence and entry into the cell cycle (Lord, 1988). Homeostasis is regulated through the control of cell proliferation mediated through cell–extracellular matrix interactions in concert with growth-promoting and inhibitory cytokines (Giancotti, 1997; Schwartz, 1997). A growth inhibitory cytokine of particular importance in tissue homeostasis is the multifunctional cytokine TGF-β (Lord, 1988). The important role of TGF-β in homeostasis is illustrated by the fact that TGF-β1–deficient mice develop epithelial hyperplasias (in addition to lethal multiorgan inflammation) within weeks after birth (Shull et al., 1992; Crawford et al., 1998).
TGF-β1 is normally maintained in a latent or inactive state by the noncovalent association of the bioactive peptide of TGF-β1 with its NH2-terminal propeptide, latency-associated peptide (LAP)*-β1 (Munger et al., 1997). Therefore, normal TGF-β function is thought to be largely controlled by its activation from the latent state, a process that is not understood completely (Munger et al., 1999). However, recent evidence suggests that cell surface molecules or secreted extracellular molecules can activate TGF-β. Specifically, the integrin αvβ6 and the secreted extracellular matrix molecule thrombospondin (TSP)-1 have been implicated in activation of TGF-β1 through nonproteolytic mechanisms (Crawford et al., 1998; Munger et al., 1999). In addition, plasmin or the cell surface localization of matrix metalloprotease MMP-9 by CD44 has been proposed to lead to activation of TGF-β through proteolytic degradation of LAP-β1 and LAP-β2, respectively (Lyons et al., 1990; Yu and Stamenkovic, 2000). Although these mechanisms may be important to activation of TGF-β, particularly in response to injury (Jirtle et al., 1991; Munger et al., 1999; Murphy-Ullrich and Poczatek, 2000) or during neoplastic progression (Yu and Stamenkovic, 2000), they individually do not explain the activation of TGF-β1 in normal tissues. Indeed, mice deficient in TSP-1 (Crawford et al., 1998), plasminogen (Bugge et al., 1995), CD44 (Protin et al., 1999), or αvβ6 (Munger et al., 1999) are all born viable and are able to reproduce, in marked contrast to the uniform lethality of TGF-β1–null mice (Shull et al., 1992).
The propeptide of TGF-β1, LAP-β1, contains an RGD motif that is recognized by a subset of integrins sharing in common the αv integrin subunit (Munger et al., 1998). Thus, three of the five αv integrins, αvβ1, αvβ5, and αvβ6, have been shown to bind to LAP-β1, and of these only αvβ6 can mediate TGF-β activation (Munger et al., 1998, 1999). Recently, evidence suggests that αvβ6-mediated activation of TGF-β1 plays an important role in response to injury (Munger et al., 1999). Of the two remaining αv integrins, αvβ3 does not bind to LAP-β1 or mediate activation of TGF-β1 (Munger et al., 1998), and αvβ8 has not been investigated since, until recently, a system has not been available to study its function (Cambier et al., 2000). The αvβ8 integrin is of particular interest, since it has been identified recently as an epithelial growth inhibitory molecule (Cambier et al., 2000). αvβ8 is expressed in the normal human airway epithelium but is lost in its malignant counterpart, suggesting a role in epithelial homeostasis (Cambier et al., 2000). Furthermore, heterologous expression of αvβ8 inhibits lung carcinoma cell growth both in vivo and in vitro (Cambier et al., 2000). Since αvβ8 and TGF-β1 are coexpressed in normal tissues, such as the human airway (Crawford et al., 1998; Cambier et al., 2000), we considered the possibility that αvβ8 may participate in the maintenance of airway homeostasis through activation of TGF-β.
In this article, we demonstrate a novel mechanism of cell growth regulation mediated by activation of TGF-β1 via the integrin αvβ8. We show that αvβ8 can bind LAP-β1 and that the consequence of this interaction is activation of TGF-β1. This mechanism of activation of TGF-β1 differs from other reported mechanisms because it is regulated through the coordinated interactions of integrins, TGF-β, and MMPs on the cell surface. Furthermore, we show that when lung cancer cells are reconstituted with αvβ8, which is normally present on the epithelial cells from which they are derived, growth is now inhibited by TGF-β1. These data provide novel insights into the mechanisms underlying cellular homeostasis.
Results
The integrin αvβ8 binds with high affinity to the RGD site of recombinant LAP-β1
To determine if αvβ8 can bind to the RGD-containing LAP-β1, we performed affinity chromatography using surface radiolabeled αvβ8 and immobilized LAP-β1 (Fig. 1 a). Two bands of the appropriate kD for the αv and β8 subunits (150 and 90 kD, respectively) bound to immobilized LAP-β1 in a divalent cation-dependent fashion as shown by elution with EDTA (Fig. 1 a, lanes 4–8). This binding was also dependent on the RGD sequence of LAP-β1 as demonstrated by the inability of RGE (Fig. 1 a, lanes 1′–3′) and the ability of RGD to elute αvβ8 (Fig. 1 a, lanes 4′–7′). The identity of the αv and β8 subunits in the elution fractions was confirmed by immunoprecipitation (Fig. 1 b). Anti-β8 antibodies immunoprecipitated 150- and 90-kD bands from the RGD and EDTA elution fractions, which comigrated exactly with the two bands immunoprecipitated with anti-αv and anti-β8 antibodies from cell lysates (Fig. 1 b, RGD, EDTA, and LYSATE). Immunoprecipitations of the elution fractions using antibodies to the other RGD binding integrins present on HT1080 cells, namely αvβ5 and α5β1, failed to detect any proteins (unpublished data). To measure the relative ability of truncated αvβ8 to bind to LAP-β1 and vitronectin (VN), the only other known ligand for αvβ8 (Nishimura et al., 1994), we developed solid phase binding assays for αvβ8–ligand interactions using alkaline phosphatase (AP)-tagged αvβ8 (AP-αvβ8). The ability of AP-αvβ8 to bind to immobilized LAP-β1 was confirmed by metabolic labeling and affinity chromatography (Fig. 1 c). Two bands of the appropriate molecular weight for the truncated αv (140 kD) and β8-AP (130 kD) subunits were eluted by RGD peptide (Fig. 1 c). No other metabolically labeled proteins from the cell supernatants were shown to bind to LAP-β1 by this method (Fig. 1 c). In solid phase assays, considerably more AP-αvβ8 bound to wells coated with LAP-β1 (10 μg/ml) than to wells coated with VN (100 μg/ml) (Fig. 1 d). AP-αvβ8 did not bind to a mutant form of LAP (LAP [RGE]) with a single amino acid substitution in the integrin recognition sequence (Fig. 1 d) and was completely eluted by either RGD (1 mg/ml) or EDTA (10 mM) (unpublished data). Furthermore, αvβ8 binding was almost completely inhibited by the monoclonal antibody 37E1, specific to β8 (Fig. 1 d). The affinity of AP-αvβ8 for LAP-β1–Sepharose was determined by saturation binding experiments. AP-αvβ8 bound to LAP-β1 with high affinity (Kd 13 pM) with binding saturation reached at ∼50% ligand occupancy (Fig. 1 e, Bmax: 0.5 fM/1.0 fM LAP-β1). Scatchard analysis revealed a single high affinity state of AP-αvβ8, consistent with other reports that secreted αv integrins are expressed in a constitutively active affinity state (Nishimura et al., 1994; Weinacker et al., 1994). In contrast to the high affinity of AP-αvβ8 for LAP-β1, the affinity of AP-αvβ8 for VN, the only other known ligand for αvβ8 (Nishimura et al., 1994), was too low to be determined using this assay (unpublished data). Thus, αvβ8 binds preferentially to LAP-β1 with high affinity, and this binding is both RGD and divalent cation dependent.
Figure 1.
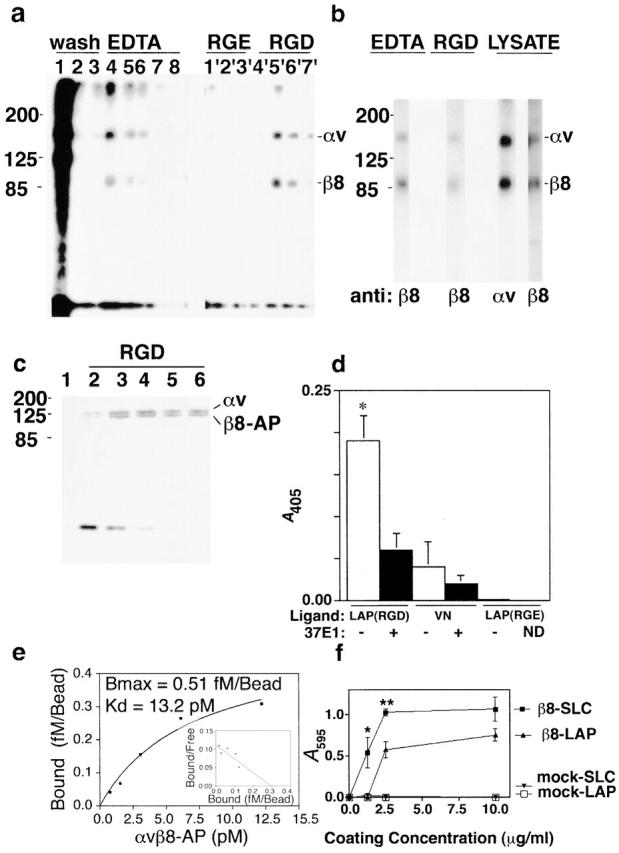
αvβ8 binds to LAP-β1 in an RGD- and cation-dependent fashion. (a) n-octylglucoside lysates from 125I-cell surface-labeled β8-expressing HT1080 cells (1 ml) were passed over two identical LAP-β1–Sepharose columns (1 ml) and washed with 12 ml of wash buffer (shown in lanes 1, 2, and 3 are the 4th, 11th and 12th wash fractions). One column was eluted with 1-ml fractions containing 10 mM EDTA (fractions 4–8), and the other column was eluted with 1-ml fractions containing 1 mg/ml GRGESNK (lanes 1'–3') or 1 mg/ml GRGDSNK (lanes 4'–7'). Samples were resolved by 7.5% SDS-PAGE under nonreducing conditions and visualized by autoradiography. (b) EDTA and RGD elution fractions were immunoprecipitated with anti-β8 (14E5) and compared with anti-αv (L230) and anti-β8 (14E5) immunoprecipitations from cell lysates. The migration of the MW markers is shown on the left, and the expected migration of the αv (150 kD) and β8 (90 kD) subunits is shown on the right. Samples were resolved by 7.5% SDS-PAGE under nonreducing conditions and visualized by autoradiography. (c) 35S metabolically labeled, (Translabel and ICN Biomedicals) truncated secreted αvβ8-AP fusion protein was applied to a 0.5-ml column of LAP-β1–Sepharose, washed sequentially with six fractions of wash buffer (lane 1, last wash fraction), and then eluted with 1 mg/ml GRGDSPK (lanes 2–6). On the left are the migrations of the MW markers, and on the right are the expected migrations of the truncated αv (140 kD) and the truncated β8-AP (130 kD) subunits. Samples were resolved by 10% SDS-PAGE under nonreducing conditions and visualized by autoradiography. (d) Supernatant containing secreted truncated αvβ8 with a COOH-terminal AP tag (AP-αvβ8) was applied to wells of a 96-well plate coated with either LAP-β1 (10 μg/ml) containing the RGD or the RGE binding motif or with VN (100 μg/ml) in the presence or absence of an anti-β8–blocking monoclonal antibody, 37E1. Specific binding was determined colorimetrically. An asterisk indicates increased binding of receptor to LAP (RGD) compared with antibody-treated or LAP (RGE) controls (p < 0.001). (e) Binding affinity of αvβ8 for LAP-β1 was determined using concentrated AP-αvβ8 and LAP-β1–Sepharose (1fM LAP-β1/bead). Receptor concentration was determined using purified placental AP (Applied Biosystems) as a standard. Dilutions of AP-β8 were incubated under equilibrium-binding conditions (overnight at 4°C) with 10 μl LAP-β1–Sepharose. Bound receptor was determined by luminescence using a CSPD substrate (Tropix; Applied Biosystems). (f) Adhesion of β8-expressing versus mock-transduced HT1080 cells to LAP-β1 (LAP) and SLC-coated wells of a 96-well plate. Cells (5 × 104/well) were applied to each well, and after incubation for 1 h at 37°C unbound cells were removed by centrifugation. Absorbance (A 595) after staining with Crystal violet is shown on the right. *p < 0.05; **p < 0.01.
To determine if αvβ8 expressed on the cell surface could bind latent TGF-β (SLC) under physiologic conditions, we performed cell adhesion assays using both SLC and LAP-β1 as immobilized ligands. β8-expressing HT1080 cells bound significantly better to SLC than to LAP-β1 at the 1.0 and 2.5 μg/ml coating concentrations, whereas mock-transduced HT1080 cells did not bind at all (Fig. 1 f). The reason for the increase in the αvβ8-dependent cell adhesion to SLC compared with LAP-β1 is unclear. However, it is likely that the presence of the mature TGF-β1 peptide in SLC results in conformational differences between SLC and LAP-β1, which could affect either receptor binding, stability, or coating efficiency. Thus, we show that SLC is the first and only known ligand capable of supporting stable αvβ8-mediated adhesion, since VN, the only other known αvβ8 ligand, does not support stable αvβ8-mediated adhesion (Nishimura et al., 1994).
The integrin αvβ8 mediates activation of SLC
To determine the functional consequence of LAP–αvβ8 interactions, we assessed the ability of αvβ8 to activate the endogenous SLC present in coculture systems. These systems consisted of β8-expressing or mock-transduced cells cocultured with reporter cell lines (TMLC [Abe et al., 1994] or HepG2-[SBE]4-Lux [Jonk et al., 1998]) responsive to active TGF-β. We found that the TMLC reporter cell system was a more specific bioassay system for TGF-β activity than the HepG2- (SBE)4-Lux system and was therefore used for most of these studies. The TMLC system consists of mink lung epithelial cells stably transfected with a TGF-β responsive fragment of the plasminogen activator inhibitor-1 promoter driving the luciferase gene (Abe et al., 1994). TMLC cells are highly responsive to TGF-β and produce a very low background of TGF-β activation. TMLC cells can thus be used in coculture with other cell lines or cell-free fractions to test for the presence of active TGF-β using luminescence as a readout.
In the HT1080, SW480, and H647 cell lines, heterologous expression of β8 had either no effect or a slight effect on the cell surface expression of the other integrin β subunits known to interact with the RGD motif (Table I). The only significant differences were a reduction of surface expression of the β5 subunit in β8-transduced compared with mock-transduced HT1080 and SW480 cells. It is possible that these slight reductions in surface expression of αvβ5- on β8-expressing HT1080 and SW480 cells could potentially reduce the magnitude of the β8 effect on adhesion to LAP-β1 or influence the activation of TGF-β. However, this is unlikely, since the αvβ5 integrin binds very weakly and does not mediate adhesion to SLC (Fig. 1 f, mock).
Table I.
Expression of integrin subunits in cell lines used in Fig. 2
| Mean fluorescence intensity (±SE) staining | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Cell line | Vector | β8 | β1 | β3 | β5 | β6 | αv | α5 | PBS |
| HT1080 | β8 | 124 ± 5*** | 1776 ± 121 | 23 ± 1 | 175 ± 4** | 8 ± 1 | 523 ± 49 | 40 ± 4 | 4 ± 0 |
| Mock | 10 ± 4 | 1372 ± 424 | 18 ± 3 | 251 ± 13 | 8 ± 1 | 427 ± 21 | 68 ± 32 | 5 ± 1 | |
| SW480 | β8 | 91 ± 13*** | 504 ± 139 | 7 ± 1 | 119 ± 17* | 7 ± 2 | 473 ± 45 | 53 ± 27 | 6 ± 1 |
| Mock | 6 ± 1 | 386 ± 109 | 25 ± 3 | 236 ± 205 | 12 ± 5 | 757 ± 70 | 25 ± 1 | 5 ± 1 | |
| H647 | β8 | 100 ± 5*** | 580 ± 52 | 11 ± 3 | 56 ± 22 | 60 ± 14 | 561 ± 35 | ND | 9 ± 4 |
| Mock | 6 ± 1 | 671 ± 106 | 9 ± 1 | 46 ± 35 | 62 ± 29 | 283 ± 144 | ND | 9 ± 3 | |
| TMLC | β8 | 30a | NDb | NDb | NDb | NDb | NDb | NDb | 4a |
| Mock | 5a | NDb | NDb | NDb | NDb | NDb | NDb | 4a | |
Representative experiment.
ND due to lack of cross reacting antibodies.
ND, not done. ***p < 0.001; **p < 0.01; *p < 0.05.
Heterologous expression of αvβ8 mediated activation of TGF-β in HT1080, SW480, and MvLu cells as determined by coculture with either TMLC (Fig. 2, a–d) or HepG2-(SBE)4-Lux reporter cells (unpublished data). TGF-β activation was specifically mediated by αvβ8, since it was substantially inhibited by the anti-β8 antibody, 37E1 (Fig. 2, a–c). The other RGD binding integrin heterodimers normally present in SW480, HT1080, and MvLu cells (Table I) did not mediate significant activation of TGF-β because mock-transduced cells did not activate TGF-β (Fig. 2, a–c). However, H647 cells, which normally express αvβ6 (Table I), activated TGF-β (note the higher activation in mock H647 cells than in other cell lines in Fig. 2 d). This activation in mock-transduced H647 cells could be completely inhibited by antibodies to β6; activation in β8-expressing H647 cells could be completely inhibited by a combination of β8 and β6 antibodies (unpublished data). These data demonstrate that when β8 and β6 are coexpressed the resulting TGF-β activation is additive. The TGF-β isoform that was primarily responsible for TGF-β activation in our system was TGF-β1, since TGF-β1 isoform-specific antibodies inhibited 80–90% of the activation in SW480, HT1080, MvLu, and H647 cells, whereas TGF-β2 and TGF-β3 isoform-specific antibodies had minimal effect (unpublished data).
Figure 2.

Cell surface expression of αvβ8 mediates activation of TGF-β. HT1080 (a), MvLu (b), SW480 (c), and H647 (d) cells either β8-transduced or mock-transduced were cocultured with TMLC reporter cells in the presence or absence of a neutralizing anti-β8 antibody (37E1) or pan–TGF-β neutralizing antibody (1D11). Relative luciferase units represent arbitrary units minus the TMLC background. Asterisks indicate increased luciferase activity of untreated β8-expressing cells compared with antibody-treated or mock controls. p < 0.001.
Evidence that the β8-cytoplasmic domain is not required for αvβ8-mediated activation of TGF-β
The mechanism of integrin αvβ6-mediated activation of TGF-β is likely to depend on the transduction of mechanical forces to induce conformational changes of SLC (Munger et al., 1999). Thus, αvβ6-mediated activation of TGF-β is critically dependent on specific sequences within the β6 cytoplasmic domain (Munger et al., 1999). However, the β8 cytoplasmic domain has no similarity with the cytoplasmic domain of β6 or any other integrin β subunit (Moyle et al., 1991). We have shown previously that the β8 cytoplasmic domain is incapable of linking to the cytoskeleton to stabilize cell adhesion (Nishimura et al., 1994; Cambier et al., 2000). Therefore, we sought to determine whether the β8 cytoplasmic domain would influence interactions with LAP-β1. We expressed and tested a series of β8 cytoplasmic deletion mutants (Fig. 3, a–c) for their ability to mediate adhesion to LAP-β1 (Fig. 3 d) and to activate TGF-β (Fig. 3, e and f). The complete (TM) cytoplasmic deletion mutant was expressed at sixfold lower surface levels than the partial (759) cytoplasmic deletion mutant or the wild-type (FL) subunit (Fig. 3 c). Low levels of surface expression of the TM mutant could be due to a decreased ability to associate with the αv subunit, alterations in intracellular transport, or increased degradation. SW480 cells expressing sixfold lower surface levels of the TM mutant compared with SW480 cells expressing the 759 mutant or the FL subunit adhered only slightly less well to LAP-β1 (TM adhesion saturation reached at 5 compared with 2.5 μg/ml coating concentration for 759 and FL) (Fig. 3 d). Unlike β6-transduced SW480 cells (Munger et al., 1999), SW480 cells transduced with full-length or mutant forms of β8 failed to spread appreciably on LAP-β1 (unpublished data).
Figure 3.

The cytoplasmic domain of β8 is not required for cell adhesion to LAP-β1 or activation of TGF-β. (a) Construction of β8 subunit cytoplasmic truncation mutants. The full-length β8 (FL) subunit, a partial truncation mutant missing the COOH-terminal 11 amino acids (759), and a complete truncation mutant missing the complete β8 cytoplasmic domain (TM) were assembled by PCR mutagenesis and subcloned into retroviral vectors. (b) Immunoprecipitation analysis of surface-labeled SW480 cells, expressing FL, 759, TM, or retroviral backbone (mock) using an anti-β8 monoclonal antibody (37E1). The results demonstrate the presence and dimerization with the αv subunit on the cell surface and absence of the cytoplasmic domain in the TM construct. Biotinylated proteins were detected by Western blotting. Note that 37E1 is specific to αvβ8 because the two immunoprecipitated bands, corresponding to the αv subunit or the β8 subunit, were not seen in mock-transduced cells. Also, note that the TM construct was expressed at lower levels on the cell surface compared with 759 and FL. To determine the absence of the intracellular epitope in TM-expressing cells, cell lysates were immunoprecipitated with 37E1 and analyzed by Western blotting using a polyclonal anti-β8 antibody directed against the entire β8 cytoplasmic domain. In b (bottom), note that no signal for β8 is seen in the β8 immunoprecipitates of the truncation mutant (TM), indicating absence of the β8 cytoplasmic domain. (c) FACS® of cytoplasmic deletion mutants (TM and 759) versus the wild type (FL) β8 subunit expressed in SW480 cells. Note the TM mutant is expressed at sixfold lower levels than the 759 or FL constructs. Histograms using arbitrary fluorescence units are shown. (d) Adhesion assays of SW480 cells expressing β8 truncation mutants demonstrate that the cytoplasmic domain of β8 is not required for adhesion to LAP-β1. Note that despite lower levels of surface expression of the TM construct, all constructs bound well to LAP-β1, whereas mock-transduced SW480 cells do not adhere to LAP-β1. (e-f) Demonstration that the β8 cytoplasmic domain is not required for activation of TGF-β. SW480 or HT1080 cells expressing the wild-type or truncation mutants were cocultured with TMLC reporter cells in the presence or absence of anti-β8 (37E1) or pan anti–TGF-β (1D11). Relative luciferase units are shown. Single and double asterisks indicate increased luciferase activity of untreated wild-type or mutant β8-expressing cells compared with antibody-treated or mock controls (*p < 0.01; **p < 0.001).
SW480 and HT1080 cells expressing the FL subunit and the TM and 759 mutants also mediated significant activation of TGF-β compared with mock-transduced cells (Fig. 3, d and e). Concordant with sixfold lower levels of surface expression, SW480 cells expressing the TM mutant did not activate TGF-β1 as well as cells expressing either 759 mutant or the FL subunit (Fig. 3 b). However, cells expressing the TM mutant adhered to LAP-β1 and activated TGF-β only twofold less than the 759 or FL constructs (Figs. 3, c–e). This discordance may be due to dependence on the ligand concentration rather than the receptor number in these assays (e.g., saturation). Alternatively, it is possible that the cytoplasmic domain of β8 actually plays a negative regulatory role in αvβ8–TGF-β interactions. This latter possibility is consistent with the hypothesis that the divergent cytoplasmic domain of β8 plays a general inhibitory role (Cambier et al., 2000).
Together these findings suggest that the cytoplasmic domain of β8 is not required for either adhesion to LAP-β1 or activation of TGF-β. Thus, it is likely that the mechanism of αvβ8-mediated activation of TGF-β1 is distinct from the mechanosignal transduction mechanism described for αvβ6 (Munger et al., 1999).
αvβ8-mediated activation of TGF-β requires localization to the cell surface and metalloprotease activity
The fact that the cytoplasmic domain of β8 is not required for activation of TGF-β1 suggests that αvβ8-mediated activation of TGF-β might be regulated extracellularly either in the extracellular space or on the cell surface. To test these possibilities, we first tested the ability of soluble secreted αvβ8 to activate TGF-β. We found no evidence, using a variety of receptor preparations (supernatant containing secreted αvβ8 or lectin- or antibody-purified receptor), that soluble secreted αvβ8 could activate TGF-β (with supernatant containing secreted αvβ8 or media control; relative luciferase units: αvβ8, 7.8 ± 0.1; media control, 11.8 ± 1.2, p > 0.05). This suggests that cell surface localization of αvβ8 is required for TGF-β activation.
Because proteolytic cleavage is a common mechanism of regulating cytokine activity, we tested the ability of protease inhibitors to block αvβ8-mediated activation of TGF-β. GM6001, a member of the hydroxamate class of protease inhibitors specific to metalloproteases, but not a control peptide lacking the metal-binding hydroxamate modification (C1006), significantly blocked αvβ8-mediated TGF-β activation in SW480 (Fig. 4 a) and HT1080 cells (59.0 ± 10.0% inhibition using 5 μM GM6001; 0.0 ± 0.9% using 5 μM C1006, p < 0.01). This inhibition was specific to αvβ8 because αvβ6-mediated activation of TGF-β was not inhibited by GM6001 (5 μM) in SW480 cells (Fig. 4 a) or HT1080 cells (1.0 ± 0.1% inhibition, p > 0.05). Finally, other peptide and chemical inhibitors of aspartyl (pepstatin A), serine (PMSF, CK-23, aprotinin, and leupeptin), or cysteine (leupeptin and E64) proteases had no effect on αvβ8-mediated activation of TGF-β1 when used at the maximal nontoxic doses (Fig. 4 b). Together these data suggest a novel mechanism of αvβ8-mediated activation of TGF-β1 requiring both the cell surface and metalloprotease activity.
Figure 4.
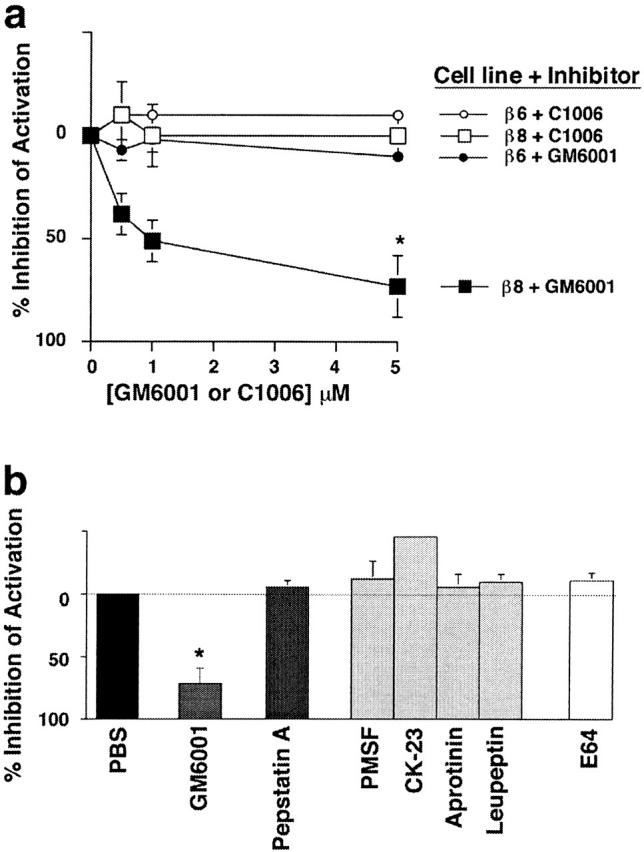
Activation of TGF-β1 by αvβ8 is dependent on metalloprotease activity. (a) GM6001, a hydroxamated metalloprotease inhibitor, but not C1006, a nonhydroxamated control peptide, inhibits αvβ8-mediated but not αvβ6-mediated activation of TGF-β in SW480 cells. The asterisk indicates significant inhibition by GM6001 of β8-mediated activation compared with the other three groups (p < 0.01). (b) Only metalloprotease inhibitors inhibit αvβ8-mediated TGF-β activation. Inhibitors to aspartyl (pepstatin A), serine (PMSF, CK-23, aprotinin, and leupeptin), or cysteine (leupeptin and E64) proteases do not inhibit αvβ8-mediated TGF-β activation in SW480 cells. β8- and β6-mediated activation was determined by neutralization with 37E1 or 10D5 in a or 37E1 in b. The asterisk indicates significant inhibition by GM6001 compared with other inhibitors (p < 0.01).
Cell surface expression of αvβ8 is associated with liberation of active TGF-β
All previously described mechanisms of cell-associated protease-dependent activation of TGF-β involve liberation of active TGF-β from SLC, presumably as a result of degradation of LAP (Lyons et al., 1988, 1990; Abe et al., 1998). Therefore, we hypothesized that if a cell surface proteolytic event was involved in αvβ8- but not αvβ6-mediated activation of TGF-β then active TGF-β should be released by β8- but not β6-expressing cells into the cell culture supernatant. To test this hypothesis, we assayed the supernatants of β8-expressing, β6-expressing, or mock-transduced HT1080 cells for active TGF-β. In HT1080 cells, β8 was expressed at lower surface levels than β6 (Fig. 5 a). Concordant with lower expression levels, αvβ8-expressing cells activated less TGF-β than αvβ6-expressing cells in coculture assays (Fig. 5 b). However, when supernatants of β8-expressing, β6-expressing, and mock-transduced HT1080 cells were tested only supernatant from β8-expressing cells contained a significant amount of active TGF-β (Fig. 5 c). The presence of active TGF-β in the cell supernatant of β8-expressing cells was confirmed by inhibition with anti–TGF-β antibodies (Fig. 5 c). Specificity was demonstrated by inhibition with anti-β8 antibodies and by the relative lack of active TGF-β in the supernatant of mock-transduced HT1080 cells (Fig. 5 c). Supernatant from β6-expressing HT1080 cells also had a slight but insignificant (p > 0.05) increase in active TGF-β compared with mock-transduced cells (Fig. 5 c). However, the presence of active TGF-β in the cell supernatant of β6-expressing cells was not β6 specific, since it was not blocked by anti-β6 antibodies. The presence of active TGF-β in the supernatant of this β6-expressing cell pool is likely due to random differences in integrin-independent release of active TGF-β by different cell pools, but we cannot exclude that it is due to an indirect effect of β6 expression in HT1080 cells. Consistent with the above HT1080 cell data, we also found that a small but significant amount of TGF-β1 was present in the cell culture supernatant of β8-expressing SW480 cells (relative luciferase units, 0.92 ± 0.23; +37E1, 0.26 ± 0.08; p < 0.05). In summary, active TGF-β is released into the supernatant of β8-expressing cells through a β8-dependent mechanism, suggesting that αvβ8 binds SLC to the cell surface, which facilitates the metalloprotease-dependent release of active TGF-β into the cell supernatant.
Figure 5.
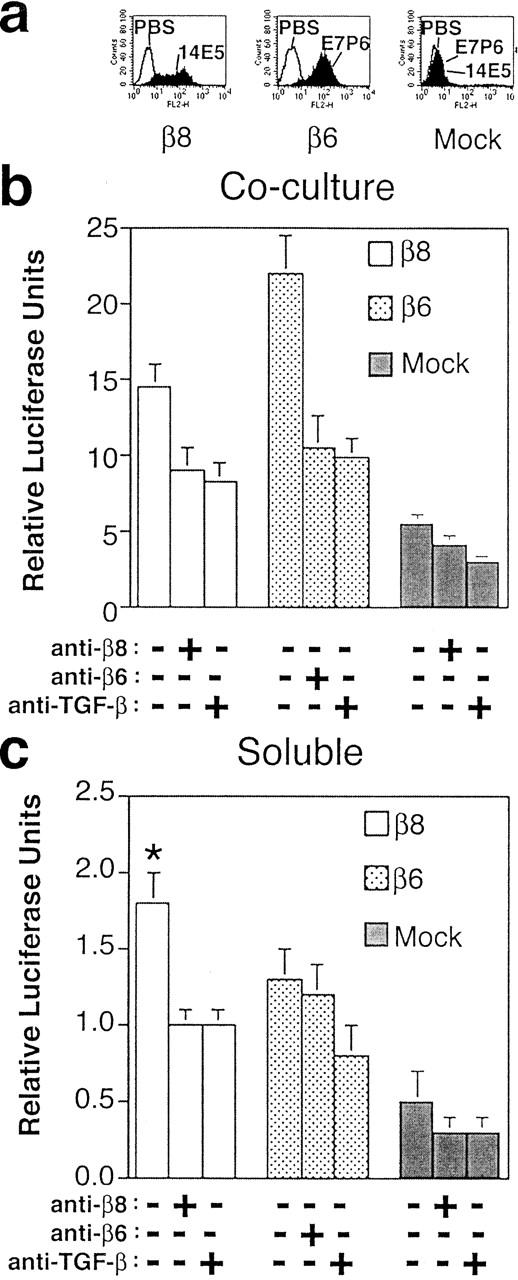
Active TGF-β is liberated into the supernatants of αvβ8-expressing cells. (a) Expression of β8 and β6 on the cell surface of β8-, β6-, and mock-transduced HT1080 cells using monoclonal antibodies specific for the β8 (14E5) or β6 (E7P6) integrin subunits. (b) Comparison of β8- and β6-mediated activation of TGF-β in cocultures with TMLC reporter cells. (c) Detection of active TGF-β liberated into the supernatant from β8-expressing but not β6- or mock-transduced HT1080 cells. Neutralizing antibodies to TGF-β were 1D11, or to β8 or β6 were 37E1 or 10D5, respectively. Relative luciferase units are shown in b and c. The asterisk indicates increased luciferase activity from supernatants of untreated β8-expressing cells compared with antibody-treated or mock controls (p < 0.01).
Expression of membrane-type 1–MMP is sufficient to support αvβ8-mediated activation of TGF-β
In the course of screening cell lines for this study, we identified one squamous lung carcinoma cell line, H1264, that expressed a low level of αvβ8 on the cell surface but did not activate TGF-β. The inability of H1264 cells to activate TGF-β was not due to low expression levels of αvβ8, since overexpression of β8 by retroviral transduction did not rescue activation (relative luciferase units; β8-transduced, 3.0 ± 0.3; +37E1, 2.2 ± 0.5; mock-transduced, 3.0 ± 0.4; ±37E1, 2.6 ± 0.2). Since our current findings suggested that cell surface expression of a metalloprotease cofactor was likely involved in αvβ8-mediated activation of TGF-β, we used H1264 cells to facilitate the identification of the specific metalloprotease(s) involved in αvβ8-mediated activation of TGF-β. Thus, we generated a metalloprotease expression profile of H1264 cells and compared it to profiles of the other tumor cell lines used in this study. We concentrated on metalloproteases known to localize to the cell surface (MMP-2 [Brooks et al., 1996], MMP-7 [Yu and Woessner, 2000], MMP-9 [Yu and Stamenkovic, 1999], membrane-type 1 [MT1]-MMP MMP [Sato et al., 1994], and a disintegrin and a metalloprotease domain [ADAM]-9, -10, and -17 [Primakoff and Myles, 2000]). Using reverse transcriptase (RT)–PCR (Fig. 6 a) and gelatin zymography (unpublished data), we found that the only metalloprotease that was deficient in H1264 cells, relative to the other tumor cell lines used in this study, was MT1-MMP. To reconstitute MT1-MMP expression in H1264 cells, we introduced either a full-length (MT1-MMP) or a transmembrane-deleted secreted form of MT1-MMP (ΔMT1-MMP). Expression or lack of expression of MT1-MMP in transduced or mock-transduced H1264 cells was verified by immunoblotting (Fig. 6 b). Immunoblots revealed an appropriate 63-kD band in cell lysates from MT1-MMP–transduced H1264 cells (Fig. 6 b) and a 54-kD band from the supernatant of ΔMT1-MMP–transduced cells (Fig. 6 b). The absence of any bands in mock-transduced cells confirmed the absence of MT1-MMP in the parental H1264 cell line (Fig. 6 b). Reconstituted MT1-MMP was active in H1264 cells as determined by the ability of MT1-MMP–transduced but not ΔMT1-MMP–transduced or mock-transduced H1264 cells to cleave pro–MMP-2 to its fully active (59 kD) form (Fig. 6 c). Secreted ΔMT1-MMP possessed gelatinolytic activity as determined by zymography (unpublished data).
Figure 6.
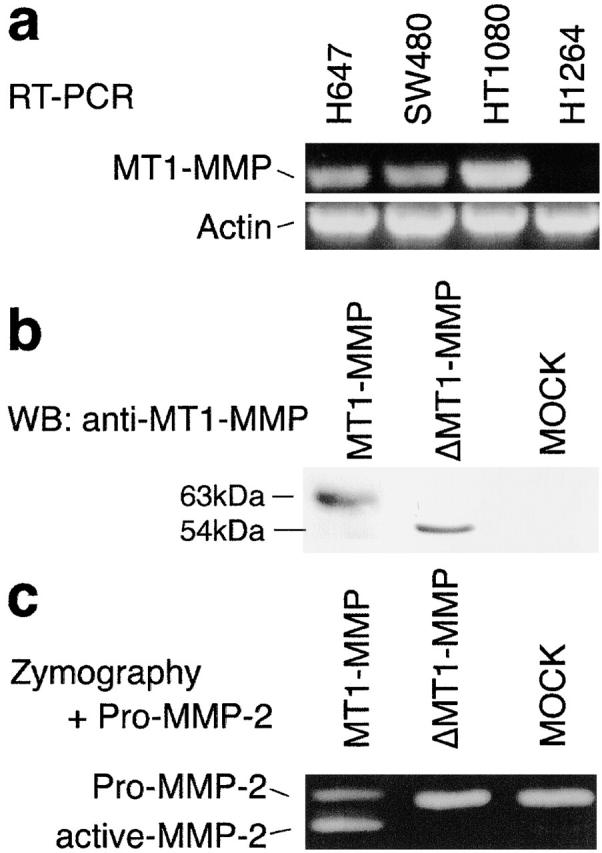
MT1-MMP is deficient and can be reconstituted in the human lung carcinoma cell line H1264. (a) RT-PCR screening of tumor cell lines with MT1-MMP–specific primers demonstrates an absence of MT1-MMP in H1264 cells. A control amplification performed in parallel using β-actin primers is shown. (b) Western blotting confirms the expression of MT1-MMP and ΔMT1-MMP in transduced H1264 cells. Cell lysate from MT1-MMP or mock-transduced H1264 cells (40 μg protein) or cell supernatant from ΔMT1-MMP–transduced H1264 cells was resolved by 10% SDS page, and proteins were detected by Western blotting using an anti–MT1-MMP monoclonal antibody. The expected migration of the full-length form of MT1-MMP is 63 kD. Note that the secreted form (ΔMT1-MMP) migrates faster (54 kD), and the mock-infected cells express no MT1-MMP. (c) MT1-MMP, ΔMT1-MMP, or mock-transduced β8-overexpressing H1264 cells were plated onto 96-well dishes in serum-free medium containing recombinant pro–MMP-2. After an overnight incubation at 37°C in 5% CO2, the supernatants were subjected to gelatin zymography (1 mg gelatin/ml, 10% SDS-PAGE). Pro-MMP2 migrates at 66 kD, and the fully activated form migrates at 59 kD. Note that only MT1-MMP–transduced H1264 cells activate MMP-2. MMP activity is shown as lucent bands against a dark Coomassie-stained background.
H1264 cells transduced with either MT1-MMP, ΔMT1-MMP, or vector alone (mock) underwent a second transduction to overexpress similar levels of αvβ8 as determined by flow cytometry using the anti-β8 antibody, 14E5 (Fig. 7 a), or SN1 (unpublished data). For β8-overexpressing H1264 cells, only those transduced with MT1-MMP and not ΔMT1-MMP or vector alone (mock) were able to support activation of TGF-β (Fig. 7 b). H1264 cells transduced with MT1-MMP alone supported a low level of αvβ8-mediated activation of TGF-β (relative luciferase units; no monoclonal antibody, 2.7 ± 0.2; +37E1, 1.3 ± 0.0; +1D11, −0.3 ± 0.01), which is consistent with their lower levels of surface expression of αvβ8. A significant portion of the TGF-β activation was specific to β8 and to metalloproteases because it was inhibited by anti-β8 antibodies (37E1), TIMP-2, and GM6001. 37E1, TIMP-2, and GM6001 were less efficient than the pan–TGF-β antibody (1D11) in inhibiting TGF-β activity. TIMP-2, a relatively specific inhibitor of MT1-MMP (Brew et al., 2000), inhibited TGF-β activation equally well as 37E1 (Fig. 7 c). The inability to completely block the β8-, MT1-MMP–dependent activation of TGF-β with anti-β8 antibodies and MMP inhibitors is likely due to a combination of factors: antibody efficacy, steric hindrance on the cell surface (Atkinson et al., 2001) and/or the high heterologous expression levels of both β8 and MT1-MMP.
Figure 7.
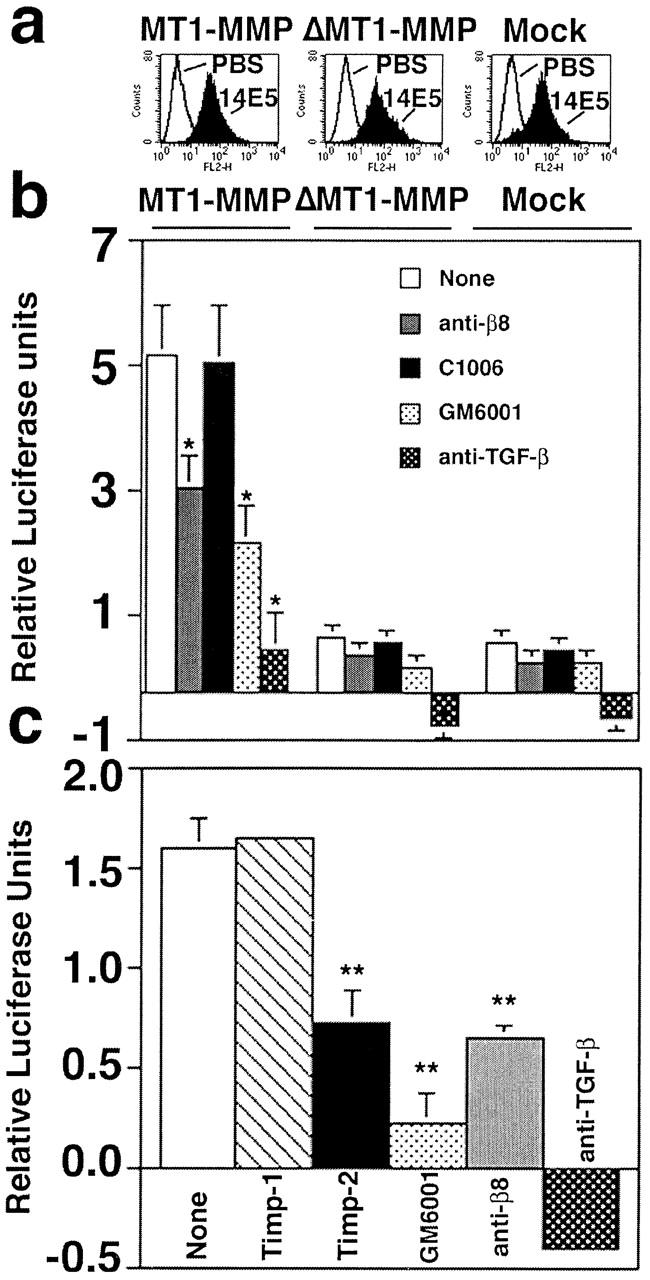
αvβ8 mediates activation of TGF-β in β8-overexpressing H1264 cells reconstituted with MT1-MMP activity. (a) Flow cytometry of β8-transduced MT1-MMP, ΔMT1-MMP, or mock-transduced H1264 cells demonstrates equivalent levels of surface expression of β8 using an anti-β8 antibody (14E5). Histograms using arbitrary units are shown. (b) β8-overexpressing H1264 cells transduced with either MT1-MMP, ΔMT1-MMP, or the retroviral vector alone (mock) (1.6 × 104) were cocultured with TMLC (1.6 × 104) reporter cells in the presence or absence of inhibitors: anti-β8 (37E1), control peptide (C1006), GM6001, or the pan–TGF-β1 antibody (1D11). Asterisks indicate significantly different than untreated MT1-MMP–expressing cells. (c) The endogenous inhibitor TIMP-2 but not TIMP-1 inhibits αvβ8-mediated activation of TGF-β in H1264s cells. β8-overexpressing, MT1-MMP–expressing H1264s cells were cocultured with TMLC in the presence or absence of TIMP-1 (1 μg/ml), TIMP-2 (1 μg/ml), GM6001 (5 μM), anti-β8 (37E1), or pan-TGF-β1 (1D11). Relative luciferase units are shown (light units of cocultured cells in the presence or absence of inhibitors minus light units of TMLC cells alone) in b and c. Negative luciferase values were occasionally observed due to a small background of TGF-β activation by the TMLC cells. Single and double asterisks indicate treated cells compared with untreated cells (*p < 0.01; **p < 0.001).
αvβ8 and MT1-MMP colocalize in substrate contacts
Our cell biologic and biochemical data indicated that αvβ8 and MT1-MMP were likely to associate on the cell surface. Thus, we hypothesized that upon ligation with LAP-β1, αvβ8 and MT1-MMP would cocluster in membrane complexes. To address this hypothesis, we expressed an MT1-MMP fusion construct with a COOH-terminal green fluorescent protein (GFP) tag. The purpose of the GFP tag was to obviate the use of available commercial anti–MT1-MMP antibodies, which we found unsuitable for immunocytochemistry (unpublished data). MT1-MMP–GFP was expressed on the cell surface of β8-expressing HT1080 cells as determined by surface labeling followed by immunoprecipitation with anti-GFP antibodies. Anti-GFP antibodies immunoprecipitated two bands from MT1-MMP–GFP–expressing cells, the upper band representing the catalytically active form of MT1-MMP–GFP and the lower band the degraded form, lacking the catalytic domain (Overall et al., 2000) (Fig. 8 a, top). Furthermore, MT1-MMP–GFP was found to be functionally active as determined by potentiation of the zymographic conversion of pro-MMP2 to active MMP2 (Fig. 8 a, bottom).
Figure 8.

αvβ8 and MT1-MMP colocalize in substrate contacts. (a, top) Immunoprecipitation of MT1-MMP–GFP from 125I cell surface-labeled MT1-MMP–GFP–expressing HT1080 cells. The catalytically active 90-kD MT1-MMP–GFP fusion protein (asterisk) was immunoprecipitated with anti-GFP antibodies from MT1-MMP–GFP–expressing HT1080 β8 cells but not mock-transduced HT1080 β8 cells. The 70-kD MT1-MMP–GFP band represents a catalytically inactive degradation product. (a, bottom) Gelatin zymography of supernatants from MT1-MMP–GFP–transduced or mock-transduced HT1080 β8 cells. The migration of Pro (Pro−) and active (Act.−) forms of MMP-2 are shown. (b–m) Confocal images of immunofluorescence microscopy. β8-expressing, MT1-MMP–GFP–expressing HT1080 cells (b–d and k–m); β8-expressing HT1080 cells (e–g); GFP-expressing HT1080 cells (h–j). Cells were allowed to attach 4 h to LAP-β1 (10 μg/ml coating concentration)-coated slides. After fixation and permeabilization, colocalization of β8 and GFP was determined using polyclonal anti-β8 and monoclonal anti-GFP antibodies. Pseudocolored confocal images of β8 (red) and GFP (green) staining taken in the plane of the substrate are shown. Bar, 7.5 μM.
We determined using confocal microscopy that β8 and MT1-MMP–GFP exactly colocalized in discrete clusters in the plane of an LAP-β1 substrate using anti-β8 and GFP antibodies (Fig. 8, b–d). The localization of β8 to these substrate contacts was not due to “bleed-over” of the MT1-MMP–GFP signal, since the GFP signal was not intense enough to visualize without staining with an anti-GFP antibody and β8 localization was seen in β8-expressing HT1080 cells not transduced with MT1-MMP–GFP (Fig. 8, e–g). As expected, antibodies against the αv subunit colocalized with antibodies against the β8 subunit in these contacts when cells were plated on LAP-β1 (unpublished data). Localization of β8 was not dependent on MT1-MMP expression, since β8 was found in substrate contacts in MT1-MMP–deficient β8-expressing H1264 cells plated on an LAP-β1 substrate (unpublished data). Localization of MT1-MMP–GFP to substrate contacts was not due to nonspecific accumulation of GFP or to a staining artifact, since no substrate contacts were found with anti-GFP antibodies in GFP-transduced non-β8–expressing HT1080 cells plated on LAP-β1 (Fig. 8, h–j). Localization of MT1-MMP to LAP-β1 substrate contacts was dependent on β8 expression, since no contacts were found in non-β8–expressing, MT1-MMP–GFP–expressing HT1080 cells (unpublished data). Finally, the colocalization of β8 and MT1-MMP to substrate contacts required ligand engagement, since no such contacts were found when β8-, MT1-MMP–GFP expressing HT1080 cells were plated on an irrelevant ligand, collagen I (Fig. 8, k–m). In conclusion, ligation of αvβ8 with LAP-β1 results in the specific colocalization of αvβ8 and MT1-MMP in substrate contacts, suggesting a biologically relevant and close physical interaction.
Overexpression of MT1-MMP is sufficient to cleave and inactivate LAP-β1
To determine if proteolysis of LAP-β1 could be a mechanism of αvβ8-mediated activation of TGF-β, we incubated recombinant LAP-β1 with β8-overexpressing H1264 cells transduced with either MT1-MMP, ΔMT1-MMP, or vector alone. After incubation of LAP-β1 with mock or ΔMT1-MMP–expressing H1264 cells, LAP-β1 remained intact (32 kD). In contrast, we found that almost all of the LAP-β1 incubated with MT1-MMP–expressing H1264 cells was smaller (26–28 kD) than intact LAP-β1, suggesting proteolytic cleavage (Fig. 9 a, lane 4). LAP-β1 cleavage was dependent on the metalloprotease activity of MT1-MMP, since the metalloprotease inhibitor GM6001 but not a control peptide C1006 completely blocked cleavage (Fig. 9 a, lanes 5 and 6). To determine if LAP-β1 cleavage was also dependent on αvβ8, we developed a peptide based on the human LAP-β1 sequence, which was a relatively specific inhibitor of αvβ8–LAP-β1 interactions. In the H1264 system, the GRRGDLATIH peptide completely blocked αvβ8–LAP-β1 interactions while having a minimal effect on the binding of other RGD-dependent integrins to VN or fibronectin (Fig. 9 b). Using these peptide inhibitors, we determined that LAP-β1 cleavage was also dependent on αvβ8, since GRRGDLATIH but not the RGE mutant peptide inhibited LAP-β1 cleavage (Fig. 9 c). Thus, in the H1264 cell system, αvβ8 and MT1-MMP together are required for LAP-β1 cleavage. Finally, αvβ8-, MT1-MMP–dependent cleavage of LAP-β1 is functionally relevant, since LAP-β1, after cleavage, loses the ability to inhibit the function of the recombinant mature TGF-β1 peptide (Fig. 9 d).
Figure 9.

Cell surface-associated MT1-MMP cleaves and inactivates LAP-β1. (a) LAP-β1 is cleaved by incubation with β8-overexpressing MT1-MMP but not ΔMT1-MMP or mock-transduced H1264 cells. LAP-β1 (10 μg/ml) was incubated overnight with either no cells (lane 1), mock-transduced β8-overexpressing H1264 cells (lane 2), ΔMT1-MMP transduced, β8-overexpressing H1264 cells (lane 3), MT1-MMP transduced, β8-overexpressing H1264 cells (lane 4), 500 μg/ml of control peptide (C1006; lane 5), or 500 μg/ml hydroxymate inhibitor GM6001 (lane 6). 20 ng of the input LAP-β1 was resolved by 12.5% SDS-PAGE under reducing conditions. After immunoblotting with an anti-LAP antibody, the migration of the cleavage products calibrated to molecular weight standards (GIBCO BRL) is shown. Note that only LAP-β1 incubated in the presence of MT1-MMP is cleaved and that this cleavage is blocked by GM6001. (b) The TGF-β1 peptide GRRGDLATIH selectively inhibits αvβ8–LAP-β1 function. Adhesion assay of β8-overexpressing, MT1-MMP expressing H1264 cells (4 × 104) to LAP-β1, VN, or fibronectin (FN) (10 μg/ml coating concentrations) in the presence or absence of 50 μg/ml of GRRGDLATIH. (c) LAP-β1 cleavage by MT1-MMP–transduced, β8-overexpressing H1264 cells is inhibited by GRRGDLATIH but not GRRGELATIH peptide (10 μg/ml). The degradation assay was performed and analyzed by immunoblotting as in a. No-cell control (lane 1); no-inhibitor control (lane 2); control GRRGELATIH peptide (lane 3); GRRGDLATIH peptide (lane 4). (d) LAP-β1 is inactivated by β8-overexpressing, MT1-MMP-expressing H1264 cells. LAP-β1 (5 μg) incubated overnight with β8-overexpressing, MT1-MMP-expressing or β8-overexpressing, mock-transduced H1264 cells was added to TMLC reporter cells in the presence of recombinant TGF-β1. As a control (white bars) TMLC reporter cells were incubated with only recombinant TGF-β and no LAP-β1. Relative luciferase units are shown. The asterisk indicates LAP-β1 incubated with mock control cells is not cleaved and decreases TGF-β activity compared with other groups (p < 0.05).
TGF-β activated by the integrin αvβ8 in human lung cancer xenografts results in growth inhibition and tumor fibrosis
To test the physiologic relevance of αvβ8-mediated activation of TGF-β1, we employed the H647 lung carcinoma cell system, which we have described recently (Cambier et al., 2000). We employed this system because β8 is expressed mainly by the basal cells of the human airway (Cambier et al., 2000), a cell type that is difficult to isolate in sufficient numbers for our studies and difficult to maintain in culture (Hicks et al., 1997). Because we have demonstrated recently that the growth of β8-expressing H647 cells was inhibited relative to mock-transduced H647 cells (Cambier et al., 2000), we asked if TGF-β might be a mediator of that growth inhibition. To address this question, we determined BrdU incorporation as a measure of DNA synthesis in β8-expressing and in mock-transduced H647 cells. We determined that the growth of β8-expressing H647 cells was potently inhibited relative to mock-transduced control cells as reflected by reduced BrdU incorporation (Fig. 10 a). In addition, since TGF-β characteristically induces a cell cycle arrest late in G1 (Laiho et al., 1990) we studied the cell cycle defect in H647 cells. Nonsynchronized β8-expressing H647 cells displayed a partial G1 arrest when compared with mock-transduced H647 cells (G1 fraction of β8-expressing H647 cells, 62% ± 3%; mock-transduced H647 cells, 47 ± 3%, p < 0.05). Furthermore, we found that this β8-dependent growth inhibition was also TGF-β–dependent because it could be reversed by TGF-β antibodies (Fig. 10 a) or by recombinant LAP-β1 (for β8-expressing H647 cells without LAP-β1, G1 fraction, 61%; with LAP-β1, 51%, in a representative experiment). Thus, TGF-β is a mediator of β8-induced growth inhibition in vitro.
Figure 10.
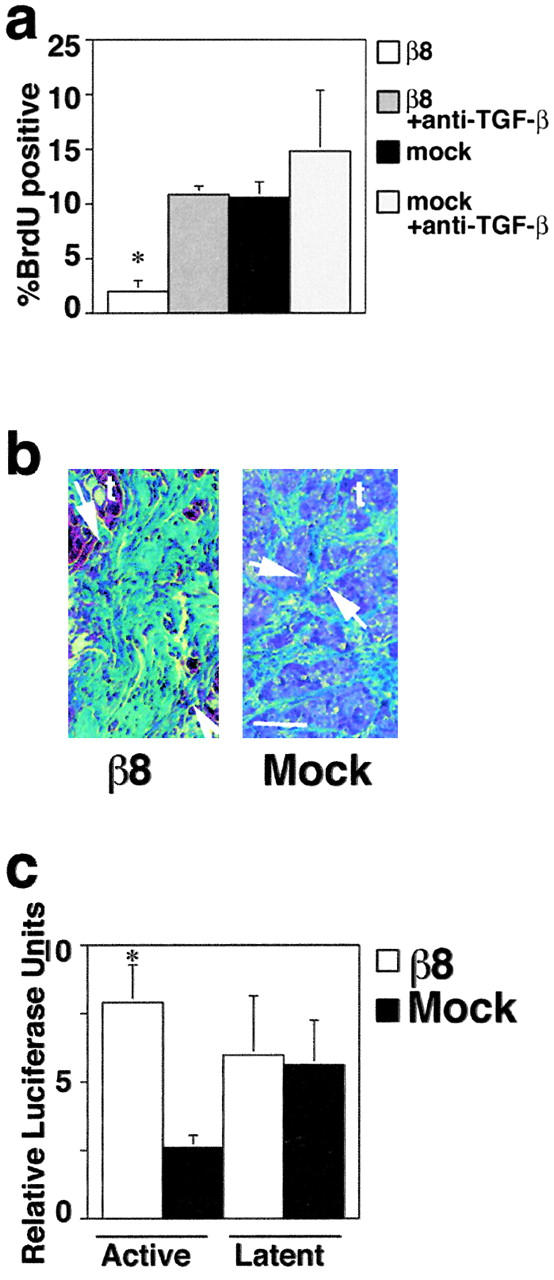
The activation of TGF-β by αvβ8 is associated both with growth inhibition and with fibrogenesis in lung cancer tumor xenografts. (a) DNA synthesis is inhibited in αvβ8-expressing H647 cells, and the inhibition is reversed by TGF-β blocking antibodies (1D11). The asterisk indicates increased BrdU incorporation of antibody-treated β8-expressing cells compared with untreated β8-expressing cell (p < 0.05). (b) Histologic analysis of tumors grown in nude mice derived from either β8-expressing or mock-transduced H647 cells. Trichrome-stained sections highlighting dense collagen (green area between arrows) between islands of tumor cells (t) are shown. Bar, 75 μm. (c) Determination of active and SLC in β8-expressing and mock-transduced lung tumor xenografts. Relative luciferase units are shown. The asterisk indicates increased active TGF-β from β8-expressing tumors compared with mock-transduced tumors (*p < 0.01).
To address if αvβ8 also mediated TGF-β activation in vivo, we used β8-expressing or mock-transduced H647 cells, which are tumorigenic in nude mice (Cambier et al., 2000). Using this system, we have found β8-expressing tumors to be significantly smaller than their mock-transduced counterparts (Cambier et al., 2000). Because fibrosis is one of the histologic hallmarks of increased TGF-β activity (Border and Noble, 1994), we examined β8-expressing tumors histologically for evidence of fibrosis. We found that β8-expressing tumors were not only smaller but were more fibrotic than tumors derived from mock-transduced cells (Fig. 10 b). To determine if the reduction in tumor size and the increase in stromal fibrosis were associated with increased active TGF-β, we used the TMLC reporter cells to determine the active and SLC activities associated with β8-expressing and mock-transduced H647 tumor xenografts. Using this bioassay, there was a significant increase in the amount of active TGF-β present in the aqueous fraction of β8-expressing tumors relative to control tumors (Fig. 10 c). In contrast, the latent component of TGF-β did not differ between tumors derived from β8-expressing or mock-transduced H647 cells (Fig. 10 c). Thus, in vivo αvβ8 can liberate physiologic levels of TGF-β activity. Moreover, these data suggest that in normal tissues αvβ8-mediated activation of TGF-β must be more tightly regulated than in the tumor xenograft model, since fibrosis is only associated with pathologic states (Blobe et al., 2000).
Discussion
We have found previously that the integrin αvβ8 inhibits epithelial cell growth (Cambier et al., 2000). Furthermore, αvβ8 is expressed in either quiescent cells or cells with a low rate of turnover (Nishimura et al., 1998; Cambier et al., 2000) and is lost in the process of neoplastic transformation (Cambier et al., 2000). These findings led us to hypothesize that β8 plays a role in the homeostatic control of normal tissues. In support of this hypothesis, we now demonstrate that the integrin αvβ8 mediates growth inhibition through a novel mechanism of activation of TGF-β1, a cytokine with a central role in homeostatic cellular processes (Blobe et al., 2000).
Two molecular mechanisms have been proposed that may lead to the activation of TGF-β1: conformational change leading to activation of the SLC complex (Crawford et al., 1998; Munger et al., 1999) or proteolysis of LAP-β1 leading to the release of active TGF-β1 (Munger et al., 1997; Yu and Stamenkovic, 2000). Our data demonstrate that a mechanism of conformational change leading to activation of TGF-β, as proposed for the αvβ6 integrin (Munger et al., 1999) or TSP-1 (Crawford et al., 1998), is not responsible for αvβ8-mediated activation of TGF-β1. Specifically, αvβ8-mediated activation of SLC does not require the β8 cytoplasmic domain in contrast to the mechanism of αvβ6-mediated activation of TGF-β, which requires the β6-cytoplasmic domain (Munger et al., 1999). Furthermore, αvβ8 is unlikely to bind directly or indirectly to LAP-β1 through a TSP-1–dependent mechanism because αvβ8 lacks the defined TSP-1 binding site for LAP-β1 (Crawford et al., 1998) and αvβ8 does not bind to TSP-1 (unpublished data). Moreover, unlike secreted TSP-1 (Crawford et al., 1998) secreted αvβ8 cannot activate TGF-β1. Thus, the mechanism by which αvβ8 activates TGF-β1 is not dependent on conformational changes, resulting from “inside-out” signal transduction as mediated by the β6 cytoplasmic domain (Munger et al., 1999) or direct physical interaction as mediated by TSP-1(Crawford et al., 1998).
Our findings support a biologically relevant mechanism whereby SLC binds with high affinity to αvβ8 on the cell surface, which results in the metalloprotease-dependent release of active TGF-β. Evidence to support this mechanism follows: (a) secreted αvβ8 binds to LAP-β1 with a high affinity with a dissociation constant similar to other TGF-β receptors (Tucker et al., 1984); (b) both synthetic and endogenous MMP inhibitors block αvβ8-mediated activation of TGF-β1; (c) reconstitution of MT1-MMP into the H1264 MT1-MMP–deficient cell line rescues αvβ8-mediated TGF-β activation; (d) αvβ8 and MT1-MMP specifically colocalize in LAP-β1 substrate contacts; (e) consistent with a proteolytic event, active TGF-β is liberated by an αvβ8-dependent mechanism into the supernatants of tumor cell lines and into the aqueous phase of lung cancer xenografts; (f) the proteolytic substrate of αvβ8-, MT1-MMP–dependent activation of TGF-β1 is likely to be LAP-β1, since β8-overexpressing, MT1-MMP–expressing H1264 cells cleave and inactivate LAP-β1, whereas β8-overexpressing, MT1-MMP–deficient H1264 cells do not; (g) cleavage of LAP-β1 requires the concomitant activity of both β8 and MT1-MMP, since β8-specific RGD inhibitors and metalloprotease inhibitors both block cleavage. Precedent for such a proteolytic mechanism is that plasmin (Lyons et al., 1990) and MMP-9 (Yu and Stamenkovic, 2000) have each been shown to activate TGF-β1 and TGF-β2, respectively, by cleavage of LAP.
It is also possible that MT1-MMP acts indirectly by proteolytically modifying the activity of αvβ8 as suggested recently for the MT1-MMP–dependent modification of the integrin αvβ3 (Deryugina et al., 2000). However, this is unlikely because of the following: (a) cell lines expressing αvβ8 attach to LAP-β1 equally well whether or not they express MT1-MMP (unpublished data), suggesting that coexpression of MT1-MMP does not modify the activity of αvβ8; (b) flow cytometry of H1264 cells overexpressing both β8 and MT1-MMP using two different anti-β8 monoclonal antibodies shows no alteration in surface expression of αvβ8, indicating that antibody epitopes are preserved along with adhesive capability; (c) immunoprecipitations or Western blots of cells coexpressing αvβ8 and MT1-MMP, using polyclonal antibodies against the cytoplasmic domain of β8, show no electrophoretic shift or proteolytic degradation products. Therefore, we have no evidence of modification of αvβ8 by MT1-MMP.
How does MT1-MMP interact with the αvβ8–TGF-β1 complex? Our data suggest that upon ligation of αvβ8 with SLC, αvβ8 and MT1-MMP become closely associated to form a complex on the cell surface. The cell surface appears to be required for productive interactions, since the secreted forms of αvβ8 and MT1-MMP do not mediate activation of TGF-β. Evidence for a physical association on the cell surface is that αvβ8 and MT1-MMP colocalize in substrate contacts specifically on LAP-β1. The nature of the MT1-MMP–β8 interaction awaits elucidation by coimmunoprecipitation and domain interaction studies. Because the localization of MT1-MMP in LAP-β1 substrate contacts is dependent on the presence of β8, it is likely that αvβ8–SLC interactions are required to initiate the recruitment of MT1-MMP. The dynamic recruitment of MT1-MMP to αvβ8–TGF-β complexes could provide a basis for the homeostatic regulation of TGF-β activity in cellular microenvironments.
Although reconstitution of wild-type MT1-MMP is sufficient to support αvβ8-mediated activation, other metalloproteases could potentially be involved. For instance, MT1-MMP binds to and is potently inhibited by TIMP-2 (Brew et al., 2000), but MT1-MMP–TIMP-2 complexes also serve as a cell surface receptor for MMP-2, and the function of this complex is activation of MMP-2 (Strongin et al., 1995). As such, it is not inconceivable that MMP-2 could also be involved in αvβ8-mediated activation of TGF-β. However, in H1264s cells MMP-2 is unlikely to be involved, since TIMP-1, a potent inhibitor of MMP-2 and weak inhibitor of MT1-MMP (Brew et al., 2000), has no effect on αvβ8-mediated activation of TGF-β. In contrast, β8-mediated TGF-β activation is inhibited by TIMP-2, suggesting that MT1-MMP may alone be sufficient to support β8-mediated activation of TGF-β. Although formally we cannot exclude additional roles for other MMPs or related metalloproteases such as ADAMs or ADAMTS, family members in αvβ8 mediated activation of TGF-β in other systems or cell types.
The β8 subunit appears to be the only integrin subunit capable of coordinating metalloprotease activity with SLC bound to the cell surface because the other LAP-β1 binding integrins are either incapable of activating TGF-β (Munger et al., 1998) or, in the case of αvβ6, activating TGF-β via a metalloprotease-independent pathway (Munger et al., 1999). Furthermore, αvβ8-mediated TGF-β activation is solely dependent on metalloproteases and not other proteases because inhibitors of aspartyl, serine, and cysteine proteases do not inhibit activation. Thus, αvβ8-mediated activation of TGF-β1 is not dependent on other proteases that have been implicated in SLC activation, including plasmin (Lyons et al., 1990), calpain (Abe et al., 1998), and cathepsin (Lyons et al., 1988).
Integrins (Brooks et al., 1996) and other cell surface molecules (Yu and Stamenkovic, 1999) have also been shown to localize MMP activity to the cell surface. For instance, the integrin αvβ3 has been shown to form an SDS stable cell surface complex with MMP-2 (Brooks et al., 1996) and to colocalize with MT1-MMP (Deryugina et al., 2001), whereas CD44 has been shown to mediate localization of MMP-9 (Yu and Stamenkovic, 2000) to the cell surface. However, αvβ3 and CD44 are unlikely to be required for αvβ8-mediated activation of TGF-β because αvβ3 is not expressed in multiple cell lines that support αvβ8-mediated activation of TGF-β (Table I) and because anti-CD44 antibodies do not inhibit αvβ8-mediated activation of TGF-β (unpublished data).
The selective MMP dependence of αvβ8- but not αvβ6-mediated activation of TGF-β1 clearly demonstrates that the mechanisms of αvβ8- and αvβ6-mediated activation of TGF-β1 are different. A structural basis for these different mechanisms may be the striking difference in the predicted secondary structure of the extracellular domains of the β8 and β6 subunits (Moyle et al., 1991). Different integrin-mediated mechanisms of TGF-β activation may have evolved to support distinct biologic functions. For instance, in the airway epithelium, a site where β8 is normally expressed (Cambier et al., 2000), a mechanism to support a low and persistent level of activation of TGF-β1 is necessary for homeostasis (Crawford et al., 1998). We speculate that αvβ8 could sequester SLC to the cell surface where, in response to an environmental cue, changes in the local balance of MMP/TIMP activity could lead to αvβ8-dependent liberation of active TGF-β1. Thus, αvβ8-mediated activation of TGF-β1 might liberate the low levels of active TGF-β1 sufficient to promote local paracrine effects but insufficient for undesirable local and systemic fibrogenic effects of TGF-β1 (Border and Noble, 1994). Conversely, if αvβ6 were to liberate TGF-β by an MMP-dependent mechanism undesirable pathologic levels of TGF-β might be released locally and into the systemic circulation because after injury expression of αvβ6 (Breuss et al., 1993; Pilewski et al., 1997) and MMPs (Holgate et al., 1999) are both strongly and rapidly induced.
In summary, abundant evidence implicates the cytokine TGF-β1, integrins, and MMPs as important mediators of homeostatic cell behaviors. This article provides the first evidence of the coordination of activity of members of these three major multigene families in the maintenance of homeostasis.
Materials and methods
Cell lines and reagents
Cell lines used include SW480, HT1080, H1264, HepG2, 293, SP2/0 (American Type Culture Collection), NCI H647 (Tsai et al., 1993), the amphotrophic retroviral packaging line Phoenix (Kinsella and Nolan, 1996), MvLu mink lung epithelial cells stably transfected with a plasminogen activator inhibitor promoter-1–luciferase construct (TMLC cells [Abe et al., 1994]), and High-Five (Invitrogen). Early passage parental H1264 cells deficient in MT1-MMP were subcloned by limiting dilution to achieve a phenotypically stable MT1-MMP–negative subclone (H1264s). LAP-β1 was produced using recombinant baculovirus-expressing wild-type simian LAP-β1 and mutant (RGE) LAP-β1 as described (Munger et al., 1998). LAP-β1–Sepharose was prepared by cross-linking 2 mg of protein A–tagged LAP-β1 to 1 ml of IgG Sepharose using 20 mM dimethylpimelimidate; VN-Sepharose was prepared as described (Nishimura et al., 1994). Human recombinant active TGF-β1, SLC, monoclonal anti-TGF-β (1D11) and affinity purified anti–TGF-β1, -β2, and -β3 were purchased from R&D Systems. The following proteases and protease inhibitors were also purchased: GM6001 (Galardy et al., 1994) and control C1006 (AMS Scientific, Inc.), PMSF, E-64 (Sigma-Aldrich), CK-23 (Enzyme Systems Products), leupeptin, aprotinin, and pepstatin A (Boehringer-Mannheim), and human recombinant TIMP-1, TIMP-2, and Pro-MMP-2 (Chemicon).
The following previously characterized antibodies were used: anti-β8, SN1 (Nishimura et al., 1994), anti-β6, 10D5 (Munger et al., 1999), E7P6 (Weinacker et al., 1994), polyclonal affinity purified rabbit anti-β8 (Nishimura et al., 1994), anti–LAP-β1 (VB3A9) (Munger et al., 1998), and anti–CD-44 (Picker et al., 1989) (Developmental Studies Hybridoma Bank). The following commercial antibodies and conjugates used were: anti-α5 (P5D10; Chemicon), anti-β1 (P5D2; Chemicon), pan-anti–TGF-β (1D11; R&D Systems), mouse or rabbit anti–MT1-MMP (Calbiochem and Chemicon), mouse anti-BrdU (Dako), phycoerythrin goat anti–mouse, rhodamine donkey anti–rabbit (Chemicon), phycoerythrin goat anti–mouse, rhodamine goat anti–mouse (Jackson ImmunoResearch Laboratories), HRP-conjugated sheep anti–mouse (Amersham Pharmacia Biotech), HRP anti–rat (Cappel), HRP–protein A (Amersham Pharmacia Biotech). VN was prepared from outdated fresh frozen human plasma (Yatohgo et al., 1988). Collagen type 1 was prepared from rat tails (Montesano et al., 1983). Peptides (GRGDSNK and GRGESNK) were purchased (BioMol) or were commercially synthesized (GRRGDLATIH and GRRGELATIH) (BioSyn). The following antibiotics were used: puromycin, chloroquine (Sigma-Aldrich), geneticin (G418; GIBCO BRL), hygromycin (Calbiochem), and Fungizone, penicillin, and streptomycin (University of California at San Francisco cell culture facility).
Retroviral vectors, constructs, and RT-PCR
Retroviral vectors used were pLXSN (CLONTECH Laboratories, Inc.), pBabe Puro (a gift from Dr. Hartmund Land, Imperial Cancer Research Fund, London, UK) (Morgenstern and Land, 1990), pBabeβ8Puro, pLXSNβ8Neo (Cambier et al., 2000), and pBabeβ6Puro. The β6 cDNA was subcloned from β6-Peak10 (a gift from R. Pytela, University of California at San Francisco) into pBabe Puro. Plasmids were purified using the QIAGEN plasmid purification system. β8 truncation mutants were constructed using a PCR strategy introducing COOH-terminal truncations (amino acids 712 and 758 of the β8 ORF [Moyle et al., 1991]), which replace the cytoplasmic domain of full-length β8 cDNA in pcDNAIneo (Invitrogen). The mutant constructs were subcloned into pLXSN (CLONTECH Laboratories, Inc.). Truncated β8 with a COOH-terminal AP tag (AP-αvβ8) was produced by in-frame blunt end ligation of the BspE1-Xho1 placental fragment from AP tag (Flanagan and Leder, 1990) into a chimeric pcDNAIβ8/3neo construct (Nishimura et al., 1994). Full length MT1-MMP or transmembrane and cytoplasmic domain deleted MT1-MMP (ΔMT1-MMP) (a gift from Stephen Weiss, University of Michigan, Ann Arbor, MI) were subcloned into pBabe Puro or pLXSN. A construct creating a MT1-MMP–GFP fusion protein (MT1-MMP–pLEGFP) was created by destroying the stop codon of MT1-MMP in PCR3.1 by PCR mutagenesis to create a unique HpaI site. The HindIII, HpaI MT1-MMP fragment was then subcloned in-frame into pLEGFP (CLONTECH Laboratories, Inc.) between a HindIII and Klenow-treated BamHI site. Sequencing in both orientations was performed to verify the fidelity of each construct. Transfection of packaging cells and retroviral transduction was performed as described (Kinsella and Nolan, 1996). Pools of β8-expressing cells were either used within 72 h for short term experiments or were sorted for uniform expression of β8 and propagated on type I collagen-coated plates for long term experiments (Cambier et al., 2000). The (SBE)4-Lux reporter, which contains 4 CAGACA repeats of the SMAD binding element of the JunB promoter (Jonk et al., 1998), was a gift of Peter ten Dijke (Ludwig Institute for Cancer Research, Uppsala, Sweden). Total cellular RNA was harvested using a commercial kit (QIAGEN), and RT-PCR was performed as described (Nishimura et al., 1994). Primers, based on published sequences (Giambernardi et al., 1998; McCulloch et al., 2000) to MMP-2, -7, and -9, MT1-MMP, ADAMS-9, -10 and -17, and β-actin were purchased (Operon Technologies). Annealing temperatures were based on published reports (Giambernardi et al., 1998; McCulloch et al., 2000).
Affinity chromatography, ligand binding, and adhesion assays
125I surface labeling and affinity chromatography was performed as described (Nishimura et al., 1994). Plasmids containing inserts for AP-tagged truncated secreted β8 and truncated secreted αv in pCDM8 (Nishimura et al., 1994) were stably expressed in 293 cells and characterized by immunoprecipitation (Nishimura et al., 1994). Serum-free supernatant containing AP-tagged αvβ8 (αvβ8-AP) was applied to 96-well plates precoated with LAP-β1 (wild-type and mutant), VN, or BSA for 1 h at 37°C in the presence and absence of monoclonal antibodies. Bound receptor was detected colorimetrically with pNPP (Sigma-Aldrich) at A405. For affinity measurements, serum-free supernatant containing secreted AP-αvβ8 was concentrated 40-fold (Vivaspin 100; Vivascience) and was applied to 10 μl of LAP-β1–Sepharose, VN-Sepharose, or IgG-Sepharose (Amersham Pharmacia Biotech) in the presence or absence of 1 mg/ml RGD peptide and incubated overnight at 4°C. The receptor concentration was determined against a standard curve generated using placental AP (Applied Biosystems). The samples were washed three times in wash buffer (Tris, 50 mM, pH 7.4, NaCl, 150 mM, CaCl2, 1 mM). Luminescence was determined using disodium 3-(4-methoxyspiro(1,2-dioxetane-3,2'-[(5′-chloro)tricyclo(3,3.1.13.7)decan]-4-yl) phenyl phosphate (CSPD) as a substrate (Tropix; Applied Biosystems) according to the manufacturer's instructions. Specific binding was defined as binding that remained after incubation with a 200-fold excess of RGD peptide. Binding curves were generated using nonlinear regression (Prism; GraphPad Software) from three independent experiments. Adhesion assays were performed as described (Nishimura et al., 1994).
Production of monoclonal and polyclonal β8 antibodies
Balb/C mice were immunized by standard protocols with truncated secreted αvβ8 according to the University of California at San Francisco Committee on Animal Research guidelines. Splenocytes were fused with SP 2/0 myeloma cells using commercial protocols (Boehringer). Clones were screened by immunoprecipitation and flow cytometry to detect αvβ8 (Nishimura et al., 1994). Antibodies were purified by ionic exchange chromatography using FPLC or used as supernatants for flow cytometry. Polyclonal anti-β8 antiserum was generated and characterized as described (Nishimura et al., 1998) by immunization of rabbits with a cytoplasmic peptide (TRAVTYRREKPEEIKMDISK) corresponding to amino acids 740–759 of the β8 ORF (BioSyn).
Fluorescence activated cell analysis, sorting, and immunocytochemistry
For FACS®, β8-expressing and mock-transduced cells were detached using 7 mM EDTA in DME, incubated with primary antibodies for 30 min at 4°C, and detected with phycoerythrin-conjugated secondary antibodies (Chemicon). Stained cells were analyzed using a FACsort® flow cytometer and CellQuest software (Becton Dickinson). Immunofluorescence microscopy was performed essentially as described with the following modifications (Cambier et al., 2000). HT1080 cells transduced with either pBabe Puro-β8 or pBabe Puro underwent a second transduction with either MT1-MMP–pLEGFP or pLEGFP and were selected with G418. LAP-β1 was used to coat glass chamber slides at a concentration of 10 μg/ml, and cells were allowed to attach to coated slides for 4 h before fixation. Antibodies used were a polyclonal anti-β8 antiserum and a monoclonal anti-GFP antibody (CLONTECH Laboratories, Inc.). Secondary reagents were AlexaFluor 595 goat anti–rabbit (Molecular Probes) and biotinylated sheep anti–mouse (Amersham Pharmacia Biotech) followed by Oregon green Streptavidin (Molecular Probes). Confocal images were obtained using a Bio-Rad Laboratories MRC-1024 laser scanning confocal system and LaserSharp2000 software (Bio-Rad Laboratories). Pseudocolored images and composites were generated in Adobe Photoshop® (v. 6.0).
Immunoprecipitation, Western blotting, and zymography
β8-expressing or mock-transduced cells in confluent 10-cm dishes were either surface labeled with 125I, biotin, or were directly lysed in PBS with 1% Triton X-100 with 1 mM PMSF as described (Milner and Ffrench-Constant, 1994; Nishimura et al., 1994). Immunoprecipitations and Western blots were performed as described (Milner and Ffrench-Constant, 1994; Nishimura et al., 1994). For LAP-β1 degradation experiments, LAP-β1 (10 μg/ml) was added to individual wells of a 96-well plate containing β8-overexpressing, MT1-MMP–, ΔMT1-MMP–, or mock-transduced H1264 cells (4 × 104) in complete medium. For GM6001 and C1006 inhibition experiments, 104 cells were added, and for RGD peptide blocking experiments 2 × 104 cells were added. After a 12–16-h incubation, the medium was collected and either added to TMLC reporter cells in the presence or absence of recombinant TGF-β1 (10 pM) or was subjected to 12.5% SDS-PAGE and Western blotting as above. For zymography, cells were incubated in serum-free media overnight in the presence or absence of pro–MMP-2 (10 ng/well). Supernatants were harvested and loaded without heating or reduction and resolved by 10% SDS-PAGE (1 mg/ml gelatin). After three washes in 2.5% Triton X-100, the gels were incubated in substrate buffer (50 mM Tris-HCL, 5 mM CaCl2, 0.01% NaN3, pH 8.0) and incubated overnight at 37°C. Lucent bands of gelatinolytic activity were revealed by Coomassie staining. Digital images were acquired using Eastman Kodak Co. 1D 3.5.3 Imaging system. Composites were assembled in Adobe Photoshop® (v. 6.0).
TGF-β bioassay
To determine TGF-β activation in a coculture assay, TMLC cells were cultured with β8-expressing or mock-transduced cells in the presence or absence of anti–TGF-β–blocking antibody (10 μg/ml, 1D11; R&D Systems), anti-β8 (100 μg/ml, 37E1) or anti-β6 (150 μg/ml, 10D5) as described (Abe et al., 1994; Munger et al., 1999). To measure active TGF-β in tumor tissue, equal weights of tumors were minced and incubated in sterile DME for 30 min at 4°C. The supernatants containing active TGF-β were harvested after centrifugation (20 g) at 4°C. The pellets were then incubated in serum-free DME for 20 min at 80°C to activate SLC after which the supernatants were harvested. The supernatants containing active or heat-activated (latent) TGF-β were then added to preplated TMLC cells with or without 1D11. For protease inhibitor assays, inhibitors were added at the initiation of the coculture. The maximal dose of each inhibitor was defined as the highest concentration that did not inhibit the ability of the TMLC cells to respond to recombinant active TGF-β. To measure soluble TGF-β activity from cultured cells, 106 HT1080 or SW480 cells, either β8-expressing, β6-expressing, or mock-transduced, were incubated in 100 μl of complete medium with or without 37E1 or 10D5 for 1 h at 37°C with gentle rotation. Cell-free supernatants were harvested by centrifugation (20 g) for 5 min at 4°C and then added to preplated TMLC cells in the presence or absence of 1D11. For soluble receptor assays, conditioned medium obtained from overnight cultures of 293 cells expressing truncated αvβ8 (Nishimura et al., 1994, 1998) was used. Relative luciferase units were defined as activity minus the background activity of the TMLC reporter cells. In some experiments, the TMLC reporter cells themselves activated a small amount of TGF-β as determined by inhibition with anti–TGF-β–blocking antibodies. In these experiments when the test cells did not activate TGF-β the relative luciferase units (sample minus the TMLC background) were less than zero.
Cell proliferation assays and lung tumor xenographs
Cell cycle analysis was performed as described previously (Cambier et al., 2000) with the exception that some cells were treated overnight with 10 μg/ml LAP-β1. BrdU incorporation assays were performed as described (Cambier et al., 2000). H647 tumor xenografts were established in nude mice as described (Cambier et al., 2000), and experiments were performed in full compliance with institutional guidelines and the University of California at San Francisco Committee on Animal Research.
Statistical analysis
Student's t test was used for comparison of two datasets; analysis of variance (ANOVA; for parametric data) or the Kruskal-Wallis test (for nonparametric data) were used for more than two datasets. Tukey's or Dunn's test was used for parametric or nonparametric data, respectively, to determine where the differences lay. Significance was defined as p < 0.05. Data are shown as means ± 1 SEM unless otherwise noted. Statistical software used was InStat v2.03 (GraphPad Software, Inc.).
Acknowledgments
This work was supported by grants from the National Institutes of Health (HL63993 and CA63143, to S.L. Nishimura, HL8985 to V.C. Broaddus, HL63786 to J.S. Munger, and HL53949 to D. Sheppard), American Cancer Society, American Lung Association, American Heart Association, University of California at San Francisco Academic Senate award, Research Evaluation and Allocation Committee, and Hellman Family (to S.L. Nishimura), Norwegian Cancer Society and the Unger-Vetlesen Legacy (to L. Fjellbirkeland), Tobacco-Related Disease Research Program (TRT-0051 to V.C. Broaddus), and the Burroughs-Wellcome Fund Career award, Pfizer award, and Howard Hughes Medical Institute (to J.L. Baron).
Footnotes
Abbreviations used in this paper: ADAM, a disintegrin and a metalloprotease domain; AP, alkaline phosphatase; GFP, green fluorescent protein; LAP, latency-associated peptide; MMP, matrix metalloprotease; MT1-MMP, membrane-type 1 MMP; RT, reverse transcriptase; SLC, latent TGF-β; TSP, thrombospondin; VN, vitronectin.
References
- Abe, M., J.G. Harpel, C.N. Metz, I. Nunes, D.J. Loskutoff, and D.B. Rifkin. 1994. An assay for transforming growth factor-β using cells transfected with a plasminogen activator inhibitor-1 promoter-luciferase construct. Anal. Biochem. 216:276–284. [DOI] [PubMed] [Google Scholar]
- Abe, M., N. Oda, and Y. Sato. 1998. Cell-associated activation of latent transforming growth factor-β by calpain. J. Cell. Physiol. 174:186–193. [DOI] [PubMed] [Google Scholar]
- Atkinson, S.J., M.L. Patterson, M.J. Butler, and G. Murphy. 2001. Membrane type 1 matrix metalloproteinase and gelatinase A synergistically degrade type 1 collagen in a cell model. FEBS Lett. 491:222–226. [DOI] [PubMed] [Google Scholar]
- Blobe, G.C., W.P. Schiemann, and H.F. Lodish. 2000. Role of transforming growth factor-β in human disease. N. Engl. J. Med. 342:1350–1358. [DOI] [PubMed] [Google Scholar]
- Border, W.A., and N.A. Noble. 1994. Transforming growth factor-β in tissue fibrosis. N. Engl. J. Med. 331:1286–1292. [DOI] [PubMed] [Google Scholar]
- Breuss, J.M., N. Gillett, L. Lu, D. Sheppard, and R. Pytela. 1993. Restricted distribution of integrin β6 mRNA in primate epithelial tissues. J. Histochem. Cytochem. 41:1521–1527. [DOI] [PubMed] [Google Scholar]
- Brew, K., D. Dinakarpandian, and H. Nagase. 2000. Tissue inhibitors of metalloproteinases: evolution, structure and function. Biochim. Biophys. Acta. 1477:267–283. [DOI] [PubMed] [Google Scholar]
- Brooks, P.C., S. Stromblad, L.C. Sanders, T.L. von Schalscha, R.T. Aimes, W.G. Stetler-Stevenson, J.P. Quigley, and D.A. Cheresh. 1996. Localization of matrix metalloproteinase MMP-2 to the surface of invasive cells by interaction with integrin αvβ3. Cell. 85:683–693. [DOI] [PubMed] [Google Scholar]
- Bugge, T.H., M.J. Flick, C.C. Daugherty, and J.L. Degen. 1995. Plasminogen deficiency causes severe thrombosis but is compatible with development and reproduction. Genes Dev. 9:794–807. [DOI] [PubMed] [Google Scholar]
- Cambier, S., D.Z. Mu, D. O'Connell, K. Boylen, W. Travis, W.H. Liu, V.C. Broaddus, and S.L. Nishimura. 2000. A role for the integrin αvβ8 in the negative regulation of epithelial cell growth. Cancer Res. 60:7084–7093. [PubMed] [Google Scholar]
- Crawford, S.E., V. Stellmach, J.E. Murphy-Ullrich, S.M. Ribeiro, J. Lawler, R.O. Hynes, G.P. Boivin, and N. Bouck. 1998. Thrombospondin-1 is a major activator of TGF-β1 in vivo. Cell. 93:1159–1170. [DOI] [PubMed] [Google Scholar]
- Deryugina, E.I., M.A. Bourdon, K. Jungwirth, J.W. Smith, and A.Y. Strongin. 2000. Functional activation of integrin αVβ3 in tumor cells expressing membrane-type 1 matrix metalloproteinase. Int. J. Cancer. 86:15–23. [DOI] [PubMed] [Google Scholar]
- Deryugina, E.I., B. Ratnikov, E. Monosov, T.I. Postnova, R. DiScipio, J.W. Smith, and A.Y. Strongin. 2001. MT1-MMP initiates activation of pro-MMP-2 and integrin αvβ3 promotes maturation of MMP-2 in breast carcinoma cells. Exp. Cell Res. 263:209–223. [DOI] [PubMed] [Google Scholar]
- Flanagan, J.G., and P. Leder. 1990. The kit ligand: a cell surface molecule altered in steel mutant fibroblasts. Cell. 63:185–194. [DOI] [PubMed] [Google Scholar]
- Galardy, R.E., M.E. Cassabonne, C. Giese, J.H. Gilbert, F. Lapierre, H. Lopez, M.E. Schaefer, R. Stack, M. Sullivan, B. Summers, et al. 1994. Low molecular weight inhibitors in corneal ulceration. Ann. NY Acad. Sci. 732:315–323. [DOI] [PubMed] [Google Scholar]
- Giambernardi, T.A., G.M. Grant, G.P. Taylor, R.J. Hay, V.M. Maher, J.J. McCormick, and R.J. Klebe. 1998. Overview of matrix metalloproteinase expression in cultured human cells. Matrix Biol. 16:483–496. [DOI] [PubMed] [Google Scholar]
- Giancotti, F.G. 1997. Integrin signaling: specificity and control of cell survival and cell cycle progression. Curr. Opin. Cell Biol. 9:691–700. [DOI] [PubMed] [Google Scholar]
- Hicks, W., Jr., L. Hall III, L. Sigurdson, C. Stewart, R. Hard, J. Winston, and J. Lwebuga-Mukasa. 1997. Isolation and characterization of basal cells from human upper respiratory epithelium. Exp. Cell Res. 237:357–363. [DOI] [PubMed] [Google Scholar]
- Holgate, S.T., P.M. Lackie, D.E. Davies, W.R. Roche, and A.F. Walls. 1999. The bronchial epithelium as a key regulator of airway inflammation and remodelling in asthma. Clin. Exp. Allergy. 29:90–95. [DOI] [PubMed] [Google Scholar]
- Jirtle, R.L., B.I. Carr, and C.D. Scott. 1991. Modulation of insulin-like growth factor-II/mannose 6-phosphate receptors and transforming growth factor-β1 during liver regeneration. J. Biol. Chem. 266:22444–22450 (erratum published 266:24860). [PubMed] [Google Scholar]
- Jonk, L.J., S. Itoh, C.H. Heldin, P. ten Dijke, and W. Kruijer. 1998. Identification and functional characterization of a Smad binding element (SBE) in the JunB promoter that acts as a transforming growth factor-beta, activin, and bone morphogenetic protein-inducible enhancer. J. Biol. Chem. 273:21145–21152. [DOI] [PubMed] [Google Scholar]
- Kinsella, T.M., and G. Nolan. 1996. Episomal vectors rapidly and stably produce high-titer recombinant retrovirus. Hum. Gene Ther. 7:1405–1413. [DOI] [PubMed] [Google Scholar]
- Laiho, M., J.A. DeCaprio, J.W. Ludlow, D.M. Livingston, and J. Massague. 1990. Growth inhibition by TGF-beta linked to suppression of retinoblastoma protein phosphorylation. Cell. 62:175–185. [DOI] [PubMed] [Google Scholar]
- Lord, B.I. 1988. Feedback regulators in normal and tumour tissues. J. Cell Sci. Suppl. 10:231–242. [DOI] [PubMed] [Google Scholar]
- Lyons, R.M., J. Keski-Oja, and H.L. Moses. 1988. Proteolytic activation of latent transforming growth factor-β from fibroblast-conditioned medium. J. Cell Biol. 106:1659–1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons, R.M., L.E. Gentry, A.F. Purchio, and H.L. Moses. 1990. Mechanism of activation of latent recombinant transforming growth factor-β1 by plasmin. J. Cell Biol. 110:1361–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCulloch, D.R., M. Harvey, and A.C. Herington. 2000. The expression of the ADAMs proteases in prostate cancer cell lines and their regulation by dihydrotestosterone. Mol. Cell. Endocrinol. 167:11–21. [DOI] [PubMed] [Google Scholar]
- Milner, R., and C. Ffrench-Constant. 1994. Developmental analysis of oligodendroglial integrins in primary cells: changes in αv-associated β subunits during differentiation. Development. 120:3497–3506. [DOI] [PubMed] [Google Scholar]
- Montesano, R., P. Mouron, M. Amherdt, and L. Orci. 1983. Collagen matrix promotes reorganization of pancreatic endocrine cell monolayers into islet-like organoids. J. Cell Biol. 97:935–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgenstern, J.P., and H. Land. 1990. Advanced mammalian gene transfer: high titer retroviral vectors with multiple drug selection markers and a complementary helper-free packaging line. Nucleic Acids Res. 18:3587–3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyle, M., M.A. Napier, and J.W. McLean. 1991. Cloning and expression of a divergent integrin subunit β8. J. Biol. Chem. 266:19650–19658. [PubMed] [Google Scholar]
- Munger, J.S., J.G. Harpel, P.E. Gleizes, R. Mazzieri, I. Nunes, and D.B. Rifkin. 1997. Latent transforming growth factor-β: structural features and mechanisms of activation. Kidney Int. 51:1376–1382. [DOI] [PubMed] [Google Scholar]
- Munger, J.S., J.G. Harpel, F.G. Giancotti, and D.B. Rifkin. 1998. Interactions between growth factors and integrins: latent forms of transforming growth factor-β are ligands for the integrin αvβ1. Mol. Biol. Cell. 9:2627–2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munger, J.S., X. Huang, H. Kawakatsu, M.J. Griffiths, S.L. Dalton, J. Wu, J.F. Pittet, N. Kaminski, C. Garat, M.A. Matthay, et al. 1999. The integrin αvβ6 binds and activates latent TGF-β1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell. 96:319–328. [DOI] [PubMed] [Google Scholar]
- Murphy-Ullrich, J.E., and M. Poczatek. 2000. Activation of latent TGF-β by thrombospondin-1: mechanisms and physiology. Cytokine Growth Factor Rev. 11:59–69. [DOI] [PubMed] [Google Scholar]
- Nishimura, S.L., D. Sheppard, and R. Pytela. 1994. Integrin αvβ8: interaction with vitronectin and functional divergence of the β8 cytoplasmic domain. J. Biol. Chem. 269:28708–28715. [PubMed] [Google Scholar]
- Nishimura, S.L., K.P. Boylen, S. Einheber, T.A. Milner, D.M. Ramos, and R. Pytela. 1998. Synaptic and glial localization of the integrin αvβ8 in mouse and rat brain. Brain Res. 791:271–282. [DOI] [PubMed] [Google Scholar]
- Overall, C.M., E. Tam, G.A. McQuibban, C. Morrison, U.M. Wallon, H.F. Bigg, A.E. King, and C.R. Roberts. 2000. Domain interactions in the gelatinase A.TIMP-2.MT1-MMP activation complex. The ectodomain of the 44-kDa form of membrane type-1 matrix metalloproteinase does not modulate gelatinase A activation. J. Biol. Chem. 275:39497–39506. [DOI] [PubMed] [Google Scholar]
- Picker, L.J., J. De los Toyos, M.J. Telen, B.F. Haynes, and E.C. Butcher. 1989. Monoclonal antibodies against the CD44 [In(Lu)-related p80], and Pgp-1 antigens in man recognize the Hermes class of lymphocyte homing receptors. J. Immunol. 142:2046–2051. [PubMed] [Google Scholar]
- Pilewski, J.M., J.D. Latoche, S.M. Arcasoy, and S.M. Albelda. 1997. Expression of integrin cell adhesion receptors during human airway epithelial repair in vivo. Am. J. Physiol. 273:L256–L263. [DOI] [PubMed] [Google Scholar]
- Potter, V.R. 1974. Probabilistic aspects of the human cybernetic machine. Perspect. Biol. Med. 17:164–183. [DOI] [PubMed] [Google Scholar]
- Primakoff, P., and D.G. Myles. 2000. The ADAM gene family: surface proteins with adhesion and protease activity. Trends Genet. 16:83–87. [DOI] [PubMed] [Google Scholar]
- Protin, U., T. Schweighoffer, W. Jochum, and F. Hilberg. 1999. CD44-deficient mice develop normally with changes in subpopulations and recirculation of lymphocyte subsets. J. Immunol. 163:4917–4923. [PubMed] [Google Scholar]
- Sato, H., T. Takino, Y. Okada, J. Cao, A. Shinagawa, E. Yamamoto, and M. Seiki. 1994. A matrix metalloproteinase expressed on the surface of invasive tumour cells. Nature. 370:61–65. [DOI] [PubMed] [Google Scholar]
- Schwartz, M.A. 1997. Integrins, oncogenes and anchorage independence. J. Cell Biol. 139:575–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shull, M.M., I. Ormsby, A.B. Kier, S. Pawlowski, R.J. Diebold, M. Yin, R. Allen, C. Sidman, G. Proetzel, D. Calvin, et al. 1992. Targeted disruption of the mouse transforming growth factor-β1 gene results in multifocal inflammatory disease. Nature. 359:693–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strongin, A.Y., I. Collier, G. Bannikov, B.L. Marmer, G.A. Grant, and G.I. Goldberg. 1995. Mechanism of cell surface activation of 72-kDa type IV collagenase. Isolation of the activated form of the membrane metalloprotease. J. Biol. Chem. 270:5331–5338. [DOI] [PubMed] [Google Scholar]
- Tsai, C.M., K.T. Chang, R.P. Perng, T. Mitsudomi, M.H. Chen, C. Kadoyama, and A.F. Gazdar. 1993. Correlation of intrinsic chemoresistance of non-small-cell lung cancer cell lines with HER-2/neu gene expression but not with ras gene mutations. J. Natl. Cancer Inst. 85:897–901. [DOI] [PubMed] [Google Scholar]
- Tucker, R.F., E.L. Branum, G.D. Shipley, R.J. Ryan, and H.L. Moses. 1984. Specific binding to cultured cells of 125I-labeled type beta transforming growth factor from human platelets. Proc. Natl. Acad. Sci. USA. 81:6757–6761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinacker, A., A. Chen, M. Agrez, R. Cone, S.L. Nishimura, E. Wayner, R. Pytela, and D. Sheppard. 1994. Role of the integrin αvβ6 in cell attachment to fibronectin, heterologous expression of intact and secreted forms of the receptor. J. Biol. Chem. 269:6940–6948. [PubMed] [Google Scholar]
- Yatohgo, T., M. Izumi, H. Kashiwagi, and M. Hayashi. 1988. Novel purification of vitronectin from human plasma by heparin affinity chromatography. Cell Struct. Funct. 13:281–292. [DOI] [PubMed] [Google Scholar]
- Yu, Q., and I. Stamenkovic. 1999. Localization of matrix metalloproteinase 9 to the cell surface provides a mechanism for CD44-mediated tumor invasion. Genes Dev. 13:35–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, Q., and I. Stamenkovic. 2000. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-β and promotes tumor invasion and angiogenesis. Genes Dev. 14:163–176. [PMC free article] [PubMed] [Google Scholar]
- Yu, W.H., and J.F. Woessner. 2000. Heparan sulfate proteoglycans as extracellular docking molecules for matrilysin (matrix metalloproteinase 7). J. Biol. Chem. 275:4183–4191. [DOI] [PubMed] [Google Scholar]