

Abstract
Prostaglandins (PGs) have been recently proven essential for parturition in mice. To dissect the contributions of the two cyclooxygenase (COX) isoforms to the synthesis of PGs during pregnancy, we have characterized the parturition phenotype of COX-1-deficient mice. We find that mice with targeted disruption of the COX-1 gene have delayed parturition resulting in neonatal death. Results of matings of COX-1-deficient females with COX-1 intact males, and blastocyst transfer of COX-1-deficient or -intact embryos into wild-type foster mothers, proved necessity and sufficiency of maternal COX-1 for the normal onset of labor. COX-1 expression is induced in gravid murine uterus and by in situ hybridization; this induction is localized to the decidua. Measurement of uterine PGs further confirmed that COX-1 accounted for the majority of PGF2α production. To evaluate the interaction of PGs with oxytocin during murine labor, we generated mice deficient in both oxytocin and COX-1. Surprisingly, the combined oxytocin and COX-1-deficient mice initiated labor at the normal time. COX-1-deficient mice demonstrated impaired luteolysis, as evidenced by elevated serum progesterone concentration and ovarian histology late in gestation, and delayed induction of uterine oxytocin receptors. In contrast, simultaneous oxytocin and COX-1 deficiency restored the normal onset of labor by allowing luteolysis in the absence of elevated PGF2α production. These findings demonstrate that COX-1 is essential for normal labor in the mouse, with a critical function being to overcome the luteotrophic action of oxytocin in late gestation.
Preterm labor occurs in 7–10% of all human pregnancies and accounts for 75% of perinatal mortality (1, 2). The physiologic mechanisms determining the timing of initiation of parturition and the subsequent progression of labor remain poorly understood despite intensive investigation. Genetic analyses of putative mediators of labor in the mouse recently have provided insight into the molecular cascade that culminates in parturition. The action of the prostaglandin (PG)F2α has proven essential for involution of the corpus luteum and labor initiation in the mouse, although is not required for parturition if a fall in progesterone is initiated by other means (3, 4). Induction of uterine oxytocin receptor expression after luteolysis has, however, been correlated with the onset of labor with or without PGF2α receptor function (4–6).
Cyclooxygenases (PG H synthases) catalyze the first committed step in the formation of PGs from arachidonic acid. Two isoforms of cyclooxygenase (COX) have been characterized. COX-1 is expressed by many cell types and in general shows constitutive activity with little regulation in synthesis (7, 8). COX-2, in contrast, shows a more limited distribution in sites of production, though is highly inducible in response to a variety of cytokines, specific inflammatory stimuli, and growth factors (7, 9, 10). To determine the necessity of COX-1-generated PGs, and the interaction between PGs and oxytocin (OT), for the initiation and progression of labor, we report here the parturition phenotypes of COX-1-deficient (KO), and combined COX-1 KO and OT KO, mice.
MATERIALS AND METHODS
Animal Husbandry.
COX-1 KO (11), OT KO (C.L. and L.J.M., unpublished data), and combined COX-1 KO/OT KO mice were maintained on an outbred genetic background, with wild-type (wt) littermates of the same outbred background used as controls. Mice were housed on a 12:12 light:dark cycle with ad libitum access to rodent chow. All mouse protocols were in accordance with National Institutes of Health guidelines and approved by the Animal Care and Use Committee of Washington University School of Medicine, St. Louis. Natural mating of estrous females with stud males was confirmed by detection of a copulation plug the morning after introduction of the female into the male cage. Females were subsequently isolated until the time of analysis to ensure accurate gestation timing, with the morning of copulation plug detection designated 0.5 d gestation. To determine the duration until the onset of labor, pregnant females were observed in the morning and evening beginning at day 18.5 of gestation. Statistical analysis was by ANOVA, with significance defined as P < 0.05.
Labor Induction.
To induce labor and assess fetal viability in n = 4 COX-1 KO pregnancies, 2.5 μg of prostaglandin F2α (Upjohn) diluted in PBS was administered in the morning of day 19.5 of gestation, with time until delivery monitored. If delivery of pups had not occurred within 8 hr of administration, a second dose of 2.5 μg of prostaglandin F2α was given.
Blastocyst Transfer.
Blastocysts at 3.5 d post coitum were harvested from uteri (12) of either COX-1 KO male × female matings (KO embryos) or wt male × COX-1 heterozygous female matings (wt and heterozygous embryos). Uteri of pseudopregnant wt foster females, generated by mating with vasectomized males, at 2.5 d post coitum were implanted with 8–12 KO or wt/heterozygous blastocysts with time until delivery determined.
RNA Hybridization Analyses.
RNA was prepared by the guanidinium thiocyanate-cesium chloride method (13). Ten micrograms of total RNA from a given gestational age and genotype were subjected to electrophoresis through 1.2% agarose-formaldehyde gels and transferred to nitrocellulose membranes. RNA probes specific for mouse COX-1 (1,298-bp ScaI fragment in pBluescript SK II+) and OT receptor (0.8-kb PCR fragment from exon 2 in pBluescript SK II+) mRNAs labeled with [α-32P]UTP were generated by transcription with T7 polymerase and then hybridized at 60°C (COX-1) or 68°C (OT receptor) in 50% formamide-containing buffer. After washing, hybridizing probes were quantitated on a Molecular Dynamics PhosphorImager. Each mRNA hybridization signal was corrected for loading and recovery by normalization to cyclophilin A or 18S ribosomal RNA hybridization on the same filter.
In Situ Hybridization Analyses.
Fetuses with surrounding membranes and uteri were fixed by immersion in 4% paraformaldehyde in PBS for 24 h at 4°C. Samples were then cryopreserved in 10% sucrose in PBS and embedded in OCT compound (Sakura Finetek, Torrance, CA) for sectioning on a cryostat. Twenty-micrometer sections were thaw mounted onto Superfrost plus slides (Fisher Scientific), and hybridized to an [α-33P]UTP-labeled COX-1 antisense riboprobe. After washing, slides were exposed to Kodak NTB-2 emulsion (Eastman Kodak) for 7–10 d. COX-1 KO uterus was used as a negative control to ensure hybridization specificity.
Ovarian Histology.
Ovaries were immersion fixed, cryopreserved, and OCT embedded as described above. Ten-micrometer sections were hematoxylin/eosin stained and evaluated by light microscopy.
Hormone Measurements.
Plasma progesterone concentration was determined by radioimmunoassay kit as described by the manufacturer (Diagnostics Products, Los Angeles) on blood samples obtained by retroorbital phlebotomy. Each measurement was performed on at least four separate pregnancies of each genotype and analyzed for significance by ANOVA. Uterine PGF2α was measured on tissue harvested from three separate pregnancies per genotype at day 19 or two separate wt pregnancies at the earlier time points and rapidly frozen in liquid nitrogen. Two wt pregnancies at day 15.5 of gestation, in addition, received indomethacin 5 mg/kg to confirm assay specificity. Ovarian PGF2α measurement was performed on three pregnancies of each genotype at day 19 of gestation. Tissue was weighed while frozen and then homogenized in 100% ethanol for extraction of prostaglandins. Debris was removed by centrifugation, and each supernatant was assayed in duplicate for PGF2α by ELISA kit per the manufacturer’s instructions (Oxford Biomedical Research, Oxford, MI). On day 19 of gestation, uterine and ovarian PGF2α measurements were analyzed for statistical significance by ANOVA.
RESULTS AND DISCUSSION
The initial description of the production and characterization of COX-1 KO mice was notable for the perinatal death, for unknown reasons, of progeny arising from matings of homozygous deficient parents (11). We established timed matings of wt and COX-1 KO males and females, with the morning of copulation plug detection designated 0.5 d of gestation. The delivery of wt litters occurred at 19.6 ± 0.2 d gestation, whereas the delivery of COX-1 KO litters was markedly delayed at 21.6 ± 0.2 d (P < 0.001, Table 1). Many of the pups of COX-1 KO matings were dead at the time of birth but were not reduced in size, degree of development, or number. The adequate pup size suggested viability through the usual time of labor and implicated post-mature gestation and/or the abnormal progression of labor as the mechanism of death. Administration of PGF2α to COX-1 KO matings at day 19.5 of gestation resulted in labor within 24 h and restoration of neonatal viability, verifying this hypothesis (Table 1). Thus, COX-1 is essential for the normal onset and progression of term labor and cannot be compensated for by unimpaired COX-2 activity. Although we have not rigorously characterized the precise timing of the initiation of labor in COX-1 heterozygous females, we routinely use matings of COX-1 KO males with COX-1 heterozygous females for propagation of our colony. There is no evidence of increased neonatal mortality from these matings, suggesting that the heterozygous females do not have a significant delay in the onset of labor.
Table 1.
Summary of parturition in COX-1, OT, and COX-1 KO/OT KO mice
| Maternal genotype | Paternal genotype | PGF2α178 | No. | Labor onset, d [range] | Avg. litter | Survival for 24 h, % |
|---|---|---|---|---|---|---|
| COX-1 KO | COX-1 KO | No | 6 | 21.6 ± 0.2† [20.5–22.0] | 7.5 ± 0.8 | 0 |
| COX-1 KO | COX-1 KO | Yes | 4 | 20.4 ± 0.1† [20.0–20.5] | 6.3 ± 0.9 | 79 |
| COX-1 KO | COX-1 Het/wt | No | 7 | 22.0 + 0.5† [20.5–24] | 8.0 ± 1.1 | 0 |
| COX-1 KO/OT KO | COX-1 KO/OT KO | No | 5 | 19.8 ± 0.5 [19–21.5] | 8.6 ± 1.0 | 0 |
| wt | wt | No | 7 | 19.6 ± 0.1 [19.5–20.0] | 8.9 ± 0.8 | 100 |
Labor onset and litter size are presented as mean ± SEM. Het, heterozygous.
Bolus injection of 2.5 μg of prostaglandin F2β beginning at 19.5 d gestation.
P < 0.001 vs. wt matings.
To determine the role of fetal COX-1 in parturition, matings of COX-1 KO females with wt or heterozygous males were established. Similar to the homozygote–homozygote matings, the heterozygous and mixed heterozygous–homozygous deficient litters from homozygous deficient females underwent delivery at day 22 ± 0.5 of gestation (P < 0.001 vs. wt, Table 1) with poor fetal viability. Thus, fetal COX-1 activity alone cannot result in the timely induction of parturition. Further, sufficiency of maternal COX-1 function to maintain normal gestational length in the setting of fetal COX-1 deficiency was explored by transfer of COX-1 intact (wt and heterozygous) or COX-1 KO blastocysts into pseudopregnant wt foster mothers. No significant difference in the timing of delivery occurred in pregnancies of transplanted COX-1 KO (19.7 ± 0.2 d, n = 3) vs. wt/heterozygous (19 ± 0.2 d, n = 4) embryos, demonstrating both the necessity and sufficiency of intact maternal COX-1 activity to promote normal labor.
Consistent with the finding that COX-1 is essential for the normal initiation of parturition in the mouse, Northern blot analysis demonstrated a 40-fold increase in COX-1 mRNA in the uterus in late gestation in comparison to nongravid expression (Fig. 1A). High level expression was found at 16.5–18.5 days of gestation, with declining mRNA expression just before delivery at 19 d and a further decrease immediately postpartum (Fig. 1A). In contrast, we found lower level, relatively unchanged COX-1 mRNA expression in the ovary during pregnancy (data not shown), suggesting that primarily uterine-derived PG determines the timing of labor. To localize COX-1 expression within the fetoplacental unit and uterus, in situ hybridization analysis was performed. Abundant expression of COX-1 mRNA was found in the decidua (uterine endometrium of pregnancy) in comparison to myometrium or fetal membranes at 18.5 days of gestation (Fig. 1B).
Figure 1.

Induction of COX-1 expression during gestation. (A) Northern blot analyses of total uterine RNA during pregnancy. Samples from nongravid (NG) or pregnant females of the gestational age in days given above the corresponding lanes were hybridized to COX-1 or cyclophilin A- (CYC) radiolabeled probes. (B) Decidual (endometrial) localization of COX-1 in gravid uterus at 18.5 d of gestation. Histologic sections of intact uterine implantation sites including uterus, fetus, placenta, and extra-embryonic membranes were hybridized to a radiolabeled COX-1 RNA probe. Shown is a representative bright field (Upper), nuclear fast red counterstained, region of uterus and amnion, along with the corresponding dark field photograph of the section (Lower), after exposure to emulsion and development. High level COX-1 expression is detected primarily within the decidua (D), as opposed to myometrium (M) or amnion, as demonstrated by deposition of silver grains.
PGF2α receptor-deficient mice, which lack the ability to respond to PGF2α generated by either COX-1 or COX-2 and fail to induce OT receptor in the uterine myometrium at term, exhibit complete failure of parturition (4). Consistent with their delay in initiation of labor, COX-1 KO females have an attenuation of OT receptor induction (Fig. 2). Despite the strong correlation of OT receptor induction with parturition, OT-KO mice have the normal onset and progression of labor (14, 15). To determine whether prostaglandin production by COX-1 was in part redundant with OT receptor activation during labor, we generated mice deficient in both COX-1 and OT (COX-1 KO/OT KO). Matings of COX-1 KO/OT KO males and females surprisingly began labor at 19.8 ± 0.5 d gestation, not differing from wt matings (P = 0.57, Table 1). In contrast to either wt, OT KO, or COX-1 KO pregnancies, which exhibited rapid progression of labor once initiated, however, the duration of labor was often extended in the double deficiency state, with some litters delivering over a 2-day period. Because of failure of the COX-1 KO/OT KO mothers to lactate normally, the pups did not survive.
Figure 2.

Impaired OT receptor mRNA induction in COX-1 KO, but not COX-1 KO/OT KO mice. Total uterine RNA from nongravid (NG) or pregnant females of the gestational age (in days) and genotype given above the corresponding lanes were hybridized to OT receptor (OTR) or 18S ribosomal RNA (18S) radiolabeled probes.
Luteolysis, and the resulting fall in progesterone, is a critical component of normal murine labor initiation (3, 4), and the administration of exogenous progesterone delays the initiation of parturition (16, 17). Therefore, the mechanism by which COX-1 acts to promote parturition, and OT deficiency restores the timing of initiation of labor in the setting of maternal COX-1 deficiency, was explored by evaluation of the pattern of progesterone and PGF2α expression in each of the single and combined deficiency pregnancies. At day 16.5 of gestation, all genotypes demonstrated a high plasma progesterone concentration indicating intact ovarian corpus luteum function (Fig. 3). In wt, OT KO, and COX-1 KO/OT KO pregnancies at day 19 of gestation, decreases in progesterone to <25% of day 16.5 levels were found. Thus, the decrease in COX-1 mRNA expression just before delivery observed above occurs after luteolysis has been initiated because, in wt mice, plasma progesterone concentration is already significantly diminished at day 19 of gestation. In contrast, COX-1 KO gravid females at day 19 of gestation demonstrated plasma progesterone concentrations significantly above those in wt, COX-1 KO, and COX-1 KO/OT KO mice. The fall in plasma progesterone resulting from involution of the corpus luteum rather than alteration of progesterone metabolism is further supported by the histologic appearance the ovary at day 19 of gestation as a function of genotype. wt and COX-1 KO/OT KO corpora lutea demonstrate an amorphous cellular appearance and disruption of luteal architecture, whereas the COX-1 corpus luteum maintains prominent vascular spaces, uniform cellular appearance, and an organized luteal architecture (Fig. 4). Hence, COX-1 KO females manifest delayed luteolysis, with simultaneous OT deficiency restoring the normal timing. OT receptor expression in uterus at day 19 was low in the COX-1 KO mice consistent with the elevated plasma progesterone, whereas induction of OT receptor was found in the COX-1 KO/OT KO females to an extent similar to wt mice (Fig. 2).
Figure 3.

Plasma progesterone concentration in COX-1 KO, OT KO, and COX-1 KO/OT KO mice. Blood was collected by retroorbital phlebotomy in n = 4–6 mice per genotype group, and plasma progesterone concentration was determined. Data are presented as mean ± SEM. ∗, P < 0.01 vs. wt, OT KO, and COX-1 KO/OT KO at day 19.
Figure 4.
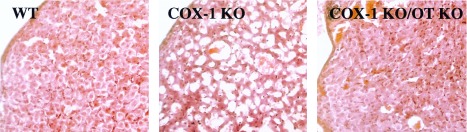
Corpus luteum histology at day 19 of gestation in wt, COX-1 KO, and COX-1 KO/OT KO mice. Luteal architecture and cell morphology in wt and COX-1 KO/OT KO mice appear similar and differ from COX-1 KO mice. (Hematoxylin/eosin-stained sections were analyzed by light microscopy at ×200 magnification.)
The restoration of the timing of luteolysis in the COX-1 KO/OT KO mice could result from OT deficiency increasing PGF2α production, possibly via induction of COX-2. Alternatively, OT may be luteotrophic in late gestation, maintaining progesterone production by the corpus luteum until a luteolytic threshold, primarily determined by PGF2α, is achieved. In this case, OT deficiency would reduce the ability of the corpus luteum to withstand the lytic action of the small amount of residual prostaglandin, or alternate luteolytic factor, production present in the COX-1 KO mice. To differentiate these hypotheses, uterine PGF2α concentration at day 19 of gestation was measured. PGF2α increased markedly at the end of gestation in wt mice, and to a similar extent in OT KO mice, demonstrating that OT is not required for induction of uterine prostaglandin synthesis at term (Fig. 5). Both COX-1 KO and COX-1 KO/OT KO mice had uterine PGF2α concentrations only 1–3% of wt or OT KO values (Fig. 5). Likewise, ovarian PGF2α was significantly elevated in wt (14.3 ± 3.7 pg/mg ovary) as compared with COX-1 KO (1.3 ± 0.4 pg/mg ovary, P < 0.05 vs. wt) and COX-1 KO/OT KO (0.9 ± 0.1 pg/mg ovary, P < 0.05 vs. wt) pregnancies at day 19 of gestation. Therefore, in the setting of OT deficiency, luteolysis occurs without the need for the marked increase in prostaglandin production found just before delivery in wt mice. A slight decrease in serum progesterone is suggested in the COX-1 KO females with advancing gestation (Fig. 3), without a significant rise in PGF2α. This finding, together with delayed rather than absent parturition in COX-1 KO females, implicates a role for low levels of PGF2α, or other hormones capable of interacting with the PGF2α receptor, in contributing to luteolysis and subsequent parturition. However, this process may be inefficient when OT is present.
Figure 5.

Impaired uterine PGF2α production in COX-1 KO and COX-1 KO/OT KO mice. Prostaglandins were extracted from uterus isolated from the indicated genotypes, and PGF2α concentration was determined by ELISA. Assay specificity was confirmed by administration of indomethacin (Indo) to a subset of wt mice at day 15.5 of gestation for suppression of prostaglandin production. Data are presented as mean, with day 19 samples ± SEM. ∗, P < 0.05 vs. wt at day 19.
Both luteotrophic and luteolytic roles for OT during pseudopregnancy in rats, the estrous cycle in sheep, and the periovulatory period in primates have been proposed (18–21). Our data suggests an unanticipated trophic role for OT in corpus luteum function in late murine gestation, either directly or via stimulation of other factors. This trophic function is not required throughout gestation, because OT KO mice do not exhibit preterm labor, but would serve to stabilize progesterone synthesis as PG levels rise before parturition. During pregnancy in the mouse, the rise in PG production occurs quite rapidly just before the onset of labor (Fig. 5), minimizing the impact of isolated OT deficiency. Thus, one critical function for COX-1 is to generate sufficient PGF2α to overcome the luteotrophic action of OT in late gestation. The marked rise in uterine PGF2α concentration occurs at least 24–48 h after high level uterine COX-1 mRNA expression is observed, and PGF2α level is maximal as the COX-1 mRNA level is declining. Explanation of this relationship requires further investigation but could be accounted for by substrate (arachidonic acid) limitation until uterine phospholipase activity is induced by other regulators of parturition, alterations in PGF2α metabolism by prostaglandin dehydrogenase, or contribution to uterine PGF2α by COX-1 activity outside of the uterus.
In summary, we have shown that COX-1 is essential for normal parturition in the mouse and undergoes a robust increase in expression during gestation in the gravid uterus especially in the decidua. COX-1 deficiency results in low level PGF2α production, which inadequately initiates luteolysis and the associated decrease in plasma progesterone. It is unclear how this mechanism translates to other species, such as humans, not dependent on the corpus luteum for maintenance of pregnancy late in gestation (22). If COX-1 serves a related function in human pregnancy, our findings suggest that maternal COX-1 inhibition by isoform-specific nonsteroidal anti-inflammatory drugs may be a therapeutic intervention for preterm labor. However, recent studies evaluating expression of COX-1 and COX-2 during human pregnancy suggest that COX-2 induction may be important for the elevated PG production seen in human labor (23–25). The prolongation of the active phase of labor we observe in the combined OT-COX-1 KO pregnancies indicates redundancy in OT and prostaglandin action for progression of labor because the duration of labor is not significantly altered in either single deficiency state. Surprisingly, we also have found that OT deficiency allows a decline in progesterone when PGF2α remains at low levels, implicating the balance of luteolytic and luteotrophic influences as defining the onset of murine labor. The induction of uterine OT receptor expression just before the onset of labor provides a mechanism by which the uterotonic effect of OT may be increased without an increase in OT expression, which could prolong gestation via effects on corpus luteum function.
Acknowledgments
We thank Dr. S. Morham for generously providing COX-1-KO mice, Dr. T. Simon for assistance with generation of pseudopregnant mice, and Drs. J. Masferrer, J. Gitlin, D. Kipnis, and A. Schwartz for helpful discussions. This work was supported by grants from the National Institutes of Health (2 P60 DK20579 and K08 DK02270), a Burroughs Wellcome Fund Career Development Award in the Biomedical Sciences (to L.J.M.), and the Searle/Monsanto Co. (to Y.S., L.M.M., and L.J.M.). L.J.M. is a Scholar of the Child Health Research Center of Excellence in Developmental Biology at Washington University School of Medicine (HD33688).
ABBREVIATIONS
- COX
cyclooxygenase
- OT
oxytocin
- PG
prostaglandin
- KO
deficient
- wt
wild type
References
- 1. Crowley P, Chalmers M J N C. Br J Obstet Gynaecol. 1990;97:11–25. doi: 10.1111/j.1471-0528.1990.tb01711.x. [DOI] [PubMed] [Google Scholar]
- 2.NIH Concensus Development Panel on the Effect of Corticosteroids for Fetal Maturation on Perinatal Outcomes. J Am Med Assoc. 1995;273:413–418. doi: 10.1001/jama.1995.03520290065031. [DOI] [PubMed] [Google Scholar]
- 3.Uozumi N, Kume K, Nagase T, Nakatani N, Ishii S, Tashiro F, Komagata Y, Maki K, Ikuta K, Ouchi Y, et al. Nature (London) 1997;390:618–622. doi: 10.1038/37622. [DOI] [PubMed] [Google Scholar]
- 4.Sugimoto Y, Yamasaki A, Segi E, Tsuboi K, Aze Y, Nishimura T, Oida H, Yoshida N, Tanaka T, Katsuyama M, et al. Science. 1997;277:681–683. doi: 10.1126/science.277.5326.681. [DOI] [PubMed] [Google Scholar]
- 5.Larcher A, Neculcea J, Breton C, Arslan A, Rozen F, Russo C, Zingg H H. Endocrinology. 1995;136:5350–5356. doi: 10.1210/endo.136.12.7588281. [DOI] [PubMed] [Google Scholar]
- 6.Kubota Y, Kimura T, Hashimoto K, Tokugawa Y, Nobunaga K, Azuma C, Saji F, Murata Y. Mol Cell Endocrinol. 1996;124:25–32. doi: 10.1016/s0303-7207(96)03923-8. [DOI] [PubMed] [Google Scholar]
- 7.Smith W L, DeWitt D L. Adv Immunol. 1996;62:167–215. doi: 10.1016/s0065-2776(08)60430-7. [DOI] [PubMed] [Google Scholar]
- 8.Simmons D L, Xie W, Chipman J G, Evett G E. In: Prostaglandins, Leukotrienes, Lipoxins, and PAF. Bailey J M, editor. New York: Plenum; 1992. pp. 67–78. [Google Scholar]
- 9.Fu J Y, Masferrer J L, Seibert K, Raz A, Needleman P. J Biol Chem. 1990;265:16737–16740. [PubMed] [Google Scholar]
- 10.Evett G E, Xie W, Chipman J G, Robertson D L, Simmons D L. Arch Biochem Biophys. 1993;306:169–177. doi: 10.1006/abbi.1993.1496. [DOI] [PubMed] [Google Scholar]
- 11.Langenbach R, Morham S G, Tiano H F, Loftin C D, Ghanayem B I, Chulada P C, Mahler J F, Lee C A, Goulding E H, Kluckman K D, et al. Cell. 1995;83:483–492. doi: 10.1016/0092-8674(95)90126-4. [DOI] [PubMed] [Google Scholar]
- 12.Hogan B, Beddington R, Constantini F, Lacy E. Manipulating the Mouse Embryo: A Laboratory Manual. Plainview, NY: Cold Spring Harbor Lab. Press; 1994. [Google Scholar]
- 13.Chirgwin J M, Przybyla A E, MacDonald R J, Rutter W J. Biochemistry. 1979;18:5294–5299. doi: 10.1021/bi00591a005. [DOI] [PubMed] [Google Scholar]
- 14.Nishimori K, Young L J, Guo Q, Wang Z, Insel T R, Matzuk M M. Proc Natl Acad Sci USA. 1996;93:11699–11704. doi: 10.1073/pnas.93.21.11699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Young W S I, Shepard E, Amico J, Henninghausen L, Wagner K-U, LaMarca M E, McKinney A, Ginns E. J Neuroendocrinol. 1996;8:847–853. doi: 10.1046/j.1365-2826.1996.05266.x. [DOI] [PubMed] [Google Scholar]
- 16.Thorburn G D, Challis J R G. Physiol Rev. 1979;59:863–917. doi: 10.1152/physrev.1979.59.4.863. [DOI] [PubMed] [Google Scholar]
- 17.Mahendroo M S, Cala K M, Russell D W. Mol Endocrinol. 1996;10:380–392. doi: 10.1210/mend.10.4.8721983. [DOI] [PubMed] [Google Scholar]
- 18.Sheldrick E L. Reprod Fertil Dev. 1992;4:505–513. doi: 10.1071/rd9920505. [DOI] [PubMed] [Google Scholar]
- 19.Einspanier A, Jurdzinski A, Hodges J K. Biol Reprod. 1997;57:16–26. doi: 10.1095/biolreprod57.1.16. [DOI] [PubMed] [Google Scholar]
- 20.Motta A B, Franchi A M, Faletti A, Gimeno M F. Prostaglandins, Leukotrienes Essent Fatty Acids. 1996;54:95–100. doi: 10.1016/s0952-3278(96)90065-4. [DOI] [PubMed] [Google Scholar]
- 21.McCracken J A, Custer E E, Lamsa J C, Robinson A G. Adv Exp Med Biol. 1998;395:133–154. [PubMed] [Google Scholar]
- 22.Challis J R G, Olson D M. In: The Physiology of Reproduction. Knobil E, Neill J, editors. New York: Raven; 1988. pp. 2177–2234. [Google Scholar]
- 23.Mijovic J E, Zakar T, Nairn T K, Olson D M. Am J Physiol. 1997;272:E832–E840. doi: 10.1152/ajpendo.1997.272.5.E832. [DOI] [PubMed] [Google Scholar]
- 24.Fuentes A, Spaziani E P, O’Brien W F. Prostaglandins. 1996;52:261–267. doi: 10.1016/s0090-6980(96)00088-3. [DOI] [PubMed] [Google Scholar]
- 25.Hirst J J, Teixeira F J, Zakar T, Olson D M. Reprod Fertil Dev. 1995;7:633–637. doi: 10.1071/rd9950633. [DOI] [PubMed] [Google Scholar]