

Abstract
Round embryonic mesenchymal cells have the potential to differentiate into smooth muscle (SM) cells upon spreading/elongation (Yang, Y., K.C. Palmer, N. Relan, C. Diglio, and L. Schuger. 1998. Development. 125:2621–2629; Yang, Y., N.K. Relan, D.A. Przywara, and L. Schuger. 1999. Development. 126:3027–3033; Yang, Y., S. Beqaj, P. Kemp, I. Ariel, and L. Schuger. 2000. J. Clin. Invest. 106:1321–1330). In the developing lung, this process is stimulated by peribronchial accumulation of laminin (LN)-2 (Relan, N.K., Y. Yang, S. Beqaj, J.H. Miner, and L. Schuger. 1999. J. Cell Biol. 147:1341–1350). Here we show that LN-2 stimulates bronchial myogenesis by down-regulating RhoA activity. Immunohistochemistry, immunoblotting, and reverse transcriptase–PCR indicated that RhoA, a small GTPase signaling protein, is abundant in undifferentiated embryonic mesenchymal cells and that its levels decrease along with SM myogenesis. Functional studies using agonists and antagonists of RhoA activation and dominant positive and negative plasmid constructs demonstrated that high RhoA activity was required to maintain the round undifferentiated mesenchymal cell phenotype. This was in part achieved by restricting the localization of the myogenic transcription factor serum response factor (SRF) mostly to the mesenchymal cell cytoplasm. Upon spreading on LN-2 but not on other main components of the extracellular matrix, the activity and level of RhoA decreased rapidly, resulting in translocation of SRF to the nucleus. Both cell elongation and SRF translocation were prevented by overexpression of dominant positive RhoA. Once the cells underwent SM differentiation, up-regulation of RhoA activity induced rather than inhibited SM gene expression. Therefore, our studies suggest a novel mechanism whereby LN-2 and RhoA modulate SM myogenesis.
Keywords: laminin-2; RhoA; SRF; smooth muscle; lung
Introduction
Visceral smooth muscle (SM)* originates from local mesenchymal cells that early in midgestation begin to elongate and synthesize SM proteins (Sawtell and Lessard, 1989; Roman and McDonald, 1992; McHugh, 1995; Li et al., 1996). Our studies indicated that this change in cell shape plays an important role in determining their myogenic fate. Using different means to control the cell's shape in cell culture (Schuger et al., 1996; Yang et al., 1998, 1999; Relan et al., 1999) and lung organ culture (Schuger et al., 1997; Yang et al., 2000), we found that essentially all undifferentiated embryonic mesenchymal cells are potential SM cell precursors but only undergo SM differentiation upon spreading/elongation. By contrast, prevention of cell elongation in vivo and in vitro prevents myogenesis even in mesenchymal cells that would normally become SM cells (Schuger et al., 1997; Yang et al., 1998, 1999, 2000; Relan et al., 1999).
Peribronchial mesenchymal cell shape is controlled, at least in part, by the surrounding extracellular matrix (ECM). We found that among the ECM components that are synthesized by the developing lung laminin (LN)-1 and mainly LN-2 promote bronchial myogenesis by stimulating peribronchial mesenchymal cell spreading/elongation (Schuger et al., 1997; Yang et al., 1998; Relan et al., 1999). LNs are a large family of basement membrane (BM)-related glycoproteins with important roles in cell adhesion, differentiation, and morphogenesis (Colognato and Yurchenco, 2000). LNs are composed of α, β, and γ chains linked together by disulfide bonds and are classified by their chain composition (Burgeson et al., 1994). LN-1 is composed of α1, β1, and γ1 chains, and LN-2 is composed of α2, β1, and γ1 chains. LNs along with collagen (CN)-IV and perlecan constitute the main scaffold of all BMs.
In the developing lung, LN-1 synthesis is induced by direct epithelial–mesenchymal interaction (Schuger et al., 1997). LN-1 is then secreted to the epithelial–mesenchymal interface where it polymerizes, thus contributing to BM assembly (Schuger et al., 1995, 1996, 1997; Yang et al., 1998). Round undifferentiated mesenchymal cells use the developing BM to spread and upon spreading/elongation begin to both synthesize LN-2 and to undergo myogenic differentiation (Relan et al., 1999). The surrounding mesenchymal cells spread/elongate on the newly deposited LN-2, differentiating into additional SM cells (Relan et al., 1999). Absence of functional LN-2 leads to skeletal muscle dystrophy, cardiomyopathy, and short hypoplastic bronchial muscle cells in dy/dy mice (Hillaire et al., 1994; Sunada et al., 1994; Xu et al., 1994a,b; Miyagoe et al., 1997; Relan et al., 1999).
To elucidate the molecular mechanisms that link cell shape with SM myogenesis, we searched a PCR-based differential expression cDNA library created from round undifferentiated mesenchymal cells subtracted with mesenchymal cells undergoing spread-induced SM myogenesis. The expression of the rho-a gene was found to be high in round undifferentiated mesenchymal cells and to decline significantly upon cell elongation. RhoA together with Rac and Cdc42 belongs to a family of small guanidine nucleotide (GTP) binding proteins (GTPases) that plays a critical role in the organization of the actin cytoskeleton (Hall, 1998; Bishop and Hall, 2000; Evers et al., 2000). These proteins cycle between an inactive (GDP-bound) and active (GTP-bound) conformation in which they interact with specific effector proteins. Active Rho is involved in cell contractility, whereas active Rac and Cdc42 induce extension of lamellipodia and filopodia, contributing to cell migration (Hall, 1998; Bishop and Hall, 2000; Evers et al., 2000).
In this study, we show that the activity and level of RhoA in lung embryonic mesenchymal cells are controlled by LN-2. More specifically, our data suggest that high RhoA activity is required to maintain the round undifferentiated mesenchymal cell phenotype; spreading on LN-2 induces a drastic down-regulation of both the activation state and level of RhoA, and this results in cell elongation and irreversible nuclear translocation of the myogenic transcription factor serum response factor (SRF). Consistent with previous studies (Takano et al., 1998; Wei et al., 1998; Mack et al., 2001), we found that once the cells differentiate into SM RhoA activation stimulates rather than inhibits myogenesis. Our data provide novel clues on the mechanism whereby LN-2 regulates SM myogenesis and suggest a surprisingly complex role for RhoA in this process.
Results
RhoA expression decreased along with bronchial myogenesis
As we showed previously (Yang et al., 1998, 1999), undifferentiated mesenchymal cells from embryonic lungs undergo spontaneous SM differentiation upon spreading in culture (Fig. 1 A). Here we used this culture system to generate a PCR-based cDNA differential expression library to search for potential suppressors of SM myogenesis. Screening of 300 transformed colonies identified the cDNA for RhoA as one of those that was significantly more abundant in undifferentiated cells compared with SM-differentiating cells. Reverse transcriptase (RT)-PCR analysis, immunoblot, and immunohistochemistry confirmed that undifferentiated lung mesenchymal cells expressed high levels of active RhoA and that both the activation state and the level of RhoA expression decreased rapidly along with SM differentiation (Fig. 1 B). Unlike RhoA, the level of Rac remained constant (Fig. 1 B). Down-regulation of RhoA expression was confirmed in vivo by immunohistochemical studies performed on E11 and E14 lung frozen sections (Fig. 1 C). These studies showed that in the developing lung, RhoA synthesis decreases in all cells between E11 and E14; however, bronchial SM cells show the most significant drop in RhoA synthesis (Fig. 1 C).
Figure 1.

RhoA expression and activity decrease during SM differentiation in vitro and in vivo. (A and B) Undifferentiated mesenchymal cells isolated from E11 mouse embryonic lungs were cultured for 1 and 18 h. (A) As shown previously (Yang et al., 1999), undifferentiated mesenchymal cells undergo spread-induced SM differentiation. This is in part indicated by the expression of SM-related proteins. In contrast, α-fetoprotein, an embryonic protein, decreases with myogenic cell differentiation. (B) RT-PCR, Western blot analysis, and immunohistochemistry demonstrating rho-a differential expression in embryonic mesenchymal cells undergoing spread-induced SM differentiation in culture. Densitometry analysis showed 8.2-, 7.5-, and 11.5-fold decrease in RhoA mRNA, protein, and activity levels, respectively, after 18 h in culture. No changes were observed in Rac protein levels. Immunohistochemistry, on the bottom left and right, shows high level of cytoplasmic RhoA in round undifferentiated mesenchymal cells and weak, almost negative, RhoA immunostaining in a confluent monolayer of spread SM-differentiating cells. Notice a single cell that remained round and strongly positive for RhoA. (C) RhoA immunolocalization in mouse embryonic lung at days 11 and 14 of gestation. RhoA is diffusely present in the cytoplasm of undifferentiated mesenchymal cells in both gestational stages, although it is more intense at day 11. The elongated bronchial SM cells (SM) show very weak RhoA immunostaining. The small insets represent parallel sections immunostained for SM α-actin to demonstrate absence of SM in E11 lung and its presence in E14 lung. Bars: (C) 10 μm; (insets) 30 μm.
Up-regulation of RhoA activity delayed SM myogenesis
Undifferentiated mesenchymal cells were isolated from the embryonic lung and treated with various agonists and antagonists of RhoA activation. The treatments were added before the cells spread (1 h after plating). Treatment of mesenchymal cell cultures with 1 μM of the agonist endothelin-1 (Fleming et al., 1996) stimulated RhoA activity and delayed SM-specific gene expression (Fig. 2 , A–C). Fig. 2 A demonstrates that the decrease in RhoA activity normally occurring in mesenchymal cell cultures undergoing SM differentiation was prevented in the first hours by endothelin-1 treatment. These cells remained round for a longer period of time (3–5 h) and exhibited a significant delay in SM myogenesis as indicated by the lower expression of SM α-actin, desmin, and SM-myosin (Fig. 2, B and C). Lysophosphatidic acid (LPA), another agonist for RhoA activation (Hirshman and Emala, 1999), stimulated RhoA activity, resulting in prolonged cell rounding and inhibition of SM myogenesis (Fig. 2, D and E). C3 exoenzyme, an antagonist of RhoA (Leonard et al., 1992), inhibited RhoA activity in SM-differentiating cultures, and this resulted in stimulation of SM myogenesis (Fig. 2 F). After 2–3 d in culture, the effect of RhoA activation on SM gene expression changed from inhibitory to stimulatory (Fig. 2 G).
Figure 2.

Up-regulation of RhoA activity delays SM myogenesis, but once the cells are fully differentiated RhoA stimulates SM gene expression. Undifferentiated mesenchymal cells were isolated from E11 mouse embryonic lungs, allowed to attach, and cultured for up to 8 h in the presence of RhoA agonists endothelin-1 and LPA (A–E) or in the presence of RhoA antagonist C3 (F). Additional cells were allowed to differentiate for 4 d and were then stimulated with endothelin-1 (G). (A) The decrease in RhoA activity normally occurring in mesenchymal cell cultures undergoing spread-induced SM differentiation is prevented in the first hours of culture by 1 μM endothelin-1. These cultures exhibit a significant delay in SM myogenesis as indicated by the lower expression of SM α-actin, desmin, and SM-myosin mRNA (18 S represents the internal control) (B) and protein (C). (D) LPA at a concentration of 10 μM stimulates RhoA activity and as endothelin-1 also inhibits SM myogenesis (E). (F) 10 μM C3 inhibits RhoA activity in the SM-differentiating cultures, and this results in stimulation of SM myogenesis. (G) Once mesenchymal cells differentiate into SM cells after 4 d in culture (Yang et al., 1999), their response to RhoA activation reverses, and then 1 μM endothelin-1 stimulates SM gene expression.
RhoA modulated SM myogenesis by modulating mesenchymal cell shape
Undifferentiated mesenchymal cells were isolated from the lung and transfected with dominant positive (RhoA+) or dominant negative (RhoA−) hemagglutinin (HA)1-tagged RhoA plasmid constructs before spreading (1 h after plating). Transfection of mesenchymal cell cultures with RhoA+ kept the cells round and blocked SM myogenesis (Fig. 3 A, left), whereas transfection of mesenchymal cell cultures with RhoA− facilitated spontaneous cell spreading/elongation (Fig. 3 A, right). The cells expressing RhoA+ remained negative for SM markers such as desmin (Fig. 3 A, bottom left), whereas the spread cells differentiated into SM cells as usual (Fig. 3 A, bottom right). Overexpression of wild-type RhoA caused an effect similar to RhoA+ (unpublished data). Approximately 25–30% of mesenchymal cells in primary culture were transfected with the RhoA constructs; therefore, immunoblots showed a corresponding decrease in SM markers in the cultures transfected with RhoA+ (Fig. 3 B). Since mesenchymal cells transfected with RhoA− spread faster than the nontransfected cells in the same culture, this resulted in an increase in SM gene expression compared with controls (Fig. 3 B). Cells transfected with RhoA+ detached relatively more easily; therefore, cultures were lightly trypsinized for 2 min, and the detached cells were collected for antitag immunostaining (to confirm selection of transfected cells) and for immunoblot analysis. This enriched population of RhoA+ cells had total RhoA levels (unpublished data) and activity similar to those found in round undifferentiated mesenchymal cells (Fig. 3 C) and did not undergo SM myogenesis (Fig. 3 D).
Figure 3.

Transfection studies with RhoA + and RhoA − plasmid constructs, showing that RhoA modulates SM myogenesis by modulating mesenchymal cell shape. Undifferentiated mesenchymal cells were isolated from E11 mouse embryonic lungs and transfected with the two different RhoA plasmid constructs before spreading (1 h after plating). (A) Cells transfected with RhoA+ and immunostained with anti-HA1 tag remain round in shape (top left). Spread nontransfected cells in the same picture cannot be visualized because they are not stained and are in a different focus. Round cells do not immunostain for desmin (bottom left; arrow points to one of these cells). In the same picture, note desmin-positive spread cells (arrowhead points to one of them). Cells transfected with RhoA− have spread (top right) and demonstrate strong immunopositivity for desmin (bottom right). (B) Immunoblots of cells transfected with RhoA+ and RhoA−. Mesenchymal cells transfected with RhoA− spread faster than the nontransfected cells in the same culture, leading to an increase in SM gene expression. For comparison, mitogen-activated protein kinases show no differences in levels. (C and D) RhoA+-transfected mesenchymal cells, which detach faster than spread nontransfected counterparts, were collected from the cultures by short trypsinization and immediately lysed. (C) After 24 h, cells expressing RhoA+ have the same RhoA activity as control undifferentiated mesenchymal cells 1 h after isolation. Here RhoA activity assay was run on 3.5 μg of protein per lane. (D) Cells expressing RhoA+ do not undergo SM myogenesis as indicated by the absence of SM myosin, desmin, and SM α-actin. Note that 3 μg protein/lane was loaded for this experiment rather than the 15 μg protein loaded in other experiments. Thus, the bands seen in the vector control sample are less intense than in other panels. Bar, 20 μm.
Mesenchymal cell spreading on LN-2 induced maximal RhoA down-regulation
Undifferentiated mesenchymal cells were trypsinized from embryonic lungs and immediately plated on poly-l-lysine, CN-I, CN-IV, LN-1, or LN-2 coated onto nontissue culture dishes. These experiments showed that RhoA levels and activity decreased in mesenchymal cells spreading on all the ECM substrata, but the decrease was most marked on LN-2 (Fig. 4 A). Rac levels were not affected (Fig. 4 A). Maximal cell spreading/elongation was seen on CN-IV followed by LN-2 (Fig. 4 B). Since cell spreading in itself induces LN-2 synthesis (Relan et al., 1999), 10 μg/ml of anti–LN-2 antibody was added to the cultures to block LN-2 (Relan et al., 1999). Under these conditions, the cells did not spread and RhoA levels did not decrease (Fig. 4 A). Reprobing of the membranes with antibodies to SM α-actin, desmin, and SM22 indicated that LN-2 represented the best stimulus for SM myogenesis among the ECM constituents studied (Fig. 4 C). To further test the relationship between LN-2 and RhoA, we compared the levels of RhoA in lungs from dy/dy, which express very low levels of functionally active LN-2 (Sunada et al., 1994; Xu et al., 1994a,b). Immunoblot studies done on three different affected mice and controls demonstrated that dy/dy lungs had significantly higher levels of RhoA protein (Fig. 4 D) and message (unpublished data).
Figure 4.

Mesenchymal cell spreading/elongation on LN-2 causes maximal decrease in the activity and level of RhoA and maximal stimulation of SM myogenesis. (A) Undifferentiated mesenchymal cells were isolated from E11 mouse embryonic lungs and immediately plated on 0.05% poly-l-lysine PLL, CN-1, CN-IV, LN-1, or LN-2 coated onto nontissue culture dishes. RhoA levels and activity decreased rapidly in mesenchymal cells spreading on all of the ECM substrata and particularly on LN-2 but remained high in round cells (plated on PLL). Rac levels were not affected. Since cell elongation per se induces LN-2 synthesis in mesenchymal cells (Relan et al., 1999), 10 μg/ml of anti–LN-2 antibody (X-LN-2) were added to the cultures to block LN-2 activity (Relan et al., 1999). Under these conditions, the cells did not spread and RhoA levels did not decrease. (B) Histogram showing the average length achieved by the cells on different substrata. UNC, uncoated. Note that cells acquired the longest diameters on CN-IV rather than LN-2. Nevertheless, reprobing the membranes shown in A with antibodies to SM myosin, desmin, and SM22 indicates that LN-2 represents the best stimulus for SM myogenesis among the ECM constituents studied (C). (D) RhoA levels in lungs from three dy/dy mice and three matched control animals, showing that dy/dy lungs have significantly higher levels of RhoA. The bottom panels show staining of the blots with Coomassie blue to demonstrate equal protein loading in each set of dy/dy and control lungs.
LN-2 down-regulated RhoA activity in the absence of cell elongation
Cell spreading/elongation was prevented by plating the cells either on 0.05% poly-l-lysine or on 10-μm diameter culture microsurfaces (Yang et al., 1999). CN-I, CN-IV, LN-1, and LN-2 were added to the culture medium once cell attachment was completed (1 h after plating). Under these conditions, only soluble LN-2 decreased RhoA activity. However, RhoA levels remained unchanged (Fig. 5 A). Similarly, mesenchymal cells plated on 10 μm microsurfaces, to keep cells round (Yang et al., 1999), showed a reduction in RhoA activity without a reduction in RhoA levels upon exposure to LN-2 (Fig. 5 B). Whether plated on poly-l-lysine or microsurfaces, the cells did not undergo myogenesis (unpublished data). To elucidate whether or not LN-2 had a distinct effect on focal adhesion kinase (FAK) activation, we performed immunoblots with three different FAK phosphospecific antibodies. These showed no major differences in FAK phosphorylation by the various ECM proteins studied. Actually, CN-I and CN-IV stimulated FAK phosphorylation slightly more than LN-1 and LN-2 (Fig. 5 C). When poly-l-lysine concentration was decreased to the minimal level required to prevent spontaneous cell spreading in culture (0.01%), only LN-2 and to a lesser extent LN-1 stimulated mesenchymal cell spreading/elongation (Fig. 5 D) and down-regulated RhoA levels (Fig. 5 E). A 10-fold reduction in LN-2 was still sufficient to induce cell elongation (unpublished data).
Figure 5.

LN-2 down-regulates RhoA activity in the absence of cell elongation. Undifferentiated embryonic mesenchymal cells were isolated from E11 mouse embryonic lungs and plated on poly-l-lysine (A, D, and E) or on 10-μm diameter culture microsurfaces (Yang et al., 1999) (B). CN-I, CN-IV, LN-1, LN-2, or no ECM (no-M) were added to the culture medium once cell attachment was completed (1 h after plating). (A) Cells plated on 0.05% poly-l-lysine, showing that soluble LN-2 decreases RhoA activity. Note that RhoA levels remain unchanged. (B) Mesenchymal cells plated on 10 μm microsurfaces also showing a reduction in RhoA activity without a reduction in RhoA levels upon exposure to LN-2. (C) Immunoblots with three different FAK phosphospecific antibodies, showing that LN-2 elicits low FAK phosphorylation compared with other ECM constituents. (D) Poly-l-lysine was reduced to 0.01% (minimal level required to prevent spontaneous cell spreading). Under these conditions, LN-2 and to a minimal extent LN-1 stimulate mesenchymal cell spreading/elongation. (E) LN-2– and LN-1–induced mesenchymal cell elongation results in down-regulation of both RhoA activity and level.
RhoA activity determined intracellular localization of SRF
Immunohistochemical studies showed that SRF is localized mostly in the cytoplasm of undifferentiated mesenchymal cells. However, upon spreading/elongation SRF translocates to the nucleus (Fig. 6 A). SRF translocation does not occur in undifferentiated mesenchymal cells expressing RhoA+ (Fig. 6 A, right). c-myc, another transcription factor present in these cells, localized to the nucleus of undifferentiated mesenchymal cells and did not translocate after changes in cell shape or RhoA activity (unpublished data). Currently, there is no antibody available that can distinguish between SRF and its truncated isoform SRFΔ5. However, the absence of SRFΔ5 in RT-PCR analysis of spread/elongated cells indicated that the nuclear immunoreactivity detected in these cells represented the full SRF isoform (Fig. 6 B). LN-2 or C3 treatments were not sufficient to induce SRF nuclear translocation in the absence of cell spreading (unpublished data). Immunohistochemical studies performed on E11 and E15 embryonic lung sections demonstrated cytoplasmic localization of SRF in undifferentiated mesenchymal cells (Fig. 6 C, top) and its nuclear localization in SM-differentiated cells (Fig. 6 C, bottom). Neither forced cell rounding (Fig. 6 D) nor constitutively active RhoA displaced SRF back to the cytoplasm (Fig. 6 E). It should be stressed that we were unable to develop a double immunostaining for SRF and HA1-tagged RhoA. However, since ∼25–30% of the cells were transfected we should have been able to detect changes in SRF localization if such changes had taken place. Screening of >1,000 cells revealed no cytoplasmic translocation of SRF after RhoA+ transfection.
Figure 6.
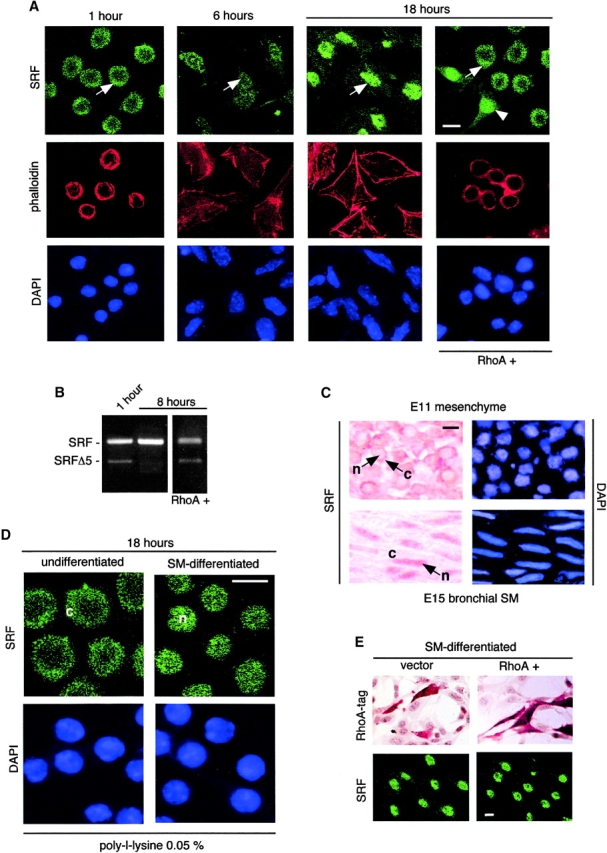
In undifferentiated and immature mesenchymal cells, RhoA activity determines the intracellular localization of SRF. (A) Undifferentiated mesenchymal cells were isolated from E11 mouse embryonic lungs and allowed to undergo spread-induced SM differentiation. Some of the cultures were transfected with RhoA+ before spreading (1 h after plating). Cells were immunostained for SRF 1, 6, and 18 h after plating. In undifferentiated mesenchymal cells, SRF is localized mainly in the cytoplasm (arrow in first panel points to cytoplasm). SRF translocates to the nucleus along with SM differentiation (arrow in second panel points to a cell with nuclear and cytoplasmic SRF, and arrow in third panel points to nuclear SRF). SRF translocation does not occur in undifferentiated mesenchymal cells expressing RhoA+ (right, arrow points to cytoplasm). In same figure, notice presence of SRF in nuclei of spread untransfected cell (arrowhead). Phalloidin stain for F-actin (red) and DAPI stain (blue) are shown to respectively highlight the cytoplasm and nucleus and thereby facilitate interpretation of the figure. (B) RT-PCR shows presence of SRF and SRFΔ5 in undifferentiated mesenchymal cells, presence of SRF but not SRFΔ5 in spread cells 8 h after plating, and presence of both in RhoA+-transfected cells. (C) Immunohistochemical studies performed on E11 and E15 embryonic lung sections, revealing cytoplasmic localization of SRF in undifferentiated mesenchymal cells in vivo (top, arrow points to cytoplasm) and its nuclear localization in SM-differentiated cells in vivo (bottom, arrow points to nucleus). n, nucleus; c, cytoplasm. (D) Undifferentiated mesenchymal cells were cultured for either 1 or 24 h. The cells were then trypsinized, replated on poly-l-lysine, and cultured for another 18 h. At the end of the second culture period, the cells were immunostained for SRF. Although cells replated on poly-l-lysine after 1 h in regular culture still have most SRF immunoreactivity in the cytoplasm (left; c, cytoplasm), cells that were allowed to spread for 24 h before replating on poly-l-lysine have all SRF immunoreactivity in the nucleus (right; n, nucleus). (E) Spread SM-differentiating mesenchymal cells transfected with RhoA+ or vector alone and immunostained with anti-HA1 tag by immunoperoxidase (top) or with anti-SRF by immunofluorescence (bottom). Immunostaining for SRF shows no translocation of SRF to the cytoplasm. Notice that although we were unable to develop a double immunostaining for SRF and HA1-tagged RhoA, since ∼25–30% of the cells were transfected we should have been able to detect scattered cells with changes in SRF localization if such changes had taken place. Bars, 10 μm.
Discussion
RhoA and SM myogenesis
We showed previously that undifferentiated mesenchymal cells from embryonic lungs undergo spontaneous SM differentiation upon spreading in culture (Yang et al., 1998, 1999). Here we used this system to generate a subtracted cDNA library to search for suppressors of SM myogenesis. RhoA was identified as a potential candidate because of its relative abundance in undifferentiated mesenchymal cells. RT-PCR, immunoblotting, immunohistochemistry, and activity assays confirmed the rapid decrease in RhoA levels and activity upon SM myogenic differentiation. Down-regulation of RhoA expression during bronchial myogenesis was confirmed in vivo by immunohistochemistry.
Functional studies indicated that the differential expression of RhoA plays a critical role in regulating SM myogenesis. Treatment of round undifferentiated mesenchymal cells with RhoA agonists endothelin-1 and LPA delayed SM-specific gene expression and prolonged cell rounding. In contrast, C3 exoenzyme, an antagonist of RhoA, caused the opposite effect.
Interestingly, once the mesenchymal cells acquired synthetic and electrical properties of mature SM cells, which happens after 2–3 d in culture (Yang et al., 1999), the effect of RhoA activation on SM gene expression changed from inhibitory to stimulatory. These last data are consistent with previous studies, which linked RhoA activity with stimulation of muscle-specific gene expression in muscle cells and myofibroblasts (Takano et al., 1998; Wei et al., 1998; Mack et al., 2001). In these cells, the effect of RhoA activation depends upon stress fiber formation (Takano et al., 1998; Wei et al., 1998; Mack et al., 2001). However, another study showed that cytochalasin D, which disrupts stress fiber formation, activates RhoA in fibroblasts (Ren et al., 1999). Similarly, Laufs et al. (2000) found that the actin cytoskeleton exerts a negative feedback regulation on rho gene transcription in endothelial cells. Therefore, the effect of the cytoskeleton on RhoA activity seems to vary depending on the cell type. It may be critical that stress fibers are not well developed in undifferentiated mesenchymal cells and mesenchymal cells undergoing SM differentiation (this can be appreciated from the phalloidin stains presented in Fig. 6). This lack of stress fibers may explain why RhoA activity is high in SM cell precursors compared with mature SM cells, which have a well-developed actin cytoskeleton.
Cell shape, RhoA, and SM myogenesis
Since endothelin-1 and LPA also act through signaling pathways other than RhoA (Swarthout and Walling, 2000), we used RhoA+ and RhoA− plasmid constructs to further evaluate the involvement of RhoA in the process of SM myogenesis. Transfection of mesenchymal cell cultures with constitutively active RhoA (RhoA+) or wild-type RhoA kept the cells round and blocked SM myogenesis, whereas expression of RhoA− facilitated spontaneous cell spreading/elongation and SM differentiation as usual.
Therefore, these studies suggested that high RhoA activity is required to maintain a round undifferentiated mesenchymal cell phenotype, whereas a decrease in RhoA facilitates cell elongation and subsequent SM myogenesis. Previous studies have shown already that high RhoA activity may cause cell rounding. The best example is represented by neurons and neuroblastoma cells in which RhoA activity also causes neurite retraction (Katoh et al., 1996; Postma et al., 1996; Tigyi et al., 1996; Kozma et al., 1997). However, our studies are the first to indicate that RhoA may play a decisive role in determining the differentiation fate of embryonic cells through the control of the cell shape.
LN-2 and RhoA
Since cells require ECM to spread/elongate, we determined the effect of some of the main ECM constituents on mesenchymal cell spreading/elongation. CN-I was selected because it represents the main non-BM ECM component, CN-IV and LN-1 because they are major BM components, and LN-2 because it is the main LN in muscle BMs (Vachon et al., 1996). In addition, our previous studies indicated the importance of LN-2 in bronchial myogenesis (Relan et al., 1999). Poly-l-lysine was used to promote cell attachment without spreading (Yang et al., 1998, 1999; Relan et al., 1999). These experiments showed that RhoA levels and activity decreased in mesenchymal cells spreading on all the ECM substrata but the decrease was most marked on LN-2. Immunoblot analysis indicated that LN-2 was also the best stimulus for SM myogenesis, supporting our previous findings.
Despite the fact that LN-2 induced maximal RhoA down-regulation, cell spreading/elongation occurred at least the same on CN-IV, indicating that the effect of LN-2 on RhoA was not correlated directly with cell length. Since cell spreading in itself induces LN-2 synthesis (Relan et al., 1999), we added LN-2 blocking antibodies (Relan et al., 1999) to the cultures. Under these conditions, the cells did not spread and RhoA levels did not decrease, demonstrating the important role of LN-2 in inducing embryonic mesenchymal cell spreading/elongation. However, studies in which cells were forced to remain round while being exposed to LN-2 showed that LN-2-induced down-regulation of RhoA was insufficient to stimulate myogenesis. Therefore, these additional studies demonstrated that cell spreading/elongation is the dominant factor in promoting SM myogenesis.
Dy/dy mice express very low levels of functionally active LN-2 due to a spontaneous genetic mutation that results in a truncated LN α2 chain. This deficiency in active LN-2 leads to muscular dystrophy (Sunada et al., 1994; Xu et al., 1994a,b). On histological examination, bronchial SM cells in dy/dy mice are shorter than control animals and express less SM-specific protein (Relan et al., 1999). Supporting our in vitro data, immunoblot studies done on three different affected mice and controls demonstrated that dy/dy lungs had significantly higher RhoA levels. Nevertheless, we were surprised by the magnitude of this difference, and part of our current effort is directed to further understand this system.
Several other ECM molecules have been shown to modulate the activity of GTPases (Giancotti and Ruoslahti, 1999). Fibronectin was the first identified and the best studied so far. Fibronectin activates RhoA while inhibiting Cdc42 and thereby supports growth factor-stimulated cell cycle progression (Bourdoulous et al., 1998; Danen et al., 2000). Our ongoing studies seem to suggest that although mesenchymal cells spread on fibronectin, they maintain relatively high RhoA levels and show delayed SM differentiation. Interestingly, a similar positive and negative regulation of SM gene expression by LN and FN has been shown in adult vascular SM cells (Thyberg and Hultgardh-Nilsson, 1994). We are currently trying to determine whether the differences between LN-2 and fibronectin are related to their ability to organize the cell's cytoskeleton. In addition to the effects of fibronectin and LN-1 and -2 to modulate GTPase activity, it has been shown recently that tenascin-C suppresses fibronectin-induced RhoA activation (Wenk et al., 2000) and thrombospondin-1 stimulates fascin spikes by activating Rac and Cdc42 (Adams and Schwartz, 2000). Therefore, these studies underline the importance of the ECM in controlling GTPase function.
RhoA and SRF cytoplasmic/nuclear translocation
The myogenic transcription factor SRF is a member of the MADS (MCM-1, agamous, and deficiens and SRF) box family of transcription factors. SRF binds to the CArG box or CArG box–like motif, an essential cis element present in muscle-specific proteins, such as SM α-actin, SM22, SM myosin, β-tropomyosin, and caldesmon, and stimulates their transcription (Belaguli et al., 1997; Kim et al., 1997; Browning et al., 1998). SRF has several truncated isoforms, which originate by alternative splicing from the same pre-mRNA (Belaguli et al., 1999; Kemp and Metcalfe, 2000). Our previous studies indicated that undifferentiated mesenchymal cells synthesize SRF and SRFΔ5 isoforms but lose SRFΔ5 early during SM myogenesis (Yang et al., 2000). This change is of functional relevance because SRFΔ5 inhibits SM myogenesis (Yang et al., 2000).
In this study, we found that SRF immunoreactivity is localized mostly in the cytoplasm of undifferentiated mesenchymal cells. However, upon spreading/elongation SRF translocates to the nucleus. SRF translocation does not occur in undifferentiated mesenchymal cells expressing RhoA+, indicating the involvement of RhoA in this process. The absence of SRFΔ5 in RT-PCR analysis of spread/elongated cells indicated that the nuclear immunoreactivity detected in these cells represented the full SRF isoform. LN-2 or C3 treatments were not sufficient to induce SRF nuclear translocation in the absence of cell spreading, suggesting a critical role for cell spreading/elongation in stimulating SRF nuclear translocation.
A similar pattern of SRF cytoplasmic/nuclear translocation was observed previously during skeletal muscle differentiation (Croissant et al., 1996). These investigators found that SRF has cytoplasmic localization in undifferentiated myoblasts but becomes enriched in nuclei of differentiated myotubes. In most cells, however, xSRF seems to be largely restricted to the nucleus, although it has been shown to translocate to the cytoplasm during prophase (Gauthier-Rouviere et al., 1995) and after prolonged serum deprivation in culture (Camoretti-Mercado et al., 2000). The cytoplasmic/nuclear translocation of SRF observed during myogenesis may serve a critical developmental function by contributing to the initiation of SM myogenesis when the right conditions are established. During the developmental time that SRF is enriched in the cytoplasm, the low levels of SRF detected in the nucleus (Fig. 6 A) may still be sufficient to ensure transcription of nonmyogenic SRF-responsive genes, such as c-fos and β-actin. Alternatively, since SRF-responsive genes also have binding sites for other transcription factors, such as AP-1 for β-actin and AP-2 for c-fos, their transcription may be controlled by the latter rather than by SRF (Berkowitz et al., 1989).
A common mechanism for intracellular translocation of proteins, including transcription factors, involves their phosphorylation/dephosphorylation (Nakabeppu and Nathans, 1989; Metz and Ziff, 1991; Norris and Manley, 1992). Although SRF has several phosphorylation sites (Fluck et al., 2000), we were unable to detect differences in SRF phosphorylation during its cytoplasmic/nuclear translocation (unpublished data). Interestingly, maintenance of high RhoA activity by RhoA+ transfection prevents the disappearance of SRFΔ5 isoform as shown in Fig. 6 B. Since SRFΔ5 has the ability to dimerize with SRF and thereby cause inhibition of SM gene transcription (Belaguli et al., 1999), we are currently investigating the possibility that SRFΔ5 may hinder SRF nuclear translocation by dimerizing with SRF in the cytoplasm.
It has been established previously that RhoA activity stimulates SRF-dependent transcription in several cell types, including mature SM cells and terminally differentiated cardiomyocytes (Hill et al., 1995; Wei et al., 1998, 2001b; Sotiropoulos et al., 1999; Mack et al., 2001). In accordance with these reports, here we have found that mesenchymal cells reverse their response to RhoA upon myogenic differentiation. Furthermore, two recent papers have shown that active RhoA can either inhibit (Wei et al., 2001a) or stimulate (Lu et al., 2001) myogenesis in different developmental models. Together, these data plus previously discussed papers (Katoh et al., 1996; Postma et al., 1996; Tigyi et al., 1996; Kozma et al., 1997; Ren et al., 1999; Laufs et al., 2000) seem to support the concept that RhoA may serve different and even opposite functions, depending on the cell type, shape, cytoskeleton, and/or differentiation status.
Based on the studies presented here, we propose that during lung development high RhoA activity contributes to maintenance of mesenchymal cell rounding and thereby restricts SRF mainly to the cytoplasm, where it cannot activate SM gene transcription. Then gradual peribronchial accumulation of LN-2 and to a lesser extent LN-1 results in RhoA down-regulation and incipient cell spreading. This change in cell shape leads to further stimulation of LN-2 production, additional decrease in RhoA activity, further cell spreading, and eventually SRF translocation to the nucleus. All of these factors contribute to the induction of SM myogenesis. Furthermore, we also propose that higher RhoA activity in the nonperibronchial areas may be one of the mechanisms put in place to maintain mesenchymal cells undifferentiated until new cues are set to allow the emergence of nonmuscular cell lineages.
Materials and methods
Construction of libraries and subtracted probe
Undifferentiated mesenchymal cells were isolated from E11 mouse lungs by differential plating as described previously (Schuger et al., 1997; Yang et al., 1998, 1999). The cells were cultured for either 1 or 18 h, the first time point representing undifferentiated embryonic mesenchymal cells and the second representing cells undergoing SM differentiation. The mRNA from the two cultures was amplified using the SMART® cDNA synthesis kit (CLONTECH, Laboratories, Inc.), and PCR-Select® (CLONTECH, Laboratories, Inc.) was then used for subtractive hybridization. Briefly, two pools of RsaI-digested cDNA from undifferentiated cells were used as tester and ligated to two different oligonucleotide adapters. RsaI-digested cDNA from differentiated SM cells was used as driver without adapters. Two hybridizations were performed between the tester population and excess driver. Only the cDNAs with different adapters at both ends were PCR amplified and produced a pool of cDNA fragments more abundant in the undifferentiated than in the differentiated cells. The subtracted cDNAs were cloned into a pGEM-T vector (Promega) and transformed into Escherichia coli. 300 transformed colonies were selected randomly for screening. Dot-blot hybridization was performed with 32P-labeled cDNA forward- and reverse-subtracted cDNAs as probes. The reverse-subtracted probe was made by subtractive hybridization performed with the original tester cDNA as a driver and the driver cDNA as a tester.
Cell culture
Crl:CD-1 (ICR) BR mice (Charles River Laboratories) were mated, and the day of finding a vaginal plug was designated as day 0 of embryonic development. Undifferentiated mesenchymal cells were isolated from E11 or E12 lungs as described previously (Schuger et al., 1997; Yang et al., 1998, 1999) and immediately plated on uncoated tissue culture dishes or on nontissue culture dishes coated with various substrata. These included CN-I, CN-IV, LN-1 (all from Collaborative Biomedical Products), LN-2 (Life Technology), and poly-l-lysine (Sigma-Aldrich). The nontissue culture dishes were coated for 3 h with 10 μg of each ECM protein dissolved in 1 ml of PBS, or poly-l-lysine (Sigma-Aldrich) at concentrations of 0.05 or 0.01% wt/vol, also in 1 ml of PBS. Poly-l-lysine was used to obtain cell attachment without spreading (Yang et al., 1998, 1999, 2000; Relan et al., 1999). The dishes were then washed with PBS and allowed to dry before use. Alternatively, the cells were plated on poly-l-lysine–coated dishes, and the various ECM constituents were added at the described concentrations to the culture medium immediately after cell attachment was completed (1 h). In some experiments, the concentration of LN-2 added to the medium was reduced by 10-fold (1 μg/ml). Cell shape was additionally controlled by plating the cells on 10- or 20-μm diameter culture microsurfaces (Relan et al., 1999; Yang et al., 1999), the first to force cell rounding and the second to allow cell spreading/elongation (Yang et al., 1999). The cells were cultured in MEM with 10 or 0.5% FCS for different time periods for up to 24 h. Our previous studies showed that 0.5% FCS arrests mesenchymal cell proliferation (Yang et al., 1999). At the end of the culture period, the cells were used for immunohistochemical studies or lysed for RNA and protein analysis.
Cell treatments
To increase RhoA activity, embryonic mesenchymal cells were treated with 1 μM of endothelin-1 (Sigma-Aldrich) (Fleming et al., 1996) or 10 μM lysophosphatidic acid (Sigma-Aldrich) (Hirshman and Emala, 1999). To reduce RhoA activity, embryonic mesenchymal cells were treated with 10 μg/ml of C3 exoenzyme (Calbiochem) after being permeabilized with 75 μg/ml of saponin for 3 min (Leonard et al., 1992). In some experiments, 10 μg/ml of blocking anti–LN-2 antibody (Alexis Biochemicals Corp.) (Relan et al., 1999) was added to the cultures after cell attachment was completed (1 h after plating). After treatment, cells were lysed and analyzed by immunoblotting for RhoA and SM markers.
Immunoblot analysis
Cell cultures and lungs from adult C57BL/6J dy/dy mice and C57BL/6J+/+ normal mice (both from Jackson ImmunoResearch Laboratories) were lysed and resolved by SDS-PAGE. Western blots were done using standard protocols. Rabbit polyclonal antibody to RhoA (Santa Cruz Biotechnology, Inc.) and mouse monoclonal antibody to RhoA (Santa Cruz Biotechnology, Inc.) were each used at a concentration of 1 μg/ml. Mouse monoclonal antibody against Rac (Upstate Biotechnology) was used at a concentration of 0.5 μg/ml. Mitogen-activated protein kinases (ERK1 and ERK2) were detected using rabbit polyclonal antibody (Zymed Laboratories) at a concentration of 0.5 μg/ml. As in previous studies (Relan et al., 1999; Yang et al., 1999, 2000), the following antibodies were used for detection of SM-specific proteins: a mouse monoclonal antibody to SM α-actin (Boehringer) at a concentration of 0.25 μg/ml, a mouse monoclonal antibody to desmin (Dako) at a concentration of 1.125 μg/ml, rabbit polyclonal antibodies to SM-myosin (Biomedical Technologies) at a concentration of 10 μg/ml, rabbit polyclonal antibodies to SM22 (provided by Dr. Rodrigo Bravo, Bristol-Myers Squibb Pharmaceutical Research Institute, Princeton, NJ) at a concentration of 0.2 μg/ml, and mouse monoclonal antibodies to HA1 tag (Santa Cruz Biotechnology, Inc.) at a concentration of 0.125 μg/ml. Anti-FAK pY861, pY397, and pY925 phosphospecific antibodies (Biosource International) were used at concentrations of 1.0, 0.36, and 0.75 μg/ml, respectively. Primary antibodies were detected with HRP-conjugated secondary antibody diluted 1:3,000 (Bio-Rad Laboratories). The bands were visualized by chemiluminescence using a commercial kit (Amersham Life Sciences) according to the manufacturer's instructions. In some experiments, the membranes were stripped of primary and secondary antibodies (Mattingly et al., 2001) and reprobed with new antibodies as described above.
Immunohistochemistry
Mesenchymal cell cultures and 5-μm thick frozen sections cut from E11 and E14 embryonic lungs were fixed for 5 min in 100% ethanol and immunostained for RhoA and SRF (tissue sections) and RhoA, SRF, and desmin (cell cultures) following published protocols (Yang et al., 1998). Antibodies to SRF were purchased from Santa Cruz Biotechnology and used at a concentration of 8 μg/ml. Rabbit polyclonal antibody to RhoA was used at a concentration of 8 μg/ml, and antibody against desmin was used at a concentration of 20 μg/ml. After incubation with the first antibody, the samples were exposed to a 1:50 dilution of biotin-conjugated goat anti–mouse or biotin-conjugated anti–rabbit IgG (Sigma-Aldrich). Cell staining was completed using a commercial peroxidase-antiperoxidase kit following the manufacturer's instructions (ABC kit; Vector Laboratories). Cell cultures were also immunostained for SRF using a FITC-conjugated secondary antibody (Molecular Probes) at a 1:100 dilution. The cells were examined with a ZEISS Laser (LSM) 310 confocal microscope.
Cytoplasmic and nuclear staining
F-actin (cytoplasm) was stained using rhodamine-phallotoxin (Molecular Probes). After fixation with 4% paraformaldehyde solution for 10 min, the mesenchymal cells were preincubated with 2% BSA dissolved in PBS with 0.1% Tween for 30 min. The cells were then stained with 5 U/ml of phallotoxin for 20 min. To highlight nuclei, cells and tissue sections were incubated for 5 min with 5 μg/ml of DAPI (Molecular Probes) dissolved in water after blocking with 2% BSA.
Transient transfections
Vectors encoding wild-type RhoA, dominant negative RhoA, mutated at residue 19 to replace threonine with asparagine (termed N19), and an active mutant of RhoA, which has a mutation of valine in place of glycine at residue 14 (termed V14), were constructed by subcloning BamHI-EcoRI fragments into pKH3 (Mattingly et al., 1994). This plasmid, which includes a cytomegalovirus promoter/enhancer and SV40 origin of replication, attaches an in-frame triple HA1 tag to the NH2 terminus of the inserts. Thus, the HA13RhoA(N19) and HA13RhoA(V14) proteins that are produced can be immunorecognized by an anti-HA1 antibody (Santa Cruz Biotechnology). As described previously (Yang et al., 1999), primary cultures of mesenchymal cells were transfected 1 h after attachment was completed using lipofectamine plus reagent (Life Technology) following the manufacturer's instructions. The plasmids or vector without insert, used as control, were mixed with the lipofectamine reagent in a 1:3.5 wt/vol proportion, and the cells were transfected for 4 h in the presence of 10% FBS. After 4 h of incubation at 37°C, the transfection medium was replaced with fresh 10% MEM, and the cultures were incubated for an additional 16–18 h. Immunohistochemical studies using anti-HA1 antibody indicated that ∼25–30% of the cells in our primary cultures were efficiently transfected.
RhoA activity assay
GTP-bound RhoA (active RhoA) was affinity isolated from cell lysates based on the capability of GST-rhotekin to bind to GTP-Rho (Ren et al., 1999). The assay was performed on mesenchymal cell cultures following the manufacturer's instructions (Upstate Biotechnology). Briefly, the cells were lysed with Rho-binding lysis buffer (50 mM Tris, pH 7.2, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 500 mM NaCl, 10 mM MgCl2 with 10 μg/ml of leupeptin, 10 μg/ml of aprotinin, and 1 mM PMSF). Lysates were cleared by centrifugation, and active RhoA was precipitated with 20 μg of a GST-tagged fusion protein corresponding to residues 7–89 of mouse rhotekin Rho binding domain expressed in E. coli and bound to agarose beads. The precipitates were washed three times in washing buffer (50 mM Tris, pH 7.2, 1% Triton X-100, 150 mM NaCl, 10 mM MgCl2, 0.1 mM PMSF, 10 μg/ml aprotinin, and 10 μg/ml leupeptin), and the bound proteins were eluted and resolved in 14% polyacrylamide gels, transferred to nitrocellulose membranes, and immunoblotted with 1 μg/ml of rabbit polyclonal antibody to RhoA.
RT-PCR
Mesenchymal cell cultures were washed with PBS, and total RNA was extracted with Trizol (Life Technology). RT-PCR was performed with the GeneAmp RNA PCR kit (PerkinElmer) following the manufacturer's instructions. The following primers were used for PCR: RhoA, 5′ forward primer, 5′-agcctcatgcggttaatttg-3′ and 3′ reverse primer 5′-tctttgaattagcgcctggt-3′; SM α-actin, 5′ forward primer, 5′-tccctggagaagagctacga-3′ and 3′ reverse primer, 5′-gggcttttaatctccttcgg-3′; desmin, 5′ forward primer, 5′-gtgaagatggccttggatgt-3′ and 3′ reverse primer, 5′-gtagcctcgctgacaacctc-3′; SM-myosin, 5′ forward primer 5′-gacaactcctctcgctttgg-3′; and SRF isoforms, 5′ forward primer 5′-atcaccaactacctggcacc-3′ and 3′ reverse primer 5′-cacctgtagctcggtgaggt-3′. All amplifications shown here represent the product of 25 cycles except for SM myosin, which is shown at 30 cycles. A primer pair (Ambion Inc.) was used to produce a 488 base pair 18S ribosomal amplicon as an internal control. Under the conditions used in these studies, plateau of internal control and most amplicons was reached at 35 cycles.
Acknowledgments
This work has been supported by National Heart, Lung, and Blood Institute grants HL-48730 and HL-67100 (to L. Schuger), a grant from The Children's Research Center of Michigan (to L. Schuger), grant CA81150 (to R.R. Mattingly), and a Faculty Development Award from the PhRMA Foundation (to R.R. Mattingly). Confocal facilities were supported in part by National Institutes of Health grants P30E306639 and P30CA22453.
Footnotes
Abbreviations used in this paper: BM, basement membrane; CN, collagen; ECM, extracellular matrix; FAK, focal adhesion kinase; HA, hemagglutinin; LN, laminin; LPA, lysophosphatidic acid; RT, reverse transcriptase; SM, smooth muscle; SRF, serum response factor.
References
- Adams, J.C., and M.A. Schwartz. 2000. Stimulation of fascin spikes by thrombospondin-1 is mediated by the GTPases Rac and Cdc42. J. Cell Biol. 150:807–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belaguli, N.S., L.A. Schildmeyer, and R.J. Schwartz. 1997. Organization and myogenic restricted expression of the murine serum response factor gene. A role for autoregulation. J. Biol. Chem. 272:18222–18231. [DOI] [PubMed] [Google Scholar]
- Belaguli, N.S., W. Zhou, T.H. Trinh, M.W. Majesky, and R.J. Schwartz. 1999. Dominant negative murine serum response factor: alternative splicing within the activation domain inhibits transactivation of serum response factor binding targets. Mol. Cell. Biol. 19:4582–4591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkowitz, L.A., K.T. Riabowol, and M.Z. Gilman. 1989. Multiple sequence elements of a single functional class are required for cyclic AMP responsiveness of the mouse c-fos promoter. Mol. Cell. Biol. 9:4272–4281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop, A.L., and A. Hall. 2000. Rho GTPases and their effector proteins. Biochem. J. 348:241–255. [PMC free article] [PubMed] [Google Scholar]
- Bourdoulous, S., G. Orend, D.A. MacKenna, R. Pasqualini, and E. Ruoslahti. 1998. Fibronectin matrix regulates activation of RHO and CDC42 GTPases and cell cycle progression. J. Cell Biol. 143:267–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning, C.L., D.E. Culberson, I.V. Aragon, R.A. Fillmore, J.D. Croissant, R.J. Schwartz, and W.E. Zimmer. 1998. The developmentally regulated expression of serum response factor plays a key role in the control of smooth muscle-specific genes. Dev. Biol. 194:18–37. [DOI] [PubMed] [Google Scholar]
- Burgeson, R.E., M. Chiquet, R. Deutzmann, P. Ekblom, J. Engel, H. Kleinman, G.R. Martin, G. Meneguzzi, M. Paulsson, J. Sanes, et al. 1994. A new nomenclature for the laminins. Matrix Biol. 14:209–211. [DOI] [PubMed] [Google Scholar]
- Camoretti-Mercado, B., H.W. Liu, A.J. Halayko, S.M. Forsythe, J.W. Kyle, B. Li, Y. Fu, J. McConville, P. Kogut, J.E. Vieira, et al. 2000. Physiological control of smooth muscle-specific gene expression through regulated nuclear translocation of serum response factor. J. Biol. Chem. 275:30387–30393. [DOI] [PubMed] [Google Scholar]
- Colognato, H., and P.D. Yurchenco. 2000. Form and function: the laminin family of heterotrimers. Dev. Dyn. 218:213–234. [DOI] [PubMed] [Google Scholar]
- Croissant, J.D., J.H. Kim, G. Eichele, L. Goering, J. Lough, R. Prywes, and R.J. Schwartz. 1996. Avian serum response factor expression restricted primarily to muscle cell lineages is required for alpha-actin gene transcription. Dev. Biol. 177:250–264. [DOI] [PubMed] [Google Scholar]
- Danen, E.H., P. Sonneveld, A. Sonnenberg, and K.M. Yamada. 2000. Dual stimulation of Ras/mitogen-activated protein kinase and RhoA by cell adhesion to fibronectin supports growth factor-stimulated cell cycle progression. J. Cell Biol. 151:1413–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evers, E.E., G.C. Zondag, A. Malliri, L.S. Price, J.P. ten Klooster, R.A. van der Kammen, and J.G. Collard. 2000. Rho family proteins in cell adhesion and cell migration. Eur. J. Cancer. 36:1269–1274. [DOI] [PubMed] [Google Scholar]
- Fleming, I.N., C.M. Elliott, and J.H. Exton. 1996. Differential translocation of rho family GTPases by lysophosphatidic acid, endothelin-1, and platelet-derived growth factor. J. Biol. Chem. 271:33067–33073. [DOI] [PubMed] [Google Scholar]
- Fluck, M., F.W. Booth, and M.N. Waxham. 2000. Skeletal muscle CaMKII enriches in nuclei and phosphorylates myogenic factor SRF at multiple sites. Biochem. Biophys. Res. Commun. 270:488–494. [DOI] [PubMed] [Google Scholar]
- Gauthier-Rouviere, C., M. Vandromme, N. Lautredou, Q.Q. Cai, F. Girard, A. Fernandez, and N. Lamb. 1995. The serum response factor nuclear localization signal: general implications for cyclic AMP-dependent protein kinase activity in control of nuclear translocation. Mol. Cell. Biol. 15:433–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giancotti, F.G., and E. Ruoslahti. 1999. Integrin signaling. Science. 285:1028–1032. [DOI] [PubMed] [Google Scholar]
- Hall, A. 1998. Rho GTPases and the actin cytoskeleton. Science. 279:509–514. [DOI] [PubMed] [Google Scholar]
- Hill, C.S., J. Wynne, and R. Treisman. 1995. The Rho family GTPases RhoA, Rac1, and CDC42Hs regulate transcriptional activation by SRF. Cell. 81:1159–1170. [DOI] [PubMed] [Google Scholar]
- Hillaire, D., A. Leclerc, S. Faure, H. Topaloglu, N. Chiannilkulchai, P. Guicheney, L. Grinas, P. Legos, J. Philpot, T. Evangelista, et al. 1994. Localization of merosin-negative congenital muscular dystrophy to chromosome 6q2 by homozygosity mapping. Hum. Mol. Genet. 3:1657–1661. [DOI] [PubMed] [Google Scholar]
- Hirshman, C.A., and C.W. Emala. 1999. Actin reorganization in airway smooth muscle cells involves Gq and Gi-2 activation of Rho. Am. J. Physiol. 277:L653–L661. [DOI] [PubMed] [Google Scholar]
- Katoh, H., M. Negishi, and A. Ichikawa. 1996. Prostaglandin E receptor EP3 subtype induces neurite retraction via small GTPase Rho. J. Biol. Chem. 271:29780–29784. [DOI] [PubMed] [Google Scholar]
- Kemp, P.R., and J.C. Metcalfe. 2000. Four isoforms of serum response factor that increase or inhibit smooth-muscle-specific promoter activity. Biochem. J. 345:445–451. [PMC free article] [PubMed] [Google Scholar]
- Kim, S., H.S. Ip, M.M. Lu, C. Clendenin, and M.S. Parmacek. 1997. A serum response factor-dependent transcriptional regulatory program identifies distinct smooth muscle cell sublineages. Mol. Cell. Biol. 17:2266–2278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozma, R., S. Sarner, S. Ahmed, and L. Lim. 1997. Rho family GTPases and neuronal growth cone remodelling: relationship between increased complexity induced by Cdc42Hs, Rac1, and acetylcholine and collapse induced by RhoA and lysophosphatidic acid. Mol. Cell. Biol. 17:1201–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laufs, U., M. Endres, F. Custodis, K. Gertz, G. Nickenig, J.K. Liao, and M. Bohm. 2000. Suppression of endothelial nitric oxide production after withdrawal of statin treatment is mediated by negative feedback regulation of rho GTPase gene transcription. Circulation. 102:3104–3110. [DOI] [PubMed] [Google Scholar]
- Leonard, D., M.J. Hart, J.V. Platko, A. Eva, W. Henzel, T. Evans, and R.A. Cerione. 1992. The identification and characterization of a GDP-dissociation inhibitor (GDI) for the CDC42Hs protein. J. Biol. Chem. 267:22860–22868. [PubMed] [Google Scholar]
- Li, L., J.M. Miano, P. Cserjesi, and E.N. Olson. 1996. SM22 alpha, a marker of adult smooth muscle, is expressed in multiple myogenic lineages during embryogenesis. Circ. Res. 78:188–195. [DOI] [PubMed] [Google Scholar]
- Lu, J., T.E. Landerholm, J.S. Wei, X.R. Dong, S.P. Wu, X. Liu, K. Nagata Ki, M. Inagaki, and M.W. Majesky. 2001. Coronary smooth muscle differentiation from proepicardial cells requires RhoA-mediated actin reorganization and p160 Rho-kinase activity. Dev. Biol. 240:404–418. [DOI] [PubMed] [Google Scholar]
- Mack, C.P., A.V. Somlyo, M. Hautmann, A.P. Somlyo, and G.K. Owens. 2001. Smooth muscle differentiation marker gene expression is regulated by RhoA-mediated actin polymerization. J. Biol. Chem. 276:341–347. [DOI] [PubMed] [Google Scholar]
- Mattingly, R.R., A. Sorisky, M.R. Brann, and I.G. Macara. 1994. Muscarinic receptors transform NIH 3T3 cells through a Ras-dependent signalling pathway inhibited by the Ras-GTPase-activating protein SH3 domain. Mol. Cell. Biol. 14:7943–7952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattingly, R.R., M.L. Milstein, and B.L. Mirkin. 2001. Down-regulation of growth factor-stimulated MAP kinase signaling in cytotoxic drug-resistant human neuroblastoma cells. Cell. Signal. 13:499–505. [DOI] [PubMed] [Google Scholar]
- McHugh, K.M. 1995. Molecular analysis of smooth muscle development in the mouse. Dev. Dyn. 204:278–290. [DOI] [PubMed] [Google Scholar]
- Metz, R., and E. Ziff. 1991. cAMP stimulates the C/EBP-related transcription factor rNFIL-6 to trans-locate to the nucleus and induce c-fos transcription. Genes Dev. 5:1754–1766. [DOI] [PubMed] [Google Scholar]
- Miyagoe, Y., K. Hanaoka, I. Nonaka, M. Hayasaka, Y. Nabeshima, K. Arahata, and S. Takeda. 1997. Laminin alpha2 chain-null mutant mice by targeted disruption of the Lama2 gene: a new model of merosin (laminin 2)-deficient congenital muscular dystrophy. FEBS Lett. 415:33–39. [DOI] [PubMed] [Google Scholar]
- Nakabeppu, Y., and D. Nathans. 1989. The basic region of Fos mediates specific DNA binding. EMBO J. 8:3833–3841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris, J.L., and J.L. Manley. 1992. Selective nuclear transport of the Drosophila morphogen dorsal can be established by a signaling pathway involving the transmembrane protein Toll and protein kinase A. Genes Dev. 6:1654–1667. [DOI] [PubMed] [Google Scholar]
- Postma, F.R., K. Jalink, T. Hengeveld, and W.H. Moolenaar. 1996. Sphingosine-1-phosphate rapidly induces Rho-dependent neurite retraction: action through a specific cell surface receptor. EMBO J. 15:2388–2392. [PMC free article] [PubMed] [Google Scholar]
- Relan, N.K., Y. Yang, S. Beqaj, J.H. Miner, and L. Schuger. 1999. Cell elongation induces laminin alpha2 chain expression in mouse embryonic mesenchymal cells: role in visceral myogenesis. J. Cell Biol. 147:1341–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren, X.D., W.B. Kiosses, and M.A. Schwartz. 1999. Regulation of the small GTP-binding protein Rho by cell adhesion and the cytoskeleton. EMBO J. 18:578–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roman, J., and J.A. McDonald. 1992. Expression of fibronectin, the integrin alpha 5, and alpha-smooth muscle actin in heart and lung development. Am. J. Respir. Cell Mol. Biol. 6:472–480. [DOI] [PubMed] [Google Scholar]
- Sawtell, N.M., and J.L. Lessard. 1989. Cellular distribution of smooth muscle actins during mammalian embryogenesis: expression of the alpha-vascular but not the gamma-enteric isoform in differentiating striated myocytes. J. Cell Biol. 109:2929–2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuger, L., A.P. Skubitz, A. de las Morenas, and K. Gilbride. 1995. Two separate domains of laminin promote lung organogenesis by different mechanisms of action. Dev. Biol. 169:520–532. [DOI] [PubMed] [Google Scholar]
- Schuger, L., A.P. Skubitz, K. Gilbride, R. Mandel, and L. He. 1996. Laminin and heparan sulfate proteoglycan mediate epithelial cell polarization in organotypic cultures of embryonic lung cells: evidence implicating involvement of the inner globular region of laminin beta 1 chain and the heparan sulfate groups of heparan sulfate proteoglycan. Dev. Biol. 179:264–273. [DOI] [PubMed] [Google Scholar]
- Schuger, L., A.P. Skubitz, J. Zhang, L. Sorokin, and L. He. 1997. Laminin alpha1 chain synthesis in the mouse developing lung: requirement for epithelial-mesenchymal contact and possible role in bronchial smooth muscle development. J. Cell Biol. 139:553–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotiropoulos, A., D. Gineitis, J. Copeland, and R. Treisman. 1999. Signal-regulated activation of serum response factor is mediated by changes in actin dynamics. Cell. 98:159–169. [DOI] [PubMed] [Google Scholar]
- Sunada, Y., S.M. Bernier, C.A. Kozak, Y. Yamada, and K.P. Campbell. 1994. Deficiency of merosin in dystrophic dy mice and genetic linkage of laminin M chain gene to dy locus. J. Biol. Chem. 269:13729–13732. [PubMed] [Google Scholar]
- Swarthout, J.T., and H.W. Walling. 2000. Lysophosphatidic acid: receptors, signaling and survival. Cell. Mol. Life Sci. 57:1978–1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano, H., I. Komuro, T. Oka, I. Shiojima, Y. Hiroi, T. Mizuno, and Y. Yazaki. 1998. The Rho family G proteins play a critical role in muscle differentiation. Mol. Cell. Biol. 18:1580–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thyberg, J., and A. Hultgardh-Nilsson. 1994. Fibronectin and the basement membrane components laminin and collagen type IV influence the phenotypic properties of subcultured rat aortic smooth muscle cells differently. Cell Tissue Res. 276:263–271. [DOI] [PubMed] [Google Scholar]
- Tigyi, G., D.J. Fischer, A. Sebok, F. Marshall, D.L. Dyer, and R. Miledi. 1996. Lysophosphatidic acid-induced neurite retraction in PC12 cells: neurite-protective effects of cyclic AMP signaling. J. Neurochem. 66:549–558. [DOI] [PubMed] [Google Scholar]
- Vachon, P.H., F. Loechel, H. Xu, U.M. Wewer, and E. Engvall. 1996. Merosin and laminin in myogenesis; specific requirement for merosin in myotube stability and survival. J. Cell Biol. 134:1483–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei, L., W. Zhou, J.D. Croissant, F.E. Johansen, R. Prywes, A. Balasubramanyam, and R.J. Schwartz. 1998. RhoA signaling via serum response factor plays an obligatory role in myogenic differentiation. J. Biol. Chem. 273:30287–30294. [DOI] [PubMed] [Google Scholar]
- Wei, L., W. Roberts, L. Wang, M. Yamada, S. Zhang, Z. Zhao, S.A. Rivkees, R.J. Schwartz, and K. Imanaka-Yoshida. 2001. a. Rho kinases play an obligatory role in vertebrate embryonic organogenesis. Development. 128:2953–2962. [DOI] [PubMed] [Google Scholar]
- Wei, L., L. Wang, J.A. Carson, J.E. Agan, K. Imanaka-Yoshida, and R.J. Schwartz. 2001. b. beta1 integrin and organized actin filaments facilitate cardiomyocyte-specific RhoA-dependent activation of the skeletal alpha-actin promoter. FASEB J. 15:785–796. [DOI] [PubMed] [Google Scholar]
- Wenk, M.B., K.S. Midwood, and J.E. Schwarzbauer. 2000. Tenascin-C suppresses Rho activation. J. Cell Biol. 150:913–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, H., P. Christmas, X.R. Wu, U.M. Wewer, and E. Engvall. 1994. a. Defective muscle basement membrane and lack of M-laminin in the dystrophic dy/dy mouse. Proc. Natl. Acad. Sci. USA. 91:5572–5576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, H., X.R. Wu, U.M. Wewer, and E. Engvall. 1994. b. Murine muscular dystrophy caused by a mutation in the laminin alpha 2 (Lama2) gene. Nat. Genet. 8:297–302. [DOI] [PubMed] [Google Scholar]
- Yang, Y., K.C. Palmer, N. Relan, C. Diglio, and L. Schuger. 1998. Role of laminin polymerization at the epithelial mesenchymal interface in bronchial myogenesis. Development. 125:2621–2629. [DOI] [PubMed] [Google Scholar]
- Yang, Y., N.K. Relan, D.A. Przywara, and L. Schuger. 1999. Embryonic mesenchymal cells share the potential for smooth muscle differentiation: myogenesis is controlled by the cell's shape. Development. 126:3027–3033. [DOI] [PubMed] [Google Scholar]
- Yang, Y., S. Beqaj, P. Kemp, I. Ariel, and L. Schuger. 2000. Stretch-induced alternative splicing of serum response factor promotes bronchial myogenesis and is defective in lung hypoplasia. J. Clin. Invest. 106:1321–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]