

Abstract
To examine the involvement of interchromatin granule clusters (IGCs) in transcription and pre-mRNA splicing in mammalian cell nuclei, the serine-arginine (SR) protein kinase cdc2-like kinase (Clk)/STY was used as a tool to manipulate IGC integrity in vivo. Both immunofluorescence and transmission electron microscopy analyses of cells overexpressing Clk/STY indicate that IGC components are completely redistributed to a diffuse nuclear localization, leaving no residual structure. Conversely, overexpression of a catalytically inactive mutant, Clk/STY(K190R), causes retention of hypophosphorylated SR proteins in nuclear speckles. Our data suggest that the protein–protein interactions responsible for the clustering of interchromatin granules are disrupted when SR proteins are hyperphosphorylated and stabilized when SR proteins are hypophosphorylated. Interestingly, cells without intact IGCs continue to synthesize nascent transcripts. However, both the accumulation of splicing factors at sites of pre-mRNA synthesis as well as pre-mRNA splicing are dramatically reduced, demonstrating that IGC disassembly perturbs coordination between transcription and pre-mRNA splicing in mammalian cell nuclei.
Keywords: interchromatin granule cluster; transcription; pre-mRNA splicing; Clk/STY; nucleus
Introduction
Pre-mRNA splicing factors localize by immunofluorescence microscopy to 20–50 irregularly shaped nuclear speckles set against a diffuse distribution within the nucleoplasm of mammalian cells (for reviews see Spector, 1993; Lamond and Earnshaw, 1998). In addition, in some cell types they also localize to Cajal bodies (Spector et al., 1992; Gall, 2000). By transmission electron microscopy (TEM),* the speckled immunofluorescence localization corresponds to interchromatin granule clusters (IGCs) and perichromatin fibrils (PFs; for review see Fakan and Puvion, 1980; Spector, 1993). IGCs are composed of particles measuring 20–25 nm in diameter, and they contain numerous factors that are involved in RNA synthesis and processing. IGC constituents include, but are not limited to, small nuclear ribonucleoprotein particles (snRNPs), arginine-serine–rich (SR) splicing factors, and the hyperphosphorylated form of the large subunit of RNA polymerase II (Bregman et al., 1995). The majority of the protein constituents of IGCs have now been identified (Mintz et al., 1999, and unpublished results), making it possible to better address the biological function of these nuclear domains.
Although splicing factors localize to IGCs, pre-mRNA synthesis does not occur within these structures, but at PFs on the IGC periphery or at some distance away from the IGCs (Fakan, 1994; Cmarko et al., 1999). One recent report indicated overlap between sites of bromo-uridine incorporation and nuclear speckles (Wei et al., 1999); however, a significant proportion of the overlap likely corresponds to PFs (transcription sites) on the periphery of speckles. The majority of nucleotide incorporation and immunocytochemistry studies have shown that IGCs are not likely to be sites of transcription because they do not contain DNA, and they are not labeled by 3H-uridine incorporation (Turner and Franchi, 1987; Spector, 1990; Fakan, 1994). Cmarko et al. (1999) performed extensive analysis of bromo-UTP incorporation at the TEM level and did not detect transcription in IGCs. Furthermore, inhibition of RNA polymerase II transcription with α-amanitin causes splicing factor recruitment to cease, and speckles become larger and more rounded (Carmo-Fonseca et al., 1992; Spector et al., 1993; Misteli et al., 1997). Therefore, enrichment of splicing factors in IGCs may be due to the fact that they are sites of complex formation and/or modification of splicing factors, or sites of splicing factor storage (Huang et al., 1994; Spector et al., 1993). In support of these possibilities, experiments in living cells revealed that hyperphosphorylation of splicing factor SF2/ASF on its RS-rich domain releases it from the IGCs for recruitment to active genes (Misteli et al., 1997). However, despite this advance, neither the structural organization nor the precise biological function of the IGCs is known.
Several SR protein kinases, including cdc2-like kinase (CLK)/STY 1, 2, 3, and 4 (Ben-David et al., 1991; Hanes et al., 1994; Howell et al., 1991) and SR protein kinases 1 and 2 (SRPK1 and SRPK2) (Gui et al., 1994; Wang et al., 1998; Yeakley et al., 1999), specifically phosphorylate RS domains. Clk/STY was isolated independently by several groups (Ben-David et al., 1991; Howell et al., 1991; Johnson and Smith, 1991) and was subsequently characterized as a LAMMER family kinase. LAMMER kinases include dual specificity kinases that can phosphorylate on tyrosine in addition to serine/threonine residues (Lindberg et al., 1992; Lee et al., 1996). When Clk/STY is overexpressed in cultured cells, SR proteins become hyperphosphorylated and the typical speckled immunolocalization of splicing factors is reorganized into a diffuse nucleoplasmic localization (Colwill et al., 1996b). In addition, Clk/STY has been shown to directly affect the activity of SR proteins; both hyper- and hypophosphorylation of SR proteins affect in vitro splicing activity (Prasad et al., 1999).
It is presently unclear why mammalian cells contain IGCs and whether their position in the nucleus reflects a spatial positioning that is essential for function. Time-lapse observations of nuclear speckles in living cells have shown that the position of IGCs is maintained over many hours (Misteli et al., 1997; Kruhlak et al., 2000), suggesting that they reside in predetermined locations. Such positioning may be a result of granules clustering upon a specific structural framework or around specific chromosomal regions. To investigate this possibility, we used overexpression of murine Clk/STY 1 as a method to completely disassemble IGCs in vivo. Ultrastructural analysis of such cells indicates that SR proteins are redistributed throughout the nucleus in small clusters, and no specific underlying structural IGC scaffold was revealed. Nascent transcripts are produced in cells without IGCs, but accumulation of splicing factors originating from an entirely nucleoplasmic pool onto pre-mRNA is significantly reduced and spliced mRNA is markedly reduced to undetectable.
Results
Overexpression of Clk/STY causes redistribution of nuclear speckle components
A-431 cells were transiently transfected with murine Clk/STY1 or catalytically inactive mutant Clk/STY1(K190R) to assess the extent of disassembly of various nuclear speckle components. Transient overexpression resulted in a population of transfected cells with variable levels of Clk/STY expression and extent of nuclear speckle disassembly. Our goal was to examine pre-mRNA synthesis and processing in cells in which nuclear speckles are no longer intact. We examined cells in which overexpression of green fluorescent protein (GFP)-Clk/STY (Fig. 1 A) induces a complete redistribution of splicing factors, such as SC35 (Fig. 1 B). Multiple SR protein family members, such as those recognized by monoclonal antibody 3C5 (Turner and Franchi, 1987), responded to hyperphosphorylation in the same manner (unpublished data). However, overexpression of GFP-Clk/STY(K190R) that is not able to phosphorylate SR proteins (Fig. 1 C) did not induce nuclear speckle disassembly of SC35 (Fig. 1 D), indicating that the release of SR proteins is dependent upon kinase activity. Next, we looked at the response of proteins that localize to nuclear speckles but are not members of the SR protein family. B” is a component of U2 snRNP that is involved in pre-mRNA splicing (Habets et al., 1986). When GFP-Clk/STY was transiently overexpressed in A-431 cells (Fig. 2 A), similar to SR proteins, B” redistributed from its typical nuclear speckle localization to a diffuse nuclear localization (Fig. 2 B). Our ongoing identification and characterization of the protein constituents of IGCs has given us the ability to sort IGC components based upon predicted functions (Mintz et al., 1999 and additional unpublished data). Among the proteins identified in our purified IGC fraction is pinin (unpublished result), which was reported previously to link intermediate filaments to the submembrane plaque of desmosomes (Ouyang and Sugrue, 1996; Ouyang et al., 1997). In contrast, pinin has also been reported to be a strictly nuclear protein that localizes in nuclear speckles (Brandner et al., 1997). Although pinin may localize to both desmosomes and nuclear speckles (Ouyang, 1999), the possibility that pinin is potentially a structural protein in the IGCs was tested by examining the response of endogenous pinin to hyperphosphorylation of SR proteins. However, cells transiently overexpressing GFP-Clk/STY (Fig. 2 C) exhibited a complete redistribution of pinin (Fig. 2 D). Several reports have indicated that populations of actin (Nakayasu and Ueda, 1984) and lamin A (Jagatheesan et al., 1999) are associated with snRNPs or nuclear speckles, respectively. Although these two proteins are very good candidates for structural IGC proteins, they also redistributed in response to Clk/STY overexpression in the same manner as splicing factors (unpublished data). It is unlikely that this redistribution is due to direct hyperphosphorylation of actin or lamins, since Clk/STY specificity is linked to phosphorylation of serines in the RS region of SR proteins (Colwill et al., 1996a,b; Nayler et al., 1997) and these proteins lack RS regions. Therefore, although actin and lamins may play some role in IGCs, they do not appear to serve as constituents of an underlying scaffold of these nuclear structures. Next, we considered that RNA, rather than protein, might provide a structural framework in IGCs. A stable population of polyadenylated (polyA+) RNA resides in nuclear speckles (Huang et al., 1994). The function of this RNA is not clear, but we reasoned that if it were a structural IGC component, it would not respond to release of SR proteins. However, similar to what was observed for IGC proteins, upon overexpression of FLAG-Clk/STY in A-431 cells (Fig. 3 A), the stable polyA+ RNA became diffusely localized throughout the nucleus (Fig. 3 B). This effect was dependent on hyperphosphorylation, because upon overexpression of FLAG-Clk/STY(K190R) (Fig. 3 C), the polyA+ RNA maintained its typical nuclear speckle localization (Fig. 3 D). We also tested IGC constituents that were recently identified by mass spectrometry analysis. KIAA0111 encodes translation initiation factor eIF4Aiii (Weinstein et al., 1997; Li et al., 1999; Holzmann et al., 2000). KIAA0801 encodes a protein of unknown function; both proteins contain DEAD/H box RNA helicase motifs. KIAA0536 is the human homologue of PRP4, a serine/threonine protein kinase in fission yeast (Alahari et al., 1993; Kojima et al., 2001). GFP fusion constructs were made for each of these cDNA clones, and cells were transiently cotransfected with the respective fusion construct plus pTetON and FLAG-Clk/STY plasmids. After allowing time for accumulation of GFP-tagged protein in nuclear speckles, expression of FLAG-Clk/STY was induced with doxycycline. In FLAG-Clk/STY–transfected cells, each of these proteins exhibited a diffuse nuclear localization, whereas in neighboring cells not overexpressing FLAG-Clk/STY they maintained a nuclear speckle localization (unpublished data). Whereas each constituent of nuclear speckles examined here redistributed upon overexpression of Clk/STY, nucleolar organization, as shown by ANA-N staining (Fig. 2, E and F), and chromatin organization (unpublished data) were not altered by overexpression of Clk/STY.
Figure 1.

SR protein SC35 redistributes to a diffuse nuclear localization upon overexpression of Clk/STY. A-431 cells transiently transfected with GFP-Clk/STY wild-type (A) show diffuse localization of SC35 (B) in contrast to untransfected cells, or cells transfected with catalytically inactive mutant GFP-Clk/STY K190R (C) in which SC35 is localized in nuclear speckles (D). Bar, 5 μm.
Figure 2.
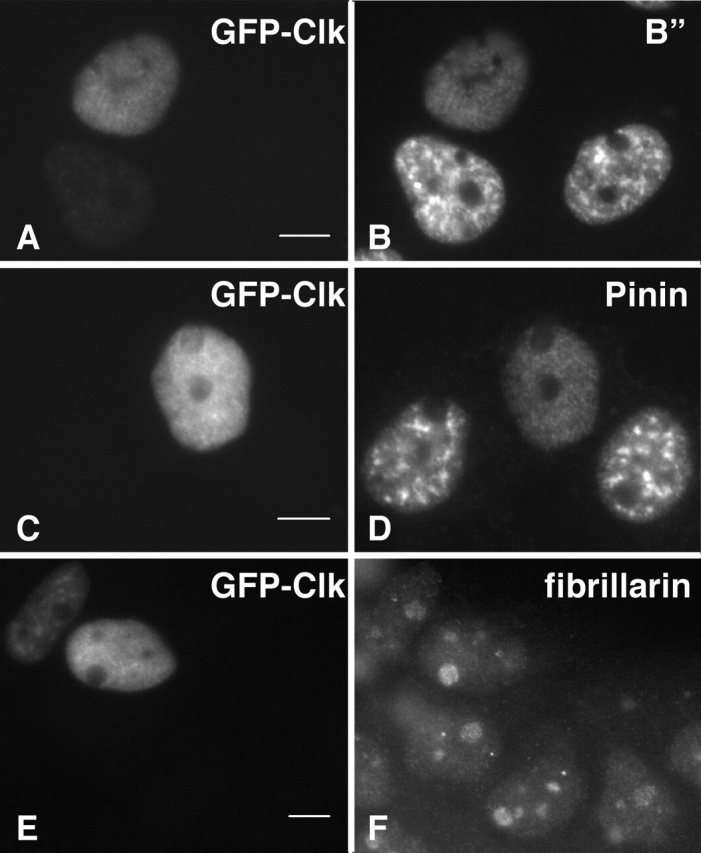
Redistribution of U2-B” and pinin upon overexpression of GFP-Clk/STY. In A-431 cells transiently transfected with GFP-Clk/STY (A, C, and E), both B” (B) and pinin (D) are redistributed to a diffuse nuclear localization. The morphology of other nuclear structures, including nucleoli (fibrillarin, F), is not affected. Bars, 5 μm.
Figure 3.

Redistribution of the nuclear speckle stable population of polyA + RNA upon overexpression of Clk/STY. A-431 cells transiently transfected with FLAG-Clk/STY wild-type (A) show redistribution of polyA+ RNA (B) to a diffuse nuclear localization. Untransfected cells and cells transfected with FLAG-Clk/STY(K190R) (C) illustrate the typical nuclear speckle localization of stable polyA+ RNA (D). Bar, 5 μm.
Cells overexpressing Clk/STY lack intact interchromatin granule clusters
Since all nuclear speckle components examined became diffusely distributed upon overexpression of Clk/STY, we were interested to determine if this redistribution would reveal a specific underlying IGC scaffold. Cells overexpressing Clk/STY and observed by immunofluorescence to have a completely diffuse distribution of splicing factor SC35 were processed for TEM. At least one neighboring untransfected cell with intact IGCs was examined in the same thin sections, serving as a control. The results shown in Fig. 4 are representative of observations from 12 Clk/STY-transfected cells. Untransfected A-431 nuclei contained large IGCs distributed throughout the nucleoplasm, and immunogold labeling for SR proteins was found in IGCs, as well as in small clusters in the surrounding nucleoplasm (Fig. 4, A and B). In cells transfected with Clk/STY, there were no intact IGCs, and immunogold labeling for SR proteins was dispersed throughout the nucleoplasm in small clusters that resemble PFs (Fig. 4, C and D). In addition, we did not detect a specific IGC substructure, nor did we observe empty nuclear regions where IGCs would previously have been located.
Figure 4.
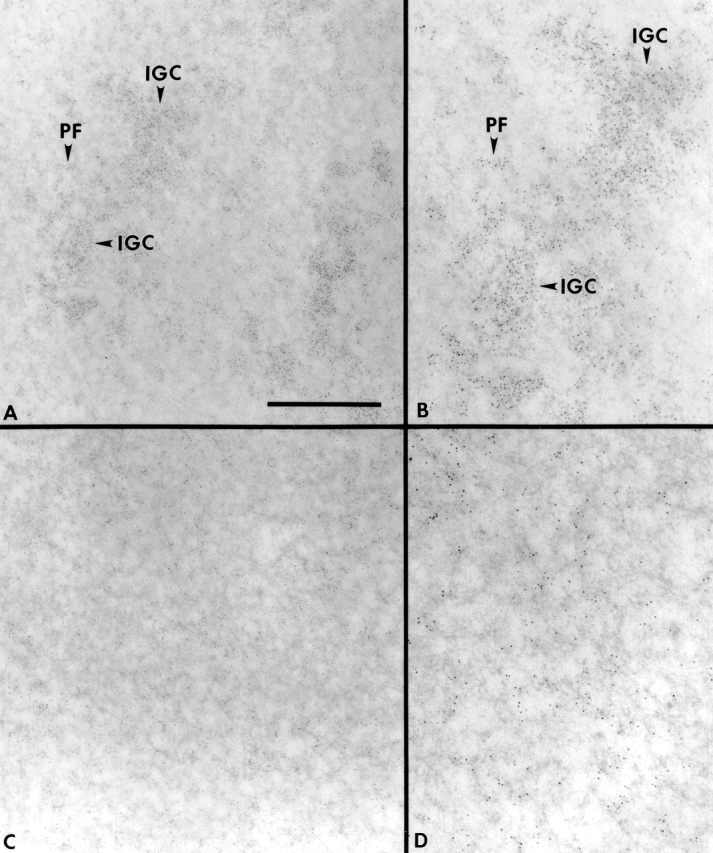
Ultrastructural analysis of nuclei in cells transfected with wild-type Clk/STY demonstrates that IGCs are no longer intact. A-431 cells were transiently transfected with GFP-Clk/STY, and transfected cells with completely disassembled SC35 nuclear speckles (C and D) and neighboring untransfected cells (A and B) were processed for TEM. IGCs in ultrathin sections were revealed using EDTA regressive staining and immunogold localization of SR proteins (3C5 antibody). Arrowheads indicate the location of intact IGCs as well as PFs in an untransfected cell (A). The granular appearance of areas labeled with 3C5 is clear in the enlargement of this region (B). In cells transfected with wild-type Clk/STY (C and D) IGCs are not observed. Gold particles, 5 nm. Bar: (A and C) 1 μm; (B and D) 0.6 μm.
Transcription is unaffected in cells lacking intact IGCs
Next, we evaluated the functional implications of complete IGC disassembly, namely the effects on transcription and pre-mRNA splicing in situ. A-431 cells transiently transfected with FLAG-Clk/STY were gently permeabilized with digitonin, and transcription buffer containing bromouridine-triphosphate (bromo-UTP) was added to the cells for 5 min at 37°C. The cells were then processed for triple-label immunolocalization of FLAG-Clk/STY, bromo-UTP, and SR proteins (Fig. 5). Cells overexpressing FLAG-Clk/STY (Fig. 5 A) exhibited a completely diffuse localization of SR proteins, as confirmed by staining with 3C5 (Fig. 5 C). Interestingly, such cells remained transcriptionally competent (Fig. 5 B), and the extent of bromo-UTP incorporation was comparable to that in surrounding untransfected cells.
Figure 5.

Bromo-uridine incorporation confirms that transcription occurs in cells overexpressing Clk/STY. A-431 cells were transiently transfected with FLAG-Clk/STY, permeabilized with digitonin (20 μg/ml) for 5 min, and transcription buffer containing bromo-UTP was added for 5 min at 37°C. The cell transfected with Clk/STY (A) exhibits completely disassembled nuclear speckles (shown by immunolabeling with 3C5 antibody; C) yet it is equally capable of incorporating bromo-UTP into nascent transcripts as neighboring untransfected cells (B). Bar, 5 μm.
Splicing factors do not accumulate at transcription sites and splicing is significantly reduced in cells without intact IGCs
A-431 cell lines stably expressing β-globin genomic DNA were generated to assess splicing capacity in cells without intact IGCs. The site of β-globin pre-mRNA synthesis was detected by RNA FISH as a single dot in each interphase nucleus (Fig. 6 A), and splicing factor SC35 was colocalized at this transcription site (Fig. 6 B) as expected from previous studies (Jiménez-García and Spector, 1993; Xing et al., 1993; Huang and Spector, 1996). Interestingly, GFP-Clk/STY was recruited to the transcription site in cells that had not yet undergone nuclear speckle disassembly (Fig. 6, C and D). The β-globin pre-mRNA transcription site (nascent transcripts) in nuclei overexpressing Clk/STY and exhibiting complete nuclear speckle disassembly was of comparable size and intensity to loci in untransfected nuclei (Fig. 6 G), confirming that transcription is not affected by Clk/STY overexpression. However, the loci in 11 of 12 cells scored without intact IGCs exhibited a largely reduced accumulation of SC35 compared with loci in untransfected nuclei (Fig. 6 F; compare regions at arrow and arrowhead). In addition, using a nonphosphoepitope antibody that recognizes the U2 snRNP B” protein, we also observed reduced accumulation of U2 snRNP at this transcription site in cells without intact IGCs (unpublished data).
Figure 6.

SC35 accumulation at transcription sites is dramatically reduced in cells without intact nuclear speckles. β-globin transcripts are detected as a single locus in the nucleus of A-431 cells stably transfected with β-globin genomic DNA (A). In these stable cell lines, SC35 accumulates at the β-globin transcription site as expected (arrows, A and B). Transiently expressed GFP-Clk/STY initially localizes to nuclear speckles (D), and in addition, accumulates at transcription sites (arrows, C and D). In cells expressing GFP-Clk/STY and with completely disassembled nuclear speckles (E), SC35 does not accumulate at the β-globin transcription site (arrow, F and G). However, in an adjacent cell that is not overexpressing GFP-Clk/STY, SC35 does accumulate at the transcription site (arrowhead, F and G). Bars, 5 μm.
Although splicing factors did not accumulate to significant levels at transcription sites in cells without intact IGCs, we directly examined the ability of β-globin pre-mRNA to be spliced at the transcription site. We used an oligonucleotide probe to specifically detect removal of intron 2 from β-globin pre-mRNA in vivo by fluorescence in situ hybridization. A probe designed to target the splice junction of exons 2/3 hybridizes to spliced β-globin mRNA in all untransfected cells scored (Fig. 7 B, arrowheads; 79/79 cells). Nearly all cells that overexpressed GFP-Clk/STY but had not yet undergone nuclear speckle disassembly also exhibited a hybridization signal with the splice junction probe (Fig. 7 B, arrow; 50 of 52 cells exhibited a hybridization signal). However, there was no hybridization signal with the splice junction probe in cells that lacked IGCs due to overexpression of GFP-Clk/STY, indicating that splicing was inhibited (Fig. 7 D; 26/26 cells) by comparison with an adjacent untransfected cell which gave a hybridization signal (Fig. 7 D, arrowhead). A probe designed to target β-globin intron 2 hybridized to β-globin pre-mRNA both in untransfected cells and in all cells transfected with GFP-Clk/STY that exhibited complete nuclear speckle disassembly (Fig. 7, E and F; 16/16 cells). Similar results were observed using a stable cell line expressing a β-tropomyosin minigene construct (unpublished data). Because we detected splicing only in cells having intact nuclear speckles, we conclude that splicing factors originating from an entirely diffuse nucleoplasmic pool (hyperphosphorylated) are not competent to perform pre-mRNA splicing in vivo.
Figure 7.

Splicing is markedly reduced to absent in cells without intact nuclear speckles. A-431 cells stably expressing β-globin genomic DNA were transiently transfected with GFP-Clk/STY (A, C, and E). In situ hybridization was performed using oligonucleotide probes to the splice junction of exons 2/3 (B and D) or to intron 2 (F). GFP-Clk/STY initially localizes in nuclear speckles and splicing of β-globin pre-mRNA is unaffected (B, arrow) by comparison with splicing in an untransfected nucleus (B, arrowhead). However, in cells with completely disassembled nuclear speckles (C, cell on the left), there is no hybridization signal in any focal plane (D, cell on the left), whereas splicing is detected in a neighboring untransfected cell (D, arrowhead). A probe that targets β-globin intron 2 hybridizes to β-globin pre-mRNA in untransfected cells (F, arrowhead) as well as cells expressing GFP-Clk/STY and exhibiting no intact nuclear speckles (F, arrow). Bar, 5 μm.
Catalytically inactive mutant Clk/STY(K190R) traps splicing factors in nuclear speckles
Since hyperphosphorylation of SR proteins is required for release of splicing factors from nuclear speckles (Misteli et al., 1997), we reasoned that we may interfere with splicing factor release by overexpression of mutant Clk/STY(K190R). A-431 cells were transfected with GFP-Clk/STY(K190R) and the effects of overexpression were analyzed in living cells. Time-lapse images of a cell overexpressing GFP-Clk/STY(K190R) are shown in Fig. 8. A sequence of 200 images (350 ms exposures) was taken every 30 min, beginning 6 h after transfection when a GFP-Clk/STY(K190R) signal was first detectable in the nuclear speckles of the cell shown. At 6.0 h (Fig. 8 A), the speckle morphology and peripheral movement was comparable to that shown previously for GFP-SF2/ASF (Misteli et al., 1997). However, in contrast to time-lapse observations of speckles in control cells, the cells transfected with Clk/STY(K190R) began to exhibit bright GFP-Clk/STY(K190R) foci on the periphery of speckles. In the example shown in Fig. 8, at 6.0 h after transfection, two speckles had bright foci (Fig. 8 A, arrows; video 1). By 6.5 h, each speckle had developed at least one bright focus (Fig. 8 B; video 2). Multiple foci appeared on all speckles by 7.0 h, often in pairs that maintained close proximity (Fig. 8 C, arrows; video 3). Finally, at 7.5 h, the speckles had multiple foci, and they no longer exhibited the typical peripheral movement. Instead, the speckles were almost completely immobilized (Fig. 8 D, arrow; video 4). Identical foci were observed in cells transfected with FLAG-Clk/STY(K190R) (unpublished data), as well as in fixed cells (see below), ruling out the possibility that the foci were merely aggregates of the GFP fusion or a result of phototoxic effects of live-cell imaging. Furthermore, immunoelectron microscopy analysis showed that foci contain interchromatin granules (unpublished data).
Figure 8.

Live-cell microscopy demonstrates accumulation of GFP-Clk/STY(K190R) in foci over time. A-431 cells were transiently transfected with GFP-Clk/STY(K190R) and live-cell observations were initiated 6 h posttransfection (A). The transfected cell exhibits small foci of GFP-Clk/STY(K190R) on the edges of two nuclear speckles at 6 h (A, arrows), although overall dynamic movement of the speckle is not affected (Video 1). These foci become brighter and more numerous over the following hour (B and C; Videos 2 and 3). At 7.5 h (D), nuclear speckles are almost completely immobilized (Video 4). Videos are available at http://www.jcb.org/cgi/content/full/jcb.200107017/DC1.
Similar to what we saw with overexpression of wild-type Clk/STY, transient overexpression of Clk/STY(K190R) resulted in a population of transfected cells expressing different amounts of kinase, except that the mutant kinase showed various stages of foci formation on the periphery of nuclear speckles. Some nuclei (without nuclear speckle foci) showed a complete colocalization of splicing factors and Clk/STY(K190R) as shown in Fig. 1, C and D. We speculate that this variation in phenotype is due to the timing of DNA entry into each cell and the lower level of mutant kinase expression, and that formation of foci might require expression of Clk/STY(K190R) to reach a certain threshold to override the activity of endogenous Clk/STY isoforms, as well as other putative SR protein kinases. We examined the focal accumulations at the periphery of nuclear speckles in cells that we interpret as being representative of the early stages of foci formation (equivalent to the stages shown in Fig. 8, A and B). Bromo-UTP incorporation indicated that global transcription is not altered in cells with this phenotype (unpublished data). RNA FISH using a probe against β-globin intron 2 verified that β-globin transcripts are being synthesized; however, the splice junction probe hybridizes in only 60% of the cells scored, suggesting that splicing is somewhat less efficient when nuclear speckles are partially immobilized (unpublished data).
Immunofluorescence localization with nonphosphoepitope antibodies against B” (Fig. 9, A–C), SF2/ASF (Fig. 9, D–I), and m3G (unpublished data) verified that these splicing factors precisely colocalized with GFP-Clk/STY(K190R) in the foci. We interpret these foci as regions of the speckles in which splicing factors accumulate because they are in a state of reduced phosphorylation and therefore cannot be released from the speckles. To confirm this hypothesis, we transfected A-431 cells with GFP-Clk/STY(K190R) and performed immunofluorescence using anti-SC35 antibody, which recognizes a phosphoepitope on SC35, and mAb104, which recognizes phosphoepitopes on a family of SR proteins (Roth et al., 1990). There was a dramatic reduction of immunolabeling with these phosphoepitope antibodies in the focal accumulations of GFP-Clk/STY(K190R), as noted by complete absence of labeling in these regions with mAb104 (Fig. 10, A–C) and with anti-SC35 (Fig. 10, D–I). This result confirms that unphosphorylated or hypophosphorylated proteins are present in the foci and might be unable to leave the speckles due to a lack of or reduced levels of phosphorylation.
Figure 9.

Splicing factors accumulate in foci on the periphery of nuclear speckles upon overexpression of GFP-Clk/STY(K190R). A-431 cells transiently transfected with GFP-Clk/STY(K190R) exhibit bright focal accumulations of mutant kinase (A and D) that also contain splicing factors U2-B” (B) and SF2/ASF (E). An enlargement of a region of the nucleus shown in D–F is shown in G–I. In merged images (C, F, and I) the foci appear yellow, indicating precise colocalization of these splicing factors and mutant kinase in the foci. Bars, 5 μm.
Figure 10.

Splicing factors that accumulate in foci upon overexpression of GFP-Clk/STY(K190R) are in a state of reduced phosphorylation. In focal accumulations containing GFP-Clk/STY(K190R) (A and D), antibodies that recognize phosphoepitopes of SR proteins, including mAb104 (B) and SC35 (E), show a lack of immunostaining. An enlargement of a region of the nucleus shown in D–F is shown in G–I. In merged images (C, F, and I), the foci appear green, indicating that phosphorylation of these proteins is reduced or absent in the foci. Bars, 5 μm.
Discussion
Observations of nuclear speckles in living cells have shown that they are highly dynamic nuclear domains (Misteli et al., 1997; Eils et al., 2000). Although there is a continuous exchange of the protein constituents of speckles over time (Kruhlak et al., 2000; Phair and Misteli, 2000), each speckle maintains its position in the nucleus, suggesting that some static component tethers the speckles to a particular location within the nucleoplasm. To investigate this possibility, we used Clk/STY overexpression as a tool for modulating the integrity of speckles in vivo. We reasoned that the release of SR proteins from speckles might allow us to reveal underlying structural elements, such as a network of filaments or SR protein receptors, hypothesizing that a specific structural component of nuclear speckles would maintain its localization while splicing factors would be released. However, all proteins examined, including some predicted to serve structural roles, like nuclear speckle populations of lamin A (Jagatheesan et al., 1999) and snRNP-associated actin (Nakayasu and Ueda, 1984), redistributed upon overexpression of Clk/STY. Furthermore, ultrastructural analysis of cells without intact nuclear speckles did not reveal empty regions or areas in the nucleoplasm that appeared to be remnants of a previously intact IGC. We conclude from this study that IGCs are likely to be maintained by protein–protein interactions, including RS domain–RS domain interactions among members of the SR protein family of pre-mRNA–splicing factors, rather than by attachment to an IGC-specific framework.
Although it does not appear that IGC components, such as lamin A or actin, form filaments that provide a scaffold for clustering of interchromatin granules, it is possible that individual interchromatin granules may require these structural proteins as monomers or very short multimers in order to assemble a large number of protein and RNA components into particles. Alternatively, G-actin or lamin A monomers may be recruited to transcription sites as members of interchromatin granules. Once at transcription sites, they may multimerize into short filaments that may act as a scaffold for the assembly/disassembly of the transcription RNA processing complex. In support of this possibility, β-actin and actin-related proteins are components of the mammalian SWI/SNF-like BAF (Brg-associated factor) complex, and binding of the BAF complex to the nuclear matrix in vitro is enhanced by phosphatidylinositol (4,5)-bisphosphate (PtdIns[4,5]P2), a lipid that regulates actin-binding proteins (Zhao et al., 1988). Furthermore, PtdIns(4,5)P2 and multiple phosphatidylinositol phosphate kinase (PIPK) isoforms have been localized to nuclear speckles in vivo by antibody labeling (Boronenkov et al., 1998). Recently, Percipalle et al. (2001) have shown that actin becomes associated with a Balbiani ring mRNA via a heterogeneous nuclear ribonucleoprotein (hrp36) at the site of transcription. Future studies will directly address the organization of individual interchromatin granules and the possible role of structural proteins in their assembly/disassembly.
We examined the effect of Clk/STY hyperphosphorylation on the release of a large number of protein constituents of IGCs, including many that do not contain the RS domain essential for phosphorylation by Clk/STY. Our finding that all proteins redistributed, regardless of the presence of an RS domain, is consistent with the proposal that transcription and RNA-processing factors may exist in the nucleus in a unitary particle called a transcriptosome (Gall et al., 1999), or alternatively, in multiple smaller complexes. However, it is currently unclear if such a particle is held together simply by protein–protein interactions or in concert with other potential mechanisms. In this regard, it is particularly intriguing that upon overexpression of Clk/STY, the stable population of poly(A)+ RNA that is present in IGCs (Huang et al., 1994) becomes diffusely distributed throughout the nucleoplasm, whereas nascent transcripts at specific transcription sites do not redistribute. This finding raises the possibility that the stable population of poly(A)+ RNA that is localized to IGCs may have a role in maintaining the organization of pre-mRNA–processing factors at these nuclear domains. These stable RNAs may represent the core organizing unit of individual interchromatin granules and the binding site for RNA-processing proteins. Studies are currently underway to purify and characterize these RNA molecules.
The release of splicing factors from IGCs by hyperphosphorylation makes these factors available for recruitment to sites of transcription and splicing (Misteli et al., 1997). Since not all interchromatin granules dissociate at once, regulatory mechanisms must influence the steady-state level of interchromatin granules within these structures as well as the rate of release of complexes into the nucleoplasmic pool. In this study, we tested whether an entirely nucleoplasmic pool of splicing factors, that presumably would be fully accessible to transcription sites, was sufficient for both recruitment and function. Interestingly, we found that disruption of IGCs did not affect synthesis of pre-mRNA on either a global or a specific level. However, IGC disassembly largely prevented accumulation of splicing factors on nascent transcripts at the site of transcription, and in doing so significantly reduced or abolished pre-mRNA splicing. Interestingly, in a previous study we have shown that microinjection of antisense oligonucleotides or antibodies to pre-mRNA splicing factors resulted in the rounding up of nuclear speckles and an inhibition of both transcription and pre-mRNA splicing (O'Keefe et al., 1994). In this study we show that this coordination can be disrupted by the break-up of nuclear speckles, suggesting that this nuclear structure plays some role in coupling these two processes.
We cannot completely exclude the possibility that reduction in splicing in vivo could be due to inactivation of splicing factors via their hyperphosphorylation rather than the loss of IGCs. For example, a recent in vitro study showed that Clk/STY directly affects the activity of SR proteins, and altering the phosphorylation state of these proteins either by hyper- or hypophosphorylation resulted in inhibition of splicing activity (Prasad et al., 1999). Overexpression of Clk/STY in vivo has also been shown to affect splice site selection on a reporter transcript (Duncan et al., 1997), although correlation with the extent of IGC disassembly was not reported. However, our data demonstrate that upon hyperphosphorylation of SR proteins, all components tested, including those that do not contain RS domains, were redistributed. Both an SR protein (SC35, Fig. 6 F) and a non-SR protein U2-B” (unpublished data) failed to accumulate at transcription sites, and there was a marked reduction in spliced product. It is therefore likely that in vivo the organization of IGCs is fundamentally linked to the phosphorylation state of SR proteins and hence to the ability of the processing machinery to perform pre-mRNA splicing. This possibility is further supported by previous studies showing that phosphorylation of SR proteins was linked to their release from IGCs and subsequent recruitment to transcription sites (Misteli et al., 1997). While pre-mRNA splicing can occur in vitro in the absence of intact IGCs, it is conceivable that nuclear extracts used for such experiments contain individual interchromatin granules that may be altered upon SR protein hyper- or hypophosphorylation, leading to decreased splicing activity.
The present study implicates IGCs in the assembly/maturation of the RNA-processing machinery into splicing-competent complexes or particles that must be maintained during transit to active genes for efficient targeting and function. Perhaps certain components of these complexes are responsible for recognizing newly synthesized pre-mRNA and/or stabilizing the association of the splicing machinery on pre-mRNA. Our finding that GFP-Clk/STY was recruited to transcription sites is consistent with the possibility that, in addition to regulating release of splicing factors from IGCs, it might also regulate/remodel splicing factor interactions during alternative or constitutive splicing.
Assuming that Clk/STY is responsible for phosphorylation events that lead to release of splicing factors from nuclear speckles, then a mutant kinase that lacks SR protein kinase activity would be expected to inhibit the release of splicing factors in vivo. As predicted, overexpression of GFP-Clk/STY(K190R) causes peripheral regions of nuclear speckles to become immobilized. Splicing factors accumulate in foci, and this ultimately leads to a reduction in splicing activity. Perhaps the mutant kinase is interacting with its substrates, but because it is unable to phosphorylate them, the result is sequestration of hypophosphorylated splicing factor complexes in foci. Examination of these regions in fixed cells confirmed that SR proteins and snRNPs are present in the foci, and that there is a depletion of phosphorylated splicing factors in the regions that become immobilized. Morphological changes of nuclear speckles in living cells overexpressing GFP-Clk/STY(K190R) and phosphoepitope depletion in foci of such nuclear speckles strongly support the idea that phosphorylation of SR proteins by Clk/STY is one of the key events that results in recruitment of splicing factor complexes from nuclear speckles to sites of transcription. Furthermore, alterations in the phosphorylation state of SR proteins is highly correlated with IGC structural reorganization, and demonstrates an important link between structure and function in the mammalian cell nucleus.
Materials and methods
cDNA constructs
PCR was used to generate restriction sites at the start codon of murine Clk/STY1 and Clk/STY1 (K190R) cDNAs for convenient subcloning into pEGFP-C3 (CLONTECH Laboratories, Inc.). Inducible Clk/STY1 overexpression was achieved by using a tetracycline responsive element FLAG-pUHD-104B (Tsukamoto et al., 2000). KIAA cDNA clones were obtained from Kazusa DNA Research Institute (Chiba, Japan) and subcloned in-frame into pEGFP-C vectors.
Cell culture and transfection
A-431 cells were grown in DME containing high glucose (GIBCO BRL/Life Technologies) supplemented with penicillin-streptomycin and 10% fetal bovine serum (Hyclone). Cells were seeded onto acid-washed coverslips in 35-mm petri dishes containing 2 ml DME, and attached cells were transiently transfected with 2 μg total DNA using FUGENE (Roche) according to manufacturer instructions. FLAG-Clk/STY was cotransfected with pTetON (CLONTECH Laboratories, Inc.), and expression of kinase was induced by addition of doxycycline (2.0 μg/ml). GFP–KIAA fusion constructs were cotransfected with FLAG-Clk/STY + pTetOn 24 h before fixation, and doxycycline was added 12–14 h before fixation. In all other experiments, cells were processed for immunofluorescence localization of nuclear speckle proteins 14–16 h after transfection.
Immunofluorescence
Cells were rinsed briefly in PBS then fixed for 15 min in 2% formaldehyde in PBS (pH 7.4) or for 5 min in methanol (−20°C) for optimal penetration of IgM antibodies into nuclei. Cells were permeabilized in PBS +0.2% Triton X-100 + 0.5% goat serum, and primary antibodies were added for 1 h at room temperature: anti-FLAG M2 (Sigma-Aldrich; 10 μg/ml), SC35 (1:1000), 3C5 ascites (1:200), B” (1:200), mAb103 anti-SF2/ASF (1:15), mAb104 anti-SR (undiluted); M3 anti-pinin guinea pig serum (1:100), AC40 antiactin (Sigma-Aldrich; 1:100), 2H10 anti-lamin (undiluted), PABII (1:200); anti-snRNA m3G (1:40); ANA-N (fibrillarin, 1:10). Cells were rinsed in PBS + 0.5% goat serum, then secondary antispecies-specific antibodies (Jackson ImmunoResearch Laboratories) were added for 1 h at room temperature: goat anti–mouse (GAM) IgG1-Texas red (1:1,000), GAM IgG H+L Texas Red (1:500), GAM IgM-Cy5 (1:1,000), donkey anti–guinea pig Cy5 (1:400), goat anti–rat IgG-fluorescein (1:1,200). Cells were examined using a ZEISS Axioplan 2i fluorescence microscope equipped with Chroma filters (Chroma Technology). OpenLab software (Improvision) was used to collect digital images.
TEM analysis
A-431 cells seeded onto gridded coverslips and transfected with GFP-Clk/STY were fixed for 15 min in 2% formaldehyde/0.5% glutaraldehyde in PBS (pH 7.4). Cells were rinsed in buffer A (PBS + 0.5% goat serum + 0.3 M glycine), then permeabilized for 20 min in PBS + 2% saponin. Anti-SC35 antibody was applied (1:1,000) in buffer B (PBS + 0.5% goat serum + 0.3 M glycine + 0.5% saponin) for 1 h at room temperature. Cells were rinsed in buffer B and Texas red GAM-IgG1 was applied (1:1,000) in buffer B. Cells were rinsed in buffer B, mounted, and sealed with rubber cement. The position of cells expressing GFP-Clk/STY and exhibiting completely disassembled SC35 nuclear speckles was documented and used later to relocate the cells for thin sectioning. Coverslips were processed as described (Huang et al., 1994); briefly, embedded cells were thin sectioned (100 nm), stained by the EDTA regressive method (Bernhard, 1969) and labeled with 3C5 antibody followed by 5 nm colloidal gold–conjugated secondary antibody. Sections were examined using a Hitachi H-7000 TEM operated at 75 kilovolts.
Bromo-UTP incorporation
A-431 cells transfected with FLAG-Clk/STY were rinsed briefly in glycerol buffer (20 mM Tris HCl, pH 7.4, 5.0 mM MgCl2, 25% glycerol, 0.5 mM PMSF, and 0.5 mM EGTA) followed by permeabilization for 5 min in glycerol buffer supplemented with digitonin (20 μg/ml). Transcription buffer (100 mM KCl, 50 mM Tris HCl, pH 7.4, 5 mM MgCl2, 0.5 mM EGTA, 25% glycerol, 1.0 mM PMSF, 2.0 mM ATP, 0.5 mM GTP, 0.5 mM CTP, 0.2 mM bromo-UTP, 1.0 μg/ml RNAsin, 5.0 μg/ml digitonin) was added for 5 min at 37°C. Cells were rinsed in PBS then fixed in 2% formaldehyde in PBS (pH 7.4) for 15 min followed by methanol (−20°C) for 5 min. Triple localization of FLAG-Clk (M2), SR proteins (3C5), and bromo-UTP (rat anti-bromo, 1:30) was performed as described above.
RNA in situ hybridization for polyadenylated (polyA+) RNA
Cells transfected with FLAG-Clk/STY were fixed in 2% formaldehyde. Hybridization and detection of digoxygenin (DIG)-labeled oligo dT(50) probe was performed according to Huang et al. (1994). FLAG-Clk/STY was detected with M2 anti-FLAG antibody (Sigma-Aldrich) followed by Texas red GAM IgG1.
Nick-translation of β-globin genomic probe
β-globin genomic DNA was subcloned into pBluescript (Stratagene). 2 μg DNA was nick translated in buffer (50 mM Tris HCl, pH 8.0; 5.0 mM MgCl2; 0.05 mg/ml BSA; 0.001 M β-mercaptoethanol; 0.5 mM each dATP, dGTP, and dCTP; 0.125 mM dTTP; 0.375 mM DIG-11-dUTP) containing 0.01 mg/ml DNaseI and 10 U E. coli DNA polymerase for 2 h at 16°C to obtain fragments of 300–500 bp. The probe was precipitated, resuspended in 20 μl water, and 2 μl of this suspension was used per hybridization sample.
RNA FISH
A-431 cells were stably transfected with β-globin genomic DNA using FUGENE and selected in 0.1 mg/ml G-418. Two stable clones were used to examine splicing factor recruitment and splicing of the reporter pre-mRNA. GFP-Clk/STY was transfected by electroporation optimized for A-431 cells. Trypsinized cells were washed in PBS, resuspended in 500 μl ice-cold cytomix buffer (10 mM K2HPO4, 10 mM KH2PO4, 25 mM Hepes, 120 mM KCl, 0.15 mM CaCl2, 5 mM MgCl2, 2 mM EGTA, 5 mM glutathione, and 2 mM ATP; pH 7.6), and added to cuvettes containing 12 μg GFP-Clk/STY. After a 5 min incubation on ice, cells were electroporated (380 mV, 950 μF), transferred to 6 ml culture medium, and plated 2 ml/coverslip coated with fibronectin (Sigma-Aldrich) to improve A-431 electroporation efficiency (Hashino et al., 1997). 16 h after transfection, cells were processed for colocalization of SC35 with β-globin pre-mRNA. Cells were fixed for 15 min in 2% formaldehyde, permeabilized 5 min in PBS + 0.5% Triton X-100, washed 3 times for 5 min each in PBS + 0.02% BSA (New England BioLabs, Inc.), incubated in anti-SC35 (1:1,000) + 0.1% BSA for 1 h at room temperature, washed 3 times for 5 min each in PBS, and incubated in Cy5-conjugated GAM IgG (1:800; Jackson ImmunoResearch Laboratories, Inc.) for 1 h at room temperature. Cells were washed 3 times for 5 min each in PBS and fixed for 5 min in 2% formaldehyde followed by washing 3 times for 5 min each in PBS and 10 min in 2XSSC. Denatured nick-translated DIG-11-dUTP–labeled β-globin probe (∼200 ng) was added in 50% deionized formamide, 10% dextran sulfate, 1 mg/ml yeast tRNA and 2XSSC, and coverslips were inverted onto slides and sealed with rubber cement for overnight hybridization at 37°C in a humidified chamber. Cells were washed 30 min in 50% formamide/2XSSC at 37°C, then 30 min each in 2XSSC and 1XSSC. Probe was detected with sheep anti-DIG Fab fragments (1:300; Roche) followed by donkey anti–sheep Texas red (1:600; Jackson ImmunoResearch Laboratories).
To detect splicing of β-globin pre-mRNA, 30-mer oligonucleotides conjugated with a single Texas red molecule at the 5′ end (GIBCO BRL) were designed to hybridize to intron 2 (i2: 5′-gacttccacactgatgcaatcattcgtctg-3′) or to the splice junction between exons 2 and 3 (e2/3: 5′-cacgttgcccaggagcctgaagttctcagg-3′). Cells were transfected with GFP-Clk/STY by electroporation as described above. Cells were extracted in CSK buffer (100 mM NaCl, 300 mM sucrose, 3 mM MgCl2, 10 mM Pipes, pH 6.8) supplemented with 0.5% Triton X-100 and 2 mM vanadyl ribonucleoside complex, and hybridization of oligo probes was performed in 25% deionized formamide, 10% dextran sulfate, 1 mg/ml yeast tRNA, and 2XSSC for 3 h at 37°C. Cells were washed for 30 min in 25% formamide/2XSSC at 37°C then 30 min in 2XSSC. Hybridization signals were scored in untransfected versus transfected cells. Multiple focal planes were examined to confirm the absence of signal in cells without intact nuclear speckles.
Live cell microscopy
Attached cells were transfected with GFP-Clk/STY using FUGENE as described above. The cells were transferred 4 h after transfection to an FCS2 live-cell chamber (Bioptechs) mounted onto the stage of an Olympus IX70 inverted fluorescence microscope (Olympus) and kept at 37°C in L-15 medium containing 10% FBS and without phenol red. Time-lapse images acquired with a 100× 1.4 NA heated objective lens were captured with a peltier-cooled IMAGO CCD camera using an SVGA interline chip (1,280 × 1,024) with a pixel size of 6.7 × 6.7 μm (Till Photonics) as soon as nuclear expression was initially detected (at ∼6.0 h). For GFP-Clk/STY(K190R), a sequence of 200 exposures (350 ms each) was recorded every 30 min.
Online supplemental material
Videos corresponding to Fig. 8 are presented. Image sequences were acquired using TillVision software (Till Photonics) and animated using QuickTime software. For each video a sequence of 200 images (350 ms each) was taken. Video speed is five times faster than real time. Video 1 shows nuclear speckle dynamics in a cell overexpressing GFP-Clk/STY(K190R) at 6.0 h posttransfection. GFP-Clk/STY localizes to nuclear speckles and exhibits rapid movement in and out of the speckles. Focal accumulations of GFP-Clk/STY are seen on several speckles. Video 2 shows the same cell at 6.5 h posttransfection. Although all of the nuclear speckles exhibit foci at this stage, nuclear speckle dynamics outside of the foci are largely unaffected. Video 3 shows the same cell at 7.0 h posttransfection. As multiple foci form on each nuclear speckle, they appear paired and larger regions of the speckles become immobilized. Video 4 shows the same cell at 7.5 h posttransfection. The nuclear speckles are almost completely immobilized. Videos are available at http://www.jcb.org/cgi/content/full/jcb.200107017/DC1.
Supplemental Material
Acknowledgments
We acknowledge Tamara Howard, Gayle Lark, and Stephen Hearn for excellent technical assistance with TEM. We thank P.S. Pendergrast and M. Izaguirre-Sierra for technical assistance. We thank the members of the Spector laboratory, E. Heard, and A. Krainer for insightful discussions and critical review of the manuscript. We thank W. Franke, A. Krainer, V. Parnaik, B. Turner, and W. van Venrooij for gifts of antibodies; and T. Pawson (Clk/STY), T. Nagase (KIAA clones), and M. Wilkinson (β-globin) for providing cDNA clones.
P. Sacco-Bubulya is funded by a postdoctoral fellowship from NIH/NIGMS. D.L. Spector is supported by NIH/NIGMS 42694.
The online version of this article contains supplemental material.
Footnotes
Abbreviations used in this paper: Clk, cdc2-like kinase; DIG, digoxygenin; GFP, green fluorescent protein; IGC, interchromatin granule cluster; PF, perichromatin fibril; RS, arginine-serine rich; SR, serine-arginine; TEM, transmission electron microscopy.
References
- Alahari, S., H. Schmidt, and N. Kaufer. 1993. The fission yeast prp4+ gene involved in pre-mRNA splicing codes for a predicted serine/threonine kinase and is essential for growth. Nucleic Acids Res. 21:4079–4083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-David, Y., K. Letwin, L. Tannock, A. Bernstein, and T. Pawson. 1991. A mammalian protein kinase with potential for serine/threonine and tyrosine phosphorylation is related to cell cycle regulators. EMBO J. 10:317–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernhard, W. 1969. A new staining procedure for electron microscopical cytology. J. Ultrastruc. Res. 27:250–265. [DOI] [PubMed] [Google Scholar]
- Boronenkov, I.V., J.C. Loijens, M. Umeda, and R.A. Anderson. 1998. Phosphoinositide signaling pathways in nuclei are associated with nuclear speckles containing pre-mRNA processing factors. Mol. Biol. Cell. 9:3547–3560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandner, J.M., S. Reidenbach, and W.W. Franke. 1997. Evidence that “pinin”, reportedly a differentiation-specific desmosomal protein, is actually a widespread nuclear protein. Differentiation. 62:119–127. [DOI] [PubMed] [Google Scholar]
- Bregman, D.B., L. Du, S. van der Zee, and S.L. Warren. 1995. Transcription-dependent redistribution of the large subunit of RNA polymerase II to discrete nuclear domains. J. Cell Biol. 129:287–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmo-Fonseca, M., R. Pepperkok, M.T. Carvalho, and A.I. Lamond. 1992. Transcription-dependent colocalization of the U1, U2, U4/U6 and U5 snRNPs in coiled bodies. J. Cell Biol. 117:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cmarko, D., P.J. Verschure, T.E. Martin, M.E. Dahmus, S. Krause, X.D. Fu, R. van Driel, and S. Fakan. 1999. Ultrastructural analysis of transcription and splicing in the cell nucleus after bromo-UTP microinjection. Mol. Biol. Cell. 10:211–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colwill, K., L.L. Feng, J.M. Yeakley, G.D. Gish, J.F. Cáceres, T. Pawson, and X.-D. Fu. 1996. a. SRPK1 and Clk/Sty protein kinases show distinct substrate specificities for serine/arginine-rich splicing factors. J. Biol. Chem. 271:24569–24575. [DOI] [PubMed] [Google Scholar]
- Colwill, K., T. Pawson, B. Andrews, J. Prasad, J.L. Manley, J.C. Bell, and P.I. Duncan. 1996. b. The Clk/Sty protein kinase phosphorylates splicing factors and regulates their intranuclear distribution. EMBO J. 15:265–275. [PMC free article] [PubMed] [Google Scholar]
- Duncan, P.I., D.F. Stojdl, R.M. Marius, and J.C. Bell. 1997. In vivo regulation of alternative pre-mRNA splicing by the Clk1 protein kinase. Mol. Cell. Biol. 17:5996–6001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eils, R., D. Gerlich, W. Tvarusko, D.L. Spector, and T. Misteli. 2000. Quantitative imaging of pre-mRNA splicing factors in living cells. Mol. Biol. Cell. 11:413–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fakan, S. 1994. Perichromatin fibrils are in situ forms of nascent transcripts. Trends Cell Biol. 4:86–90. [DOI] [PubMed] [Google Scholar]
- Fakan, S., and E. Puvion. 1980. The ultrastructural visualization of nucleolar and extranucleolar RNA synthesis and distribution. Int. Rev. Cytol. 65:255–299. [DOI] [PubMed] [Google Scholar]
- Gall, J.G. 2000. Cajal bodies: the first 100 years. Annu. Rev. Cell Dev. Biol. 16:273–300. [DOI] [PubMed] [Google Scholar]
- Gall, J.G., M. Bellini, Z. Wu, and C. Murphy. 1999. Assembly of the nuclear transcription and processing machinery: Cajal bodies (coiled bodies) and transcriptosomes. Mol. Biol. Cell. 10:4385–4402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gui, J.F., W.S. Lane, and X.-D. Fu. 1994. A serine kinase regulates intracellular localization of splicing factors in the cell cycle. Nature. 369:678–682. [DOI] [PubMed] [Google Scholar]
- Habets, W.J., M.H. Hoet, B.A.W. DeJong, A. VanDer Kemp, and W.J. Van Venrooij. 1986. Mapping of B cell epitopes on small nuclear ribonucleoproteins that react with human autoantibodies as well as with experimentally induced mouse monoclonal antibodies. J. Immunol. 143:2560–2566. [PubMed] [Google Scholar]
- Hanes, J., H. von der Kammer, J. Klaudiny, and K.H. Scheit. 1994. Characterization by cDNA cloning of two new human protein kinases. Evidence by sequence comparison of a new family of mammalian protein kinases. J. Mol. Biol. 244:665–672. [DOI] [PubMed] [Google Scholar]
- Hashino, K., H. Matsushita, and I. Kato. 1997. Effects of fibronectin fragments on DNA transfection into mammalian cells by electroporation. J. Biochem. 122:490–493. [DOI] [PubMed] [Google Scholar]
- Holzmann, K., C. Gerner, A. Poltl, R. Schafer, P. Obrist, C. Ensinger, R. Grimm, and G. Sauermann. 2000. A human common nuclear matrix protein homologous to eukaryotic translation initiation factor 4A. Biochem. Biophys. Res. Commun. 267:339–344. [DOI] [PubMed] [Google Scholar]
- Howell, B.W., D.E. Afar, J. Lew, E.M. Douville, P.L. Icely, D.A. Gray, and J.C. Bell. 1991. STY, a tyrosine-phosphorylating enzyme with sequence homology to serine/threonine kinases. Mol. Cell. Biol. 11:568–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, S., T.J. Deerinck, M.H. Ellisman, and D.L. Spector. 1994. In vivo analysis of the stability and transport of nuclear poly(A)+ RNA. J. Cell Biol. 126:877–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, S., and D.L. Spector. 1996. Intron-dependent recruitment of pre-mRNA splicing factors to sites of transcription. J. Cell Biol. 131:719–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagatheesan, G., S. Thanumalayan, B. Muralikrishna, N. Rangaraj, A.A. Karande, and V.K. Parnaik. 1999. Colocalization of intranuclear lamin foci with RNA splicing factors. J. Cell Sci. 112:4651–4661. [DOI] [PubMed] [Google Scholar]
- Jiménez-García, L.F., and D.L. Spector. 1993. In vivo evidence that transcription and splicing are coordinated by a recruiting mechanism. Cell. 73:47–59. [DOI] [PubMed] [Google Scholar]
- Johnson, K.W., and K.A. Smith. 1991. Molecular cloning of a novel human cdc2/CDC28-like protein kinase. J. Biol. Chem. 266:3402–3407. [PubMed] [Google Scholar]
- Kojima, T., T. Zama, K. Wada, H. Onogi, and M. Hagiwara. 2001. Cloning of human PRP4 reveals interaction with Clk1. J. Biol. Chem. 276:32247–32256. [DOI] [PubMed] [Google Scholar]
- Kruhlak, M.J., M.A. Lever, W. Fischle, E. Verdin, D.P. Bazett-Jones, and M.J. Hendzel. 2000. Reduced mobility of the alternate splicing factor (ASF) through the nucleoplasm and steady state speckle compartments. J. Cell Biol. 150:41–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamond, A.I., and W.C. Earnshaw. 1998. Structure and function in the nucleus. Science. 280:547–553. [DOI] [PubMed] [Google Scholar]
- Lee, K., C. Du, M. Horn, and L. Rabinow. 1996. Activity and autophosphorylation of LAMMER protein kinases. J. Biol. Chem. 271:27299–27303. [DOI] [PubMed] [Google Scholar]
- Li, Q., H. Imataka, S. Morino, G.W. Rogers, Jr., N.J. Richter-Cook, W.C. Merrick, and N. Sonenberg. 1999. Eukaryotic translation initiation factor 4AIII (eIF4AIII) is functionally distinct from eIF4AI and eIF4AII. Mol. Cell. Biol. 19:7336–7346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindberg, R.A., A.M. Quinn, and T. Hunter. 1992. Dual-specificity protein kinases: will any hydroxl do? Trends Biochem. Sci. 17:114–119. [DOI] [PubMed] [Google Scholar]
- Mintz, P.J., S.D. Patterson, A.F. Neuwald, C.S. Spahr, and D.L. Spector. 1999. Purification and biochemical characterization of interchromatin granule clusters. EMBO J. 18:4308–4320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misteli, T., J.F. Cáceres, and D.L. Spector. 1997. The dynamics of a pre-mRNA splicing factor in living cells. Nature. 387:523–527. [DOI] [PubMed] [Google Scholar]
- Nakayasu, H., and K. Ueda. 1984. Small nuclear RNA-protein complex anchors on the actin filaments in bovine lymphocyte nuclear matrix. Cell Struct. Funct. 9:317–325. [DOI] [PubMed] [Google Scholar]
- Nayler, O., S. Stamm, and A. Ullrich. 1997. Characterization and comparison of four serine- and arginine-rich (SR) protein kinases. Biochem. J. 326:693–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Keefe, R.T., A. Mayeda, C.L. Sadowski, A.R. Krainer, and D.L. Spector. 1994. Disruption of pre-mRNA splicing in-vivo results in reorganization of splicing factors. J. Cell Biol. 124:249–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang, P. 1999. Antibodies differentiate desmosome-form and nucleus-form pinin: evidence that pinin is a moonlighting protein with dual location at the desmosome and within the nucleus. Biochem. Biophys. Res. Commun. 263:192–200. [DOI] [PubMed] [Google Scholar]
- Ouyang, P., and S.P. Sugrue. 1996. Characterization of pinin, a novel protein associated with the desmosome-intermediate filament complex. J. Cell Biol. 135:1027–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang, P., Y.Y. Zhen, and S.P. Sugrue. 1997. Cloning and analysis of cDNA encoding murine pinin. Gene. 197:115–120. [DOI] [PubMed] [Google Scholar]
- Percipalle, P., J. Zhao, B. Pope, A. Weeds, U. Lindberg, and B. Daneholt. 2001. Actin bound to the heterogeneous nuclear ribonucleoprotein hrp36 is associated with balbiani ring mRNA from the gene to polysomes. J. Cell Biol. 153:229–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phair, R.D., and T. Misteli. 2000. High mobility of proteins in the mammalian cell nucleus. Nature. 404:604–609. [DOI] [PubMed] [Google Scholar]
- Prasad, J., K. Colwill, T. Pawson, and J.L. Manley. 1999. The protein kinase Clk/Sty directly modulates SR protein activity: both hyper- and hypophosphorylation inhibit splicing. Mol. Cell. Biol. 19:6991–7000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth, M.B., C. Murphy, and J.G. Gall. 1990. A monoclonal antibody that recognizes a phosphorylated epitope stains lampbrush chromosomes and small granules in the amphibian germinal vesicle. J. Cell Biol. 111:2217–2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spector, D.L. 1990. Higher order nuclear organization: three-dimensional distribution of small nuclear ribonucleoprotein particles. Proc. Natl. Acad. Sci. USA. 87:147–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spector, D.L., G. Lark, and S. Huang. 1992. Differences in snRNP localization between transformed and nontransformed cells. Mol. Biol. Cell. 3:555–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spector, D.L. 1993. Nuclear organization of pre-mRNA processing. Curr. Opin. Cell Biol. 5:442–448. [DOI] [PubMed] [Google Scholar]
- Spector, D.L., R.T. O'Keefe, and L.F. Jiménez-García. 1993. Dynamics of transcription and pre-mRNA splicing within the mammalian cell nucleus. Cold Spring Harb. Symp. Quant. Biol. 58:799–805. [DOI] [PubMed] [Google Scholar]
- Tsukamoto, T.N., Hashiguchi, S.M. Janicki, T. Tumbar, A.S. Belmont, and D.L. Spector. 2000. Visualization of gene activity in living cells. Nat. Cell Biol. 2:871–878. [DOI] [PubMed] [Google Scholar]
- Turner, B.M., and L. Franchi. 1987. Identification of protein antigens associated with the nuclear matrix and with clusters of interchromatin granules in both interphase and mitotic cells. J. Cell Sci. 87:269–282. [DOI] [PubMed] [Google Scholar]
- Wang, H.-Y., W. Lin, J.A. Dyck, J.M. Yeakley, Z. Songyang, L.C. Cantley, and X.-D. Fu. 1998. SRPK2: A differentially expressed SR protein-specific kinase involved in mediating the interaction and localization of pre-mRNA splicing factors in mammalian cells. J. Cell Biol. 140:737-750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei, X., S. Somanathan, J. Samarabandu, and R. Berezney. 1999. Three-dimensional visualization of transcription sites and their association with splicing factor-rich nuclear speckles. J. Cell Biol. 146:543–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein, D.C., E. Honore, and A. Hemmati-Brivanlou. 1997. Epidermal induction and inhibition of neural fate by translation initiation factor 4AIII. Development. 124:4235–4242. [DOI] [PubMed] [Google Scholar]
- Xing, Y., C.V. Johnson, P.R. Dobner, and J.B. Lawrence. 1993. Higher level organization of individual gene transcription and RNA splicing. Science. 259:1326–1330. [DOI] [PubMed] [Google Scholar]
- Yeakley, J.M., H. Tronchere, J. Olesen, J.A. Dyck, H.Y. Wang, and X.D. Fu. 1999. Phosphorylation regulates in vivo interaction and molecular targeting of serine/arginine-rich pre-mRNA splicing factors. J. Cell Biol. 145:447–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, K., W. Wang, O.J. Rando, Y. Xue, K. Swiderek, A. Kuo, and G.R. Crabtree. 1988. Rapid and phosphoinositol-dependent binding of the SWI/SNF-like BAF complex to chromatin after T lymphocyte receptor signaling. Cell. 95:625–636. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.