

Abstract
The mammalian Golgi complex is comprised of a ribbon of stacked cisternal membranes often located in the pericentriolar region of the cell. Here, we report that during apoptosis the Golgi ribbon is fragmented into dispersed clusters of tubulo-vesicular membranes. We have found that fragmentation is caspase dependent and identified GRASP65 (Golgi reassembly and stacking protein of 65 kD) as a novel caspase substrate. GRASP65 is cleaved specifically by caspase-3 at conserved sites in its membrane distal COOH terminus at an early stage of the execution phase. Expression of a caspase-resistant form of GRASP65 partially preserved cisternal stacking and inhibited breakdown of the Golgi ribbon in apoptotic cells. Our results suggest that GRASP65 is an important structural component required for maintenance of Golgi apparatus integrity.
Keywords: Golgi apparatus; apoptosis; GRASP65; Golgi structure; caspase
Introduction
The mammalian Golgi complex is comprised of stacked cisternal membranes that in animal cells are linked to form a compact juxtanuclear ribbon (Rambourg and Clermont, 1997). The mechanism of Golgi stacking is poorly defined but appears to involve proteins that form filamentous cross-bridges between adjacent cisternae (Mollenhauer, 1965; Franke et al., 1972; Cluett and Brown, 1992). Recent studies using an in vitro system for the postmitotic reassembly of Golgi stacks from mitotic Golgi fragments (Rabouille et al., 1995) have identified candidate stacking proteins.
The first stacking protein to be identified from in vitro studies was GRASP65 (Golgi reassembly and stacking protein of 65 kD), an N-myristoylated peripheral membrane protein localized predominantly to the cis side of the Golgi complex (Barr et al., 1997). GRASP65 is a receptor for the Golgi matrix protein GM130 (Barr et al., 1998) that in turn binds the vesicle tethering protein p115 to mediate the docking of transport vesicles with Golgi membranes (Nakamura et al., 1997; Sönnichsen et al., 1998). In addition to tethering transport vesicles, GM130 and p115 are transiently required during postmitotic stack formation, possibly for the initial alignment of cisternae that occurs before the formation of stable cross-bridges (Shorter and Warren, 1999). Importantly, GRASP65 has a function in stacking distinct from its role as a GM130 receptor; it is required for stack formation after the requirement for GM130 and p115 has passed, although the details of this later activity are poorly defined (Linstedt, 1999; Shorter and Warren, 1999). A second stacking protein related to GRASP65 has been identified recently. Although this protein, called GRASP55, is also required for postmitotic stacking in vitro, its localization to the medial-Golgi and lack of interaction with GM130 suggests it is required at a step distinct from that mediated by GRASP65 (Shorter et al., 1999).
The execution phase of apoptosis is characterized by several dramatic morphological changes including cell shrinkage, chromatin condensation, nuclear disassembly, and membrane blebbing (Kerr et al., 1972; Wyllie et al., 1980). Ultimately, these events lead to the fragmentation of the cell, generating apoptotic bodies that are engulfed rapidly by neighboring phagocytes. The execution phase requires the activation of a cascade of specific cysteine proteases termed the caspases (for reviews see Cohen, 1997; Earnshaw et al., 1999). These proteases are synthesized as inactive zymogens that undergo cleavage during apoptosis, generating large and small subunits that heterodimerize to yield the active enzyme. Caspases can be divided into two groups: the initiator caspases, whose major function is to activate the second type of caspases, and the executioner caspases, which are responsible for the cleavage of specific target proteins. Cleavage is highly selective and defined by an essential apartate residue at the site of cleavage with differential recognition by particular family members dictated by residues immediately adjacent to the site of cleavage (Thornberry et al., 1997). Although many caspase substrates have been identified, in only a few cases has the resulting cleavage been demonstrated to have a role in apoptotic execution. For example, membrane blebbing results from cleavage of Rho-associated kinase ROCK1 (Coleman et al., 2001; Sebbagh et al., 2001), whereas nuclear shrinkage and budding appears to require cleavage of nuclear lamins (Rao et al., 1996).
Recent morphological studies have shown that the Golgi complex is fragmented during apoptosis (Philpott et al., 1996; Sesso et al., 1999; Mancini et al., 2000). The molecular mechanisms underlying fragmentation are unknown. One molecule that appears to be involved is golgin-160, a member of the golgin family of coiled-coil proteins (Fritzler et al., 1993; Chan and Fritzler, 1998). Golgin-160 is cleaved by caspases 2, 3, and 7, and a mutant from of golgin-160 lacking the caspase 2 cleavage site appears to delay Golgi fragmentation in apoptosis, suggesting a role for this cleavage event in the morphological changes observed (Mancini et al., 2000). However, since the function of golgin-160 is unknown it is not clear how its cleavage contributes to the Golgi fragmentation process.
Here, we have studied the morphological changes that occur during Golgi fragmentation in apoptosis at high resolution and find that the Golgi ribbon is converted to tubulo-vesicular membrane clusters morphologically similar to those seen in mitosis. We identify GRASP65 as a substrate for caspase-3 and find that it is quantitatively cleaved early in the apoptotic execution phase. Furthermore, we show that Golgi fragmentation in apoptotic cells is due, at least in part, to GRASP65 cleavage, thereby providing the first direct evidence that GRASP65 functions as a Golgi structural protein in vivo.
Results
Morphological changes during apoptotic fragmentation of the Golgi complex
Previous studies that have addressed subcellular alterations during apoptosis have reported fragmentation and/or scattering of the Golgi complex (Philpott et al., 1996; Sesso et al., 1999; Mancini et al., 2000). At present, this change in organelle morphology remains ill defined, and the timing and its significance during apoptosis is unknown. We have used a combination of high resolution light microscopy and EM to characterize this process in more detail. HeLa cells were treated with several different stimuli to induce apoptosis and analyzed by immunofluorescence microscopy (Fig. 1 a). In healthy cells, the Golgi complex was a compact perinuclear ribbon as expected (Fig. 1 a). In contrast, in apoptotic cells, identified using an antibody specific for caspase-cleaved PARP, the Golgi complex was fragmented into punctate structures dispersed throughout the cytoplasm (Fig. 1 a, left, arrowheads). The fragmentation observed was independent of the apoptotic stimulus with morphologically similar fragments produced in cells treated with the protein synthesis inhibitor anisomycin, anti-Fas antibody, UV irradiation (Fig. 1 a), or the broad spectrum kinase inhibitor staurosporine (see Figs. 4 and 8). The extent of fragmentation varied between different apoptotic cells in a population (Fig. 1 a and Fig. 2; see Fig. 8), most likely reflecting the length of time that a given cell had been in the apoptotic execution phase. Similar changes to Golgi morphology were observed in apoptotic HeLa, A431, and NRK cells (unpublished data).
Figure 1.

Golgi fragmentation in apoptotic cells visualized by immunofluorescence microscopy. Immunofluorescence images of the Golgi complex in apoptotic HeLa cells. (a) Confocal images of HeLa cells treated for 6 h with 5 μg/ml anisomycin, 1 μg/ml anti-Fas antibody, or 100 J/m2 UV radiation and stained with antibodies to GM130 and cleaved PARP as indicated. DNA was stained with DAPI. The image on the right is a merge of GM130 (green), cleaved PARP (red), and DAPI (blue). (b) Confocal image of anisomycin-treated HeLa cells that had been preincubated with the cell permeable caspase inhibitor zVAD.fmk. Cells were stained with antibodies against GM130 (green), and the Golgi complex was imaged in rounded cells with partially condensed chromatin (DAPI, blue). Projected images of multiple confocal slices are shown. Bars, 10 μm.
Figure 4.
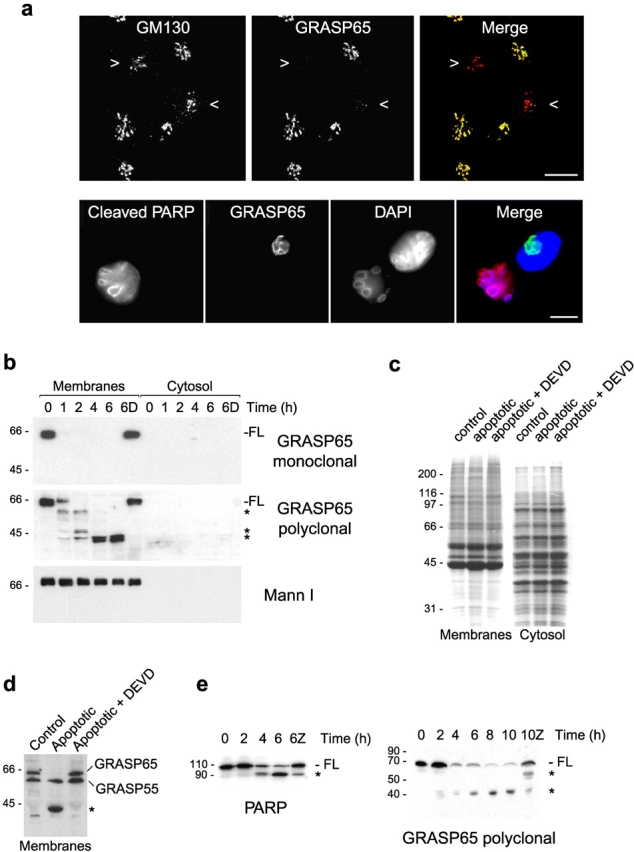
GRASP65 is a substrate for caspase-mediated proteolysis during apoptosis. (a) NRK cells treated with 1 μM staurosporine for 4 h were processed for immunofluorescence microscopy. (Top) Cells were double labeled with polyclonal antibodies to GM130 and a monoclonal antibody to GRASP65. Regions of overlap between GM130 (red) and GRASP65 (green) are shown in the merge in yellow. Apoptotic cells were identified using DAPI (unpublished data) and are indicated by arrowheads. (Bottom) Cells were labeled with polyclonal antibodies to cleaved PARP, a monoclonal antibody to GRASP65, and DAPI as indicated. A merged image showing cleaved PARP (red), GRASP65 (green), and DAPI (blue) is on the right. (b) Purified Golgi membranes were incubated at 37°C with apoptotic HeLa cytosol in the absence or presence of 2 μM Ac-DEVD-CHO (D) for the times indicated. Membranes were pelleted, and membrane and supernatant (cytosol) fractions were analyzed by immunoblotting with monoclonal or polyclonal (FBA31) antibodies to GRASP65 or antibodies to mannosidase I. (c and d) Golgi membranes were incubated with HeLa control cytosol or apoptotic cytosol in the absence or presence of 2 μM Ac-DEVD-CHO for 4 h at 37°C. Membrane and cytosol fractions were analyzed by Coomassie blue staining (c) or immunoblotting with an antipeptide antibody that recognizes both GRASP55 and GRASP65 (d). (e) NRK cells treated with 1 μM staurosporine in the absence or presence of 50 μM zVAD.fmk (Z) for the times indicated were analyzed by immunoblotting with monoclonal and polyclonal (CT) antibodies to PARP and GRASP65, respectively. (b, d, and e) Full-length (FL) and caspase cleavage products (asterisks) are marked. Bars, 10 μm.
Figure 8.

GRASP65 cleavage is required for fragmentation of the Golgi ribbon during apoptosis. The progress of Golgi fragmentation was analyzed in standard HeLa cells and cells stably expressing wild-type GRASP65-GFP or caspase-resistant mutant GRASP65-GFP. Staurosporine-treated cells (6 h) were fixed and then stained with a monoclonal antibody against GM130 (detected with Alexa 488–conjugated secondary antibody) and caspase-cleaved PARP (detected with Alexa 594 secondary antibody). (a) Cells were categorized for apoptotic progression (stage 1, PARP-positive nucleus with normal uncondensed chromatin; stage 2, PARP-positive nucleus with condensed chromatin; stage 3, highly condensed chromatin and PARP staining throughout the cell). (b) The condition of the Golgi apparatus was then assessed in apoptotic cells according to the parameters shown. (c) The proportion of cells displaying each category of Golgi morphology was quantitated in each of the three stages of apoptosis. All counting was performed blind. Means ± SEM are shown for three independent experiments. The total number of cells counted for each cell type was between 108–144 (stage 1), 358–392 (stage 2), and 35–72 (stage 3). Using the chi-square likelihood ratio contingency test, there was no significant difference at any stage between cells stably expressing wild-type GRASP65-GFP and the parental cells. For all experiments, there was a significant difference in the percentage of cells with stage 3 fragmented Golgi between cells expressing mutant GRASP65-GFP and either the parental cells (P = 0.001) or the wild-type GRASP65-GFP–expressing cells (P < 0.02). For stage 2, there was a significant difference in two out of three experiments between the mutant GRASP65-GFP cells and the parental cells and in all experiments between the mutant and wild-type GRASP65-GFP cells (P < 0.0167), whereas for stage 1 this was true in one out of three experiments.
Figure 2.

Immunofluorescence microscopy of apoptotic cells labeled with different Golgi markers. Confocal images of anisomycin-treated HeLa cells stained with antibodies to GM130 and either p115, GalNacT2, or TGN46 as indicated. DNA was stained with DAPI. Regions of overlap between GM130 (red) and either p115, GalNacT2, or TGN46 (green) are in yellow. 2× magnifications of the boxed areas in the merged images are shown on the right. Apoptotic cells were identified by their rounded morphology and characteristic condensed chromatin and are indicated by arrowheads. Projected images of multiple confocal slices are shown. Bars,10 μm (2 μm for zooms).
To determine if fragmentation requires caspase activity, cells were pretreated with the cell-permeable caspase inhibitor zVAD.fmk before apoptosis induction, and Golgi morphology was analyzed. Even though caspases were not active under these conditions (no cleaved PARP was detected; unpublished data), cells committed to undergo apoptosis continued to round-up as reported previously (McCarthy et al., 1997) (Fig. 1 b, arrowhead). However, no Golgi fragmentation was observed in these cells, demonstrating that gross changes in Golgi organization are caspase dependent (Fig. 1 b).
Apoptotic fragmentation was observed using several different Golgi markers. Golgi fragments positive for the cis-Golgi matrix protein GM130 were also positive for the cis-Golgi–associated tethering protein p115 (Fig. 2, top). GM130-containing fragments were also positive for the medial-Golgi enzyme GalNacT2, although the extent of overlap was less than that observed in nonapoptotic cells (Fig. 2, middle). In healthy cells, the trans-Golgi protein TGN46 colocalized with GM130, although a polarity could clearly be distinguished in the Golgi ribbon (Fig. 2, bottom). In apoptotic cells, TGN46 was present in fragments dispersed throughout the cytoplasm. In most cases, these appeared to be distinct from GM130, although occasionally they appeared to be adjacent to one another.
To study ultrastructural changes in Golgi morphology during apoptosis, we performed EM. HeLa cells were treated with staurosporine or anisomycin to induce apoptosis and analyzed in Epon sections or frozen cryosections labeled with antibodies to the medial-Golgi marker GalNacT2. Analysis of Epon sections showed that there were essentially no stacked cisternae remaining in the Golgi region of apoptotic cells (Fig. 3 c). Instead, there were numerous small vesiculo-tubular structures of <80 nm diameter together with a few larger electron-lucent vesicles of ∼100–200 nm diameter (Fig. 3 c). Stereological analysis confirmed the absence of stacked cisternae in apoptotic cells and revealed that nearly 80% of the membrane in the Golgi region was in the form of small vesicular structures with large vesicles accounting for the remaining 20% (Fig. 3 e). In cryosections, GalNacT2 labeling was predominantly over stacked cisternae in nonapoptotic cells as expected (Fig. 3 a). Quantitation revealed that ∼60% of the GalNacT2 was in stacked cisternae with the remaining ∼40% in small vesicles (Fig. 3 f). In contrast, in apoptotic cells there was essentially no GalNacT2 in stacks, and it was now found exclusively in large and small vesicles that accounted for ∼65 and ∼35% of the total labeling respectively (Fig. 3, b, d, and f). Interestingly, although there was little change in the amount of GalNacT2 in small vesicles between nonapoptotic and apoptotic cells, the loss of labeling in stacks could be fully accounted for by the increase of label in large vesicles (Fig. 3 f). This suggests that the large vesicles may be derived directly from GalNacT2-containing cisternae. All of the morphological changes observed were caspase dependent, since they could be prevented by the cell-permeable caspase inhibitor zVAD.fmk (unpublished data). Therefore, during apoptosis Golgi stacks are converted into vesicular membrane clusters, and this is dependent on caspase activation.
Figure 3.
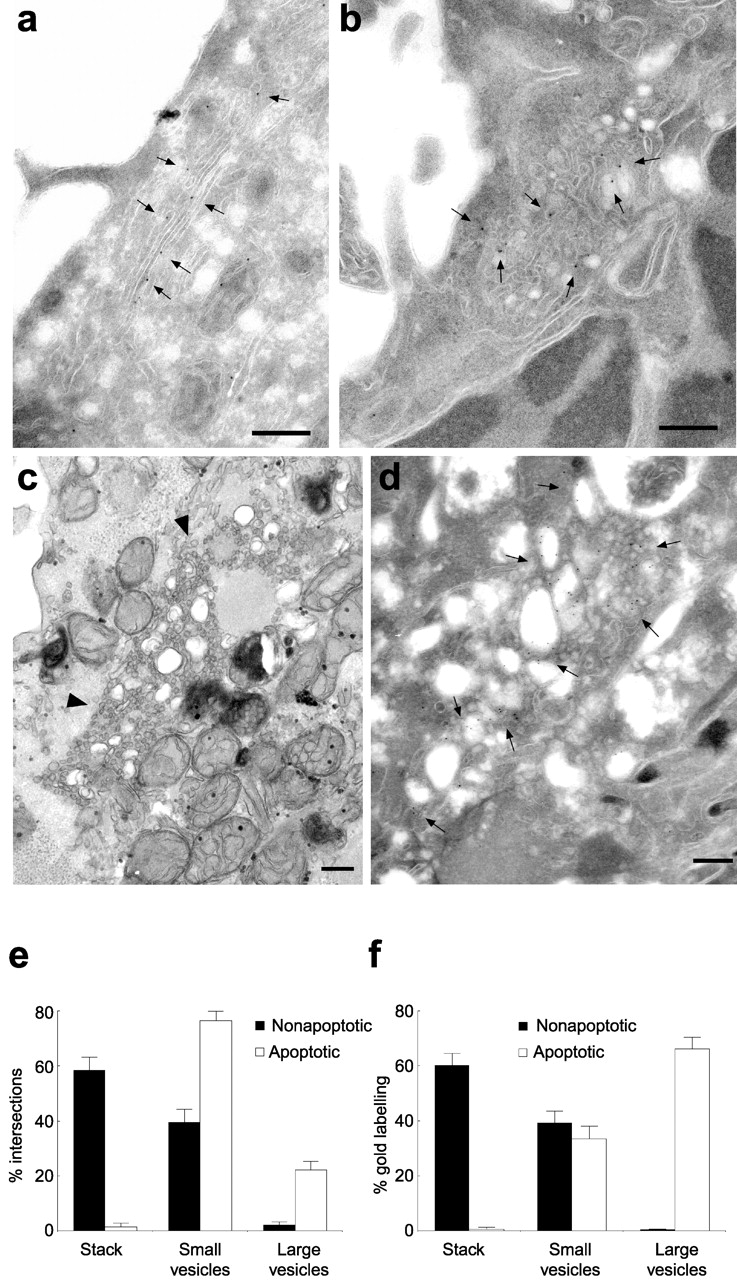
Ultrastructural changes in Golgi morphology during apoptosis. (a–d) HeLa cells were incubated with DMSO (a, control), 1 μM staurosporine (b), or 5 μg/ml anisomycin (c and d) for 6 h before fixation and preparation for Epon embedding (c) or cryo-EM and labeling with antibodies to GalNacT2 followed by secondary antibodies conjugated to 10 nm gold (a, b, and d). Arrowheads in c indicate a vesicular membrane cluster. Gold particles in a, b, and d are indicated by small arrows. (e) Quantitation of membrane profiles in the Golgi region of Epon sections of cells incubated under control conditions (nonapoptotic) or with 5 μg/ml anisomycin (apoptotic). Quantitation was performed as described in Materials and methods. Bars represent the mean ± SEM (n = 20–24). (f) Quantitation of GalNac T2 labeling of cryosections. The relative number of gold particles over stacked cisternae compared with small and large vesicular profiles was counted for cells incubated under control conditions (nonapoptotic) or with 5 μg/ml anisomycin (apoptotic). Bars represent the mean ± SEM (n = 18–29). Bar, 200 nm.
GRASP65 is cleaved by caspase-3 during apoptosis
To address the mechanism of Golgi unstacking in apoptosis, we analyzed whether known candidate stacking proteins are subjected to caspase-mediated cleavage. Immunofluorescence analysis of NRK cells suggested that GRASP65 might be cleaved during the apoptotic execution phase. Labeling of NRK cells with a monoclonal antibody to GRASP65 revealed that staining was lost in apoptotic cells (Fig. 4 a). To confirm this was due to caspase-mediated cleavage, the following experiments were performed. First, purified rat liver Golgi membranes were incubated with apoptotic cytosol, and the status of GRASP65 was monitored by immunoblotting. As shown in Fig. 4 b, reactivity to the GRASP65 monoclonal antibody was lost rapidly upon incubation with apoptotic cytosol, and this was prevented by the caspase inhibitor Ac-DEVD-CHO. Blotting with a polyclonal GRASP65 antibody showed that the protein was quantitatively cleaved to an ∼40-kD form that remained associated with the membrane and that there are at least two cleavage intermediates (Fig. 4 b). Cleavage was specific, since there was little change in the total protein profile of the membranes or the cytosol after incubation under apoptotic conditions (Fig. 4 c).
To assess whether the other candidate Golgi stacking protein GRASP55 is also cleaved during apoptosis, samples were subjected to immunoblotting with an antibody that cross-reacts with both GRASP55 and GRASP65. Unlike GRASP65, which was cleaved completely, there was no detectable cleavage of GRASP55 even after a 6-h incubation in apoptotic cytosol (Fig. 4 d). To confirm that GRASP65 is cleaved by caspases in vivo, NRK cells were treated with staurosporine for various lengths of time, and cell extracts were analyzed by immunoblotting. GRASP65 was efficiently cleaved to a ∼40-kD form in apoptotic cells (Fig. 4 e). Cleavage of GRASP65 mirrored that of PARP with little full-length protein remaining after a 4-h treatment with staurosporine (Fig. 4 e). Cleavage of both GRASP65 and PARP is inhibited by zVAD.fmk (Fig. 4 e).
Recent studies have localized caspase-2 to the Golgi complex (Mancini et al., 2000). To determine whether caspase-2 was responsible for the cleavage of GRASP65, we first performed incubations in the presence of Casputin™, a novel caspase inhibitor that specifically inhibits caspases 3 and 7 but has no effect on other known caspases including caspase-2. Cytosol was preincubated with cytochrome c for 90 min to induce caspase activation; immunoblotting with antibodies to caspases 2 and 3 confirmed that these had been cleaved from their inactive p48 and p32 proforms to the active p12 and p17 forms, respectively (Fig. 5 a, left). Preactivated apoptotic cytosol efficiently cleaved GRASP65, but when Casputin™ was present cleavage was abolished completely (Fig. 5 a, right), suggesting that GRASP65 is cleaved by caspase-3 or caspase-7. Cleavage of golgin-160 was unaffected by Casputin™ consistent with the observation that golgin-160 is a substrate for caspase-2 (Mancini et al., 2000).
Figure 5.

GRASP65 is cleaved by caspase-3. (a) HeLa cytosols preincubated in the absence (control) or presence (apoptotic) of 10 μM cytochrome c for 90 min at 37°C were immunoblotted with antibodies to caspase-3 and caspase-2 (left). Golgi membranes were incubated with control or apoptotic cytosol in the absence or presence of 80 μg/ml Casputin™ for 4 h at 37°C and analyzed by immunoblotting with polyclonal antibodies to GRASP65 and golgin-160 (right). (b) In vitro–translated 35S-labeled control proteins, GRASP65, or golgin-160 were incubated with purified recombinant caspases at the indicated concentrations for 2 h at 30°C before SDS-PAGE and autoradiography. Control proteins were PARP (for caspases 3 and 7 at 0, 0.025, 0.1, 0.25, 0.5, 1, 2, and 4 nM) and procaspase 2 (for caspase 2 at 0, 0.5, 1, 2.5, 10, 25, 100, and 250 nM). (c) In vitro–translated 35S-labeled PARP or GRASP65 were incubated with control (C) or apoptotic (A) cytosol from the parental MCF-7 cell line (WT) or MCF-7 cells stably expressing GFP–caspase-3 (C3-GFP) for 2 h at 30°C. Samples were analyzed by SDS-PAGE followed by immunoblotting with antibodies to GFP or autoradiography. Full-length (FL) and caspase cleavage products (asterisks) are indicated. The additional 27-kD band (∼) in the left panel likely corresponds to free GFP.
To determine whether GRASP65 is directly cleaved by caspase-3 or caspase-7, in vitro–translated GRASP65 was incubated with increasing concentrations of purified recombinant caspases and cleavage analyzed by SDS-PAGE. As shown in Fig. 5 b, GRASP65 was quantitatively cleaved to an ∼40-kD form by caspase-3 but was not affected by caspase-7 even though similar amounts of activity were present in each incubation. As expected, GRASP65 was also not affected by caspase-2 even though this enzyme could efficiently cleave golgin-160 and a control substrate (Fig. 5 b).
To further confirm GRASP65 is a substrate for caspase-3 only, in vitro–translated GRASP65 was incubated with cytosol prepared from caspase-3–deficient MCF-7 cells. No cleavage of GRASP65 was observed with apoptotic MCF-7 cytosol, whereas the control protein PARP, a substrate for caspase-7 and caspase-3, was completely processed under the same conditions (Fig. 5 c). Incubation with apoptotic cytosol prepared from MCF-7 cells stably expressing GFP-tagged caspase-3 restored GRASP65 cleavage (Fig. 5 c). Cleavage of GRASP65 was not complete, most likely due to the low amount of caspase-3 activation observed with this cytosol (Fig. 5 c, left). Together, our results strongly suggest GRASP65 is a specific substrate for caspase-3.
Identification of the cleavage site
Analysis of the GRASP65 sequence revealed the absence of a protypical DEXD caspase-3 cleavage site. Since our biochemical results suggested GRASP65 cleavage was occurring in the COOH-terminal half of the protein, we analyzed the sequence in this region and identified three potential cleavage sites as SLLD320S, SFPD375S, and TLPD393G (Fig. 6 a). The relevant aspartic acid residues at each of these sites were mutated to alanine either alone or in combination, and cleavage of the resulting constructs by apoptotic cytosol was analyzed. Although mutation of each site alone or in combination with one other site resulted in cleavage at the remaining nonmutated site(s), mutation of all three aspartic acid residues completely blocked cleavage (Fig. 6 b).
Figure 6.

Identification of the GRASP65 cleavage sites. (a) Schematic representation of the structure of GRASP65. (b) In vitro–translated 35S-labeled GRASP65 proteins with the indicated aspartic acid to alanine substitutions were incubated with control or apoptotic HeLa cytosol in the absence or presence of 2 μM Ac-DEVD-CHO for 4 h at 37°C and analyzed by SDS-PAGE and autoradiography. (c) HeLa cells stably expressing COOH terminally GFP-tagged wild-type and D320A/D375A/D393A triple mutant forms of GRASP65 were incubated with 1 μM staurosporine for the times indicated. Cell extracts were analyzed by immunoblotting with antibodies to GFP and PARP.
To demonstrate that the same sites are also used in vivo, wild-type and triple mutant GRASP65 were tagged at the COOH terminus with GFP, and stable cell lines expressing each construct were generated. Immunofluorescence microscopy and immunoblotting analysis confirmed that both proteins are targeted correctly to the Golgi apparatus in these cells and that they are expressed at similar levels, likely corresponding to a 5- to 10-fold overexpression relative to endogenous GRASP65 (Figs. S1 and S2). Wild-type and mutant cells were incubated with staurosporine for various lengths of time and then analyzed by immunoblotting with antibodies to GFP and PARP. As shown in Fig. 6 c, wild-type GRASP65-GFP was efficiently cleaved, generating GFP antibody-reactive cleavage products at ∼30–35 kD. In contrast, triple mutant GRASP65-GFP was completely resistant to cleavage even after a 20-h incubation in staurosporine, during which time PARP was processed completely (Fig. 6 c). Therefore, SLLD, SFPD, and TLPD are the only GRASP65 caspase cleavage sites both in vitro and in vivo.
GRASP65 cleavage visualized in living cells
To study the dynamics of GRASP65 cleavage during apoptosis, GRASP65-GFP–expressing cells were treated with anisomycin and observed by time-lapse fluorescence and phase–contrast microscopy. Alexa 594–conjugated annexin V was included in the medium to determine the onset of phosphatidylserine exposure, a hallmark of apoptotic progression (Martin et al., 1995). GFP fluorescence dissipated rapidly from the Golgi apparatus at the same time as the cells started to round up and exhibit plasma membrane blebbing (Fig. 7 a; Video 1 available at http://www.jcb.org/cgi/content/full/jcb.200110007/DC1). This was not due to Golgi membranes moving out of focus because similar analysis of cells expressing a GFP-tagged Golgi luminal protein (NAGT1) undergoing apoptosis showed that GFP fluorescence was visible on clearly discernible structures for the duration of the experiment (unpublished data). Furthermore, confocal optical sectioning of live apoptotic GRASP65-GFP cells showed that GFP was not present on any structure throughout the whole thickness of the cell (Fig. 7 c, top). Loss of GFP fluorescence from the Golgi region occurred as soon as cell rounding could be observed. The average time from cell rounding to complete loss of Golgi-associated GFP fluorescence was only 9.8 min (SD = 3.9, n = 14), significantly faster than the time from cell rounding to phosphatidylserine exposure, which took 89.7 min (SD = 16.8, n = 10). These results demonstrate that complete cleavage of GRASP65 is a rapid event that occurs very early in the execution phase of apoptosis.
Figure 7.

Real-time visualization of GRASP65 cleavage. Still images taken from time-lapse analyses of Golgi morphology in HeLa cells stably expressing wild-type GRASP65-GFP (a) or caspase-resistant mutant GRASP65-GFP (b). Cells were treated with anisomycin in the presence of Alexa 594–conjugated annexin V (red). The onset of apoptotic execution is marked by an arrow in the fluorescence panels. GFP fluorescence associated with Golgi fragments in apoptotic cells in b is indicated by arrowheads. Time elapsed from the beginning of each sequence is shown in minutes. Videos 1 and 2 are available online at http://www.jcb.org/cgi/content/full/jcb.200110007/DC1. (c) Confocal images of living anisomycin-treated wild-type and caspase-resistant GRASP65-GFP stable HeLa cells. Cells were incubated with annexin V conjugated to Alexa 594 to identify apoptotic cells. Projections of multiple confocal sections are shown. Bars, 10 μm.
Does GRASP65 cleavage occur before Golgi fragmentation? Time-lapse analysis of cells expressing GFP-tagged NAGT1 demonstrated that the Golgi ribbon starts to fragment ∼30 min after cell rounding (unpublished data), suggesting this occurs ∼20–30 min after GRASP65 cleavage. However, due to our inability to visualize GRASP65 cleavage and Golgi fragmentation in the same cells by time-lapse microscopy we decided to use the monoclonal anti-GRASP65 antibody to label fixed cells and monitor GRASP65 cleavage in combination with changes to Golgi structure. Quantitation of staurosporine-treated NRK cells revealed that 91% of cells that had cleaved GRASP65 had a fragmented Golgi (n = 144) and that there were never any cells with a fragmented Golgi that had intact GRASP65 (n = 200). This indicates that cleavage of GRASP65 occurs before Golgi fragmentation.
Time-lapse analysis of cells expressing caspase-resistant GRASP65-GFP was also performed. In this case, GFP fluorescence remained associated with Golgi membranes up to and beyond the onset of annexin V labeling (Fig. 7 b; Video 2 available at http://www.jcb.org/cgi/content/full/jcb.200110007/DC1). Confocal analysis of live cells confirmed the presence of GRASP-GFP on Golgi membranes in annexin V–positive cells (Fig. 7 c, bottom).
Cleavage of GRASP65 is required for Golgi fragmentation during apoptosis
Previous work has shown that GRASP65 is required for the in vitro reformation of Golgi stacks from mitotic Golgi fragments during postmitotic reassembly of the Golgi complex (Barr et al., 1997; Shorter and Warren, 1999). In vivo evidence supporting a role for GRASP65 in stacking is lacking currently. It is also not clear whether GRASP65 is involved in generating the higher order ribbon structure of the Golgi complex. We decided to investigate whether GRASP65 cleavage is required for loss of stacking and/or breakdown of the Golgi ribbon during apoptosis by studying fragmentation in cells expressing the noncleavable form of this protein. Should GRASP65 have a role in maintaining these structures, then expression of the caspase-resistant form might be expected to slow down the morphological changes that occur during apoptosis.
Initial experiments addressed whether GRASP65 cleavage is involved in breakdown of the Golgi ribbon. GRASP65-GFP–expressing cells were treated with staurosporine for 6 h and then fixed and stained with antibodies to GM130 and cleaved PARP to determine the extent of Golgi fragmentation and the stage of apoptosis, respectively. Three stages of apoptosis were scored: stage 1 cells had nuclei that were positive for cleaved PARP but contained normal looking uncondensed chromatin, indistinguishable from healthy cells; stage 2 cells had nuclei positive for cleaved PARP and contained condensed marginalized chromatin; and stage 3 cells had highly condensed and fragmented chromatin in combination with cleaved PARP staining distributed throughout the cytoplasm (Fig. 8 a). Golgi morphology was assessed in these cells and split into three categories. These are “wild-type ribbons” (a continuous juxtanuclear ribbon of normal appearance), “mixed ribbons and scattered Golgi fragments” (clearly discernible ribbons with a few dispersed punctate fragments of smaller size), or “scattered Golgi fragments” (no ribbon-like structure remaining and many punctate fragments dispersed throughout the cell) (Fig. 8 b). Expression of wild-type GRASP65-GFP had no effect on the progress of apoptotic Golgi fragmentation (Fig. 8 c). In contrast, the transition from mixed ribbons and scattered Golgi fragments into scattered Golgi fragments only, indicating complete breakdown of the Golgi ribbon, was reduced significantly in cells expressing caspase-resistant GRASP65 compared with the parental cells and cells expressing wild-type GRASP65 (Fig. 8 c). This was true for cells at both stages 2 and 3. Expression of the caspase-resistant GRASP65 mutant therefore significantly inhibited breakdown of the Golgi ribbon.
To assess whether GRASP65 cleavage is required for the loss of Golgi stacking observed during apoptosis, the GRASP65-GFP cells were treated with anisomycin and analyzed by EM. Apoptotic cells were identified by their condensed chromatin, and the amount of membrane in cisternal stacks and large and small vesicles was quantitated by stereology. Wild-type GRASP65 had no effect on the loss of Golgi stacking or on the accumulation of large and small vesicles during apoptosis (Fig. 9 compared with Fig. 3 e). In cells expressing caspase-resistant GRASP65, the majority of membrane in the Golgi region was also converted into large and small vesicles during apoptosis (Fig. 9). However, there was a small but significant increase in the amount of membrane remaining in stacked cisternae, and this was mirrored by a decrease in the amount of membrane in small vesicles (Fig. 9). In nonapoptotic cells, 57% of the total membrane in the Golgi region was in stacked cisternae (Fig. 3 e). In GRASP65 mutant cells, this number was 13% compared with only 1% in cells expressing wild-type GRASP65 (Fig. 9). Therefore, ∼23% (13/57) of membrane stacking was preserved in GRASP65 mutant cells compared with only 1.7% (1/57) in wild-type cells.
Figure 9.

Ultrastructural analysis of Golgi fragmentation in cells expressing caspase-resistant GRASP65. HeLa cells stably expressing wild-type (WT) or caspase-resistant GRASP65-GFP (mutant) were treated with 5 μg/ml anisomycin for 6 h, fixed, and embedded in Epon for analysis by EM. Membrane profiles were quantitated as described in Materials and methods. Bars represent the mean ± SEM (n = 20–24).
Discussion
In this paper, we have shown that the Golgi complex is fragmented into clusters of tubulo-vesicular membranes during apoptosis. Fragmentation was caspase dependent, suggesting that caspase-mediated cleavage of target proteins is required to bring about the morphological changes observed. We identified one of the caspase targets as GRASP65, a protein identified originally as a stacking factor using a cell-free assay for the postmitotic assembly of Golgi stacks from mitotic Golgi fragments (Barr et al., 1997). Although GRASP65 is clearly required for stacking in this assay, the mechanism by which stacking is promoted is not clear. It has been postulated that GRASP65 acts to promote the formation of stable cross-bridges between newly forming cisternae that have previously become tethered in a p115-dependent manner (Linstedt, 1999; Shorter et al., 1999). Whether GRASP65 is required only transiently during cross-bridge formation or has a more permanent role in maintaining stable cross-bridges once they have formed is not clear.
We found that expression of caspase-resistant GRASP65 retained a small amount of cisternal stacking in apoptotic cells, suggesting GRASP65 plays some role in maintenance of preformed stacks. The amount of cisternal stacking retained corresponded to only ∼23% of that found in a nonapoptotic cell. However, since GRASP65 is restricted to the cis-most cisterna of the Golgi stack (Shorter et al., 1999), it would only be expected to link this cisterna with its adjacent partner. The Golgi complex of our HeLa cells has on average three stacked cisternae, so even if stacking between the two cis-most cisternae was fully protected this would correspond to only ∼50% of the total stacking of a nonapoptotic cell. Therefore, caspase-resistant GRASP65 may protect ∼50% (23/50) of stacking at the cis side of the Golgi stack. The remainder of stacking was lost in cells expressing caspase-resistant GRASP65 consistent with a lack of GRASP65 involvement in stacking other Golgi cisternae. Therefore, other proteins must be responsible for stacking these cisternae, and these proteins are likely targets for apoptotic cleavage. One candidate is GRASP55, but we found that GRASP55 is resistant to caspase-mediated cleavage, suggesting additional proteins are involved.
Expression of the caspase-resistant mutant significantly inhibited breakdown of the Golgi ribbon in apoptotic cells. This suggests that GRASP65 cleavage is required for fragmentation of the Golgi ribbon during apoptosis. Importantly, it also suggests that GRASP65 plays an important role in generating the higher order ribbon structure of the Golgi apparatus. Recent work has shown that matrix proteins including GRASP65 can generate a Golgi ribbon that lacks Golgi resident enzymes (Seemann et al., 2000). This suggests that matrix proteins are sufficient for ribbon formation and is consistent with the idea that GRASP65 functions in this process. How GRASP65 might participate in ribbon formation and how this relates to a role in cisternal stacking is not clear. GRASP65 binds to the Golgi matrix protein GM130 via its NH2 terminus, and GM130 in turn binds p115 (Nakamura et al., 1997; Barr et al., 1998). However, the NH2 terminus of GRASP65 is not affected during apoptosis, and GM130 and p115 remain associated with apoptotic Golgi membranes, suggesting these proteins are not involved in the morphological changes observed. Cleavage of GRASP65 occurs in the membrane-distal COOH terminus, suggesting this part of the protein is important for maintenance of Golgi structure. The COOH terminus may be required for oligomerization of GRASP65 or for binding other stacking and/or matrix proteins. Identification of other proteins that bind GRASP65 should help unravel the roles of this protein in stacking and ribbon formation and reveal how these apparently separate processes might be linked.
Golgi fragments produced during apoptosis are morphologically similar to the Golgi clusters found in mitotic cells (Warren et al., 1995). Mitotic fragmentation is dependent on phosphorylation of Golgi proteins by mitotically active protein kinases including cyclin-dependent kinase (CDK)*1, polo-like kinase-1, and a mitotic form of mitogen-activated protein kinase kinase 1 (Lowe et al., 1998; Colanzi et al., 2000; Sutterlin et al., 2001). It has been reported that CDK1 activation can occur in apoptotic cells (Zhou et al., 1998), raising the possibility that the morphological changes we observe are a consequence of mitotic phosphorylation rather than caspase-mediated cleavage of Golgi targets. However, we think this unlikely, since we could detect no significant increase in the histone kinase activity of apoptotic cell extracts nor could we detect any increase in phosphorylation of the CDK1 substrate GM130 (unpublished data). Furthermore, in agreement with previous studies (Zhou et al., 1998) we found no increase in reactivity of apoptotic cells to the mitotic phosphoepitope-specific antibody MPM2 (unpublished data). We also found no significant increase in mitogen-activated protein kinase phosphorylation in apoptotic cells (unpublished data). Therefore, we believe the morphological changes we observe are a direct consequence of caspase-mediated cleavage of Golgi proteins.
Are similar pathways used for fragmentation during apoptosis and mitosis with common proteins targeted by the mitotic (kinases) and apoptotic (caspases) machinery? Interestingly, GRASP65 is highly phosphorylated in mitosis (Barr et al., 1997), and this is likely important for regulation of its function. Mitotic phosphorylation is mediated by polo-like kinase-1 and CDK1 and occurs in the COOH-terminal region of the protein (Lin et al., 2000; Sutterlin et al., 2001). Therefore, GRASP65 is targeted during apoptosis and mitosis. This not only supports the idea that GRASP65 is a key structural protein of the Golgi complex but also suggests that common fragmentation pathways may be used in each situation. To determine if this is the case will require further work. However, we already know of one difference: the vesicle tethering protein p115 is removed rapidly from Golgi membranes during mitosis (Lowe et al., 2000) but remains membrane bound throughout the execution phase of apoptosis consistent with a lack of GM130 phosphorylation in apoptotic cells.
Why fragment the Golgi complex during apoptosis? Presently, we can only speculate, but one reason may be to allow inclusion of the normally single copy Golgi ribbon into apoptotic bodies destined for clearance by phagocytosis. Fragmentation might also play a role in maintaining peripheral tolerance in the immune system by allowing Golgi components to be phagocytosed by antigen-presenting dendritic cells (Steinman et al., 2000). Interestingly, cytoplasmic dynein function is lost in apoptotic cells due to caspase-mediated cleavage of the dynein intermediate chain and p150glued (Lane et al., 2001). Since cytoplasmic dynein is required for maintenance of a perinuclear ribbon in healthy cells (Burkhardt et al., 1997), loss of function may account for the dispersal of Golgi fragments seen in apoptotic cells and may facilitate the inclusion of these fragments into apoptotic bodies.
It is likely that other Golgi proteins are cleaved by caspases during apoptosis. The identification of these proteins will not only lead to a greater understanding of how the Golgi complex undergoes fragmentation during apoptosis but should also lead to a more general understanding of how Golgi complex structure is controlled at the molecular level. Our aim now is to identify these proteins.
Materials and methods
Antibodies
The following antibodies were used: FBA31 and CT polyclonal anti-GRASP65, FBA19 polyclonal anti-GRASP55 and GRASP65, 7E10 monoclonal anti-GRASP65, MLO7 polyclonal anti-GM130, 4H1 monoclonal anti-p115, 3E1 monoclonal anti-GFP, monoclonal anti-GM130 from Transduction Labs, polyclonal anti-PARP p85 fragment from Promega (human specific) and Cell Signaling (rat specific), monoclonal anti-Fas (DX2) from Calbiochem, monoclonal anti-GalNac T2 (a gift from Dr. Henrik Clausen, University of Copenhagen, Copenhagen, Denmark), polyclonal anti-TGN46 (a gift from Dr. Vas Ponnambalam, Leeds University, Leeds, UK), monoclonal anti-PARP from Calbiochem and polyclonal anti–caspase 2 (C-20) and –caspase 3 (N-19) from Santa Cruz Biotechnology, Inc., polyclonal anti–golgin-160 (a gift from Dr. Ed Chan, The Scripps Research Institute, La Jolla, CA). Fluorophore-conjugated secondary antibodies were purchased from Molecular Probes. HRP-conjugated secondary antibodies were purchased from Tago Immunologicals.
Constructs
Amino acid substitutions were introduced into full-length rat GRASP65 cDNA using the QuickChange kit (Stratagene) according to the manufacturer's instructions. To generate GRASP65-GFP, full-length GRASP65 was amplified by PCR and cloned in-frame into the XmaI site of pEGFP-N2 mammalian expression vector (CLONTECH Laboratories, Inc.). All constructs were verified by DNA sequencing.
Cell culture and drug treatments
HeLa and NRK cells were grown in DME containing 10% FCS. To induce apoptosis, cells were treated with 1 μg/ml anti-Fas monoclonal antibody DX2 (Calbiochem) plus 0.1 mg/ml cycloheximide, 100 J/m2 UV radiation, 1–2 μM staurosporine (Sigma-Aldrich), or 5 μg/ml anisomycin (Calbiochem). In some cases, cells were pretreated for 15 min with 50 μM zVAD.fmk (Calbiochem) before apoptotic stimulus.
Generation of GRASP65-GFP stable cell lines
HeLa cells were transfected with wild-type or caspase-resistant GRASP65-GFP plasmids using Fugene 6 (Roche Biochemicals) according to the manufacturer's instructions. Transfected cells were grown in the presence of geneticin (G418) until individual clones could be distinguished. Clonal colonies were screened for Golgi-associated GFP fluorescence, and those clones expressing correctly localized GRASP65-GFP were selected.
Preparation of cell extracts
Floating cells were harvested by gentle aspiration of the cell medium and pelleted by centrifugation. After washing with ice-cold PBS, cell pellets were solubilized in boiling SDS sample buffer. Remaining adherent cells were washed with ice-cold PBS and pooled with the extract prepared from floating cells from the same dish to form a total cell population extract. DNA was sheared by vortexing in the presence of glass beads, and the extracts were cleared by centrifugation at 15,000 g for 10 min. Samples were analyzed by SDS-PAGE and immunoblotting with appropriate antibodies.
In vitro cleavage experiments
Cytosol was prepared from suspension HeLa cells and desalted into buffer A according to Lowe et al. (2000). MCF-7 cytosol was made in acetate/sucrose buffer containing 2 mM ATP according to Lane et al. (2001). Cytosols were supplemented with an ATP regeneration cocktail (10 mM creatine phosphate, 40 μg/ml creatine kinase). To make cytosol apoptotic, 10 μM cytochrome c was added. Where indicated, 2 μM Ac-DEVD-CHO (Calbiochem) was added. Typically, 50 μg rat liver Golgi membranes (Hui et al., 1998) were incubated with 200 μl HeLa cytosol (9 mg/ml) for various times at 37°C, chilled on ice, and centrifuged at 100,000 g for 15 min to pellet the membranes. Pellet and supernatant fractions were solubilized in SDS sample buffer and subjected to SDS-PAGE and Coomassie blue staining or immunoblotting. For the Casputin™ experiment, cytosol was preincubated for 90 min at 37°C with or without 10 μM cytochrome c before addition of the Golgi membranes. Incubations were then performed in the absence or presence of 80 μg/ml Casputin™ (BioMol Research Labs).
In vitro synthesis of 35S-labeled recombinant proteins was performed using a coupled in vitro transcription/translation kit (Promega) according to the manufacturer's instructions. Typically, 9 μl of protein translate was mixed with 45 μl of HeLa cytosol (9 mg/ml) and incubated for various times at 37°C. Incubations with MCF-7 cytosol (4.5 mg/ml) were performed for 2 h at 32°C. Alternatively, 1 μl of translate was added to 9 μl of recombinant caspase 2, 3, or 7 (Alexis Biochemicals) and incubated for 2 h at 30°C. Incubations with recombinant caspases were performed in 0.1% CHAPS, 10% (wt/vol) sucrose, 5 mM DTT, 2 mM EDTA containing either 50 mM MES, pH 5.5 (caspase 2), or 50 mM Hepes, pH 7.4 (caspases 3 and 7). Samples were diluted into SDS sample buffer and analyzed by SDS-PAGE and autoradiography.
Fluorescence microscopy
Conventional digital epifluorescence images of fixed cells were obtained using an Olympus BX60 upright microscope equipped with a MicroMax-cooled, slow scan CCD camera (Roper Scientific) driven by Metamorph software (Universal Imaging Corp.). Confocal images were obtained using a Leica NT confocal microscope. All images are projections of optical sections taken in the z-axis at 0.5-μm intervals.
For digital time-lapse analysis of live HeLa cells stably expressing wild-type or caspase-resistant GRASP65-GFP, cells were grown on coverslips in 3-cm plastic cell culture dishes (Matek). Cells were treated with 5 μg/ml anisomycin in CO2-independent, serum- and phenol red-free Leibovitz L15 medium (GIBCO BRL) supplemented with 0.5% Alexa 594–conjugated annexin V (Molecular Probes) and 2.5 mM CaCl2 and were imaged at 37°C by time-lapse fluorescence and phase–contrast microscopy using an Olympus IX-70 inverted microscope equipped with a heated stage and a PentaMax intensified cooled frame transfer CCD camera (Roper Scientific). Images were obtained over a period of 3 h with 1-min time-lapse intervals using halogen bulb illumination for both phase and fluorescence with excitation and emission filter wheels and shutters (Sutter Instrument Co.) controlled by Metamorph software.
To assess effects of GRASP65 expression on Golgi fragmentation, HeLa cells stably expressing GRASP65-GFP (wild-type and caspase-resistant) and the parental HeLa cell line were grown on coverslips for 24 h and then treated with 2 μM staurosporine for 6 h. Cells were fixed in −20°C methanol for 5 min and then stained with antibodies to GM130 (Transduction Labs) and caspase-cleaved PARP (Promega) followed by anti–mouse Alexa 488 and anti–rabbit Alexa 594 as described above. Cells were then analyzed blind for apoptotic stage and for Golgi morphology as described in Fig. 7. Golgi-associated green fluorescence derived from GRASP65-GFP plus GM130/Alexa 488 in the stable cell lines was indistinguishable from GM130/Alexa 488 staining alone in the parental HeLa cells.
Tests of association between cell type and fragmentation stage were made between each pair of cell types using a chi square likelihood ratio contingency test. To maintain a family wise type 1 error rate within each fragmentation stage, we used the highly conservative correction of dividing the family wise type I error rate by the number of test performed (0.05/3 in our case). Thus, each test within stage 1 and 2 had a type I error rate of 0.0167. Since experiments were combined for stage 3, no correction was made for this group. All analyses were run in Systat version 10 (SPSS Inc.).
EM
Cells were fixed by adding 2% glutaraldehyde in 0.4 M Hepes buffer, pH 7.2, to an equal volume of the medium for 30 min, washed in PBS, scraped with a rubber policeman, and pelleted. Cells were processed for cryosectioning and immunogold labeled for GalNac T2 as described in Farmaki et al. (1999) except the sections were picked up using the modified pick up method of Liou et al. (1996) using one part methyl cellulose and three parts 2.3 M sucrose in PBS. For structural and quantitative analysis, cells were post fixed with reduced osmium tetroxide, embedded in epoxy resin, and after ultrathin sectioning contrast, lead citrate according to Lucocq et al. (1989). For quantitation of gold labeling, cell profiles contained in a randomly selected grid square were scanned systematically, and Golgi areas were identified by the presence of cisternal stacks and/or vesicular profiles labeled for GalNac T2. Labeling was assigned to one of three categories: (1) Golgi stacks containing at least two juxtaposed elongated profiles (with a length to width ratio of greater than four); (2) small vesiculo-tubular profiles with a diameter of >80 nm; (3) large electron-lucent vesicles with a diameter of <80 nm and pale staining content. In epoxy resin sections, line intersections with the above classes of structure were counted on micrographs taken at 15,000× magnification or greater and scanned on a flat bed scanner at a resolution of at least 1,000 pixels per inch. The images were viewed and analyzed using intersection counts on membrane-bound structure with lines of a square lattice grid (spacing of 253 nm) in Adobe Photoshop® 5.5. Coefficients of error for ratio estimates were calculated according to Cochran (1977).
Online supplemental material
Video 1 shows time-lapse analysis of anisomycin-treated HeLa cells stably expressing wild-type GRASP65-GFP. Video 2 shows time-lapse analysis of anisomycin-treated HeLa cells stably expressing caspase-resistant mutant GRASP65-GFP. Fig. S1 shows the immunofluorescence pattern of stably expressed wild-type and capase-resistant mutant GRASP65-GFP relative to that of the endogenous Golgi proteins p115, GM130, GalNacT2, and TGN46. Fig. S2 shows a Western blot of NRK and wild-type and mutant GRASP65-GFP stable HeLa cells with antibodies to GM130, GRASP65, and tubulin. Videos 1 and 2 and Figs. S1 and S2 are available online at http://www.jcb.org/cgi/content/full/jcb.200110007/DC1.
Supplemental Material
Acknowledgments
We are grateful to Drs. Henrik Clausen, Vas Ponnambalam, and Ed Chan for generously providing antibodies, Dr. Yanzhuang Wang for providing antibodies and his-tagged GRASP65, and Dr. Chris Gregory for providing the MCF-7 cell lines. We would like to thank Dr. Richard Preziosi for help with the statistics and Dr. Graham Warren for critically reading the article.
This work was supported by the Medical Research Council (G120/483 to M. Lowe, G117153 to P.G. Woodman, and COG grant G9722026), The Wellcome Trust (equipment grant for confocal microscope), Biotechnology and Biological Sciences Research Council (grant 34/BI11195 to P.G. Woodman and V.J. Allan), and the Lister Institute for Preventative Medicine (to V.J. Allan).
The online version of this article contains supplemental material.
Footnotes
Abbreviation used in this paper: CDK, cyclin-dependent kinase.
References
- Barr, F.A., M. Puype, J. Vandekerckhove, and G. Warren. 1997. GRASP65, a protein involved in the stacking of Golgi cisternae. Cell. 91:253–262. [DOI] [PubMed] [Google Scholar]
- Barr, F.A., N. Nakamura, and G. Warren. 1998. Mapping the interaction between GRASP65 and GM130, components of a protein complex involved in the stacking of Golgi cisternae. EMBO J. 17:3258–3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkhardt, J.K., C.J. Echeverri, T. Nilsson, and R.B. Vallee. 1997. Overexpression of the dynamitin (p50) subunit of the dynactin complex disrupts dynein-dependent maintenance of membrane organelle distribution. J. Cell Biol. 139:469–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan, E.K.L., and M.J. Fritzler. 1998. Golgins: coiled-coil-rich proteins associated with the Golgi complex. Electr. J. Biotechnol. 1:1–10. [Google Scholar]
- Cluett, E.B., and W.J. Brown. 1992. Adhesion of Golgi cisternae by proteinaceous interactions: intercisternal bridges as putative adhesive structures. J. Cell Sci. 103:773–784. [DOI] [PubMed] [Google Scholar]
- Cochran, W.G. 1977. Sampling Techniques. John Wiley & Sons Inc., New York.
- Cohen, G.M. 1997. Caspases: the executioners of apoptosis. Biochem. J. 326:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colanzi, A., T.J. Deerinck, M.H. Ellisman, and V. Malhotra. 2000. A specific activation of the mitogen-activated protein kinase kinase 1 (MEK1) is required for Golgi fragmentation during mitosis. J. Cell Biol. 149:331–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman, M.L., E.A. Sahai, M. Yeo, M. Bosch, A. Dewar, and M.F. Olson. 2001. Membrane blebbing during apoptosis results from caspase-mediated activation of ROCK I. Nat. Cell Biol. 3:339–345. [DOI] [PubMed] [Google Scholar]
- Earnshaw, W.C., L.M. Martins, and S.H. Kaufmann. 1999. Mammalian caspases: structure, activation, substrates, and functions during apoptosis. Annu. Rev. Biochem. 68:383–424. [DOI] [PubMed] [Google Scholar]
- Farmaki, T., S. Ponnambalam, A.R. Prescott, H. Clausen, B.L. Tang, W. Hong, and J.M. Lucocq. 1999. Forward and retrograde trafficking in mitotic animal cells. ER-Golgi transport arrest restricts protein export from the ER into COPII-coated structures. J. Cell Sci. 112:589–600. [DOI] [PubMed] [Google Scholar]
- Franke, W.W., J. Kartenbeck, S. Krien, W.J. VanderWoude, U. Scheer, and D.J. Morré. 1972. Inter- and intracisternal elements of the Golgi apparatus. A system of membrane-to-membrane cross-links. Z. Zellforsch. Mikrosk. Anat. 132:365–380. [DOI] [PubMed] [Google Scholar]
- Fritzler, M.J., J.C. Hamel, R.L. Ochs, and E.K. Chan. 1993. Molecular characterization of two human autoantigens: unique cDNAs encoding 95- and 160-kD proteins of a putative family in the Golgi complex. J. Exp. Med. 178:49–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui, N., N. Nakamura, P. Slusarewicz, and G. Warren. 1998. Purification of rat liver Golgi stacks. Cell Biology: A Laboratory Handbook. Vol. 2. J. Celis, editor. Academic Press Inc., Orlando, FL. 46–55.
- Kerr, J.F., A.H. Wyllie, and A.R. Currie. 1972. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer. 26:239–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane, J.D., M.A. Vergnolle, P.G. Woodman, and V.J. Allan. 2001. Apoptotic cleavage of cytoplasmic dynein intermediate chain and p150(Glued) stops dynein-dependent membrane motility. J. Cell Biol. 153:1415–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, C.Y., M.L. Madsen, F.R. Yarm, Y.J. Jang, X. Liu, and R.L. Erikson. 2000. Peripheral Golgi protein GRASP65 is a target of mitotic polo-like kinase (Plk) and Cdc2. Proc. Natl. Acad. Sci. USA. 97:12589–12594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linstedt, A.D. 1999. Stacking the cisternae. Curr. Biol. 9:R893–R896. [DOI] [PubMed] [Google Scholar]
- Liou, W., H.J. Geuze, and J.W. Slot. 1996. Improving structural integrity of cryosections for immunogold labeling. Histochem. Cell Biol. 106:41–58. [DOI] [PubMed] [Google Scholar]
- Lowe, M., C. Rabouille, N. Nakamura, R. Watson, M. Jackman, E. Jamsa, D. Rahman, D.J. Pappin, and G. Warren. 1998. Cdc2 kinase directly phosphorylates the cis-Golgi matrix protein GM130 and is required for Golgi fragmentation in mitosis. Cell. 94:783–793. [DOI] [PubMed] [Google Scholar]
- Lowe, M., N.K. Gonatas, and G. Warren. 2000. The mitotic phosphorylation cycle of the cis-Golgi matrix protein GM130. J. Cell Biol. 149:341–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucocq, J.M., E.G. Berger, and G. Warren. 1989. Mitotic Golgi fragments in HeLa cells and their role in the reassembly pathway. J. Cell Biol. 109:463–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancini, M., C.E. Machamer, S. Roy, D.W. Nicholson, N.A. Thornberry, L.A. Casciola-Rosen, and A. Rosen. 2000. Caspase-2 is localized at the Golgi complex and cleaves golgin-160 during apoptosis. J. Cell Biol. 149:603–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin, S.J., C.P. Reutelingsperger, A.J. McGahon, J.A. Rader, R.C. van Schie, D.M. LaFace, and D.R. Green. 1995. Early redistribution of plasma membrane phosphatidylserine is a general feature of apoptosis regardless of the initiating stimulus: inhibition by overexpression of Bcl-2 and Abl. J. Exp. Med. 182:1545–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy, N.J., M.K. Whyte, C.S. Gilbert, and G.I. Evan. 1997. Inhibition of Ced-3/ICE-related proteases does not prevent cell death induced by oncogenes, DNA damage, or the Bcl-2 homologue Bak. J. Cell Biol. 136:215–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mollenhauer, H.H. 1965. An intercisternal structure in the Golgi apparatus. J. Cell Biol. 24:504–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura, N., M. Lowe, T.P. Levine, C. Rabouille, and G. Warren. 1997. The vesicle docking protein p115 binds GM130, a cis-Golgi matrix protein, in a mitotically regulated manner. Cell. 89:445–455. [DOI] [PubMed] [Google Scholar]
- Philpott, K.L., M.J. McCarthy, D. Becker, C. Gatchalian, and L.L. Rubin. 1996. Morphological and biochemical changes in neurons: apoptosis versus mitosis. Eur. J. Neurosci. 8:1906–1915. [DOI] [PubMed] [Google Scholar]
- Rabouille, C., T. Misteli, R. Watson, and G. Warren. 1995. Reassembly of Golgi stacks from mitotic Golgi fragments in a cell-free system. J. Cell Biol. 129:605–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambourg, A., and Y. Clermont. 1997. Three-dimensional structure of the Golgi apparatus in mammalian cells. The Golgi Apparatus. E.G. Berger and J. Roth, editors. Birkhauser Verlag, Basel, Switzerland. 37–61.
- Rao, L., D. Perez, and E. White. 1996. Lamin proteolysis facilitates nuclear events during apoptosis. J. Cell Biol. 135:1441–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebbagh, M., C. Renvoize, J. Hamelin, N. Riche, J. Bertoglio, and J. Breard. 2001. Caspase-3-mediated cleavage of ROCK I induces MLC phosphorylation and apoptotic membrane blebbing. Nat. Cell Biol. 3:346–352. [DOI] [PubMed] [Google Scholar]
- Seemann, J., E. Jokitalo, M. Pypaert, and G. Warren. 2000. Matrix proteins can generate the higher order architecture of the Golgi apparatus. Nature. 407:1022–1026. [DOI] [PubMed] [Google Scholar]
- Sesso, A., D.T. Fujiwara, M. Jaeger, R. Jaeger, T.C. Li, M.M. Monteiro, H. Correa, M.A. Ferreira, R.I. Schumacher, J. Belisario, et al. 1999. Structural elements common to mitosis and apoptosis. Tissue Cell. 31:357–371. [DOI] [PubMed] [Google Scholar]
- Shorter, J., and G. Warren. 1999. A role for the vesicle tethering protein, p115, in the post-mitotic stacking of reassembling Golgi cisternae in a cell-free system. J. Cell Biol. 146:57–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shorter, J., R. Watson, M.E. Giannakou, M. Clarke, G. Warren, and F.A. Barr. 1999. GRASP55, a second mammalian GRASP protein involved in the stacking of Golgi cisternae in a cell-free system. EMBO J. 18:4949–4960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sönnichsen, B., M. Lowe, T. Levine, E. Jämsä, B. Dirac-Svejstrup, and G. Warren. 1998. A role for giantin in docking COPI vesicles to Golgi membranes. J. Cell Biol. 140:1013–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinman, R.M., S. Turley, I. Mellman, and K. Inaba. 2000. The induction of tolerance by dendritic cells that have captured apoptotic cells. J. Exp. Med. 191:411–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutterlin, C., C.Y. Lin, Y. Feng, D.K. Ferris, R.L. Erikson, and V. Malhotra. 2001. Polo-like kinase is required for the fragmentation of pericentriolar Golgi stacks during mitosis. Proc. Natl. Acad. Sci. USA. 98:9128–9132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornberry, N.A., T.A. Rano, E.P. Peterson, D.M. Rasper, T. Timkey, M. Garcia-Calvo, V.M. Houtzager, P.A. Nordstrom, S. Roy, J.P. Vaillancourt, et al. 1997. A combinatorial approach defines specificities of members of the caspase family and granzyme B. Functional relationships established for key mediators of apoptosis. J. Biol. Chem. 272:17907–17911. [DOI] [PubMed] [Google Scholar]
- Warren, G., T. Levine, and T. Misteli. 1995. Mitotic disassembly of the mammalian Golgi-apparatus. Trends Cell Biol. 5:413–416. [DOI] [PubMed] [Google Scholar]
- Wyllie, A.H., J.F. Kerr, and A.R. Currie. 1980. Cell death: the significance of apoptosis. Int. Rev. Cytol. 68:251–306. [DOI] [PubMed] [Google Scholar]
- Zhou, B.B., H. Li, J. Yuan, and M.W. Kirschner. 1998. Caspase-dependent activation of cyclin-dependent kinases during Fas-induced apoptosis in Jurkat cells. Proc. Natl. Acad. Sci. USA. 95:6785–6790. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}